

Abstract
Treatment with a low dose of combined aspirin and clopidogrel, two antiplatelet drugs widely used in humans, markedly reduced the homing of virus-specific cytotoxic T lymphocytes and virus-nonspecific inflammatory leukocytes to the liver of mice acutely infected with a hepatotropic, replication-deficient, lacZ-expressing adenovirus (RAd35). Consequently, aspirin/clopidogrel-induced platelet dysfunction greatly diminished liver disease severity and inhibited viral clearance. Along with the finding that aspirin/clopidogrel caused neither bleeding nor anemia, our results suggest that antiplatelet drugs may be considered to limit excessive liver immunopathology and/or to facilitate the persistence of hepatotropic viral vectors utilized in gene therapy.
Replication-deficient adenoviruses do not directly cause hepatocyte damage, which is rather a consequence of the cytotoxic T-lymphocyte (CTL) response to viral antigens aimed at viral clearance (1, 11). Direct killing of hepatocytes by CTLs is followed by the recruitment of virus-nonspecific intrahepatic leukocytes (IHLs) that can amplify tissue damage (6). Using mouse models of acute viral hepatitis, we recently showed that platelets play a crucial role in liver immunopathology (5). Indeed, platelets are detectable within intrahepatic CTL-containing necroinflammatory foci, and their depletion greatly ameliorates disease severity by reducing the number (but not the function) of virus-specific CTLs within the liver, which secondarily diminishes IHL recruitment as well (4). These events are restored upon reconstitution of thrombocytopenic animals with normal platelets but not upon reconstitution with platelets treated with prostaglandin E1 (a potent inhibitor of platelet activation) (4). In vitro findings suggest that virus-specific CTLs tightly interact with activated platelets, and again, this process is inhibited when platelets are treated with prostaglandin E1 (4). Thus, the results of in vivo and in vitro studies are in agreement with the assumption that an initial inflammatory response within the liver may result in changes of the vessel wall that promote platelet activation and activation-dependent events resulting in the interaction with CTLs. This interaction may eventually facilitate CTLs to egress from the bloodstream, enter the liver parenchyma, and perform effector functions.
Herein, we used lacZ-immunized mice infected with RAd35 (a replication-deficient, lacZ-expressing adenovirus, which was originally provided by Pedro Lowenstein, Cedars-Sinai Medical Center, Los Angeles, CA [8-10], and grown in our laboratory) to determine whether drugs that interfere with platelet activation, namely, aspirin and clopidogrel, might reproduce the effects of platelet depletion. Aspirin affects thromboxane A2 production by irreversibly inactivating cyclooxygenase-1 (2), while clopidogrel irreversibly inactivates the ADP-receptor P2Y12 (2). We chose this experimental system because it allows monitoring of aspirin/clopidogrel-mediated events that are independent of CTL priming and also quantifying of the intrahepatic number of pathogenetic/antiviral virus-specific CTLs. Indeed, lacZ-immunized control mice develop a severe liver injury that, within 3 to 4 days of RAd35 infection, is entirely mediated by a memory CTL response specific for a single immunodominant epitope (β-Gal96) contained within the LacZ protein (4). This response precedes any other CTL response directed toward non-lacZ epitopes, and no liver disease is detected at these time points in nonimmunized mice infected with RAd35 (4).
Thus, groups of 7- to 8-week-old C57BL/6J male mice (eight mice per group) were immunized with a plasmid expressing lacZ (4) and 2 weeks later received lacZ-expressing vaccinia virus (3) (2 × 106 PFU/mouse by intravenous injection). Three weeks after developing LacZ-expressing vaccinia virus infection and 3 days before being infected with 3 × 109 PFU/mouse of RAd35 (a dose sufficient to infect all hepatocytes [4]), the animals were treated daily with either diluent or aspirin (5 mg/kg of body weight, intraperitoneally; Sigma-Aldrich, St. Louis, MO) and clopidogrel (2 mg/kg by gavage; Bristol-Myers Squibb, New York, NY) until the day before autopsy (i.e., 1, 3, or 5 days after RAd35 infection).
Compared to the RAd35-infected controls treated with diluent and killed 1 day after infection, the aspirin/clopidogrel-treated mice showed a similar percentage of lacZ+ hepatocytes and equal levels of hepatic lacZ DNA and RNA (as analyzed by β-galactosidase histochemistry, Southern, or Northern blotting, respectively, exactly as described in reference 4; data not shown). This indicates that antiplatelet treatment did not affect the capacity of RAd35 to infect the liver.
Compared to uninfected controls treated (Fig. 1A) or not (not shown) with aspirin/clopidogrel (for which we detected no evidence of liver disease throughout the experiment), diluent-treated RAd35-infected mice developed an overwhelming liver disease that, at its peak, was characterized by a marked increase in serum alanine aminotransferase (sALT) (Fig. 1A), a diffuse inflammatory infiltrate, and large numbers of hepatocytes that were either necrotic or displayed prelytic changes (Fig. 1B). Aspirin-clopidogrel drastically reduced the severity of this disease, as assessed by the ∼80% reduction of peak sALT (Fig. 1A; P < 0.01) and the detection of necroinflammatory foci scattered throughout a parenchyma in which the majority of hepatocytes were cytologically normal (Fig. 1C). Flow cytometry analysis of single-cell suspensions prepared from livers perfused by day 3 postinfection (see Materials and Methods in reference 4) revealed that a large number of intrahepatic β-Gal96-specific CTLs and IHLs accumulated within the liver of diluent-treated RAd35-infected mice (Fig. 2A and B). In keeping with the notion that by killing infected hepatocytes, CTLs not only promote liver injury but also mediate viral clearance (1, 11), only traces of lacZ DNA and RNA (not shown) and a small percentage of lacZ+ hepatocytes (Fig. 2C) were detectable in these same livers. Aspirin/clopidogrel caused a ∼70% and 50% reduction of intrahepatic β-Gal96-specific CTLs and IHLs, respectively (Fig. 2A and B; P < 0.01). The extent to which aspirin/clopidogrel reduced the homing of each subset of antigen-nonspecific inflammatory cells was proportional to the total number of IHLs, as the intrahepatic number of NK, NKT, T helper cells, B cells, lymphoid and myeloid dendritic cells, macrophages, and neutrophils decreased between 50% and 60% compared to that of controls (not shown). It is noteworthy that CTL effector functions were unaffected by aspirin/clopidogrel, since the β-Gal96-specific CTLs recovered from the livers of both groups of mice showed the identical capacity to either kill 51Cr-labeled target cells (H2b-restricted mouse lymphoma EL-4 cells pulsed with 10 μg ml−1 of β-Gal96) (Fig. 2E) or produce gamma interferon in vitro by intracellular cytokine staining (see Materials and Methods in reference 4) (data not shown). Thus, the fact that we detected ∼10-fold-higher levels of lacZ DNA and RNA (not shown) and a much higher percentage of lacZ+ hepatocytes in these livers (Fig. 2D) indicates that aspirin/clopidogrel inhibited viral clearance by reducing the number, and not the function, of virus-specific CTLs.
FIG. 1.

Aspirin/clopidogrel (Asp/Clo) treatment diminishes liver disease severity. (A) sALT activity (units/liter means ± standard deviations) was measured at the indicated time points in mice (n = 8) that received either diluent or aspirin/clopidogrel and were infected or not with RAd35. Histological analyses of representative lacZ-immunized C57BL/6J mice treated with diluent (B) or Asp/Clo (C) and sacrificed 3 days after RAd35 infection are shown. (B and C) Scale bars, 150 μm.
FIG. 2.

Aspirin/clopidogrel (Asp/Clo) treatment diminishes the intrahepatic number of CTLs and IHLs and inhibits viral clearance. Absolute numbers (means ± standard deviations) of intrahepatic β-Gal96-specific CTLs (A) and IHLs (B) analyzed 3 days after RAd35 infection are shown. Frozen liver sections from representative mice treated with diluent (C or D) and sacrificed 3 days after RAd35 infection were stained for β-galactosidase activity. (C and D) Scale bars, 150 μm. (E) In vitro cytotoxic activity (means ± standard deviations) of β-Gal96-specific CTLs isolated from the same livers shown in panel A (day 3 after RAd35 infection).
As expected, circulating platelets isolated from aspirin/clopidogrel-treated mice showed an impaired aggregation in response to either arachidonic acid (150 μM) or ADP (8 μM) (Sigma-Aldrich) (see Materials and Methods in reference 4), as opposed to those isolated from diluent-treated infected controls (Fig. 3A). Importantly, none of the mice in each group and at each time point showed either signs of hemorrhage autoptically (not shown) or a reduction in hematocrit (Fig. 3B), indicating that our antiplatelet treatment caused neither bleeding nor anemia.
FIG. 3.
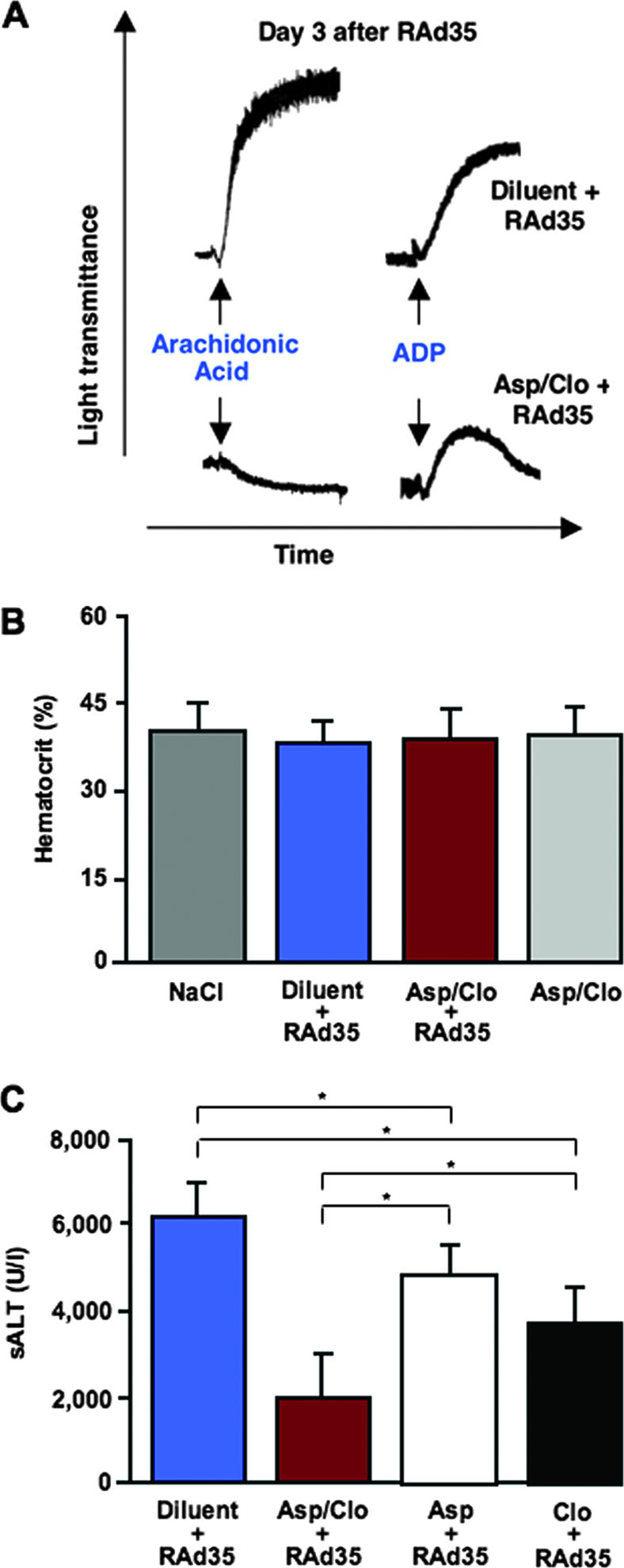
Aspirin/clopidogrel treatment impairs platelet function but causes neither bleeding nor anemia, and treatment is less efficacious when aspirin or clopidogrel is administered alone. (A) Aggregation of platelets isolated from representative diluent- or aspirin/clopidogrel-treated C57BL/6J mice infected 3 days earlier with RAd35 was measured after arachidonic acid or ADP exposure. Note that while the aggregation of platelets from aspirin/clopidogrel-treated mice was completely abrogated in response to arachidonic acid, the aggregation in response to ADP resulted, as expected, in the formation of aggregates that were smaller and unstable (i.e., they rapidly disaggregated). (B) Hematocrit values (means ± standard deviations; n = 8) were assessed individually 3 days after the indicated treatment. (C) sALT (units/liter) measured 3 days after the indicated treatment is expressed as means ± standard deviations; n = 8.
Next, we used our system to evaluate the relative efficacy of single treatments with aspirin or clopidogrel. Compared to lacZ-immunized mice treated with diluent prior to receiving an injection of a lower dose (109 PFU) of RAd35 (still sufficient to infect ∼50% of the hepatocytes; not shown), treatment with aspirin or clopidogrel alone caused an ∼25% and an ∼35% decrease in sALT, respectively, while the combined treatment reduced sALT by ∼70% (Fig. 3C; P < 0.01). Along with the fact that we observed commensurate reductions in the number of β-Gal96-specific CTLs in these livers (not shown), these observations indicate that aspirin and clopidogrel are ∼50% less efficacious when administered alone, in keeping with their capacity to affect independent antiplatelet pathways (2).
Altogether, our findings indicate that aspirin and clopidogrel, particularly when combined and without triggering side effects of bleeding, ameliorated the severity of liver disease and inhibited viral clearance in mice acutely infected with a hepatotropic, replication-deficient adenovirus. Remarkably, aspirin and clopidogrel were given at the low doses that typify the management of patients with thrombotic diseases (4 to 5 mg/kg and 1 to 2 mg/kg, respectively) (2). These doses of aspirin are too low to achieve platelet-nonspecific anti-inflammatory or analgesic/antipyretic effects (2), and additionally, they did not affect CTL function in our system. The initiation of the drug regimen (3 days before RAd35 infection) was chosen because the maximum effect of clopidogrel is achieved after approximately 3 days of daily treatment, reflecting the time needed for the liver to produce efficacious concentrations of the active metabolite (2). Note that the retarded bioavailability of clopidogrel together with the rapidity with which liver disease severity peaked in the animals (3 days after RAd35 infection) rendered a test unfeasible to determine whether this drug (alone or in combination with aspirin) is efficacious when administered after infection. The withdrawal of the drug regimen (1 day before autopsy) was chosen because the antiplatelet effect of aspirin and/or clopidogrel is still maximal 1 day after the last administration, since the irreversible effects of these drugs lasted several days, similar to the life span of human (7 to 10 days) or mouse (4 to 6 days) platelets (7).
The effects of aspirin/clopidogrel treatment are remarkably consistent with those observed for RAd35-infected mice depleted of platelets, demonstrating that platelets require a normal response to thromboxane A2 and ADP in order to promote CTL-dependent liver immunopathology and viral clearance. Notably, the role of platelets in this system is independent of their procoagulant activity, since RAd35-infected mice treated with the oral anticoagulant warfarin show no differences (compared to controls) in the homing and pathogenetic/antiviral capacities of intrahepatic CTLs, despite the very prolonged blood clotting time (4).
In conclusion, we found that antiplatelet therapy diminishes immune-mediated liver disease in a model of acute viral hepatitis. In addition to further dissecting the molecular mechanisms whereby platelets facilitate CTL effector functions in vivo, these observations may help to devise new approaches to limit excessive liver immunopathology (as it occurs during fulminant viral hepatitis in humans) or delay viral clearance (as demanded by gene therapy-based procedures with these and other hepatotropic viral vectors).
Acknowledgments
This work was supported by grants HL31950, HL42846, HL78784 (Z.M.R.) and AI40696 (L.G.G.) from the National Institutes of Health and from the VIRGIL European Network of Excellence on Antiviral Drug Resistance (LSHM-CT-2004-503359) (L.G.G.). The authors declare they have no competing financial interests.
This is manuscript number MEM-18814 from the Scripps Research Institute.
The experiments described in this report were performed according to federal guidelines and approved by the Animal Research Committee of the Scripps Research Institute.
Footnotes
Published ahead of print on 19 September 2007.
REFERENCES
- 1.Abougergi, M. S., S. J. Gidner, D. K. Spady, B. C. Miller, and D. L. Thiele. 2005. Fas and TNFR1, but not cytolytic granule-dependent mechanisms, mediate clearance of murine liver adenoviral infection. Hepatology 41:97-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cattaneo, M. 2004. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler. Thromb. Vasc. Biol. 24:1980-1987. [DOI] [PubMed] [Google Scholar]
- 3.Chakrabarti, S., K. Brechling, and B. Moss. 1985. Vaccinia virus expression vector: coexpression of β-galactosidase provides visual screening of recombinant virus plaques. Mol. Cell. Biol. 5:3403-3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iannacone, M., G. Sitia, M. Isogawa, P. Marchese, M. G. Castro, P. R. Lowenstein, F. V. Chisari, Z. M. Ruggeri, and L. G. Guidotti. 2005. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 11:1167-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iannacone, M., G. Sitia, Z. M. Ruggeri, and L. G. Guidotti. 2007. HBV pathogenesis in animal models: recent advances on the role of platelets. J. Hepatol. 46:719-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kakimi, K., T. E. Lane, S. Wieland, V. C. Asensio, I. L. Campbell, F. V. Chisari, and L. G. Guidotti. 2001. Blocking chemokine responsive to gamma-2/interferon (IFN)-gamma inducible protein and monokine induced by IFN-gamma activity in vivo reduces the pathogenetic but not the antiviral potential of hepatitis B virus-specific cytotoxic T lymphocytes. J. Exp. Med. 194:1755-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt, A., J. Guichard, J. M. Masse, N. Debili, and E. M. Cramer. 2001. Of mice and men: comparison of the ultrastructure of megakaryocytes and platelets. Exp. Hematol. 29:1295-1302. [DOI] [PubMed] [Google Scholar]
- 8.Shering, A. F., D. Bain, K. Stewart, A. L. Epstein, M. G. Castro, G. W. Wilkinson, and P. R. Lowenstein. 1997. Cell type-specific expression in brain cell cultures from a short human cytomegalovirus major immediate early promoter depends on whether it is inserted into herpesvirus or adenovirus vectors. J. Gen. Virol. 78:445-459. [DOI] [PubMed] [Google Scholar]
- 9.Southgate, T. D., P. A. Kingston, and M. G. Castro. 2001. Gene transfer into neural cells in vitro using adenoviral vectors, p. 4.23.1-4.23.40. In J. N. Crawley, C. R. Gerfen, R. McKay, M. A. Rogawski, D. R. Sibley, and P. Skolnick (ed.), Current protocols in neuroscience. John Wiley & Sons, Inc., Hoboken, NJ. [DOI] [PubMed]
- 10.Thomas, C. E., E. Abordo-Adesida, T. C. Maleniak, D. Stone, G. Gerdes, and P. R. Lowenstein. 2001. Critical parameters for gene transfer into the brain using adenoviral vectors, p. 4.24.1-4.24.36. In J. N. Crawley, C. R. Gerfen, R. McKay, M. A. Rogawski, D. R. Sibley, and P. Skolnick (ed.), Current protocols in neuroscience. John Wiley & Sons, Hoboken, NJ. [DOI] [PubMed]
- 11.Wilson, J. M. 2001. Adenovirus-mediated gene transfer to liver. Adv. Drug Deliv. Rev. 46:205-209. [DOI] [PubMed] [Google Scholar]