

Abstract
We recently showed that the pyridinylimidazoles SB203580 and SB202190, drugs designed to block human p38 mitogen-activated protein kinase (MAPK) activation, also inhibited replication of the medically important intracellular parasite Toxoplasma gondii in cultured human fibroblasts through a direct effect on the parasite. We now show that additional pyridinylimidazole and imidazopyrimidine p38 MAPK inhibitors inhibit intracellular T. gondii replication in vitro and protect mice against fatal T. gondii infection. Mice surviving infection following treatment with p38 MAPK inhibitors were resistant to subsequent T. gondii challenge, demonstrating induction of protective immunity. Thus, drugs originally developed to block human p38 MAPK activation are useful for treating T. gondii infection without inducing significant immunosuppression. MAPK inhibitors combined with either of the approved anti-Toxoplasma drugs sulfadiazine and pyrimethamine resulted in improved survival among mice challenged with a fatal T. gondii inoculum. A MAPK inhibitor also treated mice infected with the Microsporidium parasite Encephalitozoon cuniculi, suggesting that MAPK inhibitors represent a novel class of agents that may have a broad spectrum of antiparasitic activity. Preliminary studies implicate a T. gondii MAPK homologue as the target of drug action, suggesting possibilities for more-selective agents.
Toxoplasma gondii is an obligate, intracellular protozoan parasite that causes significant morbidity and mortality in hosts with defective cell-mediated immunity (11, 12). The majority of significant Toxoplasma infections in industrialized countries occur in persons infected with human immunodeficiency virus (11, 12). Although pyrimethamine and sulfadiazine are the mainstays of current anti-Toxoplasma therapy (5, 12), administration of these drugs during concomitant antiretroviral therapy for human immunodeficiency virus infection is complicated by overlapping and significant drug toxicities (12). Thus, additional agents for treating T. gondii infection are needed.
During studies of Toxoplasma-mediated immunosuppression, we made the fortuitous observation that pyridinylimidazole drugs originally designed to inhibit human p38 mitogen-activated protein kinase (MAPK) activation (17) also blocked replication of T. gondii tachyzoites in cultured human fibroblasts in vitro (19), suggesting that p38 MAPK inhibitors might be useful for treating T. gondii infection. Further, all protozoan parasites, including agents of malaria, leishmaniasis, and trypanosomiasis, encode MAPKs (9). Thus, parasite MAPKs might be useful targets for antiparasite drug development, owing to their essential functions. For example, genetic deletion of a MAPK gene in Leishmania blocked stage-specific intracellular parasite proliferation (20).
We undertook the present studies to determine whether agents originally designed to inhibit human p38 MAPK activation could treat T. gondii-infected mice. We also tested whether combining these drugs with approved anti-Toxoplasma agents enhanced their treatment efficacy in infected mice and assessed for potential immunosuppression from p38 MAPK inhibition.
MATERIALS AND METHODS
Mice.
Female CBA/J mice and CD8−/− mice (on a CJ57BL/6 background) were purchased from Jackson Laboratory (Bar Harbor, ME). Animals were housed in microisolator cages and supplied with commercial chow and water ad libitum. These studies were approved by both the Tulane and Louisiana State University Institutional Animal Care and Use Committees.
Drugs and chemicals.
The pyridinylimidazole p38 MAPK inhibitors RWJ67657 and RWJ64809 and the imidazopyrimidine p38 MAPK inhibitor RWJ68198 were provided by Johnson & Johnson Pharmaceuticals (Raritan, NJ). The pyridinylimidazole p38 MAPK inhibitors SB203580 and SB202190, as well as a control pyridinylimidazole, SB202474, having no inhibitory activity against human p38 MAPKs, were all purchased from Calbiochem (La Jolla, CA). RWJ64809 is chemically identical to SB203580. Pyrimethamine, sulfadiazine, carboxymethyl cellulose (CMC), and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemical Co. (St. Louis, MO). All p38 MAPK inhibitor drugs were reconstituted in DMSO at 1 mM and stored frozen at −80°C until use, at which point they were diluted to a working concentration in sterile phosphate-buffered saline (PBS). For in vivo studies, pyrimethamine was dissolved in sterile PBS supplemented with 0.25% CMC and was administered to mice by oral gavage at 5 mg/kg of body weight. Sulfadiazine was dissolved in drinking water at a concentration of 75 mg/liter. All p38 MAPK drugs were given either by intraperitoneal (i.p.) injection in 50 μl of DMSO with a 27-gauge needle or by oral gavage. Sham treatments involved either the i.p. administration of 50 μl DMSO or the administration of 0.25% CMC suspended in sterile PBS by oral gavage, in parallel with active treatment.
Parasites.
RH and Me49 strain T. gondii tachyzoites were obtained from Randolph Berens and Edward Krug (University of Colorado, Denver, CO) and cultured in human foreskin fibroblasts (HFFs) as described previously (4, 19). Briefly, infected HFFs were maintained in at 37°C in a humidified, 5% CO2 atmosphere in RPMI 1640 medium supplemented with 10% fetal bovine serum, 10 mM HEPES buffer, 4 mM glutamine, and antibiotics. Tachyzoites were passed to new, uninfected HFF monolayers approximately weekly as the older monolayer was destroyed by replicating parasites. SBRMe49-2 is an SB203580-resistant T. gondii strain that we clonally derived from T. gondii strain Me49-infected HFFs by incubating the culture with increasing concentrations of SB203580. HFFs were infected with Me49 at a multiplicity of infection (MOI) of 5, meaning that five T. gondii tachyzoites per HFF were introduced into the confluent HFF monolayer. The flask was incubated for 3 weeks in the presence of 0.3 μM of SB203580. Surviving tachyzoites were consecutively passed to fresh HFF monolayers at 2-week intervals following incubation with 0.5, 1.0, 3.0, 5.0, and 10 μM SB203580. The SB203580-resistant population of tachyzoites was then serially diluted to a limiting dilution in the presence of 10 μM of the drug, leading to the isolation of clonally derived parasites. SBRMe49-2 is representative of 30 clonally derived isolates with similar drug susceptibility characteristics, chosen because it was fatal to mice at the same inoculum as parental Me49, making it useful for prior in vivo studies. A rabbit isolate of Encephalitozoon cuniculi (kindly provided by Elizabeth Didier of the Tulane Regional Primate Center) was maintained by continuous passage in a rabbit kidney cell line (RK-13 cells) obtained from the American Type Culture Collection (Manassas, VA) (8).
Mouse infections.
Freshly isolated T. gondii tachyzoites were serially diluted in PBS and used to challenge mice with the indicated inocula in 200 μl sterile PBS by i.p. inoculation using a 27-gauge needle. Encephalitozoon cuniculi was prepared and used to infect susceptible CD8−/− mice by i.p. inoculation with 1 × 107 E. cuniculi spores as described previously (8).
In vitro experiments with T. gondii.
Monolayers of confluent HFFs were infected at an MOI of 1 for RH tachyzoites or an MOI of 5 for Me49 tachyzoites and were cultured in R10 medium (RPMI 1640 supplemented with 4 mM glutamine, 10 mM HEPES, 10% heat-inactivated fetal calf serum, and antibiotics) as described previously (4, 19). Four hours after T. gondii infection, extracellular tachyzoites were washed away and drugs were added to the culture at various concentrations. To quantify T. gondii tachyzoite replication, 1 μCi [3H]uracil (New England Nuclear, Cambridge, MA), which is metabolized by tachyzoites but not by mammalian cells (14), was added 16 h before harvest onto nitrocellulose filter mats (Wallac, Gaithersburg, MD). Subsequently, [3H]uracil incorporation was measured using a Microbeta Trilux liquid scintillation counter (Wallac, Turku, Finland). Proliferation assays were performed in triplicate, and the means and standard errors of the means (SEMs) were calculated. The concentration of the drug reducing the [3H]uracil incorporation into tachyzoites by 50% compared with the level for untreated cultures was defined as the 50% inhibitory concentration (IC50), and its value was estimated by a polynomial equation from dose response data from at least three separate experiments.
Statistical analysis.
Differences in proliferation were assessed using Student's two-tailed t test. Survival was estimated by the Kaplan-Meier method and compared using the log rank test. P values of <0.05 were considered significant.
RESULTS
RWJ67657 and RWJ68198 block T. gondii replication in vitro.
Both RWJ67657 and RWJ68198 inhibited T. gondii replication in human HFFs (Fig. 1A and B), which is consistent with our prior studies using the p38 MAPK inhibitors SB203580 and SB202190 (19). RWJ67657 was at least as potent as RWJ68198, SB203580, or SB202190 in reducing T. gondii replication in vitro (Table 1). Owing to the fact that Me49 tachyzoites have greater sensitivity to these p38 inhibitors than RH tachyzoites (Table 1), we chose the Me49 strain for the selection of SB203580-resistant mutants. Interestingly, SBRMe49-2, one of 30 clonally derived SB203580-resistant strains, was also resistant to pyridinylimidazole p38 MAPK inhibitor RWJ67657 (Fig. 2A and B). Likewise, SBRMe49-2 was resistant to SB202190 (not shown), suggesting a common target of action for these p38 MAPK inhibitors. The control pyridinylimidazole SB202474 had no inhibitory activity against either the SBRMe49-2 strain (Fig. 2B) or the Me49 parental strain (Fig. 2A).
FIG. 1.

RWJ67657 and RWJ68198 inhibit growth of RH or Me49 T. gondii tachyzoites in HFFs. HFFs were infected at an MOI of 1 for strain RH (A) and an MOI of 5 for strain Me49 (B) T. gondii tachyzoites. Four hours after infection, extracellular tachyzoites were washed away and the drug was added. Bars represent treatment with the indicated concentrations of RWJ67657 (black bars) or RWJ68198 (hatched bars). [3H]uracil was added 48 h later, and plates were harvested after further overnight incubation. Results for one representative experiment of three with similar results are shown. Error bars are SEMs of triplicate determinations.
TABLE 1.
Comparison of the p38 MAPK inhibitors used in this study
| p38 MAPK inhibitor | IC50 (μM)a for:
|
|||
|---|---|---|---|---|
| T. gondii RHb | T. gondii Me49b | Recombinant human p38αc | Recombinant TgMAPK-1c | |
| SB202190 | 5.0 | 1.0 | 0.3 | ND |
| SB203580 | 8.5 | 2.5 | 7.0 | 0.1 |
| RWJ67657 | 5.0 | 0.8 | 1.0 | 0.2d |
| RWJ68198 | 8.6 | 2.0 | ND | ND |
ND, not determined.
Confluent HFFs were infected with T. gondii strain RH tachyzoites (MOI = 1) or strain Me49 tachyzoites (MOI = 5). Four hours after infection, extracellular tachyzoites were washed away and drugs were added to cultures at concentrations ranging from 0.01 μM to 10 μM. [3H]uracil was added 48 h later, and plates were harvested after overnight incubation. The IC50s for inhibition of T. gondii replication are shown and were calculated according to a polynomial equation based on the results of at least three experiments.
The IC50 values for inhibition of kinase activity were determined using purified recombinant human p38α (1, 17) and TgMAPK-1 (3) treated with the indicated p38 MAPK inhibitors.
M. J. Brumlik, unpublished data.
FIG. 2.

RWJ67657 or SB203580 fails to inhibit the growth of SB203580-resistant T. gondii strain Me49 tachyzoites cultured in HFFs. HFFs were infected at an MOI of 5 with either wild-type T. gondii strain Me49 tachyzoites (A) or SBRMe49-2, a clonally derived Me49 strain tachyzoite that is resistant to SB203580 (B). Tachyzoites were treated with RWJ67657 (black bars), SB203580 (hatched bars), or the control, inactive pyridinylimidazole SB202474 (white bars), at the indicated concentrations. Error bars are SEMs of triplicate determinations.
RWJ67657 treats T. gondii infection in mice.
Although the RH (type I) and Me49 (type II) strains of T. gondii are virtually identical at the genetic level (16), there is an enormous difference in the virulence of these strains in mice (15). The lethal dose required to kill one-half of the animals exposed in mice is greater than 1,000 Me49 tachyzoites, whereas infection with a single RH tachyzoite is invariably fatal (15). Therefore, the more virulent RH strain was selected as a stringent test of the potential for p38 MAPK inhibitors to protect T. gondii-infected mice.
Groups of five CBA/J mice were infected with 1,000 RH tachyzoites and treated with RWJ67657 24 h after infection with 3.8, 7.5, 15, 30, or 60 mg/kg for seven consecutive days by i.p. injection. The highest dose (60 mg/kg) significantly improved survival (Fig. 3), whereas all lower doses proved ineffective. We then tested less stringent infection conditions by challenging groups of five CBA/J mice each with a 10-fold-lower inoculum level (100 RH tachyzoites) and treating them daily for 7 days with RWJ67657, starting 2 h after infection. Treatment with RWJ67657 resulted in 40% survival when 60 mg/kg RWJ67657 was initiated within 2 h of infection. All other mice died by day 17 (Fig. 4). Using a challenge of 20 RH tachyzoites (which is invariably fatal in CBA/J mice without therapeutic intervention) followed by treatment with RWJ67657 at either 60 mg/kg or 30 mg/kg resulted in 100% survival among CBA/J mice (Fig. 5). Oral treatments were completely ineffective, even at this comparatively low inoculum level (Fig. 5). Mice treated with RWJ67657 showed no evidence of drug toxicity except for occasional, transient fur ruffling at the 60-mg/kg dose.
FIG. 3.

RWJ67657 treatment prolongs survival of mice infected with high inoculum levels of RH strain T. gondii tachyzoites. Mice (five/group) were infected with 1,000 RH strain T. gondii tachyzoites (administered i.p. in sterile PBS) and were either sham treated 24 h after infection with sterile DMSO (□) or treated with RWJ67657 (administered i.p. using 50 μl of sterile DMSO for seven consecutive days) at the following doses: 3.8 mg/kg (○), 7.5 mg/kg (▴), 15 mg/kg (⋄), 30 mg/kg (•), or 60 mg/kg (▪).
FIG. 4.

RWJ67657 treatment enhances survival and cures some mice infected with low inoculum levels of RH strain T. gondii tachyzoites. Mice (five/group) were infected with 100 RH strain T. gondii tachyzoites (administered i.p. in sterile PBS) and treated with RWJ67657 (administered i.p. using 50 μl of sterile DMSO for seven consecutive days) starting 2 h after infection at a daily dose of 60 mg/kg once daily (▪) or 30 mg/kg twice daily (▴). Alternatively, mice were infected with 100 RH tachyzoites (administered i.p. in sterile PBS) and treated 24 h later with RWJ67657 (administered i.p. using 50 μl of sterile DMSO for seven consecutive days) at 60 mg/kg once daily (□) or 30 mg/kg twice daily (▵). Surviving mice were rechallenged with 2,000 RH strain T. gondii tachyzoites 30 days after initial infection (arrow).
FIG. 5.

Oral RWJ67657 treatment is ineffective in treating T. gondii RH strain infection. Mice (five/group) were infected with 20 RH strain T. gondii tachyzoites (administered i.p. in sterile PBS), followed by treatment 2 h later with either 60 mg/kg RWJ67657 (□) or 30 mg/kg RWJ67657 (▵), each of which was administered in 50 μl of sterile DMSO for seven consecutive days. Alternatively, 50 mg/kg RWJ67657 (○) or 17 mg/kg RWJ67657 (⋄) was administered by oral gavage (in 50 μl of sterile DMSO) or mice were sham treated with sterile DMSO by i.p. injection (⧫) for seven consecutive days. Surviving mice were rechallenged with 2,000 RH strain T. gondii tachyzoites 30 days after initial infection (arrow).
RWJ67657 treats Encephalitozoon cuniculi infection in mice.
We hypothesized that RWJ67657 and additional p38 MAPK inhibitors might be useful in treating other parasitic infections, based on recent genomic data that suggest the presence of MAPK homologues in all eukaryotic parasites (9). CD8−/− mice were infected with Encephalitozoon cuniculi spores and treated with either 36 mg/kg RWJ67657 or 6 mg/kg RWJ67657 or were sham treated with DMSO, starting on the second day after infection and continuing for 15 consecutive days thereafter. All mice treated at 36 mg/kg survived the infection (Fig. 6) and had no evidence of infection, as determined by absence of ascites fluid and sluggishness.
FIG. 6.
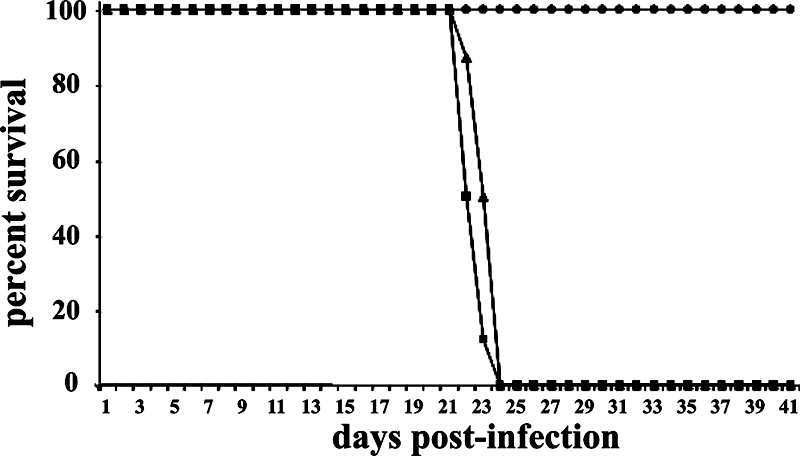
RWJ67657 treatment enhances survival of mice infected with a lethal dose of E. cuniculi. Groups of six CD8−/− CJ57BL/6 mice were infected with 1 × 107 E. cuniculi spores (administered i.p.), followed 2 days later by administration (i.p. in 50 μl of sterile DMSO) of 36 mg/kg RWJ67657 (•) or 6 mg/kg of RWJ67657 (▴) or sham treatment with sterile DMSO by i.p. injection (▪) for 15 consecutive days.
p38 MAPK inhibitors augment the activities of approved anti-Toxoplasma drugs in vivo.
Groups of 10 CBA/J mice each infected with 100 RH tachyzoites were treated with RWJ67657 (60 mg/kg i.p.) either alone or in combination with pyri methamine. Treatment started 2 h after infection and continued for 7 days. RWJ67657 treatment alone resulted in 40% survival. Pyrimethamine treatment alone resulted in only 40% survival. By contrast, the combination of both drugs protected 80% of the mice from lethal T. gondii infection (Fig. 7). Similar results were obtained using 60 mg/kg RWJ67657 in combination with sulfadiazine at 75 mg/liter (not shown).
FIG. 7.

Pyrimethamine additively augments the anti-Toxoplasma activity of RWJ67657. Mice (10/group) were infected with 100 RH strain T. gondii tachyzoites (administered i.p in sterile PBS), followed by treatment 2 h later with 60 mg/kg RWJ67657 alone (administered i.p. in 50 μl of sterile DMSO) (•), 60 mg/kg RWJ67657 (administered i.p.) plus 5 mg/kg pyrimethamine (administered by oral gavage in 200 μl of 0.25% CMC suspended in PBS) (▪), or 5 mg/kg pyrimethamine alone (administered as described above by oral gavage) (▵) or sham treatment with i.p. injection of 50 μl of sterile DMSO (□). Surviving mice were rechallenged with 2,000 RH strain T. gondii tachyzoites 30 days after initial infection (arrow).
Infected mice protected by drug treatments were immune to subsequent T. gondii infection.
p38 MAPK activation is important in cellular immunity (7), raising a concern that these agents could induce immunosuppression. To assess this possibility, mice surviving T. gondii infection following p38 MAPK inhibitor treatment were challenged with 2,000 RH T. gondii tachyzoites 30 days after initial infection. Five previously uninfected mice as well as five mice previously uninfected and treated with RWJ67657 were infected with 2,000 RH T. gondii tachyzoites as controls. All mice surviving treatment with any of the p38 MAPK inhibitor drugs used at any dose (n = 15) survived 30 days after the second T. gondii challenge (Fig. 4, 5, and 7), at which time they were euthanized. By contrast, all control mice (n = 10) died on day 8 or 9 following infection. These data demonstrate that p38 MAPK inhibitors at clinically relevant doses do not prevent generation of protective T. gondii-specific immunity.
DISCUSSION
Our previous work (18) demonstrated that the pyridinylimidazole p38 MAPK inhibitors SB203580 and SB202190 inhibited intracellular T. gondii tachyzoite replication in vitro. Here, we extend this observation to demonstrate that the pyridinylimidazole p38 MAPK inhibitors RWJ67657 and RWJ64809 (which is chemically identical to SB203580) and the imidazopyrimidine p38 MAPK inhibitor RWJ68198 also inhibit T. gondii replication in vitro. Agents inhibiting human ERK1 or ERK2 or Jun N-terminal protein kinase MAPK did not significantly inhibit T. gondii tachyzoite replication under these conditions, suggesting that these agents act on a parasite homologue of p38 MAPK (18). The cross-resistance of SB203580-resistant tachyzoite to RWJ67657 suggests that these agents may act on the same target to mediate antiparasite effects.
Our previous work suggests that the tachyzoite antiproliferative effects of these agents depend on direct action on tachyzoites and not the host p38 MAPK (18). Differences in IC50 among the various agents tested suggest differential affinities for these drugs against a common target, although action against distinct targets is not excluded. We hypothesize that these drugs affect a p38 MAPK homologue in tachyzoites that regulates their replication. In support of this hypothesis, we recently cloned the tgMAPK-1 gene, encoding a p38 MAPK homologue in T. gondii (3). Activation of the recombinant TgMAPK-1 protein expressed and purified from Escherichia coli is inhibited by SB203580 (3) and RWJ67657 (unpublished observations) in vitro, consistent with the concept that this gene product is a target of action of these p38 MAPK inhibitor drugs.
We now further demonstrate the in vivo efficacies of RWJ67657 and RWJ68198 for treating T. gondii infection in mice. Protection was observed only at high drug doses given soon after infection and directed against low levels of infectious inocula. Further, oral drug treatment was entirely ineffective even against the lowest inoculum level. Higher oral doses were not studied here, owing to their known toxicities and given the generally poor oral bioavailability of these agents (13). Nonetheless, the important concept that agents designed to inhibit human p38 MAPK may be useful for treating T. gondii infection is established. Because T. gondii MAPKs have active sites distinct from human homologues (10), it may be possible to develop more-selective and more-potent agents to inhibit T. gondii MAPK inhibition. If the targets of these drugs ultimately prove to be distinct from MAPKs, more-selective and more-potent agents can still be developed.
Pyrimethamine and sulfadiazine are currently the mainstays for treating human toxoplasmosis (5, 12). However, they are not always efficacious and are frequently discontinued due to intolerable toxicities (12). Our studies show that RWJ67657 and related agents may be useful when combined with approved anti-Toxoplasma agents to effect comparable efficacy with reduced toxicity. Additive and nonsynergistic drug effects suggest that the pathways affected by p38 MAPK inhibitors and the folate metabolic pathways inhibited by pyrimethamine and sulfadiazine (5) do not overlap significantly.
An important consideration in using p38 MAPK inhibitors is the potential for immunosuppression or immune dysfunction (7) at the doses that we studied (2, 17, 18). Our data demonstrate that protective immunity against T. gondii (a biologically meaningful endpoint) was generated in vivo despite p38 MAPK inhibitor treatment, suggesting that significant immunosuppression is not a clinically significant issue with this approach. Development of Toxoplasma-specific agents will further help in this regard.
Several lines of evidence suggest that pyridinylimidazole agents can be developed as antiparasitic agents and have the potential to treat a variety of human pathogens. We showed here that RWJ67657 was also effective for treating Encephalitozoon cuniculi infection in vivo. Genetic deletion of an extracellular signal-regulated kinase-like MAPK gene in Leishmania mexicana blocked stage-specific intracellular parasite proliferation (20). MAPK homologues are found in Plasmodium falciparum and other medically important human parasites (10).
Thus, parasite MAPKs represent important drug discovery targets, as has also been suggested elsewhere (6). In this regard, MAPKs in T. gondii and other parasites differ significantly from human homologues with respect to their active sites (10), suggesting that selective agents for inhibiting parasite MAPKs can be developed. Our data suggest that RWJ67657 defines a novel class of agents that may be useful for treating human toxoplasmosis. Clinical translation could be facilitated given the prior use of this agent in humans (13). Nonetheless, additional modifications for boosting efficacy and oral bioavailability are required for optimal use in humans. Defining the specific target of these agents in T. gondii (and other parasites) will allow development of more-selective agents.
Acknowledgments
This research was supported by R01 AI060424 and a Focused Funding grant from Johnson & Johnson (both to T.J.C.) and by R01 AI43963 (to I.A.K.).
Thanks to Suzanne Thibodeaux, Pete Mottram, and Tracey Todd for technical assistance.
Footnotes
Published ahead of print on 8 October 2007.
REFERENCES
- 1.Adachi, T., B. K. Choudhury, S. Stafford, S. Sur, and R. Alam. 2000. The differential role of extracellular signal-regulated kinases and p38 mitogen-activated protein kinase in eosinophil functions. J. Immunol. 165:2198-2204. [DOI] [PubMed] [Google Scholar]
- 2.Boehm, J. C., J. M. Smietana, M. E. Sorenson, R. S. Garigipati, T. F. Gallagher, P. L. Sheldrake, J. Bradbeer, A. M. Badger, J. T. Laydon, J. C. Lee, L. M. Hillegass, D. E. Griswold, J. J. Breton, M. C. Chabot-Fletcher, and J. L. Adams. 1996. 1-Substituted 4-aryl-5-pyridinylimidazoles: a new class of cytokine suppressive drugs with low 5-lipoxygenase and cyclooxygenase inhibitory potency. J. Med. Chem. 39:3929-3937. [DOI] [PubMed] [Google Scholar]
- 3.Brumlik, M. J., S. Wei, K. Finstad, J. Nesbit, L. E. Hyman, M. Lacey, M. E. Burow, and T. J. Curiel. 2004. Identification of a novel mitogen-activated protein kinase in Toxoplasma gondii. Int. J. Parasitol. 34:1245-1254. [DOI] [PubMed] [Google Scholar]
- 4.Curiel, T. J., E. C. Krug, M. B. Purner, P. Poignard, and R. L. Berens. 1993. Cloned human CD4+ cytotoxic T lymphocytes specific for Toxoplasma gondii lyse tachyzoite-infected target cells. J. Immunol. 151:2024-2031. [PubMed] [Google Scholar]
- 5.Derouin, F. 2001. Anti-toxoplasmosis drugs. Curr. Opin. Investig. Drugs 2:1368-1374. [PubMed] [Google Scholar]
- 6.Doerig, C. 2004. Protein kinases as targets for anti-parasitic chemotherapy. Biochim. Biophys. Acta 1697:155-168. [DOI] [PubMed] [Google Scholar]
- 7.Dong, C., R. J. Davis, and R. A. Flavell. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55-72. [DOI] [PubMed] [Google Scholar]
- 8.Khan, I. A., J. D. Schwartzman, L. H. Kasper, and M. Moretto. 1999. CD8+ CTLs are essential for protective immunity against Encephalitozoon cuniculi infection. J. Immunol. 162:6086-6091. [PubMed] [Google Scholar]
- 9.Kultz, D. 1998. Phylogenetic and functional classification of mitogen- and stress-activated protein kinases. J. Mol. Evol. 46:571-588. [DOI] [PubMed] [Google Scholar]
- 10.Lacey, M. R., M. J. Brumlik, R. E. Yenni, M. E. Burow, and T. J. Curiel. 2007. Toxoplasma gondii expresses two mitogen-activated protein kinase genes that represent distinct protozoan subfamilies. J. Mol. Evol. 64:4-14. [DOI] [PubMed] [Google Scholar]
- 11.Luft, B. J., and J. S. Remington. 1992. Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 15:211-222. [DOI] [PubMed] [Google Scholar]
- 12.Montoya, J. G., and O. Liesenfeld. 2004. Toxoplasmosis. Lancet 363:1965-1976. [DOI] [PubMed] [Google Scholar]
- 13.Parasrampuria, D. A., P. de Boer, D. Desai-Krieger, A. T. Chow, and C. R. Jones. 2003. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J. Clin. Pharmacol. 43:406-413. [DOI] [PubMed] [Google Scholar]
- 14.Pfefferkorn, E. R., and L. C. Pfefferkorn. 1977. Specific labeling of intracellular Toxoplasma gondii with uracil. J. Protozool. 24:449-453. [DOI] [PubMed] [Google Scholar]
- 15.Saeij, J. P., J. P. Boyle, and J. C. Boothroyd. 2005. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 21:476-481. [DOI] [PubMed] [Google Scholar]
- 16.Su, C., D. Evans, R. H. Cole, J. C. Kissinger, J. W. Ajioka, and L. D. Sibley. 2003. Recent expansion of Toxoplasma through enhanced oral transmission. Science 299:414-416. [DOI] [PubMed] [Google Scholar]
- 17.Wadsworth, S. A., D. E. Cavender, S. A. Beers, P. Lalan, P. H. Schafer, E. A. Malloy, W. Wu, B. Fahmy, G. C. Olini, J. E. Davis, J. L. Pellegrino-Gensey, M. P. Wachter, and J. J. Siekierka. 1999. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 291:680-687. [PubMed] [Google Scholar]
- 18.Wei, S., F. Marches, J. Borvak, W. Zou, J. Channon, M. White, J. Radke, M. F. Cesbron-Delauw, and T. J. Curiel. 2002. Toxoplasma gondii-infected human myeloid dendritic cells induce T-lymphocyte dysfunction and contact-dependent apoptosis. Infect. Immun. 70:1750-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei, S., F. Marches, B. Daniel, S. Sonda, K. Heidenreich, and T. Curiel. 2002. Pyridinylimidazole p38 mitogen-activated protein kinase inhibitors block intracellular Toxoplasma gondii replication. Int. J. Parasitol. 32:969-977. [DOI] [PubMed] [Google Scholar]
- 20.Wiese, M. 1998. A mitogen-activated protein (MAP) kinase homologue of Leishmania mexicana is essential for parasite survival in the infected host. EMBO J. 17:2619-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]