

Abstract
We have performed molecular dynamics (MD) calculations by using one of the recently solved crystal structures of a hammerhead ribozyme. By rotating the α, β, γ, δ, ɛ, and ζ torsion angles of the phosphate linkage of residue 17, the nucleobase at the cleavage site was slightly rotated out of the active site toward the solution. Unconstrained MD simulations exceeding 1 ns were performed on this starting structure solvated in water with explicit counter ions and two Mg2+ ions at the active site. Our results reveal that near attack conformations consistently were formed in the simulation. These near attack conformations are characterized by assumption of the 2′-hydroxyl to a near in-line position for attack on the -O-(PO2−)-O- phosphorous. Also during the time course of the MD study, one Mg2+ moved immediately to associate with a pro-R phosphate oxygen in the conserved core region, and the second Mg2+ remained associated with the pro-R oxygen on the phosphate linkage undergoing hydrolysis. These results are in accord with a one-metal ion mechanism of catalysis and give insight into the possible roles of many of the conserved residues in the ribozyme.
The hammerhead ribozyme is one of a small class of self-cleaving RNAs that catalyzes the hydrolysis of phosphate esters (1–4). This ribozyme consists of a conserved core of 15 nucleotides required for full activity formed by three base pairing stem regions (5). The secondary structure of the hammerhead ribozyme studied here is shown in Chart 1 by using the standard nomenclature (6), with the conserved bases shown in outlined letters and the cleavage site indicated by the arrow. Note that the hammerhead ribozyme structure of Chart 1 exists as two separate strands with functions of enzyme (E) and substrate (S) such that it cleaves in trans (4, 7). 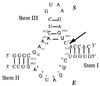
The hammerhead ribozyme catalyzes the chemically well studied RNA hydrolysis mechanism (8), which involves in-line nucleophilic attack of the 2′-hydroxyl on phosphate phosphorous at the 3′-position with elimination of 5′-hydroxyl and formation of a 2′,3′-cyclic phosphate ester with inversion of configuration at the phosphorus (Scheme 1 refs. 9–11). ![]()
Hammerhead ribozyme solvolysis is first order in HO− and requires one or more divalent metal ions (10, 12, 13). Thus, two kinetically equivalent mechanisms may be considered. The first is that metal ligated hydroxide acts as a general base to initiate the reaction by forming the 2′-O− nucleophile at residue 17. A second possibility is specific base catalysis of 2′-O− formation by lyate HO− and participation of the metal ion elsewhere. The metal ion may participate in various ways. It has been shown that a metal ion must ligate to the pro-R oxygen of the scissile phosphate group undergoing reaction (14, 15). By doing so, the negative charge on the phosphate is neutralized and facilitates nucleophilic attack on the phosphorus atom. This divalent metal ion or possibly an additional one can carry out the required stabilization of the developing negative charge on the 5′-leaving group. This could be by direct interaction of the metal ion with the developing negative charge on the leaving group oxygen or by orienting a water molecule such that a proton may be donated to the leaving group.
In a previous study conducted in this laboratory (16), a computational approach to the mechanism of self-cleavage was used to formulate a rational mechanism of self-cleavage that could be used as a working hypothesis until structural data of the hammerhead ribozyme was available. This early study, before the current x-ray crystal structures, revealed that it should be anticipated that the average structural coordinates will not reveal the self-cleavage mechanism of the hammerhead. In addition, this model proposed several features necessary for the alignment of the 2′-hydroxyl group for it’s in-line attack on the scissile phosphodiester bond and resultant catalysis. The most important involves the nucleobase at the cleavage site (C17). Not being involved in hydrogen bonding or proper stacking because of the bend at the junction of Stems I and III, this residue can turn out toward the solution (Effect A), a prediction borne out by the early model. Attainment of a near in-line attack conformation also can be assisted by Effect A in concert with a 3′-endo to 2′-endo flip of the ribose ring (Effect B).
In the crystal structures that have been solved recently (17–20), the nucleotides at the cleavage site are not arranged in a conformation that would allow for the in-line cleavage of the scissile phosphodiester bond. Many suggestions ranging from localized to global conformational changes have been offered regarding the rearrangement of the phosphodiester backbone necessary to achieve the in-line configuration for catalysis. A crystal structure depicting a much larger conformational change at the cleavage site has been solved, in accordance with the hypotheses that a greater conformational change is necessary to achieve catalysis, although an in-line attack conformation was not observed (20). Scott et al. (18, 19) observed that the ribozyme cleaved in the crystal, suggesting that slight conformational changes allow for hydrolysis. The conformational changes required to bring the hammerhead into a reactive conformation and the mechanism by which it catalyzes specific bond cleavage are not known. It appears that, minimally, torsional backbone rotations at the active site would be required to orient the 2′-hydroxyl group for in-line attack. In this study, we investigated the structural changes necessary to generate a near attack conformation (NAC) in the hammerhead ribozyme. A NAC is a kinetically essential energy minimum structure that geometrically must form before reaching a transition state in a reaction pathway (21–23). Our approach has been to perform molecular dynamics (MD) simulations by using the structure solved by Scott et al. (18) to examine the effects of rotating torsional angles of the phosphodiester backbone at the active site of the hammerhead ribozyme [Effect A (16)].
METHODS
The crystal structure [resolved to 3.1 Å (18)] containing a 2′-methoxy in place of the hydroxyl substituent on the ribose attacking residue (C17) (Protein Data Bank, Biology Department, Brookhaven National Laboratory, refcode: 1 mme) was used in this molecular dynamics study. Hydrogens were added to this crystal structure. The methyl group (which could not be seen in the x-ray structure) located on the 2′-oxygen of residue C17 was replaced by a hydrogen in this simulation. The backbone torsional angles α, β, γ, δ, ɛ, and ζ of the phosphate linkage that undergoes hydrolysis were rotated manually such that residue C17 was “turned out” slightly toward the major groove (no longer stacked in Stem I) with the 2′-hydroxyl group poised for an in-line attack of the scissile phosphodiester bond. Two magnesium ions were added to this structure. One Mg2+ ion was placed ≈2.0 Å from the pro-R phosphate oxygen of the scissile bond and the 2′-hydroxyl group of residue C17. The second was placed as given for Site 3 from Scott et al. (18) The parameters used for the Mg2+ ions were taken from Åqvist (24). TIP3P (25) water molecules were placed ≈2.0 Å from the magnesium ion to occupy the remaining coordination sites. Sodium counter ions were placed adjacent to the phosphate groups to maintain charge neutrality of the system. The RNA construct was placed in a cubic box constructed in the edit module of amber 4.1 (26) along with ≈6,100 explicit TIP3P water molecules to give the simulation system.
Energy minimization for 10,000 steps (500 steps of steepest descents followed by 9,500 steps of conjugate gradient) was performed on the system. The constant-volume system was heated to ambient temperature (300 K) within the first 20 ps. The constant-volume dynamics were continued for an additional 20 ps and then were followed by 60 ps of constant-temperature, constant-pressure dynamics by using the Berendsen temperature coupling algorithm and periodic boundary conditions to slowly relax the system. A 10-Å cutoff was used for the nonbonded interactions. The remainder of the equilibration procedure and the production MD simulations were continued by using the particle mesh Ewald summation (27–30) to calculate the electrostatic interactions as implemented in amber 5.0 (26) with a charge grid spacing of ≈1 Å. Simulations were run by using shake (31) to constrain bonds containing hydrogen. A 1-fs time step was used, and a geometric tolerance of 10−7 was set for coordinate resetting. Heating and equilibrations were complete by 100 ps. No positional restraints were placed on any of the atoms in the system during the energy minimization or MD simulations. The MD simulation was run for 1.1 ns. In order for a conformation to be considered a NAC, it must meet the following criteria for this system: the (attack) angle formed by C17 2′O—A1.1 P—A1.1 O5′ must be >150° and the (attack) distance between C17 2′O—A1.1 P must be <3.25 Å. The results were analyzed with the carnal module of amber 5.0. Structures were visualized, and pictures were made by using midasplus (32, 33). The average distances and angle data were calculated for the production dynamics period only.
RESULTS AND DISCUSSION
Conformational changes and interactions within the hammerhead ribozyme need to be studied if a detailed knowledge of the enzymatic reaction is to be understood. To understand the necessary conformational changes leading to the generation of an in-line attack of the 2′-oxygen on the scissile phosphate, an unconstrained MD study was conducted. The goal of this study was to examine the dynamic motions associated with the phosphodiester backbone and the interactions of the conserved bases in the hammerhead ribozyme. The starting structure used in this study was that of Scott et al. (18) in which the α, β, γ, δ, ɛ, and ζ torsion angles of the attacking nucleotide, residue C17, were rotated manually. The torsional angle rotations were conducted based on (i) the observations that the hammerhead ribozyme is able to undergo self-cleavage in the crystal, potentially requiring only minor conformational changes to achieve an in-line NAC (22, 23) and (ii) the conclusions reached by Mei et al. (16): C17 unstacks from Stem I, and the phosphodiester backbone rotates, pointing the pro-S and pro-R oxygens of the phosphate inward. These rotations could be achieved by turning out the C17 residue toward either the major groove or the minor groove of the ribozyme. Turning the cytosine residue out toward the major groove was reasoned to be the most energetically favorable option as the minor groove appeared to be hindered sterically by bases of the enzyme strand of Stem I in the crystal structure. The MD simulations were conducted with explicit solvent molecules, sodium counter ions, and magnesium cations without positional restraints on any of the atoms.
Active Site Structure.
The active site of the hammerhead ribozyme consists of conserved residues located (in the three dimensional structure) just opposite the cleavage site, C3U4G5A6, and those residues on either side of the bond that is cleaved, C17 and A1.1. This site also has been referred to as the “catalytic pocket” (18). The uridine turn is a sharp bend between Stems I and II in the enzyme strand of the ribozyme of Chart 1 and serves as a metal binding site much like the anticodon loop in tRNA. In addition to these roles, two of the conserved residues in the uridine turn (C3U4G5A6), residues G5 and A6, also form a stacking platform; this stacking platform has been observed in all crystal structures of the hammerhead ribozyme to date (17–20). Residues G5 and A6 appear to stabilize residue C17 in the crystal structures when it stacks with the bottom of the platform (with C17 stacked against A6) because C17 does not form a standard Watson–Crick base pair at the active site.
During the equilibration portion of the unconstrained MD simulation, it was found that the C17 residue, which had been turned out, spontaneously rotated to stack with the platform created by residues G5 and A6. This suggests that this stacking platform serves as an initial step in orienting residue C17 in the active site and guiding its motions such that a reactive conformation can be achieved. Residue G5 then unstacked from this platform and rotated out of the catalytic pocket to hydrogen bond with G16.2; N3 of G5 hydrogen bonded with H22 of G16.2, and H22 of G5 hydrogen bonded with N3 of G16.2. Formation of these two hydrogen bonds appears to be a consequence of the movement of Stems I and II toward each other and favorable interactions between additional conserved residues (vide infra). Stems I and II were free to move because they no longer experienced the lattice and blunt end packing effects, formed by two hammerhead ribozyme molecules stacking end to end, that were present in the asymmetric crystals. [The helical arms of the hammerhead ribozyme appear to possess an intrinsic flexibility (34), and it has been proposed that movement of the helical arms may be necessary to achieve catalysis.] These hydrogen bonding interactions between G5 and G16.2 were not maintained (occurring for ≈50 ps during the equilibration period), which allowed the G5 residue to rotate around further such that it stacked with residue U16.1 in the minor groove. Also during the equilibration time, the stacking platform interaction was further disrupted when A6 rotated to stack with the remaining bases of the conserved uridine turn, C3 and U4, leaving C17 free to move again. Residues C3 and U4 stacked with U2.1 to form a stable loop that persisted throughout the remainder of the simulation. Residue A1.1 moved to stack with the bases in the enzyme strand of Stem I (G2.2G2.3U2.4G2.5), effectively opening up the active site to allow for additional backbone conformational changes to occur in the substrate strand. Once the G5 and U16.1 stacking interaction was established, along with several other conserved residue interactions (vide infra), the first NACs formed immediately after 100 ps. In order for a conformation to be considered a NAC, it must have met the following criteria for this system: a C17 2′O—A1.1 P—A1.1 O5′ (attack) angle of >150° and C17 2′O—A1.1 P (attack) distance of < 3.25 Å. Based on this criteria, it can been seen from Fig. 1 a and b that NACs formed consistently throughout the simulation. The average attack angle was 140°, and the average attack distance was 3.43 Å. A snapshot representative of the initial NACs (formed within the first 200 ps of production dynamics) in the MD simulation is shown in Fig. 2. In addition, the 2′-hydroxyl of residue G5 formed hydrogen bonds with H3 of residue U7 and O2P of residue A6 that were maintained throughout the rest of the simulation (Fig. 3 a and b) with average distances of 2.45 Å and 2.47 Å, respectively. This observation is particularly exciting because it provides an explanation for the 103 decrease in activity when the 2′-hydroxyl of residue G5 is absent. Modifications of any of the exocyclic groups on G5 have been shown to result in a severe loss of activity (35). Although the precise reason that altering any of the exocyclic groups of residue G5 is not immediately evident from our simulations, it is believed that such modifications of G5 may restrict the range of motions that this residue undergoes during its interactions with other conserved core residues, preventing this base from forming the interactions mentioned above.
Figure 1.

(a) A plot over the entire MD simulation depicting the angle for nucleophilic attack of the 2′-oxygen of residue C17 on the phosphorus of residue A1.1. The average attack angle for the production dynamics portion of the simulation is 140°. (b) A plot of the attack distance between the C17 2′-oxygen and the A1.1 phosphorus. The average distance is 3.43 Å.
Figure 2.

A snapshot of the active site of the hammerhead ribozyme in a NAC that developed after 100 ps of total simulation time. In this snapshot, the attack angle is 172° and the attack distance is 3.17 Å. The arrow shows the bond that is cleaved during the reaction. The Mg2+ ion was coordinated to the pro-R phosphate oxygen and the attacking 2′-oxygen. Notice the outer sphere water molecule (Wat3701) that is poised to donate a proton to the 5′-leaving group (Wat3701 H1 to A1.1 O5′ distance is 2.32 Å). The H1 proton of the additional water molecule near the leaving group shown in this figure, Wat2821, is 2.27 Å from the 5′-oxygen but is not associated with a magnesium or counter ion.
Figure 3.

(a) A plot of the hydrogen bond that developed during the simulation time between the H2′ of residue G5 and O2P of residue A6. The average distance between these two atoms was calculated to be 2.45 Å. (b) A plot of the hydrogen bond that developed between atom O2′ of residue G5 and the H3 of residue U7. These two hydrogen bonds are believed to assist in maintaining the sharp bend in the substrate strand of this hammerhead ribozyme.
The NACs after 200 ps of production dynamics displayed slightly different features from those found earlier in the simulation with regard to base stacking interactions around the active site. The base stacking interaction between A6 and C17 reformed. Initially, the A6 and C17 interaction was stabilized by water-mediated hydrogen bonds between the 2′-oxygen of A6 and H42 of C17 and O2 of C17 and H41 of C1.2; this stacking interaction was stabilized later through direct hydrogen bonds between these nucleobases. The base stacking between G5 and U16.1 persisted for >750 ps but then was disrupted toward the end of the simulation. The sugar pucker of residue C17 remained in the C3′-endo conformation throughout the MD simulations.
Movement of Magnesium Ions.
Two magnesium ions were considered in this simulation: the magnesium ion coordinated to the pro-R phosphate oxygen, denoted as Mg1, and the magnesium ion placed consistent with Site 3 from Scott et al. (18), denoted as Mg2. Both Mg1 and Mg2 have been suggested to play key roles in catalysis (18) whereas the remaining magnesium ions appear to serve structural roles. Throughout the simulation, the Mg1 cation remained simultaneously coordinated to the pro-R phosphate oxygen of residue A1.1 and to the 2′-oxygen of residue C17, although no constraints were applied to maintain this interaction. The remaining inner-sphere coordination sites of Mg1 were occupied by the same water molecules throughout the simulation. On generation of the first NACs, a proton on water molecule Wat3701 in the outer sphere of Mg1 was 2.32 Å from the 5′-oxygen and 2.07 Å from the pro-R phosphate oxygen of residue A1.1, placing it in a favorable position for donating its proton to the 5′-leaving group (Fig. 2). This position was occupied by a few different water molecules with similar hydrogen bond distances during the formation of the initial NAC structures. As a result of base stacking interactions of residue A1.1 with bases in Stem I (vide supra), backbone atoms of residue A1.1 appeared to block outer sphere waters from this position in later NACs. Other solvent water molecules were found to form hydrogen bonds with the 5′-oxygen of residue A1.1, and it is reasoned that one would be able to donate a proton to the leaving group. A snapshot representing the NACs forming later in the simulation (after 500 ps of production dynamics) is shown in Fig. 4. Mg2 (not shown) moved from its Site 3 crystallographic position to interact with the pro-R phosphate oxygen of residue U7 during the equilibration period of the simulation and remained there for the duration of the simulation. Based on our results, it appears that Mg2 assumes the position of a counter ion and does not assist in the catalysis of hydrolysis.
Figure 4.

A snapshot of the active site of the hammerhead ribozyme after 600 ps of total simulation time displaying a NAC. The attack angle and attack distance in this snapshot are 164° and 3.23 Å, respectively. The 2′-hydroxyl and the pro-R phosphate oxygen remained coordinated to the Mg2+ ion while the stacking of the bases at the active site were altered somewhat (compare with Fig. 2). Here, the H1 of Wat2447 is 2.01 Å from the 5′-oxygen of residue A1.1. A sodium counter ion has migrated to this region and is 2.76 Å from the Wat2447 oxygen and 3.98 Å from the leaving group oxygen.
Additional Conserved Residue Interactions.
In addition to the favorable interactions that developed within the active site, our MD simulations revealed that many of the conserved core residues are necessary to form the active site and to keep the phosphodiester backbone of the substrate strand properly oriented such that NACs can consistently be generated. It was found that residue G8 participated in many hydrogen bonds during the simulation. As shown in Fig. 5 a and b, the pro-R phosphate oxygen of residue A13 formed a bifurcated hydrogen bond with H1 and H21 of the exocyclic amino group of G8, with the bond between H1 and O2P being slightly stronger of the two throughout the simulation. (The average distance for a was 1.89 Å whereas the average distance for b was 2.00 Å). These interactions provide a reasonable explanation for the 1,000-fold decrease in activity when this base was replaced with 2-aminopurine, xanthosine, or 1-methylguanosine as well as other less detrimental functional group modifications that altered the donors at positions 1 and 2 of this base. The reason for the decreased activity was unclear because it did not appear that H1 of G8 was functioning as a hydrogen bond donor in the crystal structures (36). As a result of the formation of these hydrogen bonds, the H22 of G8 formed a hydrogen bond with N7 of residue A13, as shown in Fig. 5c. Fig. 5d shows the hydrogen bond (average distance 2.39 Å) that formed between the 2′-hydroxyl of residue G8 and O5′ of residue A9 and may explain the >100-fold decrease in activity corresponding to the alteration of this group (35). The base of residue G12 stacked with G8 to assist in maintaining these hydrogen bonds. The many interactions that residue G8 participated in also served to maintain the sharp bend in the substrate strand.
Figure 5.

This figure shows plots of the hydrogen bonding interactions associated with residue G8. a and b show the distances of the bifurcated hydrogen bond that formed between O2P of residue A13 and the H1 and the H21 of residue G8 with average distances of 1.89 Å and 2.00 Å, respectively. (c) A plot of the hydrogen bond that occurred between H22 of residue G8 and N7 of residue A13. The shortest distances between these two atoms correspond with the formation of NACs. (d) Plot of the hydrogen bond between the H2′ of residue G8 and the O5′ of residue A9. The average distance between these two atoms is 2.39 Å.
Some of the additional hydrogen bonds that formed during the simulation between conserved core residues are shown in Fig. 6. The formation of the hydrogen bond between U16.1 and C17, plotted in Fig. 6a, appears to be a result of the stacking interaction G5 and U16.1 that formed early in the simulation (vide supra). The data suggest that this bond may have participated in orienting the C17 2′-hydroxyl group for attack on the P of A1.1, allowing NACs to occur consistently during the latter portion of the simulation. Fig. 6b displays a hydrogen bond that formed between residue A14 and A15.1 that appears to be the result of the disruption of Watson–Crick base pairing between U16.1 and A15.1. This hydrogen bond became established after 300 ps of production dynamics and was maintained by the stacking of the base of residue A15.1 between the surrounding bases of residues U7 and G16.2. The formation of the hydrogen bond between H41 of residue C15.2 and O2P of A14 (Fig. 6c; average distance of 2.22 Å) occurred after the Watson–Crick base pairing was disrupted somewhat between residues G16.2 and C15.2. Residue G16.2 was wedged in place by stacking with surrounding bases (A15.1, A16.3, etc.) and the formation of a nonstandard hydrogen bond between H1 of G16.2 and O2 of C15.2. This breaking of the Watson–Crick base pairing between G16.2 and C15.2 initially seemed unfavorable but resulted in the formation of new and favorable interactions. The aforementioned series of interactions of the conserved core residues, many of which acted at a distance from the active site, served to hold the ribozyme together while allowing the necessary conformation changes to occur that lead to the generation of NACs and subsequent catalysis.
Figure 6.

(a) A plot of the hydrogen bond that developed between the 2′-hydroxyl groups of residues U16.1 and C17. The 2′-hydroxyl group of U16.1 may assist in the formation of NACs by helping to orient the 2′-hydroxyl of C17 for attack. (b) Plot of the hydrogen bond that formed between A14 and A15.1. Notice that this bond formed after 400 ps total simulation time and appears to precede the formation of the hydrogen bond mentioned in a. (c) A plot of the hydrogen bond between H41 of residue C15.2 and O2P of residue A14 with an average distance of 2.22 Å.
CONCLUSIONS
In this study, we examined the flexibility of the hammerhead ribozyme by using unconstrained MD simulations. We used the crystal structure solved by Scott et al. (18) and performed torsional backbone rotations such that the nucleobase at the cleavage site, C17, was slightly turned out toward the major groove (Effect A) and the nonbridging phosphate oxygens were directed toward the center of Stem I. This starting structure was placed in a box of ≈6,100 TIP3P (25) water molecules with sodium counter ions and two Mg2+ ions at the active site to give a neutral simulation system. Energy minimizations and MD simulations exceeding 1 ns then were conducted without constraints by using the particle mesh Ewald summation method (27–30) and periodic boundary conditions as implemented in amber (26). NACs (21–23), conformations in which the 2′-hydroxyl oxygen is positioned for in-line attack and departure of the 5′-oxygen, were formed in the picosecond time range. RNA model studies (14, 15) have shown metal ion catalysis of hydrolysis to be first order in [HO−] and first and second order in [Mn+]. In these studies, it was established that the primary catalytic effect of metal ions are to ligate to a nonbridging phosphate oxygen to cancel the negative charge (act as a Lewis acid) and provide a 104-105 rate enhancement in RNA hydrolysis. Throughout our MD simulations, one Mg2+ ion (Mg1) was associated with the pro-R phosphate oxygen. This metal ion also was coordinated to the 2′-hydroxyl oxygen. Ligation of Mg1 to the 2′-hydroxyl group would lower its pKa, thus assisting specific base ionization with [HO−] or [Mn+(HO)]. Also, an outer-sphere water on Mg1 was in position that it may act as a general acid to donate a proton to the 5′-leaving group in several of the NAC structures. Thus, it appears that one Mg2+ ion is able to perform the necessary catalytic functions at the active site. The sugar pucker of residue C17 remained in the C3′-endo sugar puckering mode throughout the MD simulations; therefore, Effect B was not examined in this simulation. The cooperative tertiary interactions of the conserved nucleotides, many acting at a distance, appeared to function in holding the ribozyme strands together while allowing necessary conformational changes until the rate-determining (37, 38) cleavage of the P-O5′ bond of the leaving group occurred. The current results represent a possible way that catalysis of hydrolysis by the hammerhead ribozyme can occur that is consistent with experimental results. Of interest, many of these structural features (hydrogen bonding and base stacking between conserved core residues and the formation of NACs) also have been observed in unconstrained MD simulations using the freeze-trapped intermediate crystal structure in which Effects A and B act in concert to form NACs (R.A.T. and T.C.B., unpublished results).
Acknowledgments
We thank Dr. David A. Case for his helpful suggestions and for providing us with the amber 5.0 program before its release. This work was supported by grants DK09171-34 and DK 09171-34S1 from the National Institutes of Health. Some of the calculations performed for this study were conducted at the National Center for Supercomputing Applications, University of Illinois at Urbana-Champaign, under Grant CHE980041N.
ABBREVIATIONS
- NAC
near attack conformation
- MD
molecular dynamics
References
- 1.Prody G A, Bakos J T, Buzayan J M, Schneider I R, Bruening G. Science. 1986;231:1577–1580. doi: 10.1126/science.231.4745.1577. [DOI] [PubMed] [Google Scholar]
- 2.Buzayan J M, Gerlach W L, Bruening G. Proc Natl Acad Sci USA. 1986;83:8859–8862. doi: 10.1073/pnas.83.23.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buzayan J M, Gerlach W L, Bruening G. Nature (London) 1986;323:349–353. [Google Scholar]
- 4.Uhlenbeck O C. Nature (London) 1987;328:596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 5.Ruffner D E, Stormo G D, Uhlenbeck O C. Biochemistry. 1990;29:10695–10702. doi: 10.1021/bi00499a018. [DOI] [PubMed] [Google Scholar]
- 6.Hertel K J, Pardi A, Uhlenbeck O C, Koizumi M, Ohtsuka E, Uesugi S, Cedergren R, Eckstein F, Gerlach W L, Hodgson R, Symons R H. Nucleic Acids Res. 1992;20:3252. doi: 10.1093/nar/20.12.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasseloff J, Gerlach W L. Nature (London) 1988;334:585–591. doi: 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- 8.Westheimer F H. Acc Chem Res. 1968;1:70–78. [Google Scholar]
- 9.van Tol H, Buzayan J M, Feldstein P A, Eckstein F, Bruening G. Nucleic Acids Res. 1990;18:1971–1975. doi: 10.1093/nar/18.8.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slim G, Gait M J. Nucleic Acids Res. 1991;19:1183–1188. doi: 10.1093/nar/19.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koizumi M, Ohtsuka E. Biochemistry. 1991;30:5145–5150. doi: 10.1021/bi00235a005. [DOI] [PubMed] [Google Scholar]
- 12.Dahm S A, Uhlenbeck O C. Biochemistry. 1991;30:9464–9469. doi: 10.1021/bi00103a011. [DOI] [PubMed] [Google Scholar]
- 13.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 14.Dempcy R O, Bruice T C. J Am Chem Soc. 1994;116:4511–4512. [Google Scholar]
- 15.Bruice T C, Tsubouchi A, Dempcy R O, Olson L P. J Am Chem Soc. 1996;116:9867–9875. [Google Scholar]
- 16.Mei H Y, Kaaret T W, Bruice T C. Proc Natl Acad Sci. 1989;86:9727–9731. doi: 10.1073/pnas.86.24.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pley H W, Flaherty K M, McKay D B. Nature (London) 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 18.Scott W G, Finch J T, Klug A. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 19.Scott W G, Murray J B, Arnold J R P, Stoddard B L, Klug A. Science. 1996;274:2065–2069. doi: 10.1126/science.274.5295.2065. [DOI] [PubMed] [Google Scholar]
- 20.Murray J B, Terwey D P, Maloney L, Kerpelsky A, Usman N, Beigelman L, Scott W G. Cell. 1998;92:665–673. doi: 10.1016/s0092-8674(00)81134-4. [DOI] [PubMed] [Google Scholar]
- 21.Lightstone F C, Bruice T C. J Am Chem Soc. 1994;116:10789–10790. [Google Scholar]
- 22.Lightstone F C, Bruice T C. J Am Chem Soc. 1996;118:2595–2605. [Google Scholar]
- 23.Lightstone F C, Bruice T C. J Am Chem Soc. 1997;119:9103–9113. [Google Scholar]
- 24.Åqvist J. J Phys Chem. 1990;94:8021–8024. [Google Scholar]
- 25.Jorgensen W L, Chandrasekhar J, Madura J D. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 26.Pearlman D A, Case D A, Caldwell J W, Ross W S, Cheatham T E I, Ferguson D M, Seibel G L, Singh U C, Weiner P K, Kollman P A. AMBER. San Francisco: Univ. of California Press; 1995. [Google Scholar]
- 27.Cheatham T E I, Miller J L, Fox T, Darden T A, Kollman P A. J Am Chem Soc. 1995;117:4193–4194. [Google Scholar]
- 28.Essmann U, Perera L, Berkowitz M L, Darden T A, Lee H, Pedersen L G. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 29.Darden T A, York D, Pedersen L G. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 30.Chen Z M, Çagin T, Goddard W A., III J Comp Chem. 1997;18:1365–1370. [Google Scholar]
- 31.van Gunsteren W F, Berendsen H J C. Mol Phys. 1977;34:1311–1327. [Google Scholar]
- 32.Huang C C, Pettersen E F, Klein T E, Ferrin T E, Landridge R. J Mol Graph. 1991;9:230–236. doi: 10.1016/0263-7855(91)80016-s. [DOI] [PubMed] [Google Scholar]
- 33.Ferrin T E, Huang C C, Jarvis L E, Landridge R. J Mol Graph. 1988;6:13–27. [Google Scholar]
- 34.McKay D B. Catalytic RNA: Nucleic Acids and Molecular Biology. New York: Springer; 1996. pp. 161–172. [Google Scholar]
- 35.McKay D B. RNA. 1996;2:395–403. [PMC free article] [PubMed] [Google Scholar]
- 36.Thomson J B, Tuschl T, Eckstein F. Catalyic RNA: Nucleic Acids and Molecular Biology. New York: Springer; 1996. pp. 173–196. [Google Scholar]
- 37.Zhou D M, Usman N, Wincott F E, Matulic-Adamic J, Orita M, Zhang L H, Komiyama M, Kumar P K R, Taira K. J Am Chem Soc. 1996;118:5862–5866. [Google Scholar]
- 38.Uchimaru T, Uebayasi M, Hirose T, Tsuzuki S, Yliniemelä A, Tanabe K, Taira K. J Org Chem. 1996;61:1599–1608. doi: 10.1021/jo951517+. [DOI] [PubMed] [Google Scholar]