

Abstract
Deregulation of cell proliferation is a hallmark of cancer. In many transformed cells, the cyclin A/CDK2 complex that contains S-phase kinase associated proteins 1 and 2 (SKP1 and SKP2) is highly induced. To determine the roles of this complex in the cell cycle regulation and transformation, we have examined the composition of this complex. We report here that this complex contained an additional protein, human CUL-1, a member of the cullin/CDC53 family. The identification of CUL-1 as a member of the complex raises the possibility that the p19SKP1/p45SKP2/CUL-1 complex may function as the yeast SKP1-CDC53-F-box (SCF) protein complex that acts as a ubiquitin E3 ligase to regulate the G1/S transition. In mammalian cells, cyclin D, p21CIP1/WAF1, and p27KIP1 are short-lived proteins that are controlled by ubiquitin-dependent proteolysis. To determine the potential in vivo targets of the p19SKP1/p45SKP2/CUL-1 complex, we have used the specific antisense oligodeoxynucleotides against either SKP1, SKP2, or CUL-1 RNA to inhibit their expression. Treatment of cells with these oligonucleotides caused the selective accumulation of p21 and cyclin D proteins. The protein level of p27 was not affected. These data suggest that the human p19SKP1/p45SKP2/CUL-1 complex is likely to function as an E3 ligase to selectively target cyclin D and p21 for the ubiquitin-dependent protein degradation. Aberrant expression of human p19SKP1/p45SKP2/CUL-1 complex thus may contribute to tumorigenesis by regulating the protein levels of G1 cell cycle regulators.
To ensure the faithful duplication and passage of genetic information during cell division, the transitions between different phases of the cell cycle are precisely coordinated and controlled by the cyclin-dependent kinases (CDKs) (1, 2). The sequential activation of each CDK in the cell cycle primarily is achieved by its association with a specific regulatory subunit, a cyclin. In mammals, the G1 cell cycle progression is controlled by the activity of cyclin D/CDK4 or CDK6, which specifically phosphorylates and thus inactivates the growth suppression activity of retinoblastoma susceptibility gene product, pRb (2). In late G1, both cyclin E/CDK2 and cyclin A/CDK2 play a role during the G1/S transition, although their critical targets are mostly undefined.
The cyclin/CDK activities are further regulated by both positive and negative phosphorylation and the association of CDK inhibitors such as p21CIP1/WAF1, p27KIP1, and p16INK4A (1, 2). The CDK inhibitors usually act as checkpoint control proteins to prevent the unscheduled entry of the S phase. p21 has been shown to be regulated by p53 tumor suppressor during the DNA damage response (2). p16, a specific CDK4 or CDK6 inhibitor, is itself encoded by a tumor susceptibility gene located on the chromosome 9p21 locus, the loss of which is associated with a wide variety of human cancer, including familial melanomas (2).
Previously we found that in normal human fibroblasts, a substantial fraction of cyclin A/CDK2 was associated with p21 and the proliferating cell nuclear antigen (PCNA) (3). However, in many DNA viral oncoprotein transformed or other established tumor cells that are deficient in p53 expression, p21 and PCNA disappeared and cyclin A/CDK2 was prominently complexed with two novel proteins, S-phase kinase associated proteins 1 and 2 (SKP1 and SKP2) (4). To investigate the significance of this change, we have isolated the p19SKP1/p45SKP2/cyclin A/CDK2 complex and cloned the genes encoding p19SKP1 and p45SKP2 (4). Our studies indicate that p45SKP2 expression is highly induced in many transformed cells (4). In addition, ablation of p45SKP2 by antibody microinjection into G1 cells prevented these cells from entry into the S phase, suggesting that p45SKP2 and probably p19SKP1, is required for the G1/S transition (4). p45SKP2 contains a leucine-rich-repeated domain at its carboxyl terminus and a small, but relatively conserved, motif, an F-box, at its amino terminal region, which was later found to mediate the interaction with p19SKP1 (5).
Recently, a yeast SKP1 homologue was isolated as a high copy suppressor of certain cdc4 mutants (5). It is now clear that in yeast SKP1, CDC53, and CDC4 form a protein complex (5–7) that functions as a ubiquitin E3 ligase to target the yeast CDK inhibitor, p40SIC1, for the ubiquitin-dependent degradation during the G1/S transition (8, 9). Another yeast SKP1/CDC53 binding protein, GRR1, is implicated in the yeast G1 cyclin stability, which may coordinate the availability of carbon source with the timing of S-phase entry (9). Both CDC4 and GRR1 have been shown to be F-box-containing proteins (5).
At least three enzymatic activities are required for protein ubiquitination (9). The ubiquitin is activated by forming a thioester bond with the ubiquitin activation enzyme, E1, at the expense of ATP. The activated ubiquitin then is transferred from E1 to the ubiquitin-conjugating enzyme, E2. The E2 enzyme mediates the transfer of ubiquitin to protein substrates either directly or with the help of the E3 ligase. In many cases, the E3 enzyme functions to confer the substrate specificity. The polyubiquitinated protein substrates are eventually degraded by the 26S proteosome.
In this report, we found that the human p19SKP1/p45SKP2/cyclin A/CDK2 complex also contained CUL-1, a member of the cullin/CDC53 family. Our studies suggest that the p19SKP1/p45SKP2/CUL-1 complex is likely to function as a conserved ubiquitin E3 enzyme that regulates the mammalian G1/S transition by specifically targeting mammalian G1 cell cycle regulators, such as p21 and cyclin D proteins, for the ubiquitin-dependent degradation.
MATERIALS AND METHODS
Cell Culture.
The HaCat (human keratinocyte), RKO (human colorectal carcinoma cells), and 293 (adenovirus-transformed human embryonic kidney) cells were gifts from David Beach (Cold Spring Harbor Laboratory, Plainview, NY). HCT116 (human colon carcinoma), DLD1 (human colon adenocarcinoma) cells, and the early passages of IMR90 human fibroblasts were purchased from the American Type Culture Collection. The mouse ts20 cells were kindly provided by Harvey Ozer (University of Medicine and Dentistry of New Jersey-New Jersey Medical School). All cells were grown in DMEM supplemented with 10% fetal bovine serum at 37°C as previously described (4). For metabolic labeling, cells were cultured in methionine-deficient medium supplemented with 10% dialyzed fetal bovine serum (GIBCO/BRL) in the presence of 250 μCi/ml of [35S]methionine for 6 hr. The protein complexes were isolated by immunoprecipitation as described previously (4).
Plasmids, Chemicals, and Antibodies.
The cDNA encoding full-length human CUL-1 was kindly provided by Edward Kipreos (University Of Georgia). For transfection assays using the calcium phosphate method (10), human CUL-1 cDNA was fused in-frame with a T7 epitope tag in pCGTVP16 vector (a gift from Hong Sun, Yale University) under the cytomegalovirus promoter control. The cDNAs encoding p19SKP1 and p45SKP2 were cloned into pcDNA3 expression vector, respectively (Invitrogen). The anti-T7 epitope tag was purchase from Novagen. For human CUL-1 antibodies, the DNA fragment containing nucleotides 1–1408 was amplified by the PCR method and cloned in-frame between the BamHI–HindIII sites in the bacterial expression vector pGEX-KG to create a fusion protein (GST-CUL-N1) between the glutathione S-transferase (GST) and the amino-terminal region of human CUL-1. The recombinant protein was produced in bacteria and affinity-purified on the glutathione Sepharose beads as described (11). This protein was used to raise rabbit polyclonal antibodies against the human CUL-1 protein, and the specificity of the antisera was tested by using the purified recombinant GST-CUL-1 protein by immunoblotting assay. The rabbit anti-cyclin A, p21, p27, and cyclin D antibodies were gifts from David Beach and were described in detail before (11). The polyclonal rabbit anti-ubiquitin antibody was purchased from Sigma. N-acetyl-l-leucinyl-l-leucinal-l-norleucinal (LLNL), a specific inhibitor of 26S proteosome, and N-acetyl-l-leucinyl-l-leucinyl-methional (LLM), a structurally similar but not an inhibitor of 26S proteosome, were purchased from Sigma and dissolved in dimethyl sulfoxide. Aprotinin, leupeptin, trypsin inhibitor, and benzamidine also were purchased from Sigma. The protein A Sepharose CL-4B beads were purchased from Pharmacia. Enhanced chemiluminescence (ECL) reagents were obtained from Amersham and used according to the attached instructions.
Antisense Experiments.
Four oligodeoxynucleotides that contain phosphorothioate backbone and C-5 propyne pyrimidines were synthesized by using an automated synthesizer by the Keck Biotechnology Resource Laboratory located in the Boyer Center for Molecular Medicine at Yale University: (i) 5′-CCGCGTTGTTGTTTATG-3′ (the antisense direction of human CUL-1 cDNA nucleotides 1134–1150); (ii) 5′-TTCCCAAATCTTCCAACA-3′ (the antisense direction of human p19SKP1 cDNA nucleotides 88–106); (iii) 5′-CCTGGGGGATGTTCTCA-3′ (the antisense direction of human p45SKP2 cDNA nucleotides 180–196); and (iv) 5′-CTACCTCCCCTCCATTA-3′, the control oligonucleotide that contains similar pyrimidines as the CUL-1 and p19SKP1 oligonucleotides. For ablation experiments, cells were seeded to 15% confluence the night before lipofection. The antisense oligonucleotides were delivered into cells by using 2.5 μg/ml of GS 2888 cytofectin (kindly provided by Michael Flanagan, Gilead Sciences, Foster City, CA) and 30–75 nM of the oligonucleotides according to the described protocols (12). The cells were harvested between 16 and 18 hr posttransfection. Antibodies against p19SKP1, p45SKP2, and other cell cycle regulatory proteins and the procedures for immunoprecipitation and immunoblotting were the same as described previously (4).
Immunological Procedures.
Cells were harvested and immunoprecipitated in a buffer containing 0.5% Nonidet P-40, 150 mM NaCl, 50 mM Tris (pH 7.4), 100 mM NaF, and 10 μg/ml each of the following protease inhibitors: trypsin inhibitor, aprotinin, and leupeptin as well as 1 mM benzamidine for 3 hr at 4°C (4). To minimize the protein degradation by cysteine proteases and the 26S proteosome, in some experiments (see Fig. 5), LLNL and N-ethylmaleimide were added to the lysates during immunoprecipitation. Western blot analysis of various proteins was conducted by using the ECL method (Amersham) as described previously (4).
Figure 5.

Human CUL-1 protein associates with cyclin D and p21. Cell lysates from actively growing HaCat cells were immunoprecipitated with specific antibodies as indicated on the top of each lane. The immunoprecipitated protein complexes were blotted with CUL-1 (Left), p27 and p21 (Middle), or CDK2 (Right) antibodies, respectively, as indicated on the right side of each panel.
RESULTS
Human CUL-1 Interacts with p19SKP1 and p45SKP2.
To determine the roles of p45SKP2/p19SKP1 complex in the cell cycle, we have analyzed the composition of this complex isolated from 35S-methionine-labeled 293 cells, an adenovirus-transformed human embryo kidney cell line. Immunoprecipitation of p45SKP2 complex using an affinity-purified p45SKP2 antibody indicated that p19SKP1, CDK2, and cyclin A were the major proteins that associated with p45SKP2 (Fig. 1A), as characterized previously (4). A careful examination of the p45SKP2-associated proteins also revealed the specific presence of a novel protein, p86, in the p45SKP2 complex (Fig. 1A, indicated as CUL-1, see below). Recently, it has been shown that the yeast SKP1 homologue can bind to CDC53, a cullin family member, and CDC4, an F-box containing protein (5, 9). In mammals and Caenorhabditis elegans, there are at least six cullin family members, with CUL-1 and CUL-2 being more closely related to CDC53 (13). Because p86 had an electrophoretic mobility similar to the calculated molecular weight of human CUL-1 protein (13), we tested whether the p45SKP2-associated p86 is human CUL-1.
Figure 1.

Association between human CUL-1 and p45SKP2/p19SKP1. (A) 293 cells were metabolically labeled with [35S]methionine. The CUL-1, p45SKP2, and cyclin A complexes were isolated by immunoprecipitation using their specific antibodies from labeled 293 cell lysates. The proteins in each protein complex were resolved in an SDS/polyacrylamide gel and compared. The positions of p45SKP2 associated p19SKP1, cyclin A, CDK2, and CUL-1/p86 are indicated on the right. The molecular mass standard (in kDa) are shown on the left. Preimmune serum is the serum from the CUL-1 rabbit before immunization and is used as a control. Peptide, the CUL-1 antigen was included for competition during immunoprecipitation. (B) Human CUL-1 gene was tagged with T7 epitope tag (T/Cul-1) and cloned under the cytomegalovirus promoter control. This construct was cotransfected with expression constructs containing either p19SKP1, p45SKP2, or both into 293 cells as indicated. Twenty-four hours after transfection, the cells were [35S]-methionine-labeled, and the protein complexes were isolated with either T7-tag, p19SKP1-, or p45SKP2-specific antibodies as indicated.
Two alternative approaches were taken to determine whether p86 is human CUL-1. To directly examine whether human CUL-1 is p86, a rabbit polyclonal antibody was raised against human CUL-1. This antibody could specifically recognize either the bacterially produced or the in vitro-translated full-length human CUL-1 protein in the immunoprecipitation and Western blot analyses (data not shown). This antibody was used to immunoprecipitate the CUL-1 protein complex from cell lysates prepared from 35S-methionine-labeled 293 cells. As shown in Fig. 1A, the immunoprecipitated CUL-1 migrated exactly as the p45SKP2-associated p86 during the protein gel electrophoresis (Fig. 1A). In addition, the CUL-1 immunocomplex contained a 19-kDa protein that had the same mobility as the p45SKP2-associated p19SKP1, suggesting that CUL-1 is likely to complex with p45SKP2 and p19SKP1 in vivo.
In addition, we have examined whether CUL-1 could complex directly with p19SKP1 and p45SKP2 in vivo by using transfection assays. The cDNA encoding human CUL-1 was epitope-tagged with T7-tag and cloned into a mammalian expression vector. When this CUL-1 protein was coexpressed with either p19SKP1, p45SKP2, or both in 293 cells, an association between these proteins was detected (Fig. 1B). Such an interaction appears to be specific because human CUL-2 did not complex with p45SKP2 and p19SKP1 in such an assay (data not shown). Furthermore, although CUL-1 could complex with p45SKP2 or p19SKP1, the presence of all three proteins enhanced the association between them, suggesting that these proteins form a ternary complex in these assays.
To confirm that the endogenous CUL-1 is indeed associated with p19SKP1 and p45SKP2, Western blot analyses were performed. Immunoprecipitated p19SKP1 and p45SKP2 complexes from 293 and other human cell lines were immunoblotted with the human CUL-1 antibody (Fig. 2A). Reciprocal examination by blotting the CUL-1 immunoprecipitates with p19SKP1 or p45SKP2 antibodies was also performed (Fig. 2B). These experiments revealed that the endogenous CUL-1 was indeed associated with p19SKP1 and p45SKP2. In addition, because p19SKP1 and p45SKP2 were isolated as proteins that are associated with cyclin A/CDK2 (4) and it apeared that p86 was also associated with cyclin A immunoprecipitation (Fig. 1A), we tested whether the CUL-1 complex contained cyclin A and CDK2. Western blotting studies indicated that CUL-1 was also present in the cyclin A and CDK2 immunocomplexes (Fig. 2A).
Figure 2.

Association between the endogenous CUL-1 and p19SKP1, p45SKP2, cyclin A, and CDK2. (A) Presence of CUL-1 in the p19SKP1, p45SKP2, cyclin A, and CDK2 complexes. The p19SKP1, p45SKP2, CUL-1, cyclin A, and CDK2 complexes were immunoprecipitated from lysates prepared from actively growing HCT116 (Upper) or 293 (Lower) cells by specific antibodies as indicated on the top of each lane, respectively. The presence of CUL-1 is detected by immunoblotting with a CUL-1 specific antibody. The preimmune serum is the same as described in Fig. 1. (B) Presence of p45SKP2 and p19SKP1 in CUL-1 complexes. The CUL-1, p45SKP2, or p19SKP1 complex was immunoprecipitated from HCT116 cell lysates and immunoblotted with either p45SKP2 (Upper) or p19SKP1 (Lower) specific antibodies. NRS, normal rabbit serum from an unrelated rabbit.
Thus, by analogy to the yeast SCFCDC4 complex, our studies indicated that human CUL-1, p19SKP1, and p45SKP2 form a conserved protein complex that is likely to function as an E3 enzyme in the cell cycle control. In addition, our data suggest that the CUL-1/p19SKP1/p45SKP2 complex also contains cyclin A/CDK2, a cell cycle kinase that is required for both the entry and progression of the S phase. This finding raises the possibility that cyclin A/CDK2 may regulate the activity of CUL-1/p19SKP1/p45SKP2 complex, which is consistent with our previous observation that p45SKP2 could be phosphorylated by cyclin A/CDK2 in vitro (4).
Regulation of G1 Cell Cycle Regulators by the Ubiquitin-Dependent Pathway.
We previously have observed that in vivo ablation of p45SKP2 by p45SKP2 antibody microinjection prevented the G1 cells from entry into S phase (4). Based on our present data and the possibility that the p19SKP1/p45SKP2/CUL-1 complex may act as a conserved ubiquitin E3 enzyme, it is likely that one function of this complex is to target a negative G1 cell cycle regulator(s), such as a CDK inhibitor, which normally functions to prevent the unscheduled entry of the S phase for the ubiquitin-dependent proteolysis. In mammalian cells, a number of CDK inhibitors, such as p21CIP1/WAF1, p27KIP1, and p16INK4a, have been identified (2). To determine whether these proteins are ubiquitinated under our experimental conditions and thus are the potential substrates of the CUL-1/p19SKP1/p45SKP2 complex, we treated log-phase growing human cells with LLNL, a specific inhibitor of the 26S proteosome that degrades polyubiquitinated proteins (14). Consistent with previous findings (10, 15, 16), cells treated with LLNL resulted in the accumulation of p21 and p27, as well as cyclin D1 proteins (Fig. 3). In addition, accumulation of these proteins was also observed in a mouse cell line, ts20, that contained a temperature-sensitive ubiquitin E1 enzyme (17), when the E1 activity was inactivated by shifting cells from permissive temperature (34°C) to the nonpermissive temperature (39°C) (data not shown). Western blotting of p21, p27, and cyclin D1 immunoprecipitates with an ubiquitin antibody also revealed the presence of high molecular weight protein conjugates only in cells treated with LLNL but not in the control (dimethyl sulfoxide-treated) cells, suggesting that these protein conjugates are likely the polyubiquitinated forms of p21, p27, and cyclin D1 (data not shown).
Figure 3.

Regulation of cyclin D, p21, and p27 by the ubiquitin-dependent proteolysis. (A) Log-phase RKO or HaCat cells were treated with either dimethyl sulfoxide (DMSO) (0.05%), 10 μg/ml of LLNL, or N-acetyl-l-leucinyl-l-leucinyl-methional (LLM) for 6 hr. The cyclin D, p21, and p27 protein were isolated and detected by their respective antibodies by using immunoprecipitation and Western blot analyses.
Ablation of p19SKP1, p45SKP2, or Human CUL-1 Selectively Stabilizes p21 and Cyclin D Proteins.
The ubiquitination of G1 regulators raised the possibility that CUL-1/p19SKP1/p45SKP2 may target these cell cycle regulators for the ubiquitin-dependent degradation. However, it is quite difficult to use a genetic approach to mutate the genes encoding these ubiquitination proteins and test such a possibility by using cultured mammalian cells. To overcome these difficulties, we have attempted to inhibit the expression of either p19SKP1, p45SKP2, or CUL-1 by antisense oligodeoxynucleotides to examine their effects on the cell cycle. To enhance the stability of the oligonucleotides, antisense oligonucleotides that contain the phosphorothioate backbone and C-5 propyne pyrimidine substitutions were synthesized. The pyrimidine modification has been shown to facilitate the base-stacking effect of RNA/antisense oligonucleotide hybrids (18). In these experiments we also used the recently developed transfection agent, cytofectin (GS2888), which delivers the antisense oligonucleotides into the cells at nanomolar concentration and thus help to increase the specificity of the antisense effects (12).
Cytofectin-mediated delivery of each antisense oligonucleotide against either SKP1, SKP2, or human CUL-1 RNA resulted in the substantial decreases in the corresponding levels of p19SKP1, p45SKP2, or human CUL-1 protein in many cell lines, including HCT116, HaCat, and RKO (Fig. 4), as assayed by Western blot analysis. We next tested whether ablation of p19SKP1, p45SKP2, or human CUL-1 might affect the protein levels of the known G1 cell cycle regulators. The protein levels of cyclin D, p21, p27, as well as CDK2 and cyclin A were examined by immunoprecipitation and Western blotting with their respective antibodies from lysates prepared from the cells treated with either a control or an antisense oligonucleotide against either SKP1, SKP2, or CUL-1 RNA. Although p21, p27, and cyclin D all are stabilized by the LLNL treatment (Fig. 3), specific ablation of either p19SKP1, p45SKP2, or CUL-1 caused the selective stabilization of p21 and cyclin D proteins. The protein levels of p27 appeared not to be affected by such a treatment (Fig. 4A). In these experiments, we did not observe alterations in the protein levels of other cell cycle regulators such as CDK2 or cyclin A (Fig. 4A, and data not shown). These results suggest that the effects of ablation of p19SKP1, p45SKP2, and human CUL-1 are quite specific. Because the p19SKP1/p45SKP2/CUL-1 complex is likely to function as an E3 enzyme, these data suggest that this complex may target p21 and cyclin D for ubiquitin-dependent degradation, although a rigorously conducted biochemical analysis is required to further address this issue.
Figure 4.

Selective stabilization of the cellular cyclin D and p21 proteins by antisense oligodeoxynucleotides against SKP1, SKP2, or CUL-1. (A, Upper Left) HCT116 (HCT) cells were transfected with antisense oligonucleotides against SKP1, SKP2, CUL-1, or a control oligonucleotide (Ctrl) by using cytofectin GS2888 as indicated on top of each lane. Eighteen hours after transfection, each protein was immunoprecipitated and quantitated by immunoblotting analysis using the specific antibody as indicated on the right, respectively. (A, Upper Right and Lower) HCT116 or RKO cells were treated with control or antisense oligonucleotides against CUL-1, SKP2, or SKP1 RNA as indicated on the top of each panel. The relative protein levels of cyclin D, p27, CDK2, and p21 from the treated HCT116 or RKO cells (0.5 × 106 each) were compared by immunoprecipitation and immunoblotting analyses using their respective antibodies. (B, Left) The relative levels of p21, cyclin D, and p27 from HCT116 (p53 positive) and DLD1 (p53 negative) cells (0.5 × 106 each) were compared by using immunoprecipitation and Western blotting analyses. (B, Right) DLD1 cells were transfected with antisense oligonucleotides against SKP1, SKP2, CUL-1, or a control oligonucleotide (Ctrl) as indicated on the top of the lanes. The antisense effects on the protein levels of p21, cyclin D, and p27 were examined by the Western blot analysis as described in A.
p21 is transcriptionally controlled by p53. In p53-positive cells, such as RKO, HCT116, and HaCat, both cyclin D and p21 accumulate in response to the ablation of CUL-1/p19SKP1/p45SKP2 complex. Because the elevated level of p21 could lead to its binding to cyclin/CDK complexes and inhibit their kinase activity, it is possible that the increased level of cyclin D in these cells could be an indirect effect of p21 accumulation. To determine whether the accumulation of cyclin D depends on p21, we performed the antisense ablation experiments in cells that are defective in p53 function. The DLD1 human colon adenocarcinoma cells were chosen because they are well characterized as the p53 negative human cancer cells, which express very low levels of p21 (19). As shown in Fig. 4B, although we could not detect p21 in either control or treated cells, the accumulation of cyclin D was observed in DLD1 cells treated with antisense oligonucleotide against SKP1, SKP2, or CUL-1, suggesting that the increase in the cyclin D protein level is independent of p21 accumulation.
Human CUL-1 Is Associated with Cyclin D and p21.
To further analyze how the p19SKP1/p45SKP2/human CUL-1 complex may selectively target cyclin D and p21 for degradation, we tested the possibility that the components of this complex might interact with their substrates directly. Immunoprecipitated cyclin D, p21, or p27 complexes from HaCat cells were blotted with p19SKP1, p45SKP2, or human CUL-1 specific antibodies. In these experiments, although the presence of p19SKP1 and p45SKP2 was not detected, human CUL-1 was found to specifically associate with both cyclin D and p21, but not with p27 (Fig. 5). One possible explanation for the failure to detect p19SKP1 and p45SKP2 in these complexes is that they might associate with the CUL-1/cyclin D or p21 complex transiently during the ubiquitin conjugation reaction, which may result in the rapid degradation of p21 and cyclin D. To rule out the possibility that p27 expression might be low in HaCat cells, several control experiments were conducted. Our data indicated that the relative protein levels of p21 and p27 did not differ substantially in the log-phase growing HaCat cells. In addition, both p21 and p27 antibodies were capable of immunoprecipitating similar amounts of p21- or p27-associated CDK2 (Fig. 5). These data indicate that the association between CUL-1 and cyclin D or p21 is likely to correlate with the observed CUL-1 targeting specificity.
p45SKP2, But Not CUL-1 or p19SKP1, Is Overexpressed in Transformed Cells.
Previously we have isolated the p19SKP1/p45SKP2/cyclin A/CDK2 complex based on its prominent presence in many transformed cells (4). To determine the cause for such an induction, we have examined and compared the levels of each component of this complex between transformed and nontransformed human cells. Comparison between 293 cells, the adenovirus transformed cells, and IMR90, a nontransformed fibroblast, revealed that p45SKP2, but not p19SKP1 or CUL-1 protein, is highly induced in 293 cells (Fig. 6). This finding suggests that the elevated level of p45SKP2 is likely to be responsible for the high-level presence of this cyclin A/CDK2 complex in many transformed cells. This observation is also consistent with our previous observation that SKP2 RNA is induced in many transformed cells (4).
Figure 6.
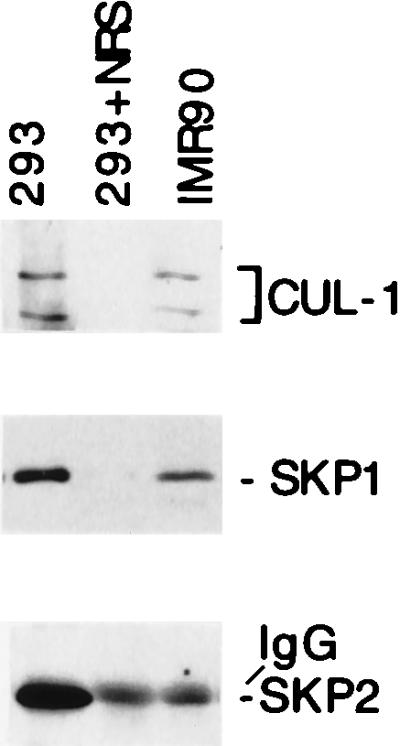
p45SKP2 is highly induced in transformed cells. Cell lysates (0.5 mg proteins) were made either from actively growing 293, an adenovirus transformed human embryo cell, or from IMR90, a nontransformed human fibroblast. The relative amounts of CUL-1, p19SKP1, and p45SKP2 were determined by immunoprecipitation and Western blot analyses.
DISCUSSION
Our data indicate that the human CUL-1 is associated with p19SKP1 and p45SKP2 (Fig. 1). This complex also contains cyclin A and CDK2. Although the exact role of cyclin A/CDK2 in this complex remains unclear, our previous study suggests that cyclin A/CDK2 could phosphorylate p45SKP2 in vitro (4). It has been shown that the activity of cyclin A/CDK2 is required for the G1 cells to enter the S phase (20). The association between p19SKP1/p45SKP2/CUL-1 complex and cyclin A/CDK2 raises the possibility that cyclin A/CDK2 may regulate the activity of p19SKP1/p45SKP2/CUL-1 complex during the G1/S transition.
The complex formation between human CUL-1, p19SKP1, and p45SKP2 suggests that this complex is likely to function as a conserved ubiquitin E3 ligase complex, similar to the activity of the SKP1-CDC53-F-box protein complex in yeast, to regulate mammalian cell cycle progression. Our in vivo studies suggest that the potential targets of this complex may be G1 cell cycle regulators such as p21 and cyclin D1. This result is consistent with our previous observation that ablation of cellular p45SKP2 by p45SKP2 antibody injection caused the G1 cells to arrest at the G1/S border (4). In the present study, we also found that treatment of cells with either p19SKP1, p45SKP2, or CUL-1 antisense oligonucleotides reproducibly resulted in a moderate G1 cell cycle arrest (data not shown), suggesting that this complex plays a role during the G1/S transition.
Our results are also consistent with the observation that both cyclin D and p21 were absent from replicating nuclei (21, 22). Such an observation indicates that the S-phase nucleus contains an activity that removes p21 and cyclin D. p21 can bind to various CDKs, including cyclin D/CDK4, cyclin E, and cyclin A/CDK2, and inhibits their kinase activity. p21 also is associated with the PCNA, a replication and repair factor. The binding of p21 to PCNA leads to the inhibition of PCNA replication activity. Thus the removal of p21 from the replicating nuclei may be necessary for both the CDK and PCNA replication activities. Overexpression of cyclin D also has been shown to inhibit DNA replication in many cells (22). One possibility is that the ability of cyclin D to bind to CDK2 may prevent CDK2 from associating with other essential S-phase cyclins (3). Because the levels of p45SKP2 increase at late G1 (4), the disappearance of both cyclin D1 and p21 from the replicating nuclei is consistent with the notion that p19SKP1/p45SKP2/human CUL-1 complex may regulate the removal of cyclin D and p21 from the nucleus during G1/S transition and possibly in S phase as well. However, because p45SKP2 expression is relatively late in the G1 cell cycle, we could not rule out the possibility that there are additional pathways that regulate p21 and cyclin D proteins in the early G1 phase of the cell cycle.
Although p27 is also shown to be controlled by the ubiquitin-dependent degradation (Fig. 3) (15), we found that the level of p27 is not affected by the treatment of antisense oligonucleotides against SKP1, SKP2, or CUL-1 (Fig. 4). It has been shown that p27 accumulates in G0 cells but becomes degraded once cells re-enter the cell cycle (23). Because the p19SKP1/p45SKP2/CUL-1 complex is likely to function in the late G1 (4), p27 may not be the main target of this complex during the G1/S transition. However, several reports indicate that p27 degradation can occur in extracts from S-phase cells and such an activity depends on cyclin E/CDK2 in transfection assays (24, 25). At present we cannot rule out the possibility that p27 can be ubiquitinated by the p19SKP1/p45SKP2/CUL-1 complex. It is possible that in our experiments only a small fraction of p27 is affected by the down-regulation of the p19SKP1/p45SKP2/CUL-1 complex, which might have escaped our detection. If this is true, there should be additional major ubiquitination or proteolysis pathways that control p27 stability.
The induction of the p19SKP1/p45SKP2/cyclin A/CDK2 complex in many transformed cells raises the possibility that overexpression of this complex may provide a growth advantage for these cells. From our study, it appears that this induction is caused by the high-level expression of p45SKP2. These observations also suggest that p45SKP2 is likely to be the rate-limiting step for the assembly and the activity of this complex. The altered activity of this complex may affect the levels of G1 cell cycle regulator to deregulate the entry of S phase, thus contributing to the tumorigenic process.
Acknowledgments
We thank Laura Manjarrez for help during the preparation of the paper. We also thank Dr. Michael Flanagan (Gilead Sciences) for cytofectin GS 2888 and Dr. Edward Kipreos (University Of Georgia) for the human CUL-1 cDNA. This work is supported by a grant from the Patrick and Catherine Weldon Donaghue Medical Research Foundation (DF96-130) and American Cancer Society Institutional Research Grant IN31-37.
ABBREVIATIONS
- CDK
cyclin-dependent kinase
- SKP1
S-phase kinase associated protein 1, p19
- SKP2
S-phase kinase associated protein 2, p45
- PCNA
proliferating cell nuclear antigen
- LLNL
N-acetyl-l-leucinyl-l-leucinal-l-norleucinal
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Hunter T, Pines J. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 2.Sherr C J. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Xiong Y, Beach D. Mol Biol Cell. 1993;4:897–906. doi: 10.1091/mbc.4.9.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Kobayashi R, Galaktionov K, Beach D. Cell. 1995;82:915–925. doi: 10.1016/0092-8674(95)90271-6. [DOI] [PubMed] [Google Scholar]
- 5.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper J W, Elledge S J. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 6.Feldman R M, Correll C C, Kaplan K B, Deshaies R J. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 7.Skowyra D, Craig K L, Tyers M, Elledge S J, Harper J W. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 8.Nasmyth K. Trends Genet. 1997;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- 9.King R W, Deshaies R J, Peters J-M, Kirschner M W. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 10.Diehl J A, Zindy F, Sherr C J. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Hannon G J, Beach D. Genes Dev. 1994;8:1750–1758. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]
- 12.Lewis J G, Lin K Y, Kothavale A, Flanagan W M, Matteucci M D, DePrince R B, Mook R A J, Hendren R W, Wagner R W. Proc Natl Acad Sci USA. 1996;93:3176–3181. doi: 10.1073/pnas.93.8.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kipreos E T, Lander L E, Wing J P, He W W, Hedgecock E M. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- 14.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 15.Pagano M, Tam S W, Theodoras A M, Beer-Romero P, Del Sal G, Chau V, Yew P R, Draetta G F, Rolfe M. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 16.Maki C G, Howley P M. Mol Cell Biol. 1997;17:355–363. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdary D R, Dermody J J, Jha K K, Ozer H L. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner R W, Matteucci M D, Lewis J G, Gutierrez A J, Moulds C, Froehler B C. Science. 1993;260:1510–1513. doi: 10.1126/science.7684856. [DOI] [PubMed] [Google Scholar]
- 19.Polyak K, Waldman T, He T C, Kinzler K W, Vogelstein B. Genes Dev. 1996;10:1945–1952. doi: 10.1101/gad.10.15.1945. [DOI] [PubMed] [Google Scholar]
- 20.Girard F, Strausfeld U, Fernandez A, Lamb N J. Cell. 1991;67:1169–1179. doi: 10.1016/0092-8674(91)90293-8. [DOI] [PubMed] [Google Scholar]
- 21.Li R, Hannon G J, Beach D, Stillman B. Curr Biol. 1996;6:189–199. doi: 10.1016/s0960-9822(02)00452-9. [DOI] [PubMed] [Google Scholar]
- 22.Pagano M, Theodoras A M, Tam S W, Draetta G F. Genes Dev. 1994;8:1627–1639. doi: 10.1101/gad.8.14.1627. [DOI] [PubMed] [Google Scholar]
- 23.Nourse J, Firpo E, Flanagan W M, Coats S, Polyak K, Lee M H, Massague J, Crabtree G R, Roberts J M. Nature (London) 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- 24.Brandeis M, Hunt T. EMBO J. 1996;15:5280–5289. [PMC free article] [PubMed] [Google Scholar]
- 25.Sheaff R J, Groudine M, Gordon M, Roberts J M, Clurman B E. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]