

Abstract
A prolipase from Rhizopus oryzae (proROL) was engineered in order to increase its stability toward lipid oxidation products such as aldehydes with the aim of improving its performance in oleochemical industries. Out of 22 amino acid residues (15 Lys and 7 His) prone to react with aldehydes, 6 Lys and all His residues (except for the catalytic histidine) were chosen and subjected to saturation mutagenesis. In order to quickly and reliably identify stability mutants within the resulting libraries, active variants were prescreened by an activity staining method on agar plates. Active mutants were expressed in Escherichia coli Origami in a 96-well microtiterplate format, and a stability test using octanal as a model deactivating agent was performed. The most stable histidine mutant (H201S) conferred a stability increase of 60%, which was further enhanced to 100% by combination with a lysine mutant (H201S/K168I). This increase in stability was also confirmed for other aldehydes. Interestingly, the mutations did not affect specific activity, as this was still similar to the wild-type enzyme.
In the last several years, a growing number of industrial applications have required the use of lipases in the presence of partly oxidized triglycerides. Biodiesel (fatty acid monoalkyl esters) has attracted considerable attention during the past decade as a renewable, biodegradable, and nontoxic fuel (36). It is produced from various vegetable oils and has a viscosity close to that of diesel, but it is derived from triglycerides by transesterification with alcohol. Recently, enzymatic transesterification using lipases has become more attractive for biodiesel production, since the glycerol produced as a by-product can easily be recovered, and the purification of fatty methyl esters is simple to accomplish. Owing to the higher price of biodiesel relative to petroleum fuel, waste triglycerides (e.g., frying oils) are often used as cheaper feedstock for the biodiesel synthesis (6, 8, 26), thus helping also to reduce the cost of wastewater treatment in sewage systems and generally assisting in the recycling of resources (12, 19, 23, 24).
With enzymatic technology it is also possible to produce new fats according to specific requirements. Special triglycerides of the ABA-type containing medium-chain fatty acids (e.g., C8) in the sn-1,3 positions and long-chain unsaturated fatty acids (e.g., C16 to C22) in the sn-2 position are effective energy sources for patients with malabsorption, e.g., pancreatic insufficiency. Polyunsaturated fatty acids like eicosapentaenoic acid (C20:5) and docosohexaenoic acid (DHA; C22:6) have been reported to have several advantages compared to conventional fatty acids, such as reduction of blood pressure and plasma triglyceride levels and control of overactive immune functions. DHA is recognized as being important for brain and eye development in infants. Though crude triglycerides can be directly added to commercial products, a growing industrial interest is devoted to eicosapentaenoic acid/DHA-enriched triglycerides. Fish oils are the best-exploited natural source of ω-3 fatty acids and are often used as starting material to produce fats and oils with high nutritional value (19, 32, 33).
Unfortunately, a serious limitation in lipase utilization in oleochemical industries is represented by their poor stability in the presence of commercial triglycerides. In particular, the products generated by the oxidation of triglycerides strongly affect enzyme stability (28). During storage and use, oils can easily be subjected to conditions that promote oxidation of their components. The process of lipid peroxidation is a free radical-mediated deterioration of fatty acids in the presence of air, and the fatty acid composition of a fatty material is an important factor influencing oxidation: the higher the concentration of unsaturated components, the lower the stability against oxidation (24). During lipid peroxidation, hydroperoxides are formed, which then undergo decomposition and yield fatty acyloxy free radicals and hydroxyl radicals. Subsequently, a wide variety of secondary lipid peroxidation products are formed, including aldehydes, ketones, and other carbonyl-containing compounds (11). Among all carbonyl compounds produced, aldehydes were shown to decrease lipase stability to the greatest extent (28). The mechanism of aldehyde formation during the decomposition of lipid hydroperoxides involves primarily the homolytic cleavage of carbon-carbon bonds of lipid acyloxy radicals. The main aldehydes found in peroxidizing biological samples are hexanal, malonaldehyde, propanal, and 4-hydroxynonenal. Minor amounts of many other carbonyls are also generated, such as acrolein, trans-2 nonenal, trans-2 heptenal, pentenal, octanal, and butanal (10, 12).
The interaction of an aldehyde with an enzyme involves specific residues of the protein, such as cysteine, lysine, and histidine, typically via covalent modifications involving an attack by nucleophilic amino acids on the unsaturated β-carbon, e.g., the sulfhydryl group of cysteine, imidazole group of histidine, and the ɛ-amino group of lysine (2, 23, 37). A cross-linking action among proteins causing their polymerization has been also reported (5, 35). The protein activity decay of the polymers is related to the alteration of functional groups directly affecting enzymatic activity but, of course, also to a restriction for substrate diffusion.
Microbial lipases, such as lipases from the genus Rhizopus, are particularly attractive as potential catalysts in lipid modification processes (1, 20), especially as they exhibit an usual sn1,3-regiospecificity required for the synthesis of structured triglycerides. The lipase from Rhizopus oryzae (ROL) serving as model enzyme in this study is initially synthesized as a pre-proenzyme (17), and the role of the pre- and prosequences has already been deeply investigated (2, 33, 34, 36) using heterologous expression in Escherichia coli and Saccharomyces cerevisiae. Recently, we could reduce the problem of inclusion body formation in E. coli using the strain Origami (7), thus creating the molecular basis for the investigation of ROL stability in the presence of aldehydes. As the prolipase (proROL) is produced more efficiently, the protein engineering studies described here were performed using this enzyme. The key strategy to improve enzyme stability was the identification of amino acid residues potentially prone to the interaction with aldehydes as derived from the three-dimensional structure of ROL, followed by saturation mutagenesis. For the identification of more stable—but still highly active variants—a microtiterplate (MTP)-based assay was designed to determine inactivation kinetics.
MATERIALS AND METHODS
Materials.
Octanal, heptenal, hexanal, malonaldehyde bis-(diethylacetal) (MDA), and acrolein were purchased from Sigma. 4-Hydroxynonenal (HNE) was provided by Cayman Chemical, as a 10-mg/ml solution in ethanol. All chemicals used were of analytical grade.
Lipase origin.
The lipase utilized comes from the fungus R. oryzae. The gene coding directly for the proform of this lipase, cloned in pET11-d(+), was kindly provided by M. Haas (U.S. Department of Agriculture, Eastern Regional Research Center, ARS, Wyndmoor, PA).
Bacterial strains, plasmids, and growth conditions.
E. coli strain DH5α [λ− φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1] was used as host for genetic manipulation of plasmids. E. coli Origami (DE3) [Δara-leu7697 ΔlacX74 ΔphoAPvuII phoR araD139 ahpC galE galK rpsL (Smr)4F1[lac+(lacIq) pro] gor522::Tn10 (Tcr) trxB::kan (DE3)] strain was used for the functional expression of proteins. E. coli was grown in Luria-Bertani (LB) (17) medium containing 100 mg/liter ampicillin, 30 mg/liter kanamycin, and 10 mg/liter tetracycline, as required. The construct pET22-proROL has been previously described (7).
Mutagenesis strategy.
Site-directed saturation mutagenesis of specific residues of the target protein was performed on pET22-proROL according to the Quikchange protocol (Stratagene). The primers designed to introduce the desired mutations in the protein are given in Table 1.
TABLE 1.
Primers used for mutations in this study
| Primera | Sequenceb |
|---|---|
| Primers for His mutations | |
| His201 FW | CCACCGGTATCCCTTTCCAACGTACCGTTNNNAAGAGAGATATCGTTCC |
| His201 RV | GGAACGATATCTCTCTTNNNAACGGTACGTTGGAAAGGGATACCGGTGG |
| His144 FW | CTTATAAGGTCATCGTTACCGGTNNNTCACTCGGTGGTGCACAAGC |
| His144 RV | GCTTGTGCACCACCGAGTGANNNACCGGTAACGATGACCTTATAAG |
| His218 FW | CCTCAATCCTTCGGATTCCTTNNNCCCGGTGTTGAATCTTGG |
| His218 RV | CCAAGATTCAACACCGGGNNNAAGGAATCCGAAGGATTGAGG |
| His134 FW | CTGTCGTCCAAGAACAATTGACCGCCNNNCCTACTTATAAGGTCATCG |
| His134 RV | CGATGACCTTATAAGTAGGNNNGGCGGTCAATTGTTCTTGGACGACAG |
| His109 FW | CTGTCAAGGGCGCCAAAGTTNNNGCTGGTTTCCTTTCCTCTTATG |
| His109 RV | CATAAGAGGAAAGGAAACCAGCNNNAACTTTGGCGCCCTTGACAG |
| His208 FW | CCGTTCACAAGAGAGATATCGTTCCTNNNGTTCCTCCTCAATCCTTCG |
| His208 RV | CGAAGGATTGAGGAGGAACNNNAGGAACGATATCTCTCTTGTGAACGG |
| Primers for Lys mutations | |
| Lys5 FW | CAGCGCCTCTGATGGTGGTNNNGTTGTTGCTGCTACTACTGC |
| Lys5 RV | GCAGTAGTAGCAGCAACAACNNNACCACCATCAGAGGCGCTG |
| Lys37 FW | GTCGTTCTGTTGTCCCTGGTAACNNNTGGGATTGTGTCCAATGTC |
| Lys37 RV | GACATTGGACACAATCCCANNNGTTACCAGGGACAACAGAACGAC |
| Lys45 FW | GGGATTGTGTCCAATGTCAANNNTGGGTTCCTGATGGCAAG |
| Lys45 RV | CTTGCCATCAGGAACCCANNNTTGACATTGGACACAATCCC |
| Lys101 FW | CGTCTTCAACTTTTCTGACTACNNNCCTGTCAAGGGCGCCAAAGTTC |
| Lys101 RV | GAACTTTGGCGCCCTTGACAGGNNNGTAGTCAGAAAAGTTGAAGACG |
| Lys104 FW | CTTTTCTGACTACAAGCCTGTCNNNGGCGCCAAAGTTCATGCTG |
| Lys104 RV | CAGCATGAACTTTGGCGCCNNNGACAGGCTTGTAGTCAGAAAAG |
| Lys168 FW | ACGTGAACCAAGATTGTCTCCCNNNAATTTGAGCATCTTCACTGTCG |
| Lys168 RV | ACGTGAACCAAGATTGTCTCCCNNNAATTTGAGCATCTTCACTGTCG |
| Lys 202 FW | CCCTTTCCAACGTACCGTTCACNNNAGAGATATCGTTCCTCACGTTC |
| Lys202 RV | GAACGTGAGGAACGATATCTCTNNNGTGAACGGTACGTTGGAAAGGG |
| Primers for H201S/K168X double mutations | |
| H201S FW | CCACCGGTATCCCTTTCCAACGTACCGTTTCGAAGAGAGATATCGTTCC |
| H201S RV | GGAACGATATCTCTCTTCGAAACGGTACGTTGGAAAGGGATACCGGTGG |
FW, forward; RV, reverse.
Underlining in the primers for the double mutations indicates the codon for the introduced Ser residue.
Library generation in E. coli Origami. (i) Protocol I.
Two hundred E. coli DH5α colonies containing the mutated plasmid were picked, resuspended in 200 μl of LB medium containing ampicillin, and grown in 96-well MTPs for 24 h at 37°C. Purification in 96-well MTPs of the mutated plasmid DNA was performed with a DirectPrep 96 kit from QIAGEN (Hilden, Germany). Using a high-throughput transformation procedure, the previously purified plasmid DNA was added to PCR tubes containing 25 μl of E. coli Origami competent cells. The tubes were chilled and then subjected to heat shock according to standard protocols (18) but performed on a high-throughput scale using a thermocycler. One hundred microliters of SOC medium (LB medium supplemented with 0.2 g/liter KCl, 2 g/liter MgCl2, 2 g/liter MgSO4, and 4 g/liter glucose) was then added to the cells, and a 1-h incubation at 37°C followed. The transformants were then selected by plating on solid medium containing the appropriate antibiotic.
(ii) Protocol II.
Two hundred E. coli DH5α colonies containing the mutagenized plasmid were picked, resuspended in 1 ml of LB medium containing ampicillin, and grown for 4 h. Plasmid isolation and transformation in E. coli Origami followed. A rapid selection of active clones on agar plates was performed with an activity staining assay. Master plates were generated by considering only clones showing activity.
Activity staining on agar plates (prescreening).
Plates containing mutant libraries of proROL in E. coli Origami were replicated with a sterile cloth onto a plate containing the appropriate antibiotics and 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to induce protein expression. The replicated plates were incubated at 20°C. After 2 days the grown colonies were overlaid with 10 ml of soft agar (0.5%, wt/vol) containing 160 μl of Fast blue (89 mg/ml in dimethyl sulfoxide) and 80 μl of α-naphthyl acetate (40 mg/ml in dimethyl formamide). Active clones (stained in red) were selected for the master plates.
Expression in MTPs.
Two hundred microliters of LB medium containing the necessary antibiotics was dispensed into each well of a 96-well MTP. Each transformant was picked with a sterile toothpick and used to inoculate each well. The plates were grown for 24 h at 37°C; afterwards, 100 μl of sterile 60% glycerol (vol/vol) was added, and the plates were mixed briefly and stored at −80°C as master plates. With a 96-spike replicator, new MTPs containing 200 μl of LB medium with antibiotics were inoculated and grown for 24 h. From these preinoculum plates, new plates containing 100 μl of LB medium with antibiotics were inoculated with 100 μl and incubated at 37°C for 6 h. IPTG was added to each well up to a final concentration of 0.1 mM, and the MTPs were incubated for approximately 20 h until harvested by centrifugation at 213 × g and 4°C for 30 min.
Cell disruption in MTP scale.
In the case of expression in MTPs, cell disruption was carried out via enzymatic lysis. The culture was centrifuged (213 × g for 30 min), and after supernatant elimination, the pellets were resuspended with 150 μl of 50 mM sodium phosphate buffer-300 mM NaCl (pH 8) containing 0.1% DNase. The cells were lysed by incubating the plates for 30 min at 4°C, freezing for 1 h at −80°C, and thawing for 30 min at 37°C. The lysates were clarified by centrifugation at 213 × g for 30 min.
Activity assay.
Lipase activity was assayed in vitro by monitoring the amount of p-nitrophenol released upon hydrolysis of a 1 mM solution of p-nitrophenyl butyrate (pNPB) in 50 mM phosphate buffer, pH 7.5, at room temperature with a FLUOstar Optima spectrofluorometer (BMG Labtechnologies, Offenburg, Germany). Aliquots (10 μl) of the cell fraction assayed were added to 190 μl of the reaction mixture, and the increase in absorbance at 410 nm was measured for 1 min using an apparent extinction coefficient of 1.33 × 104 M−1. One unit of hydrolase activity was defined as the amount of enzyme required to transform 1 μmol of pNPB to p-nitrophenol per min at room temperature.
Stability assay. (i) Semimicro scale (1.5 ml) procedure.
After cell disruption, the amount of lysate containing 50 U of hydrolase activity was incubated with octanal in phosphate buffer (pH 7.5; 50 mM) in a total volume of 1.3 ml at 20°C. As a control, the lipase-containing lysate was incubated without aldehyde. At defined time intervals, the residual activity was measured using pNPB as described above.
(ii) MTP scale procedure.
After cell disruption, two screening plates were generated per each MTP: the incubation with aldehyde was performed in one, and the other was kept as a control. Each screening plate produced included both the wild-type proROL and a negative control, represented by the buffered solution. The incubation was performed in phosphate buffer (pH 7.5; 50 mM) in a total volume of 200 μl at 20°C. At defined time intervals, the residual activity was measured using pNPB as described above.
Lipase production in flasks.
E. coli Origami harboring the pET22-proROL construct was grown in 100 ml of LB medium supplemented with the required antibiotics at 20°C. IPTG was used as an inducer, to a final concentration of 0.1 mM. At 20 h postinduction, the culture was centrifuged for 10 min at 800 × g to harvest the cells. The cells were then resuspended in 10 ml of phosphate buffer (pH 7.5; 50 mM) and disrupted by sonication (10 min; 50% pulse). The protein concentration of the samples was determined according to the method of Bradford (3).
Protein purification.
Cells obtained from a 100-ml culture of E. coli Origami harboring pET22-proROL were resuspended in 3 ml of phosphate buffer (pH 7; 50 mM), sonicated (10 min; 50% pulse; 50% power), and centrifuged at 800 × g. The total sample volume was then added to a 1-ml bed volume of the cobalt-based Talon cellThru immobilized metal affinity chromatography resin (BD Biosciences, Palo Alto, CA), previously equilibrated with the same buffer, and lightly shaken for 20 min. The suspension was then centrifuged at 700 × g for 5 min and washed twice with 10 ml of sodium phosphate buffer (pH 7; 50 mM). Elution was then carried out in a column with 4 ml of phosphate buffer containing 150 mM imidazole. Imidazole was removed, and the purified protein was concentrated by filtration using Amicon Ultra devices (Millipore, Billerica, MA) with a 10-kDa nominal molecular weight limit. Glycerol was added to the protein preparation to a final concentration of 50% (vol/vol), and the purified protein was stored at −20°C.
Mutant recombination.
Recombination of stable mutants by site-directed mutagenesis was performed according to the Quikchange protocol (Stratagene). The primers used to create H201S/K168X double mutants are given in Table 1.
Molecular dynamics of lipase structure.
A model of the H201S, H201A, and H201G mutants was built on the basis of the crystal structure of lipase from Rhizopus niveus (Protein Data Bank [PDB] code 1LGY). Amino acid changes were introduced using the O graphic program (22) running on a Silicon Graphics workstation. Side chain rotamers were chosen from a database of more common conformers (29). Models for each Ser201 conformer was energy minimized using the minimizer algorithm implemented in the CNS (for crystallography and NMR [nuclear magnetic resonance] system) package (4). The Engh and Huber (9) force field was used in all energy minimization calculations. Finally, the conformation exhibiting the least variation with respect to the native structure was chosen. The stereochemical quality of the model was checked with the program PROCHECK (25).
RESULTS
Visualization of target residues.
The residues susceptible to oxidation were identified using the three-dimensional structure of the R. niveus lipase (PDB 1LGY), since the PDB file corresponding to the crystal structure of the R. oryzae lipase (PDB 1TIC) contained only the α-carbon skeleton. The lipase from R. niveus differs from ROL only by the substitution M1A in the prosequence (21). The structural characteristics of the ROL are as follows: preprolipase (amino acids [aa] 1 to 25), prolipase (aa 26 to 86), mature lipase (aa 87 to 393), and lid (aa 208 to 215). The active site is characterized by the triad composed of S145, D204, and H257. The protein is characterized by three disulfide bonds: C-29—C-268, C-40—C-43, and C-235—C-244 (from hereon, numbering of residues throughout the text corresponds to the PDB file which contains only the mature lipase).
After analysis of the protein structure, the following residues possibly involved in the interaction with aldehydes were located: six cysteine groups involved in the three disulfide bonds; 15 lysine residues distributed on the protein surface, some of them involved in H-bonds (Fig. 1A); and 7 histidine residues, all but one (His134) in the vicinity of the active site (Fig. 1B). We decided to direct the mutagenesis toward all of the His residues and to all of the Lys groups involved either in no H-bonds (K168, K101, and K104) or forming only one H-bond (K37 and K202), except for Lys5, which forms two H-bonds. All of the selected lysine residues are on the surface and show no salt bridge or polar interactions involving the side chain amino group. Only Lys202 is located slightly deeper in the protein, but its side chain faces outward and does not exhibit any interactions either. The cysteines were not mutagenized, as all are involved in disulfide bonds considered to be important to maintain the tertiary structure of the lipase. In fact, it has been already reported that the functional proROL expression occurring in E. coli Origami is related to the capacity of this mutated strain to assist in the proper formation of the protein sulfhydryl bonds in the cytoplasm (7).
FIG. 1.
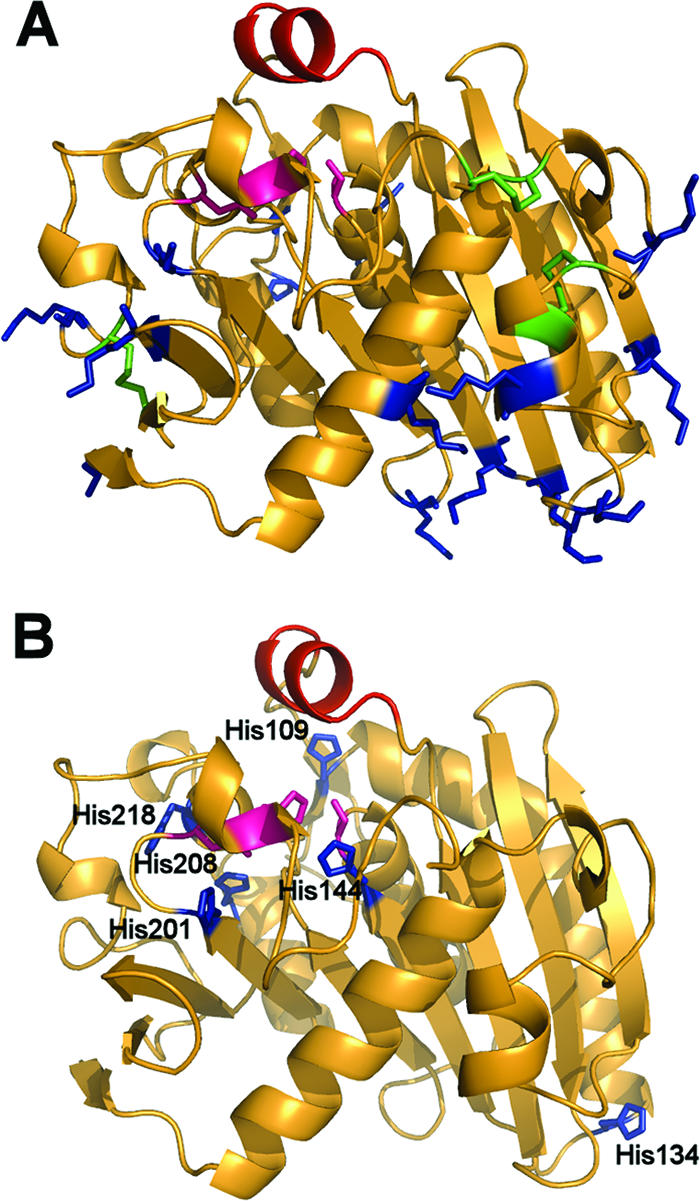
(A) Visualization of the target residues on the ROL. The lid (red), the active site (pink), the lysine groups (blue), and the cysteine groups forming disulfide bonds (green) are highlighted. (B) The lid (red), the active site (pink), and the histidine groups (blue) are highlighted.
Mutagenesis strategy and library generation.
The technique utilized to generate variants with single site mutations was based on random mutagenesis at a defined position and control of multiplicity in the resulting mutant library, which resulted in focused libraries. For each position to be mutated, a 10-fold oversampling was used to ensure that all possible variants were present in the library (200 clones per single saturated position). It is clear that the quality of the genomic libraries generated for each mutated position had to be high; i.e., each clone of each library should be different. A lower library quality (multiple copies of every different individual) implies that a higher number of clones would have to be screened to consider all the possible cases. Another requirement was that the strain utilized for the expression of proROL variants necessarily had to be E. coli Origami, according to the protein expression conditions previously optimized for the expression of the wild-type protein in flask (7). Unfortunately, when the E. coli Origami strain was used for the direct transformation of the mutated DNA after the mutagenesis reaction, no transformants were obtained. On the other hand, when E. coli DH5α was used as a cloning strain, enough transformants could be obtained. Two different strategies were then considered in order to generate the genomic library in the Origami strain, as described in the Materials and Methods section. We decided to perform protocol I in the case of mutation of the lysine residues and the histidine residue far from the active site (H134) and protocol II in the case of mutation of the other histidine residues. In the case of the histidine residues close to the active site, it was very likely that their mutation would affect protein activity negatively, and, thus, a prior activity staining test carried out in solid agar plates effectively allowed us to ascertain which clones were still active, reducing the number of variants to be screened. Additionally, the percentage of clones per plate that retained activity provided a rough impression of the influence of the position mutated on lipase activity. The results showed the highest percentage of active clones for histidine 134, while positions 218 and 144 were shown to be essential for the enzymatic activity since the number of active clones per plate in both cases was less than 10% (Fig. 2). It may be concluded that owing to the prior activity staining performed, a high number of clones was rapidly screened, and the libraries were smaller when protocol II was used for library generation.
FIG. 2.

Percentage of active clones revealed by the activity staining test. The percentages have been calculated by considering an average of 10 plates per each position mutated. In the case of His134, the percentage was based on the two MTPs obtained from protocol II.
Stability screening assay.
Generally, the method utilized for the high-throughput screening of enzyme libraries is of critical importance. Prior to the generation of genetic diversity, the screening procedure best suited to handling capacity, sensitivity, and achievement of the envisioned goal must be determined. It is important to ensure that the screening procedure directly measures the property that one wishes to improve, particularly for directed evolution applications, where it is well known that “you only get what you screen for” (34).
Before performing a high-throughput screening of the mutants, the method was previously developed on a semimicro scale (1.5 ml) with cell lysates containing the overexpressed wild-type proROL. As shown in a prior work, control lysates without the overexpressed enzyme did not exhibit any measurable lipase activity (7).
It was crucial to identify conditions under which (i) a rapid selection of stable variants could be performed, (ii) the control (i.e., enzyme without deactivator) presented a minimum deactivation during the assay, (iii) deactivation was due only to the deactivating agent introduced, (iv) deactivation was slow enough as to allow for several intermediate activity measurements during the screening time considered, and (v) deactivation could be measured quantitatively.
The deactivating effect of the triglyceride oxidation products on proROL was investigated by considering only aldehydes as typical secondary oxidation products. Obviously, the effect of oxidized oils on lipases is much more complex since several oxidation products are produced at concentration levels that depend on oil or fat (24). MDA, 4-HNE, and acrolein are among the many different aldehydes that can be formed during lipid peroxidation, the most intensively studied (11). The poor stability of MDA and acrolein during the screening conditions considered and the high price of HNE led to the search for another aldehyde better suited for the high-throughput screening of the proROL variants generated, such as octanal. The experiments demonstrated that the proROL deactivation in the presence of HNE was the same observed with octanal in a 50-fold higher concentration (Fig. 3).
FIG. 3.

Time-dependent residual activity decay of the wild-type proROL incubated with several aldehydes at different concentrations. Inactivation was studied as described in Materials and Methods using 50 U of enzyme (crude cell lysate).
The incubation temperature was fixed to 20°C in order to avoid thermal protein inactivation. Because of the simplicity and throughput required for library screening, an aqueous system was considered, and the reaction investigated in the screening was the hydrolysis of pNPB.
The amount of cell lysate used played a critical role in enzyme stability during the assay as other proteins in the cell lysate might react with the aldehyde and/or the lipase might form aggregates. Thus, the amount of lysate in the assay was set to 2% (vol/vol). Under the experimental conditions used, the protein activity decrease followed a first-order kinetic. The deactivation process was therefore accounted for by one single parameter, the deactivation factor kd. Therefore, it was possible to determine a kd value characteristic of wild-type proROL (for each concentration value of octanal) with a very low iteration variability. When, in particular, proROL was incubated with 10 mM octanal, it had a half-life of 22 h and a kd value of 0.031 ± 0.002 h−1.
High-throughput screening.
The expression and screening in MTPs of the variants generated by both protocol I and protocol II was performed as described in Materials and Methods. In order to define a variant as a positive hit, the largest variation of the wild-type kd value was considered to define an uncertainty area. The variants furthest from this area were chosen and confirmed in two replications. The positive hits resulting from the high-throughput screening of the histidine and lysine libraries were subsequently sequenced in order to analyze the nature of the substitution. In the case of His144 and His218, no hits more stable than the wild-type proROL could be found (data not shown). These positions had already been shown, with the activity staining test performed on agar plates, to be essential for the proROL activity (Fig. 2).
The positive hits were confirmed by proROL expression in flask and stability tests toward octanal on a semimicro scale. The results obtained are shown in Fig. 4. Some selected variants appear to be less stable or as stable as the wild-type enzyme. This was not especially surprising because of the intrinsic variability of the high-throughput screening. However, in most of the cases the stability increase was confirmed: the best results were obtained for histidine substitutions and, in particular, for position 201. For mutants H201A and H201S, in fact, a stability increase close to 82% could be achieved. For lysine residues, the mutagenesis effect on protein stability was much lower. An increase of 20% in residual activity was achieved only for the K101I substitution.
FIG. 4.

Expression in flask and screening on a semimicro scale of the hits found with the high-throughput screening assay. The following parameters were used: enzyme dilution, 2% (vol/vol); octanal concentration, 10 mM; temperature of incubation, 20°C. Error bars refer to an average of 10 replicate experiments. Stability increase was calculated as the ratio between the differences in half-life of each variant and the wild type, divided by the half-life of the wild type, and multiplied by 100.
Screening test of the purified enzyme.
The wild-type proROL was purified as described in Materials and Methods, and the stability after purification was investigated. The purified enzyme was shown to be less stable, with a twofold half-life decrease. This could most likely be attributed to the removal from the cell lysate of all other macromolecules that were able to react with the aldehyde, including other proteins. The stabilizing effect of macromolecules on enzymes is well known and has been extensively studied (14-16).
The purification of H201X variants was performed as well. In Table 2 the stability increases of these variants before and after purification are compared. Once again, the results shown refer to an average of 10 replicates. Lower stability due to purification was also observed for the H201X variants and, in particular, for the variants H201A and H201N. An explanation could be that the stabilizing effect of other macromolecules was more critical for some of the proROL variants. In contrast, for the variants H201S and H201G a good stability increase was still observed. Both the wild-type enzyme and the mutants had specific activities ranging between 76 to 80 U/mg; i.e., no differences were observed in terms of activity loss as a result of the mutations.
TABLE 2.
Half-life and stability of the H201X mutants as crude extract and in purified forma
| Variant | Half-life (h) of:
|
Stability increase (%) relative to wild type ofb:
|
||
|---|---|---|---|---|
| Crude extract | Purified form | Crude extract | Purified form | |
| Wild type | 22 | 10 | NA | NA |
| H201S | 40 | 16 | 82 | 60 |
| H201A | 42 | 12 | 91 | 20 |
| H201G | 38 | 15 | 72 | 50 |
| H201N | 32 | 11 | 45 | 10 |
Mutants were incubated with 10 mM octanal. Average initial activity was 50 U.
Stability increase was calculated as the ratio between the difference in half-life of each variant and the wild type, divided by the half-life of the wild type, and multiplied by 100 (relative standard deviation amounted to less than 5%; n = 10). NA, not applicable.
Analysis of double recombinants.
In order to further improve proROL stability, recombination of some of the mutants showing higher stability than the wild-type proROL was considered. Basically, we wanted to investigate the combination effect of close mutations with distant ones. The results from the recombination of the variant H201S with the K168X variants and with the variant H134P are shown in Table 3. The best result was obtained in the case of the variant H201S/K168I, which was twice as stable as the wild-type enzyme. On the other hand, the stability of the variants H201S/K168X and H201S/H134P was approximately the same as that of the single variant H201S. To avoid having selected mutants with enhanced stability only towards octanal, the improvement in stability was confirmed with other aldehydes, as shown in Fig. 5.
TABLE 3.
Properties of purified double mutantsa
| Variant | Half-life (h) | Stability increase relative to the wild type (%)b |
|---|---|---|
| Wild type | 10 | NA |
| H201S/K168P | 13 | 30 |
| H201S/K168L | 14 | 40 |
| H201S/K168I | 20 | 100 |
| H201S/K168S | 15 | 50 |
| H201S/H134P | 14 | 40 |
The data refer to the average value from 10 replicates, with a maximum deviation among replicates of 0.002 for kd. The octanal concentration was 10 mM; average initial activity was 50 U.
Stability increase was calculated as the ratio between the difference in half-life of each variant and the wild type, divided by the half-life of the wild type, and multiplied by 100. NA, not applicable.
FIG. 5.

Effect of several aldehydes on the stability increase of the H201S/K168I proROL mutant relative to the wild-type enzyme. Error bars refer to an average of 10 replicate experiments. The following parameters were used: initial activity, 50 U; final concentration of each aldehyde, 10 mM; time of incubation, 7 h. Stability increase was calculated as the ratio between the differences in half-life of each variant and the wild type, divided by the half-life of the wild type and multiplied by 100.
DISCUSSION
Evolutionary design approaches have enjoyed considerable success in recent years. Numerous enzymes have been improved by directing their evolution in the laboratory, which usually involves iterations of random mutagenesis or recombination followed by screening or selection (13). In particular, we adopted a mutation strategy which combines the principles of directed evolution with rational design. Some authors already refer to the production of focused libraries in order to reduce the number of variants to be screened by choosing from a prior analysis of the protein structure the site of mutation most likely to improve the target enzyme's properties. This concept of spatially predetermined saturation mutagenesis has already been successfully employed with the aim of enhancing substrate selectivity or influencing enantioselectivity (30, 31). On the other hand, many examples of physical and chemical modification of lipases with the aim of improving their native properties and endowing them with useful new functions have been reported (6, 21, 25). In particular, chemical modification of lysine residues with pyridoxal-5′-phosphate and of disulfide bonds with dithiothreitol in order to increase lipase stability in the presence of oxidized oils was considered in our working group. The mutated lipase showed a small stability improvement for long-term incubation with oxidized oils but a large decrease in initial activity. The great advantage of the protein engineering strategy considered is that it led to improvements in protein stability without affecting the initial activity. The single-site-mutated variants selected from the high-throughput screening were, in fact, as active as the wild-type enzyme.
All of the His positions were tolerant of substitution to different extents except for H144 and H218, which could not tolerate substitution. As shown in Fig. 6A, His144 lies adjacent to the catalytic serine, and thus it is logical that any substitution might disrupt distances and electronic effects within the catalytic center. With regard to His 218, Fig. 6B illustrates that this residue is tightly packed between two neighboring Arg residues (Arg179 and Arg198), which, in turn, are within reasonable distance from two glutamate residues that establish a salt bridge. Therefore, replacement of His218 would destabilize this complex network of interactions, destabilizing the loop it is located on, and rendering the lipase inactive. It may be concluded that the substitution of histidine residues close to the active site is critical in terms of protein activity. It has been reported that in many cases mutations closer to the active site are more effective than distant ones. Therefore, depending on the enzyme property to be improved, focusing mutation near the substrate-binding site might increase the success rate in many directed-evolution experiments and avoid the screening of large libraries (27). With respect to the H201X substitutions, energy minimization was performed on the modified crystal structure in order to explain the stability enhancement found experimentally. The results of the molecular dynamics simulation are shown in Fig. 6C to E. The substitutions H201S, H201A, and H201G contribute to protein stability not only by replacing a reactive residue but also by introducing residues smaller than His. In all cases the distance between Lys202 and Asp256 is reduced from 5 Å in the wild type to 3.6 Å so that a salt bridge may be formed, thus helping stabilize the whole loop. In the case of the H201S substitution (Fig. 6C), the most likely conformer of the Ser residue introduced is shown to be within H-bond distance of Tyr260, which may explain the fact that this mutant exhibits higher stability than the other two. In summary, replacing sensitive residues not only solved the problem of oxidation by aldehydes but also increased stability.
FIG. 6.

(A) His144 (in red) is adjacent to the catalytic serine. The catalytic triad Ser145, Asp204, and His257 is shown in green. (B) His218 (shown as red spheres) is “sandwiched” between Arg179 and Arg198 (shown as green spheres), which, in turn, establish salt bridge interactions with Glu222 and Glu240 (shown in yellow). (C to E) Local environment of H201X mutants. The ribbon diagrams show the ROL structure orange. The arrangement of the mutated His201 residues for Ser, Ala, or Gly (shown in green) is presented with the remaining residues defining the region (shown in light gray). (F) Wild-type ROL.
The replacement of Lys residues proved to be less effective. Indeed, the mutation of just one lysine cannot prevent covalent interactions between the other 14 lysine residues distributed on the proROL surface and the deactivating molecules. We could conclude that in this case one single mutation is not enough to insure higher stability; variants with more than one lysine residue substitution should be generated. The relative lysine content of a lipase was previously related to its stability against aldehydes. A higher level of stability was observed for lipases having lower lysine content, such as Mucor javanicus, Mucor miehei, and Pseudomonas species lipases (37). Moreover, the single mutation of all the lysine residues considered did not markedly affect the enzyme activity, as in the case of the histidine libraries; generally, all the mutants of a lysine library were active. It is likely, therefore, that the mutation of more than one lysine may not be critical for protein activity. However, it should be considered that the lysine groups distributed on the protein surface may play a role in determining protein solubility. An evaluation of the number of lysine substitutions allowed should then be performed to avoid undesired effects on enzyme solubility and also on the tertiary structure, since some of the proROL lysine residues are involved in H-bonds.
The effect of the combination of two mutations was investigated by analyzing the stability of the double recombinants generated. The double recombinants kept the enhanced stability of variant H201S, but a further stability increase due to the additional Lys replacement was observed only for the variant H201S/K168I. This good result is promising and demonstrates that, by properly acting on the protein in order to produce variants with more than one substitution, it is possible to obtain greater improvements in stability.
Acknowledgments
M.D.L. thanks G. Greco for helpful advice and discussion and Claudia Cerbone for her help in screening and purifying the proROL variants. A.H. thanks J. Berenguer for fruitful discussion.
A.H. and U.T.B. acknowledge financial support provided through the European Community's Human Potential Programme under contract HPRN-CT2002-00239. A.H. acknowledges financial support from the Spanish Ministry of Education through the Ramon y Cajal Programme.
Footnotes
Published ahead of print on 21 September 2007.
REFERENCES
- 1.Beer, H. D., J. E. G. McCarthy, U. T. Bornscheuer, and R. D. Schmid. 1998. Cloning, expression, characterization and role of the leader sequence of a lipase from Rhizopus oryzae. Biochim. Biophys. Acta 1399:173-180. [DOI] [PubMed] [Google Scholar]
- 2.Bolgar, M. S., and S. J. Gaskell. 1996. Determination of the sites of 4-hydroxy-2-nonenal adduction to protein by electrospray tandem mass spectrometry. Anal. Chem. 68:2325-2330. [Google Scholar]
- 3.Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 4.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D 54:905-921. [DOI] [PubMed] [Google Scholar]
- 5.Buko, V. U., A. A. Artsukevich, and K. V. Ignatenko. 1999. Aldehydic products of lipid peroxidation inactivate cytochrome P-450. Exp. Toxicol. Pathol. 51:294-298. [DOI] [PubMed] [Google Scholar]
- 6.Cvengros, J., and Z. Cvengrosová. 2004. Used frying oils and fats and their utilisation in the production of methyl esters of higher fatty acids. Biomass Bioenerg. 27:173-181. [Google Scholar]
- 7.Di Lorenzo, M., A. Hidalgo, M. Haas, and U. T. Bornscheuer. 2005. Heterologous production of functional forms of Rhizopus oryzae lipase in Escherichia coli. Appl. Environ. Microbiol. 71:8974-8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Encinar, J. M., J. F. Gonzalez, and A. Rodríguez-Reinares. 2007. Ethanolysis of used frying oil. Biodiesel preparation and characterization. Fuel Processing Technol. 88:513-522. [Google Scholar]
- 9.Engh, R. A., and R. Huber. 1991. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. Sect. A 47:392-400. [Google Scholar]
- 10.Esterbauer, H., P. Eckl, and A. Ortner. 1990. Possible mutagens derived from lipids and lipid precursors. Mutat. Res. 238:223-233. [DOI] [PubMed] [Google Scholar]
- 11.Esterbauer, H., R. J. Shaur, and H. Zollner. 1991. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol. Med. 11:81-128. [DOI] [PubMed] [Google Scholar]
- 12.Esterbauer, H., and H. Zollner. 1989. Methods for determination of aldehydic lipid peroxidation products. Free Radical Biol. Med. 7:197-203. [DOI] [PubMed] [Google Scholar]
- 13.Farinas, E. T., T. Bulter, and F. H. Arnold. 2001. Directed enzyme evolution. Curr. Opin. Biotechnol. 12:545-551. [DOI] [PubMed] [Google Scholar]
- 14.Gianfreda, L., A. M. Livolsi, M. R. Scarfi, and G. Greco, Jr. 1982. β-d-Glucosidase stabilization in a polarized ultrafiltration membrane reactor. Enzyme Microb. Technol. 4:322-326. [Google Scholar]
- 15.Gianfreda, L., M. Modafferi, and G. Greco, Jr. 1985. Enzyme stabilization towards thermal chemical and proteolytic deactivation. Enzyme Microb. Technol. 7:78-82. [Google Scholar]
- 16.Greco, G., Jr., and L. Gianfreda. 1981. Enzyme stabilization by linear chain polymers in ultrafiltration membrane reactors. Biotechnol. Bioeng. 23:2199-2210. [Google Scholar]
- 17.Haas, M. J., D. G. Bailey, W. Baker, T. R. Berka, D. J. Cichowicz, Z. S. Derewenda, R. R. Genuario, R. D. Joerger, R. R. Klein, K. Scott, and D. Woolf. 1999. Biochemical and molecular characterization of a lipase produced by the fungus Rhizopus delemar. Fett Lipid 101:364-370. [Google Scholar]
- 18.Hanahan, D. 1983. Studies on the transformation of E. coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 19.Harwood, J. L., and B. Caterson. 2006. Dietary omega-3-polyunsaturated fatty acids and inflammation. Lipid Technol. 18:7-10. [Google Scholar]
- 20.Holzwarth, H.-C., J. Pleiss, and R. D. Schmid. 1997. Computer-aided modelling of stereoselective triglyceride hydrolysis catalyzed by Rhizopus oryzae lipase. J. Mol. Catal. B 3:73-82. [Google Scholar]
- 21.Joerger, R. D., and M. J. Haas. 1993. Overexpression of a Rhizopus delemar lipase gene in Escherichia coli. J. Am. Oil Chem. Soc. 28:81-88. [DOI] [PubMed] [Google Scholar]
- 22.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 23.Kaga, H., B. Siegmund, E. Neufellner, and K. Faber. 1994. Stabilization of Candida lipase against acetaldehyde by adsorption onto celite. Biotechnol. Technol. 8:369-374. [Google Scholar]
- 24.Knothe, G., and R. O. Dum. 2003. Dependence of oil stability index of fatty compounds on their structure and concentration and presence of metals. J. Am. Oil Chem. Soc. 80:1021-1026. [Google Scholar]
- 25.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 26.Lebedevas, S., A. Vaicekaukas, G. Lebedeva, V. Makariciene, P. Janulis, and K. Kazancev. 2006. User of waste fats of animal and vegetal origin for the production of biodiesel fuel: quality, motor properties and emissions of harmful components. Energy Fuels 20:2274-2280. [Google Scholar]
- 27.Morley, K. L., and R. J. Kazlauskas. 2005. Improving enzyme properties: when are closer mutations better? Trends Biotechnol. 23:231-237. [DOI] [PubMed] [Google Scholar]
- 28.Pirozzi, D. 2003. Improvement of lipase stability in the presence of commercial triglycerides. Eur. J. Lipid Sci. Technol. 105:608-613. [Google Scholar]
- 29.Ponder, J. W., and F. M. Richards. 1987. Tertiary templates for proteins: use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 193:775-791. [DOI] [PubMed] [Google Scholar]
- 30.Reetz, M. T., M. Bocola, J. D. Carballeira, D. Zha, and A. Vogel. 2005. Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angew. Chem. Int. Ed. Engl. 44:4192-4196. [DOI] [PubMed] [Google Scholar]
- 31.Reetz, M. T., L.-W. Wang, and M. Bocola. 2006. Directed evolution of enantioselective enzymes: iterative cycles of casting for probing protein-sequence space. Angew. Chem. Int. Ed. Engl. 45:1236-1241. [DOI] [PubMed] [Google Scholar]
- 32.Schmid, U., U. T. Bornscheuer, M. M. Soumanou, G. P. McNeill, and R. D. Schmid. 1999. Highly selective synthesis of 1,3-oleoyl-2-palmitoylglycerol by lipase catalysis. Biotechnol. Bioeng. 64:678-684. [PubMed] [Google Scholar]
- 33.Schmid, U., U. T. Bornscheuer, M. M. Soumanou, G. P. McNeill, and R. D. Schmid. 1998. Optimization of the reaction conditions in the lipase-catalyzed synthesis of structured triglycerides. J. Am. Oil Chem. Soc. 75:1527-1531. [Google Scholar]
- 34.Schmidt-Dannert, C., and F. H. Arnold. 1999. Directed evolution of industrial enzymes. Trends Biotechnol. 17:135-136. [DOI] [PubMed] [Google Scholar]
- 35.Uchida, K., M. Kanematsu, Y. Morimitsu, T. Osawa, N. Noguchi, and E. Niki. 1998. Acrolein is a product of lipid peroxidation reaction. J. Biol. Chem. 273:16058-16066. [DOI] [PubMed] [Google Scholar]
- 36.Varese, R., and M. Varese. 1996. Methyl ester biodiesel: opportunity or necessity? INFORM 7:816-824. [Google Scholar]
- 37.Weber, H. K., H. Srecher, and K. Faber. 1995. Sensitivity of microbial lipases to acetaldehyde formed by acyl-transfer reactions from vinyl esters. Biotechnol. Lett. 17:803-808. [Google Scholar]