

Abstract
The hrcA and hspR genes of Helicobacter pylori encode two transcriptional repressor proteins that negatively regulate expression of the groES-groEL and hrcA-grpE-dnaK operons. While HspR was previously shown to bind far upstream of the promoters transcribing these operons, the binding sites of HrcA were not identified. Here, we demonstrate by footprinting analysis that HrcA binds to operator elements similar to the so-called CIRCE sequences overlapping both promoters. Binding of HspR and HrcA to their respective operators occurs in an independent manner, but the DNA binding activity of HrcA is increased in the presence of GroESL, suggesting that the GroE chaperonin system corepresses transcription together with HrcA. Comparative transcriptome analysis of the wild-type strain and hspR and hrcA singly and doubly deficient strains revealed that a set of 14 genes is negatively regulated by the action of one or both regulators, while a set of 29 genes is positively regulated. While both positive and negative regulation of transcription by HspR and/or HrcA could be confirmed by RNA primer extension analyses for two representative genes, binding of either regulator to the promoters could not be detected, indicating that transcriptional regulation at these promoters involves indirect mechanisms. Strikingly, 14 of the 29 genes which were found to be positively regulated by HspR or HrcA code for proteins involved in flagellar biosynthesis. Accordingly, loss of motility functions was observed for HspR and HrcA single or double mutants. The possible regulatory intersections of the heat shock response and flagellar assembly are discussed.
The heat shock proteins of the gastric pathogen Helicobacter pylori have been studied in some detail both because of their general role in protection of the bacteria from the hostile environment of the human stomach and because of their involvement in specific pathogenic processes. The H. pylori GroEL homologue (18, 32) has been proposed to play a role in regulating the activity of the nickel-dependent urease enzyme, which generates ammonia ions from the hydrolysis of urea and therefore protects the bacterium from the low pH of the stomach lumen (8, 10). Its cochaperone, GroES, is thought to contribute to this regulation by controlling the availability of nickel ions by means of its intrinsic metal binding activity (17, 32). Although relatively controversial, it has been reported that GroEL, as well as another major heat shock protein, DnaK (Hsp70), can be found in association with the outer membrane, and this surface localization has been suggested to modify the glycolipid binding specificity of H. pylori cells at low pH (9, 16, 23).
Because of their functions in the general stress response as well as in specific pathogenic mechanisms, the H. pylori heat shock proteins are expected to be tightly regulated in the level of expression. We have previously demonstrated that transcription of the groESL, hrcA-grpE-dnaK, and cbpA-hspR-orf operons encoding the major chaperones of H. pylori is negatively regulated by the HspR and/or HrcA repressor protein (28, 31). HspR is a homologue of the repressor of the dnaK operon of Streptomyces coelicolor that has been shown to bind to inverted repeats in the promoter region designated HAIR (HspR-associated inverted repeat) (4, 12). HrcA is a homologue of the repressor of a set of heat shock genes of Bacillus subtilis that binds to an inverted repeat in the promoter region designated CIRCE (controlling inverted repeat of chaperone expression) (20, 27, 37). While HrcA is widely distributed in the prokaryotic kingdom, HspR is found in a restricted number of bacteria (20). Genetic and biochemical studies with different microorganisms have revealed that the chaperone systems directly influence the transcriptional control exerted by HspR and HrcA. For example, in the pathogenic bacterium Chlamydia trachomatis, the GroEL protein is able to increase the ability of HrcA to bind to the CIRCE element and to repress transcription (35). In B. subtilis the activity of the HrcA repressor is modulated by the GroE chaperonin system (19, 24). Furthermore, detailed in vitro and in vivo studies have provided evidence that DnaK functions as a transcriptional corepressor by binding to HspR at its operator sites in S. coelicolor (2, 3).
In H. pylori, HspR alone represses transcription of the cbpA-hspR-helicase operon, while both HspR and HrcA regulators are required to repress transcription of the hrcA-grpE-dnaK and groES-groEL heat shock operons (28, 31). Whether HrcA and HspR control transcription of additional genes is unknown. Initial studies indicated that while transcription of the groESL and cbpA-hspR-orf operons was strongly inducible by treatment with 300 mM NaCl, no induction was observed when cultures were incubated at 45°C (31). Subsequently, Homouth and coworkers (15) showed that transcription of these operons is strongly inducible by a mild heat shock at 42°C, suggesting that HspR can indeed mediate the transcriptional response to a sudden temperature increase, characterized by fast and strong induction of transcription and a subsequent shutoff phase, whose onset is determined largely by the stability of the respective mRNAs (29). Until recently, the study of HrcA-dependent regulation was hampered by difficulties in purifying a recombinant protein to map the HrcA binding sites on the promoters (25, 31). Thus, so far the interplay between HspR and HrcA at the level of the coregulated promoters, as well as the possible involvement of chaperone proteins, has not been explored. Specifically, HrcA localizes in the inner membrane of H. pylori and shows toxic or insoluble properties when it is expressed in Escherichia coli (25). However, these effects could be alleviated by expression at 42°C, allowing purification of a recombinant protein suitable for biochemical analyses (25). In the present study we determined the binding sites of HrcA on the coregulated promoters by performing footprinting experiments and showed that under in vitro conditions the binding of HspR and HrcA occurs in an independent manner. The genome-wide regulatory functions of HspR and HrcA were further investigated by transcriptome and phenotypic trait analysis of singly or doubly deficient strains. The results indicate that there is an intimate, although indirect, interconnection between the stress response and motility functions in H. pylori.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
H. pylori strains (Table 1) were recovered from frozen stocks by growth on Columbia agar plates containing 5% horse blood, 0.2% cycloheximide, and Dent's or Skirrow's antibiotic supplement (Oxoid) for 2 to 3 days. After passage on fresh plates, bacteria were cultured in a 9% CO2-91% air atmosphere at 37°C and 95% humidity. Liquid cultures were grown in modified brucella broth supplemented with 5% fetal calf serum, 0.2% cycloheximide, and Dent's or Skirrow's antibiotic supplement. Motility of H. pylori strains was assayed by stab inoculating bacteria with a pipette tip into 0.3% agar plates containing brucella broth supplemented with 10% fetal calf serum and Dent's or Skirrow's antibiotic supplement and culturing them as described above.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference(s) or source |
|---|---|---|
| Strains | ||
| E. coli DH5α | supE44 ΔlacU169 (φ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1β | 14 |
| E. coli BL21(DE3) | hsdS gal (λcIts857 ind1 Sam7 nin5 lacUV5-T7 gene 1) | 13 |
| H. pylori G27 | Clinical isolate; wild type | 36 |
| H. pylori G27(hrcA::km) | G27 derivative; bp 156 to 375 of hrcA replaced by a km cassette | 28 |
| H. pylori G27(hspR::km) | G27 derivative; bp 66 to 334 of the hspR coding sequence replaced by a km cassette | 28 |
| H. pylori G27(hrcA::km hspR::cm) | G27(hrcA::Km) derivative; bp 66 to 334 of the hspR coding sequence replaced by a cat cassette | 28 |
| H. pylori G27(hrcA-HA) | G27(hrcA::Km) derivative; hrcA-HA complementing strain obtained by double homologous recombination of pVAC-cat-hrcA-HA | 25, 28 |
| Plasmids | ||
| pGEM-T Easy | Cloning vector, Ampr | Promega |
| pGEM-T-Easy-Phrc | pGEM-T Easy derivative containing a 308-bp PCR fragment (oligonucleotides hrcA and hrcA1) encompassing the Phrc promoter | 25 |
| pGEM-T-Easy-Pgro | pGEM-T Easy derivative containing a 294-bp PCR fragment (oligonucleotides gro1 and groFP) encompassing the Pgro promoter | This study |
| pGEM-T-Easy-Pmda66 | pGEM-T Easy derivative containing a 250-bp PCR fragment (oligonucleotides mda66PE and mda66rev2) encompassing the Pmda66 promoter | This study |
| pGEM-T-Easy-PflaB | pGEM-T Easy derivative containing a 323-bp PCR fragment (oligonucleotides fla and fla2) encompassing the PflaB promoter | 30 |
| pET22b | Expression vector allowing C-terminal histidine-tagged gene fusion; Ampr | Novagen |
| pET22b-GroEL | pET22b derivative containing the groEL coding sequence amplified by PCR with oligonucleotides groEL-fwd and groEL-rev on chromosomal DNA of H. pylori, digested with restriction enzymes NdeI and XhoI | This study |
| pET22b-GroES | pET22b derivative containing the groES coding sequence amplified by PCR with oligonucleotides groES-fwd and groES-rev on chromosomal DNA of H. pylori, digested with restriction enzymes NdeI and XhoI | This study |
See Table 2 for oligonucleotide sequences.
DNA techniques.
DNA manipulations were performed routinely as described by Sambrook and colleagues (26). All restriction and modification enzymes were used according to the manufacturer's instructions (New England Biolabs). Nucleic acid purification was carried with QIAGEN kits (QIAGEN, Inc.). PCRs were carried out using 50 ng of H. pylori chromosomal DNA, 40 pmol of each specific primer, and Taq DNA polymerase (New England Biolabs) in a 50-μl (final volume) mixture containing 200 μM of each deoxynucleotide in 1× PCR buffer containing Mg2+. A total of 33 cycles consisting of denaturation of the DNA at 95°C for 1 min, annealing at the appropriate temperature for 1 min, and extension at 72°C for 1 min were performed.
RNA preparation.
Total H. pylori RNA was extracted using a hot-phenol extraction procedure described previously (6). Briefly, 10 ml of exponentially growing cells was added to 1.25 ml of ice-cold ethanol-phenol stop solution (5% water-saturated phenol [pH < 7.0] in ethanol), harvested, and resuspended in 800 μl of a 0.5-mg/ml lysozyme solution in Tris-EDTA (10 mM Tris, 1 mM EDTA; pH 8.0). Then 50 μl of 10% sodium dodecyl sulfate was added, and samples were incubated for 2 min at 64°C. After incubation, 88 μl of 1 M sodium acetate (pH 5.2) and 1 ml of water-saturated phenol (pH < 7.0) were added, and samples were incubated at 64°C for 6 min with occasional shaking on ice for 5 min, spun at 13,000 × g at 4°C for 10 min, extracted with 1 volume of chloroform, ethanol precipitated, and stored at −80°C. Prior to use, an aliquot of RNA samples was collected by centrifugation, quantified, and loaded on a 1% agarose gel to assess RNA purity and integrity.
Transcriptome analysis.
Prior to reverse transcription, RNA samples were treated with 1 U/μg RQ1 RNase-free DNase (Promega) at 37°C for 30 min, phenol-chloroform extracted, and ethanol precipitated. cDNA synthesis and labeling were carried out with a thermal cycler by combining 25 to 50 μg RNA with 150 pmol random hexamers (Invitrogen) in 28-μl reaction mixtures. After denaturation for 3 min at 94°C and annealing for 5 min at 37°C, 22 μl of a reverse transcriptase labeling mixture containing 25 U of avian myeloblastosis virus reverse transcriptase (Promega), [α-33P]dATP (2,500 Ci/mmol; Amersham), and 80 U of the RNase inhibitor RNasin (Promega) was added and reverse transcribed at 42°C for 3 h. The reaction was stopped by addition of 2 μl of 0.5 M EDTA, and RNA was degraded by alkaline treatment with 0.15 N NaOH for 15 min at 37°C and then neutralized with 17.5 μl of 1 M Tris-Cl (pH 7.5). cDNA was purified from unincorporated radioactive nucleotides using Chromaspin-TE10 spin columns (Clontech) and was hybridized to H. pylori Panorama open reading frame arrays (Sigma-Genosys) according to the manufacturer's instructions. Images were acquired with a Storm phosphorimager (Molecular Dynamics). RNAs were extracted from two independent cultures. Spot intensities on arrays were quantified with ImageQuant 5.2 software (Molecular Dynamics), processed with Microsoft Excel, and normalized by expressing values as percentages of the total gene specific intensity. To avoid background noise, spots with levels of intensity of <0.005% were not considered. For data analysis the statistical significance of expression ratios was determined by running the Web-based Cyber-T application program (http://visitor.ics.uci.edu/genex/cybert), which was specifically developed for array data analysis, based on a Bayesian probabilistic framework. In particular, given our sample size of 1,671 genes, we adjusted the sliding window size to 81 and determined a Bayesian confidence estimate value corresponding to three times the number of experimental replicates. These settings were previously shown to suit transcriptome analysis using macroarrays in H. pylori (6). Genes with mutant strain/wild-type strain expression ratios of ≥1.5 or ≤−1.5 and Bayesian P values of ≤0.05 were considered to be significantly deregulated.
Primer extension analysis.
Transcription from the Pmda66 and PflaB promoters was assayed by primer extension analysis using oligonucleotides mda66PE and fla, respectively (Table 2). An oligonucleotide (5 pmol) was 5′ end labeled in the presence of [γ-32P]ATP (5,000 Ci/mmol; Amersham) and T4 polynucleotide kinase. The labeled oligonucleotide (0.4 to 1.0 pmol) was coprecipitated with 15 μg of H. pylori total RNA and resuspended in 7.5 μl of H2O, 3.5 μl of 2 mM deoxynucleoside triphosphates, and 3 μl of 5× reverse transcription buffer (Promega). The reaction mixtures were incubated for 3 min at 95°C and for 1 min at 42°C, and then 1 μl of avian myeloblastosis virus reverse transcriptase (10 U/μl; Promega) was added to each sample and reverse transcription was carried out by incubating the samples at 42°C for 45 min. Samples were incubated for 10 min at room temperature with 1 μl of RNase A (1 mg/ml), extracted with phenol-chloroform (1:1), ethanol precipitated, and resuspended in 10 μl of sequencing loading buffer. After denaturation at 95°C for 2 min, samples were subjected to 6% urea-polyacrylamide gel electrophoresis and autoradiographed. To map the Pmda66 promoter, plasmid pGEM-T-Easy-Pmda66 (Table 1) was sequenced in parallel with oligonucleotide mda66PE, using a T7 sequencing kit (USB).
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′-3′)a | Restriction recognition site |
|---|---|---|
| hrc1 | ATTATTGAATTCTTGGGTTAGGGGGATTTTAAGGG | EcoRI |
| hrcA | CAAACGCATCTAACAAACTCTC | None |
| gro1 | ATTATTGGATCCAGGGATGATGATGCCTGAACTGG | BamHI |
| groFP | ATAAGGTTTGTTAATAACGCCCCTTTCTC | None |
| groEL-fwd | ATTACATACCATATGGCAAAAGAAATCAAATTTTC | NdeI |
| groEL-rev | ATATATCTCGAGCATCATGCCACCCATGCCTC | XhoI |
| groES-fwd | ATTACATACCATATGAAGTTTCAGCCATTAGGAG | NdeI |
| groES-rev | ATATATCTCGAGGTGTTTTTTGTGATCATGAC | XhoI |
| mda66PE | TGGTCAGTCAAGGTTTCATTG | None |
| mda66rev2 | ATCGTAGAACATGACCACTCCTTA | None |
| fla | GCATGAGAAGTTAAAGCGGC | None |
| fla2 | ATTATAGAATTCCCTAACATGCCCTTTAGAGGC | EcoRI |
Restriction sites added for cloning purposes are underlined.
Overexpression and purification of recombinant proteins.
His6-tagged recombinant HrcA and HspR proteins were overexpressed in freshly transformed E. coli BL21(DE3) cells and affinity purified as previously described (25, 31). For overexpression of His6-tagged recombinant GroES and GroEL proteins, the expression vectors pET22b-GroES and pET22b-GroEL (Table 1) were transformed separately into E. coli strain BL21(DE3). Overnight bacterial cultures were diluted 1:50 in 250 ml of LB medium, grown to an optical density at 600 nm of 0.5, induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 37°C, and then centrifuged. Cells were resuspended in 25 ml of ice-cold lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole; pH 8.0), incubated with 1 mg/ml lysozyme at 4°C for 30 min on a Tilt-Roll, disrupted with a French pressure cell (two cycles), incubated on ice for 15 min with 10 μg/ml of DNase I and 10 μg/ml of RNase A, and centrifuged to remove cellular debris (6,000 × g, 30 min, 4°C). The soluble fractions were mixed with 750 μl of 50% Ni2+-nitrilotriacetic acid slurry (QIAGEN, Inc.) and incubated for 90 min at 4°C on a Tilt-Roll. Two 10-ml polypropylene columns were then packed with the samples and washed twice with 7.5 ml of wash buffer 20 (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole; pH 8.0) and once with 7.5 ml of wash buffer 50 (50 mM NaH2PO4, 300 mM NaCl, 50 mM imidazole; pH 8.0). Recombinant proteins were eluted by applying 1 ml of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole; pH 8.0) three times, dialyzed against storage buffer (50 mM Tris [pH 8.0], 50 mM KCl, 10 mM MgCl2, 0.01% NP-40 Igepal) containing 50% glycerol, and stored at −20°C.
DNase I footprinting.
The promoter regions of the groESL, hrcA-grpE-dnaK, mda66, and flaB genes were PCR amplified with oligonucleotide pairs gro1/groFP, hrcA/hrcA1, mda66PE/mda66rev2, and fla2/fla (Table 2), respectively, from chromosomal DNA of H. pylori G27 and cloned into the pGEM-T Easy vector, resulting in the plasmids listed in Table 1. Promoter DNA fragments obtained by NcoI (for Pgro, Pmda66, and PflaB) or SalI (for Phrc) digestion were 5′ end labeled with [γ-32P]ATP and T4 polynucleotide kinase at one extremity and gel purified, and approximately 10,000 cpm of each probe was used for footprinting experiments. Labeled DNA probes were incubated with a purified protein(s) in 50 μl of footprinting buffer (10 mM Tris-HCl [pH 8.0], 50 mM NaCl, 10 mM KCl, 5 mM MgCl2, 50 mM dithiothreitol, 0.01% NP-40, 10% glycerol) containing 250 ng of sonicated salmon sperm DNA as a nonspecific competitor for 15 min at room temperature. Two microliters of DNase I (0.01 U/μl), freshly diluted in footprinting buffer containing 5 mM CaCl2, was added, and incubation was continued for 75 s at room temperature. DNase I digestion was stopped by addition of 140 μl of stop buffer (192 mM sodium acetate, 32 mM EDTA, 0.14% sodium dodecyl sulfate, 64 μg/ml sonicated salmon sperm DNA). Samples were phenol-chloroform extracted, ethanol precipitated, resuspended in 10 μl of sequencing loading buffer, denatured at 95°C for 2 min, subjected to 6% polyacrylamide-urea gel electrophoresis, and autoradiographed.
RESULTS
HrcA binds CIRCE-like sequences mapping within the Pgro and Phrc promoter regions.
The structural organization of the three HspR-regulated operons and the respective promoters is schematically depicted in Fig. 1. While transcription of the Pcbp promoter is controlled solely by HspR, transcriptional repression of Pgro and Phrc requires both the HspR and HrcA repressors (28). In vitro DNase I footprinting experiments with a purified recombinant protein allowed identification of the HspR binding sites within the three promoters (31). In contrast, the binding sites of the HrcA protein have not yet been determined, due to difficulties in obtaining a recombinant purified protein. However, very recently we purified a recombinant HrcA protein from heat-shocked E. coli cells and used it in filter binding assays (25).
FIG. 1.

Structural organization of H. pylori chaperone genes. The gray arrows indicate chaperone genes and an open reading frame with a putative helicase-like function, and the black arrows indicate regulatory genes. All genes are labeled based on the genome sequence published by Tomb et al. (33). Pgro transcribes one bicistronic mRNA encoding the GroESL chaperonin machinery (32); Phrc transcribes a tricistronic mRNA encoding the membrane-associated repressor HrcA, the chaperone DnaK, and its cochaperone GrpE (15, 25, 31); and Pcbp transcribes a tricistronic mRNA encoding the DnaJ homologue CbpA (34), the HspR repressor, and a putative DNA helicase (4, 31).
To define the architectural organization of the HrcA and HspR binding sites at coregulated promoters, we purified both recombinant His6-tagged proteins from E. coli as previously described (25, 31) and used them in DNase I footprinting assays for the Pgro and Phrc promoters. Figure 2 shows that after addition of HspR (20 nM), large protected regions and bands of enhanced DNase I sensitivity appeared for the Pgro (Fig. 2A, lane 4) and Phrc (Fig. 2C, lane 4) promoter probes. In accordance with previous observations (31), the protected regions extended from position −43 to position −120 and from position −78 to position −149 with respect to the transcriptional start sites of the Pgro and Phrc promoters, respectively. In contrast, in the presence of HrcA (at a concentration of 18 nM or higher), three bands of DNase I hypersensitivity and proximal regions of weak protection were detected for the Pgro promoter probe (Fig. 2B, lanes 4 to 8). While two bands of DNase I hypersensitivity mapped within the area of protection spanning from position −13 to position 16, the other band mapped at position 58 with respect to the transcriptional start site (Fig. 2B). Addition of HrcA to the Phrc promoter probe resulted in a single band of DNase I hypersensitivity in the middle of a protected region spanning from position −34 to position −59 (Fig. 2D, lanes 4 to 8).
FIG. 2.

DNase I footprinting of HspR and HrcA on Pgro and Phrc promoters. Specific DNA probes for Pgro (A and B) and Phrc (C and D) fragments, end labeled in their noncoding strands, were incubated with increasing amounts of recombinant HspR and HrcA proteins. (A and C) Lanes 1 to 8, 0, 10, 15, 20, 30, 40, 60, and 100 nM HspR added, respectively. (B) Lanes 1 to 8, 0, 5, 10, 20, 30, 60, 90, and 180 nM HrcA added, respectively. (D) Lanes 1 to 8, 0, 1.5, 3, 6, 9, 12, 18, and 24 nM HrcA added, respectively. The open boxes on the right indicate the regions of DNase I protection, while the arrowheads indicate bands of hypersensitivity to DNase I digestion. On the left in each panel, the −10 and −35 regions and the transcriptional start site (bent arrow) are indicated, and the open reading frames are indicated by vertical open arrows. Higher-resolution mapping of HrcA binding at the Pgro promoter was carried out by using the same probe end labeled at the opposite extremity (data not shown).
Figure 3 shows the sequences of the DNA regions that are protected from DNase I digestion by HspR and HrcA and the positions of the DNase I-hypersensitive sites on the two promoters. The HrcA binding sites map to regions with sequence similarity to the B. subtilis CIRCE consensus motif (TTAGCACTC-N9-GAGTGCTAA) proposed by Narberhaus and Bahl (21). Sequences with similarities to the HAIR consensus motif were found in the middle of the HspR binding regions (2, 7, 11). We thus concluded that while the HspR repressor binds upstream of the Pgro and Phrc promoter elements, the HrcA regulator binds to regions overlapping the corresponding −10 and −35 hexamers. Most likely, HrcA represses transcription by interfering directly with the binding of RNA polymerase to these promoter elements. Notably, the distances between the HspR and HrcA binding sites on the Pgro and Phrc promoters are 27 and 18 bp, respectively.
FIG. 3.

Features of the Pgro and Phrc promoter sequences. For each promoter, the numbers refer to the positions with respect to the transcriptional start site (position 1), and the −10 and −35 promoter elements are in boldface type and underlined. HspR and HrcA binding sites in the Pgro and Phrc promoters are enclosed in boxes with solid and dashed lines, respectively. The shaded boldface type indicates sites of hypersensitivity to DNase I digestion after binding of HspR on the coding (Fig. 2) and noncoding DNA strands (31). Sites of DNase I hypersensitivity after binding of HrcA are indicated by a black background (Fig. 2). Known HAIR and CIRCE-like sequences are shown, and nucleotide similarities in the Pgro and Phrc promoters are shaded and double underlined and are indicated by converging arrows, respectively. At the bottom, the consensus sequences of HAIR and CIRCE elements are compared. The H. pylori CIRCE consensus sequence has been defined by alignment of the two HrcA binding sites on Pgro and Phrc.
To study possible interactions of HspR and HrcA, we assayed the DNA binding activities of both proteins under competitive conditions. Addition of increasing amounts of HrcA after binding of HspR to the Pgro and Phrc promoter probes resulted in footprinting patterns similar to those shown in Fig. 2 (data not shown). Similar results were obtained when HspR was added to HrcA bound to the same promoter probes. Consequently, we concluded that under the in vitro conditions used by us, the two regulatory proteins bind to their operators in an independent manner.
GroESL enhances binding of HrcA and HspR to the Phrc promoter in vitro.
Early attempts to purify HrcA from E. coli cells were severely hampered by toxicity and/or insolubility of the overexpressed protein. However, these effects were alleviated by induction of HrcA expression at 42°C, suggesting that chaperone proteins were necessary for proper expression and folding of the recombinant HrcA (25). In addition, we observed that the DNA binding activity of the purified recombinant HrcA declined rapidly (within a few days) during storage at −20°C, indicative of loss of folding (not shown). Moreover, as mentioned above, in other bacterial species, the binding activity of the heat shock repressors is stimulated by the chaperone systems that they control. Consequently, we decided to assess the ability of the purified H. pylori GroESL chaperone machinery to influence binding of HrcA and HspR to the Phrc promoter.
To do this, we first assessed by footprint analysis the effect of GroES and GroEL on the binding activity of HrcA on the Phrc promoter probe. The results showed that while addition of increasing amounts of each of these proteins in the absence of ATP resulted in no changes in the binding of HrcA on the Phrc promoter, addition of GroEL and ATP resulted in slight enhancement of HrcA binding (data not shown). Finally, the binding of HrcA and HspR was assessed by footprint analysis using both the GroES and GroEL chaperones in the presence of ATP (Fig. 4). With the addition of HrcA, the DNase I-hypersensitive site at position −45 of the Phrc promoter, indicative of HrcA binding, was detected with 30 nM protein and clearly established with 120 nM protein (Fig. 4A, lanes 4 to 6). In the presence of GroESL and ATP, the same hypersensitive site was clearly detected with 7.5 and 15 nM HrcA (Fig. 4A, lanes 8 and 9), indicative of at least a 10-fold increase in the affinity of HrcA for its binding site. Moreover, the intensity of the band increased with increasing amounts of HrcA, also showing the two expected flanking areas of DNase I protection. It is likely that interactions between HrcA and GroESL improve the folding of HrcA and increase its affinity for DNA. Similarly, binding of HspR to the same promoter probe in the presence of GroESL and ATP revealed a slight increase in the patterns of DNase I protection (Fig. 4B, compare lanes 5 and 11), suggesting that binding of HspR could be improved by the action of GroESL. By contrast, incubation of the promoter probes with only GroESL resulted in no changes in the pattern of DNase I digestion (Fig. 4, compare lanes 7 and lanes 1). These data suggest that GroESL directly interacts with HrcA and possibly with HspR to increase their DNA binding affinities for the operators, contributing to the transcriptional repression of the regulated promoters.
FIG. 4.

Effect of GroESL chaperonin on the binding of HrcA and HspR to the Phrc promoter. (A) DNase I footprinting analysis of HrcA on the Phrc promoter in the absence (left panel) and in the presence (right panel) of purified recombinant GroESL complex. A specific end-labeled Phrc fragment was incubated with increasing amounts of purified His-HrcA. Lanes 1 to 6, 0, 7.5, 15, 30, 60, and 120 nM His-HrcA, respectively (in each reaction 240 nM bovine serum albumin was added); lanes 7 to 12, 0, 7.5, 15, 30, 60, and 120 nM His-HrcA, respectively (in each reaction 240 nM GroESL complex and 500 μM ATP were added). (B) DNase I footprinting analysis of HspR on the Phrc promoter in the absence (left panel) and in the presence (right panel) of purified recombinant GroESL complex. A specific end-labeled Phrc fragment was incubated with increasing amounts of purified His-HspR. Lanes 1 to 6, 0, 25, 50, 100, 200, and 400 nM HspR-His, respectively (in each reaction 800 nM bovine serum albumin was added); lanes 7 to 12, 0, 25, 50, 100, 200, and 400 nM HspR-His, respectively (in each reaction 800 nM GroESL complex and 500 μM ATP were added). The open boxes on the right indicate the regions of DNase I protection, while the arrowheads indicate bands of hypersensitivity to DNase I digestion.
HrcA and HspR transcriptome analyses.
To identify genes regulated by HrcA, HspR, and both regulators, we employed DNA macroarray analysis of RNA isolated from exponentially growing wild-type and mutant cells. The ΔhrcA/wild-type, ΔhspR/wild-type, and ΔhrcA-ΔhspR/wild-type ratios from three hybridization experiments were evaluated to determine statistical significance (P ≤ 0.05) and compared as described in Materials and Methods. Overall, 43 genes were up- or down-regulated at least 1.5-fold in the double-mutant strain (ΔhrcA ΔhspR) or in one of the single-mutant strains (ΔhrcA or ΔhspR), and the results are summarized in Table 3. Fourteen of 43 genes were up-regulated, while 29 genes were down-regulated.
TABLE 3.
Results of the DNA macroarray hybridization experiments
| Genome open reading framea | Fold change in ΔhrcA | Fold change in ΔhspR | Fold change in ΔhrcA-ΔhspR | Regulationb | Annotation (gene name) |
|---|---|---|---|---|---|
| Repressed by HrcA | |||||
| HP0229 | 1.64c | −1.21 | 2.49 | Outer membrane protein (omp6) | |
| HP0630 | 1.65 | −1.56 | 1.54 | Modulator of drug activity (mda66) | |
| HP0916 | 1.57 | 1.01 | 1.51 | Iron-regulated outer membrane protein (frpB) | |
| HP1177 | 1.29 | −1.56 | 1.61 | Outer membrane protein (omp27) | |
| Repressed by HspR | |||||
| HP0692 | −1.24 | 1.53 | −1.67 | 3-Oxoadipate coenzyme A transferase subunit B (yxjE) | |
| HP1024 | −1.10 | 10.09 | 4.12 | Cochaperone-curved DNA binding protein A (cbpA) | |
| HP1025 | 1.15 | −5.50 | −3.79 | Putative heat shock protein (hspR) | |
| HP1026 | 1.10 | 3.58 | 2.35 | Conserved hypothetical helicase-like protein | |
| Repressed by HrcA/HspR | |||||
| HP0010 | 1.28 | 2.14 | 1.76 | Chaperone and heat shock protein (groEL) | |
| HP0011 | 1.47 | 2.26 | 1.98 | Cochaperone (groES) | |
| HP0109 | −1.39 | 3.87 | −1.22 | Chaperone and heat shock protein 70 (dnaK) | |
| HP0110 | −1.39 | 4.91 | −1.23 | Cochaperone and heat shock protein (grpE) | |
| HP0722 | 3.45 | 1.84 | 2.98 | Outer membrane protein (omp16) | |
| HP0724 | 4.88 | 2.49 | 3.56 | Anaerobic C4-dicarboxylate transport protein (dcuA) | |
| Induced by HrcA | |||||
| HP0295 | −1.96 | −1.23 | −3.40 | σ54 | Flagellin B homolog (fla) |
| HP0367 | −1.66 | −1.33 | −2.52 | Predicted coding region | |
| HP0472 | −3.77 | 1.03 | −2.62 | σ28 | Outer membrane protein (omp11) |
| HP0601 | −3.31 | −1.22 | −1.88 | σ28 | Flagellin A (flaA) |
| HP0751 | −2.56 | −1.06 | −2.51 | σ28 | Polar flagellin (flaG) |
| HP0752 | −2.96 | −1.10 | −2.86 | σ28 | Flagellar hook-associated protein 2 (fliD) |
| HP0753 | −1.75 | −1.02 | −1.75 | σ28 | Flagellar protein (fliS) |
| HP0868 | −1.68 | −1.33 | −2.20 | σ54 | Predicted coding region |
| HP0869 | −1.58 | −1.05 | −1.89 | σ54 | Hydrogenase expression/formation protein (hypA) |
| HP0907 | −1.75 | −1.14 | −1.87 | σ54 | Hook assembly protein, flagella (flgD) |
| HP0908 | −1.47 | −1.12 | −2.03 | σ54 | Flagellar hook (flgE) |
| HP1001 | −1.55 | −1.24 | −1.50 | Predicted coding region | |
| HP1052 | −1.87 | 1.05 | −1.45 | σ28 | UDP-3-O-acyl-N-acetylglcosamine deacetylase (envA) |
| HP1120 | −1.52 | −1.12 | −1.91 | σ54 | Predicted coding region |
| HP1122 | −1.60 | −1.01 | −1.46 | Anti-sigma factor (flgM) | |
| HP1243 | −2.20 | 1.08 | −1.54 | Outer membrane protein (omp28) | |
| HP1440 | −1.55 | −1.21 | −1.46 | Predicted coding region | |
| Induced by HspR | |||||
| HP0009 | −1.24 | −1.77 | −1.46 | Outer membrane protein (omp1) | |
| HP0556 | 1.12 | −1.53 | −1.44 | Predicted coding region | |
| Induced by HcrA/HspR | |||||
| HP0115 | −3.08 | −1.32 | −22.92 | σ54 | Flagellin B (flaB) |
| HP0119 | −1.90 | −1.43 | −1.40 | Predicted coding region | |
| HP0366 | −2.05 | −1.59 | −3.86 | Spore coat polysaccharide biosynthesis protein C | |
| HP0870 | −2.78 | −1.45 | −9.71 | σ54 | Flagellar hook (flgE) |
| HP0906 | −2.85 | −1.80 | −61.08 | σ54 | Flagellar hook filament, fliK |
| HP1076 | −2.27 | −1.84 | −2.65 | σ54 | Predicted coding region |
| HP1119 | −3.08 | −1.57 | −11.65 | σ54 | Flagellar hook-associated protein 1 (flgK) |
| HP1188 | −1.69 | −1.48 | −1.51 | Predicted coding region | |
| HP1233 | −2.51 | −2.00 | −5.96 | σ54 | Predicted coding region |
| HP1559 | −1.53 | −1.55 | −1.85 | Flagellar basal body rod protein (proximal rod protein) (flgB) |
As expected, most of the genes previously shown to be under transcriptional control of HrcA and/or HspR were detected in this analysis. For instance, transcription of the cbpA-hspR-helicase operon (HP1024 to HP1026) was derepressed in both the ΔhspR and ΔhspR-ΔhrcA mutant strains but not in the ΔhrcA mutant, confirming the transcriptional repression of this operon by HspR (28, 31). Similarly, transcription of the groES-groEL operon (HP0010 and HP0011) and of the grpE and dnaK genes (HP0109 and HP0110) was clearly derepressed in the ΔhspR and ΔhspR-ΔhrcA mutants. Transcription of these genes was apparently not affected in the ΔhrcA mutant. The latter observation appears to contrast with previous studies which demonstrated that repression of transcription of these genes is dependent on both HrcA and HspR (28). This discrepancy might have resulted from different experimental designs. While the macroarray technique employed in this study uses open reading frames to measure steady-state levels of cellular transcripts, previous studies focused on primer extension and S1 nuclease mapping analyses which specifically detect RNA 5′ regions. In the case of groESL mRNA, the particularly high stability of this RNA (29) might make it more difficult to detect significant differences in the RNA amounts. Nevertheless, there seems to be slight up-regulation of the groESL transcript in the ΔhrcA mutant. However, transcription of two new genes, the dcuA (HP0724; coding for an anaerobic C4-dicarboxylate transport protein) and omp16 (HP0722; coding for an outer membrane protein) genes, was clearly derepressed in all three mutant strains, suggesting that repression of transcription is exerted by both regulatory proteins, HspR and HrcA. By contrast, transcription of the omp6 and omp27 (coding for putative membrane proteins), mda66 (coding for a putative NADPH-quinone reductase), and frpB (coding for an outer membrane protein) genes was found to be specifically derepressed in the ΔhrcA and ΔhrcA-ΔhspR mutant strains, indicating that there is negative regulation by HrcA alone. Surprisingly, the same analysis highlighted the finding that transcription of 29 genes was decreased in the ΔhspR-ΔhrcA double mutant and/or in the ΔhrcA and ΔhspR mutants, suggesting a positive role of HrcA and HspR in transcription of these genes (Table 3). Specifically, transcription of two genes (omp1 and HP0556) was down-regulated in the absence of HspR, transcription of 17 genes was down-regulated in the absence of HrcA, and transcription of 10 genes was down-regulated in the absence of both HrcA and HspR. Intriguingly, the majority of these positively regulated genes belong to the class of alternative σ54 and σ28 transcribed promoters, and 14 of the 29 down-regulated genes code for proteins involved in regulation and assembly of the flagellar apparatus.
Primer extension analysis of novel HrcA- and HspR-regulated genes.
Since suppressive as well as enhancing effects of HspR and/or HrcA on transcript abundance were revealed by transcriptome analyses, we selected the mda66 and flaB genes as opposite representative cases to study in detail the transcription regulation exerted by HspR and HrcA. Transcription was assessed by primer extension analysis with RNA extracted from wild-type strain G27 and ΔhrcA, ΔhspR, and ΔhspR-ΔhrcA mutant strains grown at 37°C.
The mda66 transcriptional start site was mapped at a position 25 nucleotides upstream of the ATG translation start codon and is preceded by a putative −10 region (TAAAAT), suggesting that mda66 is transcribed from a promoter (Pmda66) recognized by the RNA polymerase containing the vegetative sigma factor σ80 (Fig. 5A). In comparison to the wild-type strain (Fig. 5A, lane 1), the amount of transcript was increased in both the ΔhrcA and ΔhrcA-ΔhspR mutants (Fig. 5A, compare lane 1 to lanes 2 and 4). Interestingly, mda66 transcription appeared to be down-regulated in the ΔhspR mutant (lane 3), possibly due to increased HrcA and GroESL levels arising from transcriptional derepression of Phrc and Pgro, which are known to be under negative control of HspR (28).
FIG. 5.

Primer extension analysis of the promoters of the mda66 and flaB genes. Total RNAs isolated from H. pylori strains G27 (lane 1), G27(hrcA::km) (lane 2), G27(hspR::km) (lane 3), and G27(hrcA::km hspR::cm) (lane 4) were hybridized to the radiolabeled oligonucleotides mda66PE (A) and fla (B) (Table 2) and elongated with reverse transcriptase. The positions of elongated products are indicated on the right by arrows. The corresponding cloned promoters were sequenced in parallel with the primers used in the primer extension reactions, and the nucleotide sequences upstream of the transcriptional start sites are indicated on the left; the −10 (A) and −12 and −24 (B) motifs are indicated by the vertical bars, and the nucleotides corresponding to position 1 initiation sites are indicated by bent arrows.
It was previously reported that transcription of the flaB gene starts 25 nucleotides upstream from the ATG start codon at a σ54-dependent promoter (30). Transcription from this promoter resulted in a marked reduction in the amount of transcript in the ΔhrcA and ΔhspR mutant strains (Fig. 5B, lanes 2 and 3) and was essentially undetectable in the double mutant (lane 4).
We concluded that while transcription from the Pmda66 promoter appears to be repressed by HrcA, transcription from the PflaB promoter appears to be positively controlled by both regulators (HrcA and HspR), thus confirming the differential regulation patterns observed in the transcriptome analysis. To test whether HspR and HrcA interact with these promoters directly, we carried out DNase I footprinting with labeled DNA fragments encompassing the Pmda66 and PflaB promoters. Surprisingly, after addition of increasing amounts of HrcA and HspR, no evidence for DNA binding was obtained (data not shown), suggesting that neither of the two proteins binds to these promoters. Therefore, the regulation of these genes is likely to be due to indirect mechanisms.
HrcA and HspR are required for H. pylori motility.
It has been reported that a ΔhspR mutant strain of H. pylori is nonmotile (30) and that the same phenotype was observed in the hspR-deficient strain of the closely related microorganism Campylobacter jejuni (1). We also tested the motility of the ΔhrcA mutant strain of H. pylori by assaying the ability of the cells to spread on soft agar plates. To do this, cells were spotted onto low-concentration agar plates and incubated for 72 h at 37°C under microaerophilic conditions. Figure 6 shows that the areas of spreading of the ΔhrcA, ΔhspR and ΔhrcA-ΔhspR strains were severely reduced compared with the area covered by the wild-type strain, thus showing a loss of motility functions. Complementation of the HrcA function restored the spreading phenotype to a level similar to that of the wild-type strain. Consequently, we concluded that both heat shock regulators, HspR and HrcA, are required for H. pylori motility functions.
FIG. 6.
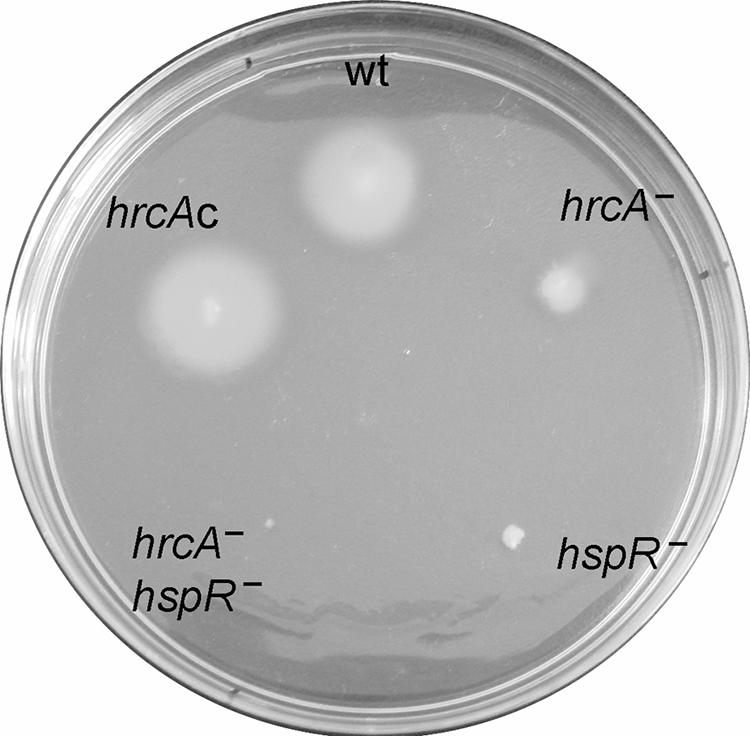
Bacterial motility assay. Bacteria were stab inoculated with a pipette tip into semisolid agar plates and incubated for 72 h at 37°C under microaerophilic conditions. The strains used in this assay are indicated as follows: wt, G27; hrcA−, G27(hrcA::km); hspR−, G27(hspR::km); hrcA− hspR−, G27(hrcA::km hspR::cm); and hrcAc, G27(hrcA-HA).
DISCUSSION
Bacteria respond to stress conditions by synthesizing chaperones, which protect the cells from damage by preventing protein denaturation, aggregation, or misfolding. E. coli and most other gram-negative bacteria use specialized σ factors, which become activated after exposure to stress and direct the RNA polymerase to their target promoters, whereas a subgroup of gram-negative bacteria and all gram-positive bacteria use specialized repressors which become inactivated under stress conditions, leading to derepression of target promoters. We have previously demonstrated that the major chaperone-encoding operons of H. pylori are negatively regulated by HspR, the homologue of the repressor of the dnaK operon of Streptomyces species (31). In addition, two of the HspR-regulated operons, groESL and hrcA-grpE-dnaK, are also regulated by HrcA (28), the homologue of the repressor of the groESL operon of B. subtilis. The presence of both regulators is therefore necessary for maintaining Pgro and Phrc in the repressed state. HspR binds to large operators located far upstream from these promoters (31) (Fig. 2A and 2C), while the HrcA operators overlap the core promoter regions (Fig. 2B and 2D). In agreement with a previous hypothesis (28), HrcA binds to sequences that include the CIRCE-related inverted repeats centered at positions 9 and −42 of the Pgro and Phrc promoters, respectively (Fig. 3). It is likely that binding of HrcA to these DNA elements represses transcription by steric interference of RNA polymerase binding. Furthermore, the discovery that the affinity of HrcA for its operator is considerably increased in the presence of GroESL (Fig. 4) parallels observations with HrcA proteins from B. subtilis and C. trachomatis, which showed a positive influence of GroE on the DNA binding activity of the repressor (24, 35). According to a “titration model” proposed for the B. subtilis HrcA repressor (19), GroE might interact with H. pylori HrcA to aid its folding and enhance its DNA binding activity, thereby efficiently assisting in the repression of transcription of the target promoters. In the presence of stress stimuli, the GroE chaperonin would be titrated away by increasing levels of misfolded proteins, relieving HrcA transcriptional repression of the heat shock promoters. However, HrcA-mediated regulation depends on the presence of HspR, as demonstrated by deletion of the hspR gene and deletion of the HspR binding site, both of which lead to promoter deregulation (28). It should therefore be assumed that binding of HrcA to its target sequences is not efficient in vivo for repressing transcription in the absence of a functional HspR repressor. The reason for this dependence might be found in chaperone-mediated protein-protein interactions between the two repressors, which may be a prerequisite for the formation of a stable repression-competent complex. While GroE chaperonin stimulates binding of HrcA to its target, no effects on the binding of HspR have been detected (Fig. 4). The possibility that the DnaK-DnaJ-GrpE chaperone system is involved in the formation of a stable HspR-HrcA repression complex should be considered.
As shown by transcriptome analysis, HspR and HrcA affect transcription of 43 genes in either a positive or negative fashion (Table 3). Of the 29 positively regulated genes, 14 code for proteins involved in regulation and assembly of the flagellar apparatus. Accordingly, loss of motility functions was observed for both mutants (Fig. 6), and transcription of the flaB gene was down-regulated both in single mutants and in the HspR-HrcA double mutant (Fig. 5). No binding of HrcA and/or HspR was observed on the promoter, suggesting that positive regulation of this gene is due to indirect mechanisms. Although the possibility was not investigated further, we speculated that induction of chaperone proteins alters the assembly of the flagellar apparatus and/or increases the activity of specialized anti-sigma factors, such as FlgM (5), which in turn establishes negative feedback for the programmed transcription of flagellar and motility genes (22, 30). In H. pylori, motility is associated with pathogenesis, and colonization of the gastrointestinal tract depends on the presence of flagellins and heat shock proteins (5, 22). Furthermore, interconnections between the heat shock response and motility have been observed in the closely related bacterium C. jejuni (1). In fact, in this bacterium, HspR also controls the expression of genes involved in oxidative stress and motility functions in an indirect fashion (1). Indirect mechanisms might also be responsible for the transcriptional control of other genes of the HspR/HrcA regulon, as highlighted by analysis of the Pmda66 promoter. Transcription from this promoter appeared to be repressed by HrcA, although the purified protein failed to bind to the promoter region (Fig. 5). Whether similar regulatory feedback mechanisms like those involved in the control of flagellar gene expression or other dedicated systems are active at this promoter has to be established. In this context it is interesting that mda66, coding for an NADPH-quinone reductase involved in the oxidative stress response, and the genes coding for proteins that localize to the outer membrane of the bacterium (Omp6, Omp27, and FrpB) appear to be coregulated by the inner membrane-associated protein HrcA, suggesting a putative link between heat shock and oxidative stress responses throughout the bacterial membranes.
Our results support a model in which, either independently or cooperatively, HspR and HrcA control transcription of chaperone genes by binding to the corresponding promoter regions (Fig. 7). Of crucial importance is the maintenance of chaperone protein homeostasis, as its alteration determines changes in transcription of several genes, including genes involved in motility and flagellar functions. For instance, enhanced synthesis of one or more components of the HspR/HrcA regulon(s), such as the GroESL and/or DnaK chaperones, might alter the programmed assembly of the flagella or other cellular structures, which in turn establishes a proper transcriptional response.
FIG. 7.

Regulatory circuit of HspR and HrcA. The dashed lines indicate hypothetical interactions, and the solid lines indicate experimentally supported interactions.
Acknowledgments
We thank S. Romagnoli for critical reading of the manuscript and C. Mallia for editing the manuscript.
This work was supported in part by Novartis Vaccines (formerly Chiron) and by grants from the University of Bologna (ex60% and Strategic project) to V.S.
Footnotes
Published ahead of print on 10 August 2007.
REFERENCES
- 1.Andersen, M. T., L. Brondsted, B. M. Pearson, F. Mulholland, M. Parker, C. Pin, J. M. Wells, and H. Ingmer. 2005. Diverse roles for HspR in Campylobacter jejuni revealed by the proteome, transcriptome and phenotypic characterization of an hspR mutant. Microbiology 151:905-915. [DOI] [PubMed] [Google Scholar]
- 2.Bucca, G., A. M. Brassington, G. Hotchkiss, V. Mersinias, and C. P. Smith. 2003. Negative feedback regulation of dnaK, clpB and lon expression by the DnaK chaperone machine in Streptomyces coelicolor, identified by transcriptome and in vivo DnaK-depletion analysis. Mol. Microbiol. 50:153-166. [DOI] [PubMed] [Google Scholar]
- 3.Bucca, G., A. M. Brassington, H. J. Schonfeld, and C. P. Smith. 2000. The HspR regulon of Streptomyces coelicolor: a role for the DnaK chaperone as a transcriptional co-repressordagger. Mol. Microbiol. 38:1093-1103. [DOI] [PubMed] [Google Scholar]
- 4.Bucca, G., G. Ferina, A. M. Puglia, and C. P. Smith. 1995. The dnaK operon of Streptomyces coelicolor encodes a novel heat-shock protein which binds to the promoter region of the operon. Mol. Microbiol. 17:663-674. [DOI] [PubMed] [Google Scholar]
- 5.Colland, F., J. C. Rain, P. Gounon, A. Labigne, P. Legrain, and H. De Reuse. 2001. Identification of the Helicobacter pylori anti-sigma28 factor. Mol. Microbiol. 41:477-487. [DOI] [PubMed] [Google Scholar]
- 6.Danielli, A., D. Roncarati, I. Delany, V. Chiarini, R. Rappuoli, and V. Scarlato. 2006. In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. J. Bacteriol. 188:4654-4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delany, I., G. Spohn, R. Rappuoli, and V. Scarlato. 2002. In vitro selection of high affinity HspR-binding sites within the genome of Helicobacter pylori. Gene 283:63-69. [DOI] [PubMed] [Google Scholar]
- 8.Dunn, B. E., R. M. Roop II, C. C. Sung, S. A. Sharma, G. I. Perez-Perez, and M. J. Blaser. 1992. Identification and purification of a cpn60 heat shock protein homolog from Helicobacter pylori. Infect. Immun. 60:1946-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn, B. E., N. B. Vakil, B. G. Schneider, M. M. Miller, J. B. Zitzer, T. Peutz, and S. H. Phadnis. 1997. Localization of Helicobacter pylori urease and heat shock protein in human gastric biopsies. Infect. Immun. 65:1181-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans, D. J., Jr., D. G. Evans, L. Engstrand, and D. Y. Graham. 1992. Urease-associated heat shock protein of Helicobacter pylori. Infect. Immun. 60:2125-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandvalet, C., V. de Crecy-Lagard, and P. Mazodier. 1999. The ClpB ATPase of Streptomyces albus G belongs to the HspR heat shock regulon. Mol. Microbiol. 31:521-532. [DOI] [PubMed] [Google Scholar]
- 12.Grandvalet, C., P. Servant, and P. Mazodier. 1997. Disruption of hspR, the repressor gene of the dnaK operon in Streptomyces albus G. Mol. Microbiol. 23:77-84. [DOI] [PubMed] [Google Scholar]
- 13.Grodberg, J., and J. J. Dunn. 1988. ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol. 170:1245-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 15.Homuth, G., S. Domm, D. Kleiner, and W. Schumann. 2000. Transcriptional analysis of major heat shock genes of Helicobacter pylori. J. Bacteriol. 182:4257-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huesca, M., S. Borgia, P. Hoffman, and C. A. Lingwood. 1996. Acidic pH changes receptor binding specificity of Helicobacter pylori: a binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect. Immun. 64:2643-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kansau, I., F. Guillain, J. M. Thiberge, and A. Labigne. 1996. Nickel binding and immunological properties of the C-terminal domain of the Helicobacter pylori GroES homologue (HspA). Mol. Microbiol. 22:1013-1023. [DOI] [PubMed] [Google Scholar]
- 18.Macchia, G., A. Massone, D. Burroni, A. Covacci, S. Censini, and R. Rappuoli. 1993. The Hsp60 protein of Helicobacter pylori: structure and immune response in patients with gastroduodenal diseases. Mol. Microbiol. 9:645-652. [DOI] [PubMed] [Google Scholar]
- 19.Mogk, A., A. Volker, S. Engelmann, M. Hecker, W. Schumann, and U. Volker. 1998. Nonnative proteins induce expression of the Bacillus subtilis CIRCE regulon. J. Bacteriol. 180:2895-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narberhaus, F. 1999. Negative regulation of bacterial heat shock genes. Mol. Microbiol. 31:1-8. [DOI] [PubMed] [Google Scholar]
- 21.Narberhaus, F., and H. Bahl. 1992. Cloning, sequencing, and molecular analysis of the groESL operon of Clostridium acetobutylicum. J. Bacteriol. 174:3282-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niehus, E., H. Gressmann, F. Ye, R. Schlapbach, M. Dehio, C. Dehio, A. Stack, T. F. Meyer, S. Suerbaum, and C. Josenhans. 2004. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol. Microbiol. 52:947-961. [DOI] [PubMed] [Google Scholar]
- 23.Phadnis, S. H., M. H. Parlow, M. Levy, D. Ilver, C. M. Caulkins, J. B. Connors, and B. E. Dunn. 1996. Surface localization of Helicobacter pylori urease and a heat shock protein homolog requires bacterial autolysis. Infect. Immun. 64:905-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reischl, S., T. Wiegert, and W. Schumann. 2002. Isolation and analysis of mutant alleles of the Bacillus subtilis HrcA repressor with reduced dependency on GroE function. J. Biol. Chem. 277:32659-32667. [DOI] [PubMed] [Google Scholar]
- 25.Roncarati, D., G. Spohn, N. Tango, A. Danielli, I. Delany, and V. Scarlato. 2007. Expression, purification and characterization of the membrane-associated HrcA repressor protein of Helicobacter pylori. Protein Expr. Purif. 51:267-275. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 27.Schulz, A., and W. Schumann. 1996. hrcA, the first gene of the Bacillus subtilis dnaK operon encodes a negative regulator of class I heat shock genes. J. Bacteriol. 178:1088-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spohn, G., A. Danielli, D. Roncarati, I. Delany, R. Rappuoli, and V. Scarlato. 2004. Dual control of Helicobacter pylori heat shock gene transcription by HspR and HrcA. J. Bacteriol. 186:2956-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spohn, G., I. Delany, R. Rappuoli, and V. Scarlato. 2002. Characterization of the HspR-mediated stress response in Helicobacter pylori. J. Bacteriol. 184:2925-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spohn, G., and V. Scarlato. 1999. Motility of Helicobacter pylori is coordinately regulated by the transcriptional activator FlgR, an NtrC homolog. J. Bacteriol. 181:593-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spohn, G., and V. Scarlato. 1999. The autoregulatory HspR repressor protein governs chaperone gene transcription in Helicobacter pylori. Mol. Microbiol. 34:663-674. [DOI] [PubMed] [Google Scholar]
- 32.Suerbaum, S., J. M. Thiberge, I. Kansau, R. L. Ferrero, and A. Labigne. 1994. Helicobacter pylori hspA-hspB heat-shock gene cluster: nucleotide sequence, expression, putative function and immunogenicity. Mol. Microbiol. 14:959-974. [DOI] [PubMed] [Google Scholar]
- 33.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzegerald, N. Lee, M. D. Adams, E. K. Hickey, D. E. Berg, J. D. Gocayne, T. R. Utterback, J. D. Peterson, J. M. Kelley, M. D. Cotton, J. M. Weidman, C. Fujii, C. Bowman, L. Watthey, E. Wallin, W. S. Hayes, M. Borodovsky, P. D. Karp, H. O. Smith, C. M. Fraser, and J. C. Venter. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 34.Ueguchi, C., M. Kakeda, H. Yamada, and T. Mizuno. 1994. An analogue of the DnaJ molecular chaperone in Escherichia coli. Proc. Natl. Acad. Sci. USA 91:1054-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson, A. C., C. C. Wu, J. R. Yates III, and M. Tan. 2005. Chlamydial GroEL autoregulates its own expression through direct interactions with the HrcA repressor protein. J. Bacteriol. 187:7535-7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang, Z., S. Censini, P. F. Bayeli, J. L. Telford, N. Figura, R. Rappuoli, and A. Covacci. 1995. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect. Immun. 63:94-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuber, U., and W. Schumann. 1994. CIRCE, a novel heat shock element involved in regulation of heat shock operon dnaK of Bacillus subtilis. J. Bacteriol. 176:1359-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]