

Abstract
We determined the pharmacokinetic-pharmacodynamic (PK-PD) measure most predictive of gatifloxacin efficacy and the magnitude of this measure necessary for survival in a murine Bacillus anthracis inhalation infection model. We then used population pharmacokinetic models for gatifloxacin and simulation to identify dosing regimens with high probabilities of attaining exposures likely to be efficacious in adults and children. In this work, 6- to 8-week-old nonneutropenic female BALB/c mice received aerosol challenges of 50 to 75 50% lethal doses of B. anthracis (Ames strain, for which the gatifloxacin MIC is 0.125 mg/liter). Gatifloxacin was administered at 6- or 8-h intervals beginning 24 h postchallenge for 21 days, and dosing was designed to produce profiles mimicking fractionated concentration-time profiles for humans. Mice were evaluated daily for survival. Hill-type models were fitted to survival data. To identify potentially effective dosing regimens, adult and pediatric population pharmacokinetic models for gatifloxacin and Monte Carlo simulation were used to generate 5,000 individual patient exposure estimates. The ratio of the area under the concentration-time curve from 0 to 24 h (AUC0-24) to the MIC of the drug for the organism (AUC0-24/MIC ratio) was the PK-PD measure most predictive of survival (R2 = 0.96). The 50% effective dose (ED50) and the ED90 and ED99 corresponded to AUC0-24/MIC ratios of 11.5, 15.8, and 30, respectively, where the maximum effect was 97% survival. Simulation results indicate that a daily gatifloxacin dose of 400 mg for adults and 10 mg/kg of body weight for children gives a 100% probability of attaining the PK-PD target (ED99). Sensitivity analyses suggest that the probability of PK-PD target attainment in adults and children is not affected by increases in MICs for strains of B. anthracis to levels as high as 0.5 mg/liter.
Bacillus anthracis, the causative agent of anthrax, is a pathogen primarily of animals and can produce a fatal disease in humans when introduced by the inhalation or ingestion of spores (19). Of concern is the potential use of this organism as a biological weapon, as demonstrated in the 2001 bioterrorism-related distribution of anthrax spores through the U.S. mail (7, 8, 22). Beginning on 18 September 2001 and continuing for several weeks thereafter, terrorists mailed letters containing B. anthracis to news media offices and U.S. senators. In total, 22 people were infected, of whom 5 died. For citizen and soldier alike, it is critical to identify effective treatments against agents, such as B. anthracis, that can be used as weapons of terror. While conducting clinical trials evaluating treatment modalities for bacterial agents of bioterrorism is impossible, in many instances it is possible to identify regimens likely to be effective. Effective treatment regimens can be identified through the use of Monte Carlo simulation to integrate exposure-response relationships obtained from pharmacokinetic-pharmacodynamic (PK-PD) animal infection models with information about drug disposition in human populations.
Animal infection models are fundamental tools in the evaluation of the therapeutic efficacy of antimicrobial agents (16). From these models, one can gain important information about the time course of the antimicrobial effect, which can be used to construct exposure-response relationships and to determine PK-PD target measures that are predictive of clinical efficacy in humans (10). Over the past 5 years, Monte Carlo simulation has been used to integrate human pharmacokinetic data and PK-PD targets derived from nonclinical or clinical data in order to provide support for dose regimen selection (6, 14, 15). This approach to evaluating antimicrobial regimens may be especially valuable under circumstances in which formal exposure-ranging clinical trials are unlikely, as in the case of agents of bioterrorism.
The objectives of this study were threefold: (i) to identify the PK-PD measure (i.e., the ratio of the area under the concentration-time curve from 0 to 24 h [AUC0-24] to the MIC of the drug for the organism [AUC0-24/MIC ratio], the ratio of the maximum concentration of the drug in serum [Cmax] to the MIC [Cmax/MIC ratio], or the percentage of time during the dosing interval that the drug concentration remains above the MIC [%t>MIC]) that best predicts gatifloxacin efficacy against B. anthracis; (ii) to determine the magnitude of the PK-PD measure associated with the 50% effective dose (ED50) and the ED90 and ED99; and (iii) to utilize Monte Carlo simulation, human pharmacokinetic data, and PK-PD magnitude targets in an effort to determine the adequacy of adult and pediatric dosing regimens for postexposure prophylaxis of pulmonary B. anthracis infection.
MATERIALS AND METHODS
Bacteria, media, and antibiotics.
Spores were prepared with the Ames strain of B. anthracis by using the medium and growth conditions defined by Leighton and Doi (21) as described previously (24). Before aerosol challenge, spore preparations were heated at 65°C for 30 min and diluted in sterile water. Bacteria were counted by serially diluting preparations or samples 1:10 in sterile water and plating the appropriate dilutions in triplicate onto sheep blood agar plates. Plates were incubated at 35°C for 18 h, and colonies were counted. Gatifloxacin was supplied by the Bristol-Myers Squibb Company (Princeton, NJ).
In vitro susceptibility studies.
The MICs of gatifloxacin for 30 strains of B. anthracis, including the Ames strain, were determined by the microdilution methods approved by the Clinical and Laboratory Standards Institute (formally known as the National Committee for Clinical Laboratory Standards or NCCLS) (9).
Humanization of murine pharmacokinetics.
The drug exposure profiles for mice were humanized as follows. The once-daily doses needed to achieve the desired steady-state AUC0-24 values in humans were computed. Then these human regimens were densely simulated to determine the appropriate plasma drug concentrations and AUC values throughout the planned duration of the murine studies. The doses for the murine studies were computed to mimic, as closely as possible, the AUC values obtained in the human simulations. For the studies in which mice were dosed every 6 h, for example, the murine dose at time zero was that which would provide the same AUC0-6 that was simulated for humans. Given that dose at time zero, the 6-h dose was that which was computed to provide a murine AUC0-12 similar to that simulated for humans. This process was continued for the entire planned duration of the murine studies. For each regimen, the doses were all computed in a single step, by using a module we produced in ADAPT II, release 4 (12). This approach is depicted in Fig. 1.
FIG. 1.
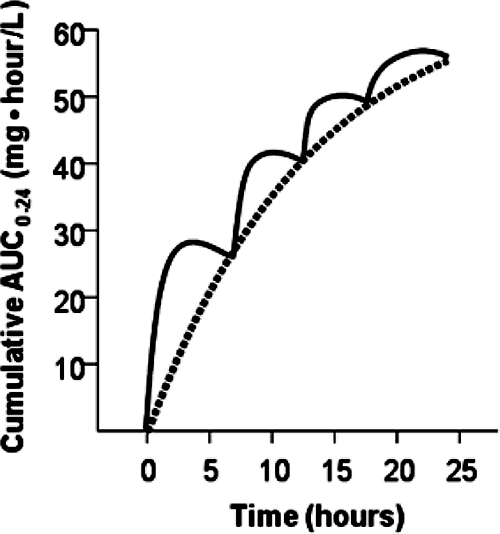
Typical human cumulative AUC0-24 following a 400-mg dose of gatifloxacin (dashed line) and murine cumulative AUC0-24 (solid line) representative of those for mice given a regimen designed to mimic the cumulative exposure in humans.
Murine pharmacokinetic studies and pharmacokinetic modeling.
For pharmacokinetic determinations, three groups of mice each received a single intraperitoneal dose of gatifloxacin (10, 50, or 100 mg/kg of body weight). Sera from three mice in each dosing group were collected by exsanguinations under anesthesia at 0.25, 0.5, 1, 2, 3, 4, 6, and 12 h postdose. Samples were then centrifuged for 5 min at 10,000 × g, and sera were removed. Sera were stored at −70°C until assayed for gatifloxacin concentrations. Gatifloxacin concentrations in sera were determined according to a modified bioassay using Staphylococcus aureus ATCC 29213 as the indicator organism and were compared to a standard curve for gatifloxacin in control mouse serum (17, 20). All samples were assayed in triplicate. The lower and upper limits of detection of the assays were 0.12 and 64 μg/ml, respectively. Intraday variation was one twofold dilution. All pharmacokinetic studies were performed on the same day. All serum drug concentration data, regardless of dosing group, were comodeled using maximum likelihood (ADAPT II) (12). Observations were weighted by the estimated variance of the measurement. The model discrimination was determined by Akaike's information criteria (1).
Murine inhalational anthrax challenge model.
Animals were maintained in accordance with the criteria of the American Association for Assessment and Accreditation of Laboratory Animal Care. All animal studies were approved by the Animal Research Committee of the U.S. Army Medical Research Institute of Infectious Diseases, Fort Detrick, MD.
In efficacy studies, 6- to 8-week-old nonneutropenic female BALB/c mice were challenged by aerosol spray with between 50 and 100 times the established 50% lethal dose (3.4 × 104 CFU) of the Ames spore preparation (20). Antibiotic treatment (0.2 ml intraperitoneally) was administered to mice (10 per group) to emulate human regimens, with several daily doses divided by 6- or 8-h intervals beginning 24 h postchallenge and continuing for 21 days. As mentioned above (see “Humanization of murine pharmacokinetics”), doses of various sizes were administered to mice in order to better emulate dosing in humans. For instance, a mouse in the every-12-h fractionation cohort received two doses over 12 h, of which the first was larger and the second was smaller, while a mouse in the every-24-h cohort received three doses over 24 h, each decreasing in size. Total daily doses corresponded to AUC0-24 values ranging from 1.875 to 100 mg·h/liter. Control mice received phosphate-buffered saline. Mice were evaluated daily for survival.
PK-PD modeling and Monte Carlo simulation.
Fitted pharmacokinetic functions for each dosage regimen were used to compute the candidate PK-PD exposure measures (%t>MIC and AUC0-24/MIC and Cmax/MIC ratios). Hill-type models were fitted to survival data (percentage of mice surviving the study) regressed to %t>MIC and AUC0-24/MIC and Cmax/MIC ratios.
Mean pharmacokinetic parameter estimates and dispersion measures from previously derived population pharmacokinetic models were used for simulations (equations 1 and 2) (2, 23a). Log-normal distributions were assumed.
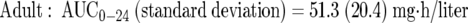 |
(1) |
 |
(2) |
Five thousand individual adult and pediatric patient exposure estimates (400 mg once daily and 10 mg/kg of body weight/day, respectively) were generated using SYSTAT, version 11.
The level of plasma protein binding by gatifloxacin is 20% in both mice and humans (TEQUIN gatifloxacin tablet and injection package insert; Bristol-Myers Squibb Company, Princeton, NJ, 2003). Thus, in all the analyses described herein, no correction for protein binding was made. Except where free drug is specified, all drug exposures detailed herein refer to the total drug.
RESULTS
In vitro susceptibility testing.
The MIC of gatifloxacin for the Ames strain of B. anthracis was 0.125 mg/liter. The range for the diverse 30-strain set was 0.06 to 0.5 mg/liter.
Pharmacokinetic modeling.
Gatifloxacin concentration-time data for mice were fit to a two-compartment model with first-order absorption and a lag time (the delay between the time of administration and the onset of absorption). Over the dose range studied, gatifloxacin exhibited linear and dose-proportional pharmacokinetics in mice (data not shown). Table 1 shows the final fitted gatifloxacin pharmacokinetic parameter estimates for mice. As expected, drug clearance was faster and the half-life was shorter in mice than in humans (2.7 versus 0.1 liter/h/kg and 0.6 versus 9 h, respectively) (TEQUIN package insert).
TABLE 1.
Final fitted gatifloxacin pharmacokinetic parameter estimates for mice
| Parametera | Units | Estimate |
|---|---|---|
| Vc/F | Liters/kg | 0.20 |
| Vp/F | Liters/kg | 1.6 |
| CLD/F | Liters/h/kg | 7.7 |
| CLT/F | Liters/h/kg | 2.7 |
| Ka | h−1 | 11.0 |
| tlag | h | 0.23 |
| t1/2 | h | 0.60 |
Vc/F, apparent volume of distribution in the central compartment; Vp/F, apparent volume of distribution in the peripheral compartment; CLD/F, apparent distributional clearance; CLT/F, apparent total clearance; Ka, absorption rate constant; tlag, lag time; t1/2, half-life.
PK-PD modeling.
The relationships between exposure and response for each of the PK-PD measures, the AUC0-24/MIC ratio, the Cmax/MIC ratio, and %t>MIC, for gatifloxacin used against the Ames strain of B. anthracis are presented in Fig. 2. As for members of other bacterial genera and other fluoroquinolones, the strongest relationship was observed when results were correlated with the AUC0-24/MIC ratio (R2 = 0.96). However, the Cmax/MIC ratio (R2 = 0.78) and %t>MIC (R2 = 0.88) were also reasonably well correlated with animal survival. The use of bound- or unbound-gatifloxacin concentrations did not appreciably impact the relationship between efficacy and %t>MIC (data not shown). Table 2 shows the final parameter estimates for each PK-PD model. The ED50, ED90, and ED99 corresponded to AUC0-24/MIC ratios of 11.5, 15.8, and 30, respectively, where the maximum effect was 97% survival.
FIG. 2.

Relationship between three PK-PD measures (AUC0-24/MIC ratio, Cmax/MIC ratio, and %t>MIC) and survival of nonneutropenic mice challenged with aerosolized B. anthracis (Ames strain) after 21 days of therapy with gatifloxacin. Observed data for individual regimens stratified by dosing regimen are shown by different symbols. Q8hr, Q12hr, and Q24hr, every 8, 12, and 24 h, respectively.
TABLE 2.
Final parameter estimates of the model for each of the PK-PD measures
| Parametera | AUC0-24/MIC ratio | Cmax/MIC ratio | %t>MIC |
|---|---|---|---|
| Emax | 0.97 | 0.83 | 1.00 |
| ED50 | 11.8 | 8.10 | 27.5 |
| E0 | 0.10 | 0.07 | 0.08 |
| Gamma | 7.03 | 10.0 | 10.0 |
Emax, maximum effect; E0, effect at time zero; gamma, Hill's constant.
Monte Carlo simulation.
The shapes of simulated AUC0-24 distributions for adult and pediatric patients closely resembled those previously described for patient populations (TEQUIN package insert). For instance, the mean (standard deviation) AUC0-24 values for simulations and previous measurements were 51.3 (20.4) versus 51.3 (20.4) mg·h/liter for adult patients, respectively, and 34.4 (12.0) versus 32.9 (11.5) mg·h/liter for pediatric patients. A target AUC0-24/MIC ratio of 30 was chosen based on the association of this ratio with greater than 99% of the maximum effect. Figure 3 shows the probability of PK-PD target attainment for adult and pediatric dosing regimens. With a MIC of 0.125 mg/liter, that for the Ames strain of B. anthracis, the probability of PK-PD target attainment was 1.0 for both the adult (400-mg once-daily) and pediatric (10-mg/kg/day) dosing regimens. The probability of PK-PD target attainment was 0.95 or greater for MICs of 0.5 mg/liter or lower. For MICs of 1.0 mg/liter or higher, the performance of standard dosing regimens was degraded markedly.
FIG. 3.
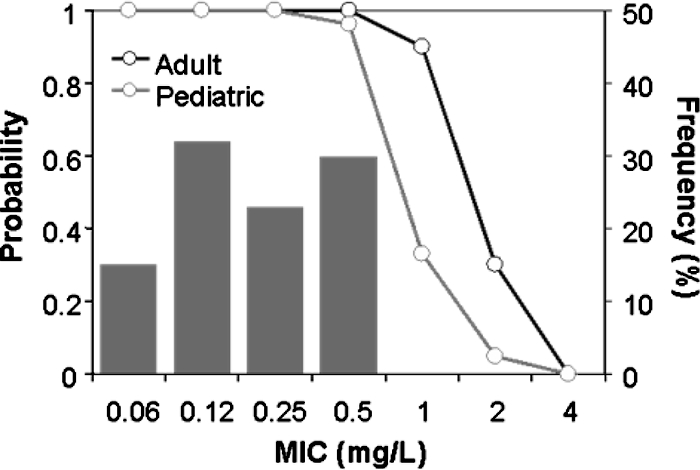
Probabilities of attaining the PK-PD target (AUC0-24/MIC ratio of 30) by following a 400-mg once-daily adult and a 10-mg/kg/day pediatric gatifloxacin dosing regimen. The gray bars represent the distribution of the MICs of gatifloxacin for B. anthracis.
DISCUSSION
The objectives of this study were (i) to identify the PK-PD measure that best predicts gatifloxacin efficacy against B. anthracis; (ii) to determine the magnitude of the PK-PD measure required for maximal efficacy; and (iii) to utilize Monte Carlo simulation to integrate human pharmacokinetic data and PK-PD targets in an effort to determine the adequacy of adult and pediatric dosing regimens for postexposure prophylaxis of pulmonary B. anthracis infection.
We identified the AUC0-24/MIC ratio as the PK-PD measure that best predicts gatifloxacin efficacy against B. anthracis. To our knowledge, the in vivo exposure-response relationships described herein represent the first time such relationships have been identified for an agent of bioterrorism. Previous studies conducted with B. anthracis and ciprofloxacin in primates, which resulted in U.S. Food and Drug Administration approval of an indication, were based on a successful empirical twice-daily regimen (18). Levofloxacin was granted U.S. Food and Drug Administration approval based upon results of studies conducted with mice and primates. The dosage regimens used in these animal studies simulated the shape and magnitude (AUC0-24) of the levofloxacin concentration-time profile achieved in humans following a 500-mg once-daily regimen (13). The regimens were effective in both species, and thus, the drug gained an indication. In neither circumstance was an exposure- or dose-response relationship identified.
In this study, the AUC0-24/MIC ratio necessary for the maximum effect (97% survival) in nonneutropenic mice was 30. This PK-PD target is much lower than that observed for the maximum effect of gatifloxacin or other fluoroquinolones against gram-positive bacteria in nonneutropenic mice (5, 11). For instance, for ciprofloxacin, gatifloxacin, gemifloxacin, levofloxacin, moxifloxacin, and sitafloxacin against Streptococcus pneumoniae, near-maximum effect was observed when free-drug AUC0-24/MIC ratios ranged from 50 to more than 100 (11).
Since patients with pulmonary anthrax will most likely die without prompt and effective therapy, we should choose exposure targets near the maximum possible effect (i.e., an AUC0-24/MIC ratio of 30). It should be noted that choosing an exposure target near the maximum effect is unusual. Oftentimes, the exposure targets in animal systems that correlate with positive clinical responses in patients are not near the maximum effect (3). For instance, the animal-derived exposure target (AUC0-24/MIC ratio of 83) for linezolid against Staphylococcus aureus was associated with net bacterial stasis (4), which correlated well with exposure thresholds predictive of clinical responses in patients with staphylococcal or enterococcal bacteremia (AUC0-24/MIC ratio of 85) (23).
Monte Carlo simulation results indicate that standard gatifloxacin dosing regimens would be effective for postexposure prophylaxis of pulmonary B. anthracis infection. Gatifloxacin regimens of 400 mg once daily for adult patients and 10 mg/kg/day for pediatric patients provide a greater than 95% probability of attaining the exposure target for strains for which MICs are up to 0.5 mg/liter, which covers essentially the entire distribution of gatifloxacin MICs for B. anthracis (Fig. 3). Given that the gatifloxacin MIC for the Ames strain is 0.125 mg/liter and that a fourfold increase in MIC would be expected after a ParC mutation or efflux pump overexpression (13), the dosing regimens described above should be adequate for single-step mutants that may emerge during therapy or that are created by those who would use this strain of B. anthracis as a weapon of terror.
In conclusion, we successfully utilized the lethal murine B. anthracis inhalation infection model to identify the PK-PD measure that best predicts gatifloxacin efficacy against B. anthracis. Moreover, we were able to identify the magnitude of gatifloxacin exposure needed to protect against pulmonary exposure to B. anthracis. Finally, Monte Carlo simulation was used to evaluate potentially efficacious adult and pediatric dosing regimens. Given that it is impossible to conduct clinical trials with bacterial agents that can be used as biological weapons, the approach described herein should become the standard for the identification of likely effective treatment regimens.
Acknowledgments
The opinions, interpretations, conclusions, and recommendations expressed herein are those of the authors and are not necessarily endorsed by the U.S. Army.
The research described herein was sponsored by a grant from Bristol-Myers Squibb to P.G.A. and the Defense Threat Reduction Agency, grant no. 02-4-2C-013 to H.S.H. and 1PO1-AI060908-01A1 to G.L.D.
Footnotes
Published ahead of print on 17 September 2007.
REFERENCES
- 1.Akaike, H. 1974. A new look at the statistical model identification. IEEE Trans. Automatic Control 19:716-723. [Google Scholar]
- 2.Ambrose, P. G., D. M. Grasela, T. H. Grasela, P. Passarell, H. B. Mayer, and P. F. Pierce. 2001. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections Antimicrob. Agents Chemother. 45:2793-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambrose, P. G., S. M. Bhavnani, C. M. Rubino, A. Louie, T. Gumbo, A. Forrest, and G. L. Drusano. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin. Infect. Dis. 44:79-86. [DOI] [PubMed] [Google Scholar]
- 4.Andes, D., M. L. van Ogtrop, J. Peng, and W. A. Craig. 2002. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob. Agents Chemother. 46:3484-3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andes, R., and W. A. Craig. 2002. Pharmacodynamics of the new fluoroquinolone gatifloxacin in murine thigh and lung infection models. Antimicrob. Agents Chemother. 46:1665-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhavnani, S. M., J. P. Hammel, B. B. Cirincione, M. A. Wikler, and P. G. Ambrose. 2005. Use of pharmacokinetic-pharmacodynamic target attainment analyses to support phase 2 and 3 dosing strategies for doripenem. Antimicrob. Agents Chemother. 49:3944-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. 2001. Update: investigation of anthrax associated with intentional exposure and interim public health guidelines. Morb. Mortal. Wkly. Rep. 50:889-897. [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. 2001. Investigation of bioterrorism-related anthrax and interim guidelines for exposure management and antimicrobial therapy. Morb. Mortal. Wkly. Rep. 50:909-919. [PubMed] [Google Scholar]
- 9.Clinical and Laboratory Standards Institute. 2006. Performance standards for antimicrobial susceptibility testing. Standard M100-S16. Clinical and Laboratory Standards Institute, Wayne, PA.
- 10.Craig, W. A. 2002. Pharmacodynamics of antimicrobials: general concepts and applications, p. 1-22. In C. H. Nightingale, T. Murakawa, and P. G. Ambrose (ed.), Antimicrobial pharmacodynamics in theory and clinical practice. Marcel Dekker, Inc., New York, NY.
- 11.Craig, W. A., and D. A. Andes. 2000. Correlation of the magnitude of the AUC24/MIC for six fluoroquinolones against Streptococcus pneumoniae with survival and bactericidal activity in an animal model, abstr. 289. Abstr. 40th Intersci. Conf. Antimicrob. Chemother. American Society for Microbiology, Washington, DC.
- 12.D'Argenio, D. Z., and A. Schumitzky. 2003. ADAPT II user's guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, CA.
- 13.Deziel, M. R., H. Heine, A. Louie, M. Kao, W. R. Byrne, J. Basset, L. Miller, K. Bush, M. Kelly, and G. L. Drusano. 2005. Effective antimicrobial regimens for use in humans for therapy of Bacillus anthracis infections and postexposure prophylaxis. Antimicrob. Agents Chemother. 49:5099-5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drusano, G. L., K. H. P. Moore, J. P. Kleim, W. Prince, and A. Bye. 2002. Rational dose selection for a nonnucleoside reverse transcriptase inhibitor through use of population pharmacokinetic modeling and Monte Carlo simulation. Antimicrob. Agents Chemother. 46:913-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drusano, G. L., S. L. Preston, C. Hardalo, R. Hare, C. Banfield, D. Andes, O. Vesga, and W. A. Craig. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eagle, H., R. Fleischman, and A. D. Musselman. 1950. The effective concentrations of penicillin in vitro and in vivo for streptococci, pneumococci, and Treponema pallidum. J. Bacteriol. 59:625-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edberg, S. C. 1986. The measurement of antibiotics in human body fluids: techniques and significance, p. 382-399. In V. Lorian (ed.), Antibiotics in laboratory medicine, 2nd ed. Williams and Wilkins, Baltimore, MD.
- 18.Friedlander, A. M., S. L. Welkos, M. L. Pitt, J. W. Ezzell, P. L. Worsham, K. J. Rose, B. E. Ivins, J. R. Lowe, G. B. Howe, P. Mikesell, et al. 1993. Post-exposure prophylaxis against experimental inhalation anthrax. J. Infect. Dis. 167:1239-1243. [DOI] [PubMed] [Google Scholar]
- 19.Friedlander, A. M. 2000. Anthrax: clinical features, pathogenesis, and potential biological warfare threat. Curr. Clin. Top. Infect. Dis. 20:335-349. [PubMed] [Google Scholar]
- 20.Heine, H. S., J. Bassett, L. Miller, J. M. Hartings, B. E. Ivins, M. L. Pitt, D. Fritz, S. L. Norris, and W. R. Byrne. 2007. Determination of antibiotic efficacy against Bacillus anthracis in a mouse aerosol challenge model. Antimicrob. Agents Chemother. 51:1373-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leighton, T. J., and R. H. Doi. 1971. The stability of messenger ribonucleic acid during sporulation in Bacillus anthracis. J. Biol. Chem. 246:3189-3195. [PubMed] [Google Scholar]
- 22.Pile, J. C., J. D. Malone, E. M. Eitzen, and A. M. Friedlander. 1998. Anthrax as a potential biological warfare agent. Arch. Intern. Med. 158:429-434. [DOI] [PubMed] [Google Scholar]
- 23.Rayner, C. R., A. Forrest, A. K. Meagher, M. C. Birmingham, and J. J. Schentag. 2003. Clinical pharmacodynamics of linezolid in seriously ill patients treated in a compassionate use programme. Clin. Pharmacokinet. 42:1411-1423. [DOI] [PubMed] [Google Scholar]
- 23a.Rubino, C. M., P. Ambrose, B. Cirincione, A. Arguedas, L. Sher, E. Lopez, X. Sáez-Llorens, and D. M. Grasela. 2007. Pharmacokinetics and pharmacodynamics of gatifloxacin in children with recurrent otitis media: application of sparse sampling in clinical development. Diagn. Microbiol. Infect. Dis. 59:67-74. [DOI] [PubMed] [Google Scholar]
- 24.Welkos, S. L., T. J. Keener, and P. H. Gibbs. 1986. Differences in susceptibility of inbred mice to Bacillus anthracis. Infect. Immun. 51:795-800. [DOI] [PMC free article] [PubMed] [Google Scholar]