

Abstract
A typical molecular ion mass spectrum consists of a sum of signals from species of various possible isotopic compositions. Only the monoisotopic peak (e.g., all carbons are 12C; all nitrogens are 14N, etc.) has a unique elemental composition. Every other isotope peak at approximately integer multiples of ∼1 Da higher in nominal mass represents a sum of contributions from isotope combinations differing by a few mDa (e.g., two 13C vs. two 15N vs. one 13C and one 15N vs. 34S, vs. 18O, etc., at ∼2 Da higher in mass than the monoisotopic mass). At sufficiently high mass resolving power, each of these nominal-mass peaks resolves into its isotopic fine structure. Here, we report resolution of the isotopic fine structure of proteins up to 15.8 kDa (isotopic 13C,15N doubly depleted tumor suppressor protein, p16), made possible by electrospray ionization followed by ultrahigh-resolution Fourier transform ion cyclotron resonance mass analysis at 9.4 tesla. Further, a resolving power of m/Δm50% ≈8,000,000 has been achieved on bovine ubiquitin (8.6 kDa). These results represent a 10-fold increase in the highest mass at which isotopic fine structure previously had been observed. Finally, because isotopic fine structure reveals elemental composition directly, it can be used to confirm or determine molecular formula. For p16, for example, we were able to determine (5.1 ± 0.3) the correct number (five) of sulfur atoms solely from the abundance ratio of the resolved 34S peak to the monoisotopic peak.
A molecular ion mass spectrum typically consists of a sum of signals from species of various possible isotopic compositions (e.g., one or more 13C or 15N, etc.). At unit (nominal) mass resolution, the isotopic distribution consists of isotope peaks spaced ∼1 Da apart. However, only the monoisotopic peak formed from the lowest-mass stable isotope of each element (e.g., all carbons are 12C; all nitrogens are 14N, etc.) has a unique elemental composition, whereas other isotope peaks include contributions from different elemental combinations (e.g., two 13C vs. two 15N vs. one 13C and one 15N vs. 34S, etc., at ∼2 Da higher in mass than the monoisotopic mass). These species differ from each other in mass by a few mDa. At sufficiently high mass resolving power, these peaks resolve into isotopic fine structure. Such fine structure has been resolved for small (∼1 kDa) peptides (1, 2) but has proved elusive for larger species because of the tendency of closely spaced peaks to coalesce into a single resonance (3, 4). However, the coalescence tendency scales inversely with magnetic field strength, so one can hope to resolve isotopic fine structure at sufficiently high magnetic field strength.
Because ion cyclotron resonance (ICR) is a frequency-based mass measurement, it provides the potentially highest-resolution and highest-accuracy mass analysis, as demonstrated by numerous previous determinations of masses of individual atomic and small molecular ions to ppb precision and accuracy (5). The ICR principle may be extended to simultaneous detection of ions of multiple mass-to-charge ratios by pulsed excitation followed by Fourier transformation (FT) of the digitized response (6–8). FT-ICR mass spectrometry has produced ppb precision in atomic ion masses (9–13). Mass resolving power, m/Δm50% ≈ 200,000,000, has been achieved for 40Ar+, where Δm50% is the full FT-ICR mass spectral peak width at half-maximum peak height (14).
Although the highest previously reported protein mass resolving power, m/Δm50% of 2,000,000 was achieved for bovine ubiquitin (8.6 kDa) (15), protein isotopic fine structure was not resolved. Rather, the (many) resonances at each nominal mass coalesced into a single narrow resonance, because closely spaced ion cyclotron frequencies can “lock” together by a Coulomb repulsion mechanism when enough ions are present (3, 4). In other experiments, FT-ICR mass spectral peak width for peptides has been narrowed by postacquisition frequency drift correction to yield m/Δm50% ≈1,500,000 (16) or even 2,500,000 (17); however, we have observed that whenever enough ions are present to produce frequency drift during detection that number of ions is also sufficient to collapse the isotopic fine structure. As a result, isotopic fine structure has been observed previously only for small peptides (1, 2).
Here, we report experimental observation of isotopic fine structure of proteins up to 15.8 kDa by combining several techniques: high-field (9.4 T) external accumulation electrospray ionization (ESI) FT-ICR mass spectrometry (18), digital quadrature heterodyne detection (19), minimization of the number of trapped ions, frequency-domain averaging of multiple data sets, large-diameter (4-inch) ion trap, and 13C, 15N isotopic double depletion (20). In this way, we have achieved a mass resolving power of up to 8,000,000 for (natural-abundance) ubiquitin, with complete elimination of peak coalescence, to reveal the full isotopic fine structure. Finally, we show that resolution of that fine structure makes it possible to “count” the number of sulfur atoms in a protein, simply by “weighing” it.
EXPERIMENTAL PROCEDURES
Sample Preparation.
Bovine insulin and ubiquitin were purchased from Aldrich. 13C, 15N doubly isotopically depleted p16 tumor supressor protein was kindly provided by Ming-Daw Tsai (Ohio State University). A 10 μM solution of each protein was prepared in 50:50 methanol/water containing 2% (vol/vol) acetic acid for ESI and FT-ICR mass analysis.
Micro-ESI and ICR Time-Domain Data Acquisition.
FT-ICR experiments were performed with a passively shielded 9.4 Tesla home-built FT-ICR mass spectrometer (18, 21) controlled by a midas data station (22) and featuring an open cylindrical three-section ion trap of 10 cm diameter and 30 cm total length. The ions generated from a micro-electrospray source (23) were accumulated outside the magnet (18) in a 45 cm long rf-only octopole for 5–10 s, and then transferred through a 200 cm long rf-only octopole into the Penning trap. Each octopole has an i.d. of 4.7 mm and is operated at 1.5 MHz at a rf amplitude of 400 Vp-p. The pressure reading (noncalibrated) on the ion gauge was below 7 × 10−9 torr (1 torr = 133 Pa) in the Penning trap.
Ions of specific charge state were isolated by stored waveform inverse FT (SWIFT) (24, 25) radial dipolar mass-selective ejection. After ion transfer and SWIFT isolation, the applied trapping potential was dropped stepwise from 2 V to 1 V and then to a much lower value (0.20 V for p16, 0.18 V for ubiquitin and 0.12 V for insulin) over a period of approximately 60 s.
Ions were excited by frequency-sweep (chirp) radial dipolar excitation (150 Hz/μs at 100 Vp-p). A 20-MHz VXI digitizer (E1437A, Hewlett–Packard) with digital heterodyne/digital quadrature/digital filtering capability (19) sampled the time-domain ICR signal for 52–104 s (16–32 KWord complex time-domain data) at a Nyquist bandwidth of 312.5 Hz. The midas data station was modified to acquire, store, and process the complex transient generated by the digital quadrature heterodyne process.
Processing of ICR Time-Domain Data.
Conventional heterodyne detection followed by analog-to-digital conversion yields a single time-domain data set that is treated as real numbers. In contrast, the present discrete quadrature heterodyne detection generates two time-domain data sets, each of which is treated as real and imaginary parts of a mathematically complex time-domain data set. The discrete complex time-domain data was either not apodized (p16 and ubiquitin) or Hanning-apodized (insulin), and then zero-filled once before fast FT (to yield real and imaginary frequency-domain data) and magnitude calculation. Mass calibration (26) was based on a lower-resolution (m/Δm50% = 100,000) ESI FT-ICR mass spectrum of the same data set.
For better signal-to-noise ratio (SNR), nine (for p16 data) or 11 (for insulin data) spectra obtained under identical conditions were coadded in the frequency domain, taking care to align the monoisotopic peak in each frequency-domain spectrum before coaddition. The coadded spectrum then was transformed from frequency domain to mass domain. These operations were performed in a spreadsheet (Excel 97, Microsoft). We observed scan-to-scan variation in ICR frequency of up to 0.06 Hz (i.e., three times the linewidth at half-maximum peak height), or 0.5 ppm of the cyclotron frequency (146 kHz). We selected the spectrum closest to the average offset as the standard and aligned others with it before frequency-domain coaddition of spectra. As noted at the end of Results and Discussion, ultraprecise mass calibration is not essential to the present analysis. Of course, as shown in this paper, an internal calibration may be performed on the selected standard spectrum to provide accurate mass determination.
Mass Spectral Simulation.
Each theoretical isotope distribution shown in the figures was generated by isopro, version 3.03 (ref. 27; http://members.aol.com/msmssoft/). For each isotopic peak distribution spectrum (i.e., isotopic fine structure not resolved), a Gaussian line shape was assumed. For simulated isotopic fine structure spectra, the simulated peak width was set equal to the experimental peak width, and the line shape was either Gaussian (for apodized spectra) or Lorentzian (for unapodized spectra).
RESULTS AND DISCUSSION
Experimental Observation of Protein Isotopic Fine Structure.
Fig. 1 shows the isotopic fine structure of the isotope peak ∼5 Da higher than the monoisotopic mass of bovine insulin (M.W.monoisotopic = 5729.6 Da), for the 5+ charge state of the protein. Five different isobars are clearly identified in this mass spectrum obtained from a single time-domain data acquisition. Mass resolving power >106 is needed to observe isotopic fine structure for this species. For example, a mass resolving power, m/Δm50% > 2,300,000 is required to resolve the two closely spaced species with five 13Cs vs. three 13Cs and one 18O, differing by only 2.5 mDa. The experimental mass resolving power is ∼5,000,000.
Figure 1.

ESI FT-ICR mass spectrum (Upper Left), from a single time-domain data acquisition, of bovine insulin. Theoretical (Upper Right) and experimental (Lower) isotopic fine structure is shown for the isotopic peak (★) ∼5 Da above the monoisotopic mass. Individual elemental compositions are clearly resolved at approximately correct relative abundances.
Fig. 2 compares theoretical and experimental mass spectra for bovine ubiquitin (M.W.monoisotopic = 8559.6 Da) for the 9+ charge state of the protein. Note that the mass spectra in Fig. 2 are charge state deconvolved to the mass domain, whereas the spectrum in Fig. 1 is shown in the mass-to-charge ratio (m/z) domain. The theoretical mass spectrum for the complete isotope distribution (shown above the dotted line in Fig. 2) was generated at an assumed mass resolving power, m/Δm50% = 100,000 and a Gaussian line shape. Theoretical mass spectra for individual isotopic peaks (shown below the dotted line in Fig. 2) were generated at an assumed resolving power of 10,000,000 and a Lorentzian line shape. The experimental mass spectrum of the complete isotope distribution was obtained by FT of just the first 1/32 of the time-domain data for the 9+ charge state of the protein. The experimental mass resolving power ranged from 6,400,000 (the most abundant component of the M+6 isotope peak in Fig. 2) to 10,600,000 (the peak adjacent to the most abundant M+6 component). Because the experimental spectra are in such good agreement with the theoretical spectra at a resolving power of 10,000,000, we feel safe in claiming an average experimental mass resolving power of 8,000,000. The reported mass resolving power varies from peak to peak for two reasons. First, although our software interpolates to find the peak maximum position, the software computes the peak width simply from the separation between the data points closest to the half-maximum peak height. Thus, when (as in this case), there are only ≈5 points per peak width (20% threshold), the reported peak width (and thus reported mass resolving power) will necessarily vary from peak to peak. Second, even at the present ultrahigh resolving power, some peaks still consist of a sum of unresolved components that affect the peak shape and thus the mass resolving power.
Figure 2.

Theoretical (Left) and experimental (Right) mass spectra of bovine ubiquitin. The isotopic distribution (m/Δm50% ≈ 100,000) is shown above the dotted line, and the isotopic fine structure (m/Δm50% ≈8,000,000) for each of several isotopic peaks (at ≈2, 3, 4, 5, and 6 Da above the monoisotopic mass) appears below the dotted line. The experimental data were obtained from a single time-domain data set for ions of the isolated 9+ charge state. Note the good match between experimental and theoretical fine structure.
Fig. 3 shows the fine structure of an even larger protein, tumor suppressor protein p16 (M.W.monoisotopic = 15792.1 Da), for the 16+ charge state of the protein, at an experimental mass resolving power, m/Δm50% ≈ 5,000,000. Although p16 is much larger than ubiquitin, a lower resolving power suffices to observe isotopic fine structure in this single-scan spectrum, because 13C, 15N double isotopic depletion (20) greatly simplifies the distribution.
Figure 3.

Mass scale-expanded spectra of one isotopic peak for 13C,15N doubly depleted p16 tumor suppressor protein, from a single ESI FT-ICR mass spectrum. The theoretical isotopic distribution is shown at upper left, in which ★ denotes the isotopic peak ≈2 Da above the monoisotopic mass. The theoretical isotopic fine structure for the starred peak is shown at upper right. Double-depletion improves the SNR of the monoisotopic, 34S, and 18O species, by reducing the abundances of species containing 13C and/or 15N.
Techniques for Resolving Isotopic Fine Structure.
Figs. 1–3 clearly demonstrate resolution of isotopic fine structure in proteins. We attribute our success to several factors. First, we reduce the tendency of close-spaced ion cyclotron resonances to coalesce (3, 4) by use of high magnetic field (9.4 T), large ion cyclotron orbital radius (10 cm diameter cylindrical Penning trap), and low number of trapped ions. The number of trapped ions was first reduced by stored waveform inverse FT ejection of all but one charge state of a given protein.
Second, we reduce, stepwise over a minute or so, the electrostatic end-cap electrode potential to a very low value (a few tenths of a volt), so as to allow ions of initially high axial velocity to escape, while translationally “evaporatively” cooling the remaining ions (28). Low trapping potential also allows ions to spread out axially to minimize Coulomb repulsion (see below). Interestingly, we achieved optimal mass resolution over a very narrow range (as little as 10%) in trapping potential. We infer that optimal resolution results when the space-charge potential of the ion packet itself (an inverted parabolic potential; ref. 29) effectively cancels the (parabolic) potential from the ion trap end cap electrodes, allowing even greater axial spread of the ions. Finally, the low trapping potential also reduces frequency drift (16, 17) during the necessarily long detection period, resulting in narrower mass spectral peaks.
Third, we use quadrature heterodyne detection, in which the heterodyne
reference frequency is placed in the center (rather than at one end) of
the Nyquist frequency band, allowing for a 2-fold reduction in the
filter bandwidth and a 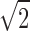 reduction in noise (30). Reducing
noise allows for detection of fewer ions, further reducing the tendency
for closely spaced resonances to coalesce. Moreover, unlike previous
analog quadrature FT-NMR (31) or FT-ICR (32, 33) detection, we use
digital quadrature heterodyne detection, in which the time-domain ICR
signal is first sampled, and then multiplied by (discrete) cosine or
sine waveforms and digitally filtered, both in real time. The two
discrete waveforms then become the real and imaginary parts of a
discrete complex waveform, which then is subjected to complex fast FT,
magnitude calculation, and frequency-to-mass conversion (26) to yield a
magnitude-mode FT-ICR mass spectrum. Digital quadrature heterodyne
detection eliminates analog noise from the previous mixing/filtering
steps, and also automatically scales the filter bandwidth to the
Nyquist bandwidth, obviating the need for a bank of analog filters of
various bandwidths.
reduction in noise (30). Reducing
noise allows for detection of fewer ions, further reducing the tendency
for closely spaced resonances to coalesce. Moreover, unlike previous
analog quadrature FT-NMR (31) or FT-ICR (32, 33) detection, we use
digital quadrature heterodyne detection, in which the time-domain ICR
signal is first sampled, and then multiplied by (discrete) cosine or
sine waveforms and digitally filtered, both in real time. The two
discrete waveforms then become the real and imaginary parts of a
discrete complex waveform, which then is subjected to complex fast FT,
magnitude calculation, and frequency-to-mass conversion (26) to yield a
magnitude-mode FT-ICR mass spectrum. Digital quadrature heterodyne
detection eliminates analog noise from the previous mixing/filtering
steps, and also automatically scales the filter bandwidth to the
Nyquist bandwidth, obviating the need for a bank of analog filters of
various bandwidths.
Given that the 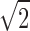 noise reduction advantage of quadrature
detection does not apply to direct-mode detection, one might ask why
heterodyne detection is used at all. The reason is the need for a long
time-domain data acquisition period to achieve high resolution.
Direct-mode detection for 104 s for insulin (Fig. 1) at 9.4 T (ICR
frequency of 125.8 kHz, for a minimum sampling rate of 251.6 kHz) would
yield a 32 Mword time-domain data set. Although our midas
data system (22) could generate such a large data set (if equipped with
a Hewlett–Packard E1437A digitizer with 64 Mbyte memory option),
storage and processing become problematic.
noise reduction advantage of quadrature
detection does not apply to direct-mode detection, one might ask why
heterodyne detection is used at all. The reason is the need for a long
time-domain data acquisition period to achieve high resolution.
Direct-mode detection for 104 s for insulin (Fig. 1) at 9.4 T (ICR
frequency of 125.8 kHz, for a minimum sampling rate of 251.6 kHz) would
yield a 32 Mword time-domain data set. Although our midas
data system (22) could generate such a large data set (if equipped with
a Hewlett–Packard E1437A digitizer with 64 Mbyte memory option),
storage and processing become problematic.
Finally, it is interesting to note that the frequency-domain spectral peak width in the present experiments is ≈0.02 Hz, or at least an order of magnitude smaller than typical high-resolution NMR peak widths. Thus, although FT-NMR spectra exhibit higher relative precision (because Larmor frequencies are typically several orders of magnitude higher than ICR frequencies for ions of, say, m/z ≈ 1,000), the present FT-ICR spectra actually provide an order of magnitude higher absolute frequency-domain precision than typical high-resolution FT-NMR spectra! The reason is that FT-ICR line width varies directly with background gas pressure (34), and we can lower the pressure to produce the equivalent of T2 ≈ 100 s transverse NMR relaxation time for gas-phase biomacromolecular ions.
Isotopic Fine Structure Numerology.
Once the monoisotopic mass has been accurately calibrated, it is convenient to refer the mass of an individual isotopic fine structure component to the mass of the monoisotopic species. It then becomes useful to tabulate the mass differences (Fig. 4) resulting from replacement of 1H by 2H, 12C by 13C, 14N by 15N, 16O by 18O, and 32S by 33S or 34S. Thus, although 15N has a positive mass defect (relative to 15.0000 Da), the mass of 15N is actually slightly less than 1 Da higher than that of 14N. In this way, one can quickly identify each fine structure component by matching its experimental mass difference from the monoisotopic mass, to that for a putative fine structure elemental composition (e.g., 13C34S). For example, at ∼2 Da above the monoisotopic mass of a protein, the species having two 13C is 2.0000 + 2 × 0.0034 = 2.0068 Da, and the index for one 18O is 2.0000 + 0.0042 = 2.0042 Da above the monoisotopic mass. The mass separation between the two isotopic combinations is 0.0026 Da. Also, note that for proteins with natural isotopic abundances, the all-13C peak is generally the most abundant component of each isotopic peak, and thus serves as a convenient starting point for identifying other isotopic fine structure components.
Figure 4.

Relative abundance (ordinate) and mass difference (abscissa) resulting from substitution of a single heavy isotope for the most abundant (lower-mass) isotope of that element. (The abundance for deuterium is magnified by a factor of 10 to make it more visible.) For ease in computation, each mass difference has been adjusted for the nominal mass difference between the two isotopes. Each displayed mass difference is thus like a mass defect (the difference between exact and nominal mass), in the sense that the mass shift is a small fraction of 1 Da. These values facilitate the identification (by abundance and accurate mass) of various fine structure components at a given nominal mass (see text).
Counting Sulfur Atoms in a Protein.
The ability to resolve and measure the relative abundance of each individual isotopic composition makes possible (in principle) the determination of elemental composition. For example, accurate measurement of the ratio of the abundance of the (resolved) species containing one 13C to the monoisotopic abundance would afford determination of the number of carbons in the molecule. Similar measurements could be made for each of the other elements.
In practice, direct elemental determination is difficult for several reasons. First, to resolve the fine structure, the number of ions in the cell must be low to minimize peak coalescence, resulting in low spectral SNR and corresponding loss in peak measurement accuracy (35). Further, many elements require ultraprecise and accurate measurement of the peak ratios, because of the relatively low abundance of the heavy isotope. For example, deuterium is incorporated in only ∼0.015% of the hydrogens in a naturally occurring molecule, so that a large number of hydrogens must be present to yield an observable deuterated peak. Moreover, unattainably high abundance measurement accuracy (by FT-ICR at any SNR) would be needed to “count” the atoms. Finally, resolution of some of the individual elemental components is exceedingly difficult. Any interference from neighboring species will skew the abundance measurement. For example, the 34S mass differs from the 15N33S mass by only 0.6 mDa! Unresolved 15N interference will contribute a ∼5% abundance measurement error for every 75 nitrogen atoms in the molecule.
Another concern in reducing the number of ions in the ICR cell is that the number of ions may be so small that the apparent isotopic abundance distribution may deviate from the statistical distribution because of scan-to-scan fluctuation in the number of ions of each elemental composition. We therefore attempted a χ2 analysis (36) by comparing the experimental and theoretical ion relative abundances for each of the nine individual low-resolution (i.e., isotopic fine structure not resolved) spectra (obtained by truncation of the time-domain signal before FT), and obtained 120 ± 125 total ions of the 9+ charge state. That SD is obviously too large to match the χ2 determined number of ions; moreover, some spectra with higher SNR actually yield a lower calculated number of ions. Thus, the observed fluctuation in ion relative abundances must arise primarily from random noise in the detected signal and cannot be used to estimate the number of ions.
FT spectral SNR can be enhanced by a factor of 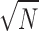 by coherent
addition of N data sets (30). Unfortunately, our present heterodyne
digitizer is not phase-synchronized with the experimental event
sequence, so that traditional time-domain coaddition is incoherent from
scan to scan, and SNR will not be enhanced by coaddition. We therefore
use frequency-domain signal averaging, in which the monoisotopic (and
thus monodisperse) peak in each spectrum serves as a frequency anchor
for the coadding process (to avoid loss in both SNR and resolution).
SNR enhancement is clearly evident by comparison of multiply coadded
mass spectra in Figs. 5 and
6 to their single-scan
counterparts in Figs. 1 and 3.
by coherent
addition of N data sets (30). Unfortunately, our present heterodyne
digitizer is not phase-synchronized with the experimental event
sequence, so that traditional time-domain coaddition is incoherent from
scan to scan, and SNR will not be enhanced by coaddition. We therefore
use frequency-domain signal averaging, in which the monoisotopic (and
thus monodisperse) peak in each spectrum serves as a frequency anchor
for the coadding process (to avoid loss in both SNR and resolution).
SNR enhancement is clearly evident by comparison of multiply coadded
mass spectra in Figs. 5 and
6 to their single-scan
counterparts in Figs. 1 and 3.
Figure 5.

Mass scale-expanded spectra of three isotopic peaks of bovine insulin, obtained by coadding 11 frequency-domain ESI FT-ICR spectra. The theoretical isotopic distribution is shown at upper left, in which ★ denote the monisotopic peak (Left), and isotopic peaks ∼2 Da (middle) and ∼4 Da (Right) above the monoisotopic mass. Note the significant improvement in SNR (compare with Fig. 1) because of frequency-domain signal averaging.
Figure 6.

Mass-scale expanded spectra of two isotopic peaks of 13C, 15N doubly depleted p16 tumor suppressor protein, obtained by coadding nine frequency-domain ESI FT-ICR spectra. The theoretical isotopic distribution is shown at upper right, in which ★ denote the monisotopic peak (Left), and isotopic peak (Right) ∼2 Da above the monoisotopic mass. From the abundance ratio of the (resolved) 34S to the monoisotopic species, we determined the number of sulfur atoms in the protein to be 5.1 ± 0.3 without use of any other information about the protein.
For example, counting sulfur atoms requires measuring the abundance of ions with a single 34S heavy atom relative to monoisotopic ions. That measurement is greatly facilitated by 13C, 15N double isotopic depletion (99.95% 12C and 99.99% 14N), which shifts (toward the monoisotopic mass) and compresses the isotopic distribution for a protein (20). Double depletion increases the monoisotopic abundance at mass, M, and reduces interference (especially by 15N33S at M+2), leaving 34S and 18O as the principal M+2 species. At 0.01% 15N, interference by 15N33S is essentially eliminated (∼5% error for every 2,800 nitrogen atoms in the molecule).
Isotope double depletion obviously also simplifies the isotopic distribution by reducing the number of isotopic species. Because peak coalescence limits the maximum number of ions that may be trapped simultaneously, a reduction in the number of species translates into more ions of given (desired) species, thereby further increasing isotopic fine structure SNR.
Finally, from the resolved isotopic fine structure (Fig. 6) of 13C, 15N doubly depleted p16 tumor suppressor protein, we determine the number of sulfur atoms to be 5.1 ± 0.3 (consistent with the value of 5 obtained by conventional methods), independent of any other prior information about the protein. In other words, we show that the number of sulfur atoms in a protein can be accurately determined solely by measurement of the relative abundance of the isotopic fine structure peaks. Total sample consumption for sulfur determination based on isotopic fine structure is currently on the microgram level but could be reduced further.
As discussed above, the relative abundance fluctuation originates from noise in the spectrum. We therefore calculated the error in sulfur atom counting based on the noise level of the coadded spectra. Note that for statistical analysis of the relative abundances for individual spectra, we observed an error of 28%, or 1.5 sulfurs for p16. The precision of the average result of the nine measurements is expected to be three times better (9% or 0.5 sulfurs). The actual result for the coadded spectra (6% or 0.3 sulfur) is better than a simple average because coadding spectra is a weighted average that favors spectra of high SNR.
Mass Resolution and Mass Accuracy.
Although the present data can, with appropriate internal mass calibration, yield ultrahigh mass accuracy, the main point here is that high mass accuracy is not required for identification and quantitation of the number of atoms of a given element in a molecule. Rather, one requires ultrahigh mass resolution (to distinguish different elemental compositions) and accurate ion relative abundances (here, to within a few percent).
CONCLUSION
The spectra shown here represent a significant advance for mass resolving power (e.g., m/Δm50% 8,000,000 for bovine ubiquitin at 8.6 kDa achieved on biological polymers by FT-ICR mass spectrometry (or any other mass analyzer). Equally important, that resolving power has been achieved without coalescing isotopic fine structure, for proteins of molecular mass up to 15.8 kDa, or about 10 times the highest mass at which isotopic fine structure previously had been observed experimentally. Isotopic fine structure reveals elemental composition directly and can be used to confirm or determine molecular formula. For example, for p16 tumor suppressor protein, we have been able to determine the number of sulfur atoms (5.1 ± 0.3) from the ratio of the 34S to monoisotopic abundance, without use of any other information about the protein.
Acknowledgments
We thank Dr. Jared J. Drader and Greg T. Blakney for programming the Hewlett–Packard digitizer, Weiqun Li for providing the program to calculate a fine structure peak list, and Thomas Selby and Dr. Ming-Daw Tsai for providing the 13C,15N doubly depleted p16 tumor suppressor protein sample. This work was supported by grants from the National Science Foundation (CHE-93-22824), the National Science Foundation National High Field FT-ICR Mass Spectrometry Facility (CHE-94-13008), the National Institutes of Health (GM-31683), Florida State University, and the National High Magnetic Field Laboratory in Tallahassee, FL.
ABBREVIATIONS
- ICR
ion cyclotron resonance
- FT
Fourier transformation
- ESI
electrospray ionization
- SNR
signal-to-noise ratio
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1. Stults J T. Anal Chem. 1997;69:1815–1819. [Google Scholar]
- 2.Solouki T, Emmett M R, Guan S, Marshall A G. Anal Chem. 1997;69:1163–1168. doi: 10.1021/ac960885q. [DOI] [PubMed] [Google Scholar]
- 3.Naito Y, Inoue M. J Mass Spectrom Soc Jpn. 1994;42:1–9. [Google Scholar]
- 4.Mitchell D W, Smith R D. Phys Rev E. 1995;52:4366–4386. doi: 10.1103/physreve.52.4366. [DOI] [PubMed] [Google Scholar]
- 5.DiFillip F, Natarajan V, Bradley M, Palmer F, Pritchard D E. Physica Scripta T. 1995;59:144–154. [Google Scholar]
- 6.Comisarow M B, Marshall A G. Chem Phys Lett. 1974;25:282–283. [Google Scholar]
- 7.Comisarow M B, Marshall A G. Chem Phys Lett. 1974;26:489–490. [Google Scholar]
- 8.Marshall, A. G., Hendrickson, C. L. & Jackson, G. S. (1998) Mass Spectrom. Rev., in press. [DOI] [PubMed]
- 9.Talrose V L, Nikolaev E N. Adv Mass Spectrom. 1985;10A:343–357. [Google Scholar]
- 10.Lippmaa E T, Pikver R, Suurmaa E, Past J, Puskar J, Koppel I, Tammik A. Phys Rev Lett. 1985;54:285–288. doi: 10.1103/PhysRevLett.54.285. [DOI] [PubMed] [Google Scholar]
- 11.Alber G M, Marshall A G, Hill N C, Schweikhard L, Ricca T. Rev Sci Instrum. 1993;64:1845–1852. [Google Scholar]
- 12.Gorshkov M V, Guan S, Marshall A G. Int J Mass Spectrom Ion Processes. 1993;128:47–60. [Google Scholar]
- 13.Schweikhard L, Alber G M, Marshall A G. J Am Soc Mass Spectrom. 1993;4:177–181. doi: 10.1016/1044-0305(93)85074-8. [DOI] [PubMed] [Google Scholar]
- 14.Bamberg M, Allemann M, Wanczek K-P. Proceedings of the 35th American Society for Mass Spectrometry Annual Conference on Mass Spectrometry and Allied Topics. Denver, CO: Am. Soc. Mass Spectrom.; 1987. pp. 1116–1117. [Google Scholar]
- 15.Beu S C, Senko M W, Quinn J P, Wampler F M, III, McLafferty F W. J Am Soc Mass Spectrom. 1993;4:557–565. doi: 10.1016/1044-0305(93)85017-R. [DOI] [PubMed] [Google Scholar]
- 16.Guan S, Wahl M C, Marshall A G. Anal Chem. 1993;65:3647–3653. doi: 10.1021/ac00072a019. [DOI] [PubMed] [Google Scholar]
- 17.Bruce J E, Anderson G A, Hofstadler S A, Winger B E, Smith R D. Rapid Commun Mass Spectrom. 1993;7:700–703. [Google Scholar]
- 18.Senko M W, Hendrickson C L, Emmett M R, Shi S D-H, Marshall A G. J Am Soc Mass Spectrom. 1997;8:970–976. [Google Scholar]
- 19.Drader J J, Shi S D-H, Blakney G T, Hendrickson C L, Marshall A G, Laude D A. Proceedings of the 46th American Society for Mass Spectrometry Annual Conference on Mass Spectrometry and Allied Topics. Orlando, FL: Am. Soc. Mass Spectrom.; 1998. , Poster WP100. [Google Scholar]
- 20.Marshall A G, Senko M W, Li W, Li M, Dillon S, Guan S, Logan T M. J Am Chem Soc. 1997;119:433–434. [Google Scholar]
- 21.Senko M W, Hendrickson C L, Pasa-Tolic L, Marto J A, White F M, Guan S, Marshall A G. Rapid Commun Mass Spectrom. 1996;10:1824–1828. doi: 10.1002/(SICI)1097-0231(199611)10:14<1824::AID-RCM695>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 22.Senko M W, Canterbury J D, Guan S, Marshall A G. Rapid Commun Mass Spectrom. 1996;10:1839–1844. doi: 10.1002/(SICI)1097-0231(199611)10:14<1839::AID-RCM718>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 23.Emmett M R, White F M, Hendrickson C L, Shi S D-H, Marshall A G. J Am Soc Mass Spectrom. 1998;9:333–340. doi: 10.1016/S1044-0305(97)00287-0. [DOI] [PubMed] [Google Scholar]
- 24.Marshall A G, Wang T-C L, Ricca T L. J Am Chem Soc. 1985;107:7893–7897. [Google Scholar]
- 25.Guan S, Marshall A G. Int J Mass Spectrom Ion Processes. 1996;157/158:5–37. [Google Scholar]
- 26.Ledford E B, Jr, Rempel D L, Gross M L. Anal Chem. 1984;56:2744–2748. doi: 10.1021/ac00278a027. [DOI] [PubMed] [Google Scholar]
- 27.Senko, M. W. (1998) isopro, version 3.03a.
- 28.Adams C S, Lee H J, Davidson N, Kasevich M, Chu S. Phys Rev Lett. 1995;74:3577–3580. doi: 10.1103/PhysRevLett.74.3577. [DOI] [PubMed] [Google Scholar]
- 29.Jeffries J B, Barlow S E, Dunn G H. Int J Mass Spectrom Ion Processes. 1983;54:169–187. [Google Scholar]
- 30.Marshall A G, Verdun F R. Fourier Transforms in NMR, Optical, and Mass Spectrometry: A User’s Handbook. Amsterdam: Elsevier; 1990. [Google Scholar]
- 31.Stejskal E O, Schaefer J. J Magn Reson. 1974;14:160–169. [Google Scholar]
- 32.Verdun, F. R., Mullen, S. L., Ricca, T. L. & Marshall, A. G. (1987) FACSS XIV, Abstr. 41.
- 33.Wachter E A, Farrar T C, Kontney M J. Int J Mass Spectrom Ion Processes. 1991;103:169–179. [Google Scholar]
- 34.Guan S, Li G-Z, Marshall A G. Int J Mass Spectrom Ion Processes. 1998;167/168:185–194. [Google Scholar]
- 35.Chen L, Cottrell C E, Marshall A G. Chemometrics Intelligent Lab Syst. 1986;1:51–58. [Google Scholar]
- 36.Senko M W, Beu S C, McLafferty F W. J Am Soc Mass Spectrom. 1995;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]