

Abstract
The Escherichia coli cell division protein FtsQ is a central component of the divisome. FtsQ is a bitopic membrane protein with a large C-terminal periplasmic domain. In this work we investigated the role of the transmembrane segment (TMS) that anchors FtsQ in the cytoplasmic membrane. A set of TMS mutants was made and analyzed for the ability to complement an ftsQ mutant. Study of the various steps involved in FtsQ biogenesis revealed that one mutant (L29/32R;V38P) failed to functionally insert into the membrane, whereas another mutant (L29/32R) was correctly assembled and interacted with FtsB and FtsL but failed to localize efficiently to the cell division site. Our results indicate that the FtsQ TMS plays a role in FtsQ localization to the division site.
Bacteria are capable of localizing integral membrane proteins (IMPs) to specific sites in the cytoplasmic membrane. A wealth of studies has shown specific localization patterns for IMPs involved in cell division, chemotaxis, sporulation, and cell wall synthesis in model organisms, such as Escherichia coli, Bacillus subtilis, Caulobacter crescentus, Staphylococcus aureus, and Streptococcus pneumoniae (14, 20, 21, 25, 31). It is not known how these IMPs find their destination: the Sec machinery that inserts IMPs into the cytoplasmic membrane is localized along the lateral wall in E. coli and B. subtilis, and proteins seem to insert in a random fashion into the membrane before being localized to their destination (5, 34). A diffusion and capture model has been proposed in which an IMP diffuses randomly through the membrane until it is captured at its destination by an as-yet-unknown factor (30). Such a factor could be the lipid composition of the membrane, as the distribution of lipids throughout the bacterial membrane is not homogeneous, with cardiolipin-enriched domains at the poles and the cell division site (23, 27, 28).
One of the best-studied sets of localizing IMPs consists of IMPs involved in E. coli cell division. These IMPs need to be targeted to mid-cell, where they will form part of the divisome, a complex consisting of 14 cytoplasmic (FtsZ, FtsA, and ZapA), inner membrane (ZipA, FtsE, FtsX, FtsK, FtsQ, FtsL, FtsB, FtsW, FtsI, and FtsN), and periplasmic (AmiC) proteins that form a ring and mediate cell constriction, synthesis of the septal cell wall, and cell separation (14). The divisome forms in two steps, starting with the assembly of the FtsZ ring followed by a well-defined time gap after which all the essential cell division IMPs, such as FtsK, FtsQ, FtsL, FtsB, FtsW, FtsI, and FtsN, assemble (1). The assembly of these proteins was long thought to occur in a hierarchical manner observing the order of proteins as listed. Recent studies using a premature targeting assay have revealed that various downstream proteins can recruit upstream proteins, which has led to a more comprehensive model in which FtsK recruits a subcomplex consisting of FtsQ, -L, and -B, which in turn recruits an FtsW/FtsI subcomplex, after which FtsN and AmiC are recruited (15, 16).
FtsQ plays a central role in the assembly of the divisome. FtsQ is a bitopic membrane protein consisting of a short cytoplasmic domain (residues 1 to 24), a fairly long transmembrane segment (TMS; residues 25 to 49), and a periplasmic domain (residues 50 to 276) (Fig. 1). Two-hybrid analyses have suggested that FtsQ interacts with itself, FtsA, -K, -X, -L, -B, -W, -I, and -N, and YmgF, a protein of unknown function (9, 11, 22). This number of interactions, 10, is greater than for any other division protein tested (a maximum of five interaction partners for FtsI, -N, and -L). The interaction with FtsA and FtsK depends on the N-terminal domain of FtsQ, whereas all other interactions require only the periplasmic domain of FtsQ (11, 22). This is in accordance with domain swap experiments that showed that the periplasmic domain of FtsQ is essential for FtsQ localization and recruitment of downstream proteins (2, 7, 19) but that the cytoplasmic domain and the TMS can be exchanged for unrelated domains. The proteolytically sensitive and unstructured γ-subdomain of the periplasmic domain (29) plays an important role in the interaction of FtsQ with FtsB and FtsL (17). This interaction is independent of localization to the cell division site and can be identified by coimmunoprecipitation (co-IP) experiments (3). Mutants in the α-subdomain of the periplasmic domain are affected in localization to the cell division site but are still capable of recruiting downstream proteins (17).
FIG. 1.

Schematic domain architecture of FtsQ. The cytoplasmic N-terminal domain (cyto), the TMS, and the periplasmic domain are indicated, with the subdomains α, β, and γ indicated in gray (subdomain assignments are according to reference 29). The amino acid sequence of the FtsQ TMS is shown, and the TMS sequences of the mutants studied with mutated amino acids are shown with changed amino acids in bold. wt, wild type.
Since the original finding that the FtsQ TMS can be swapped for the unrelated TMS from MalF (19), it has been assumed that the TMS functions as a rather inert membrane anchor but does not fulfill any FtsQ-specific role. However, a closer inspection of the results of the swap experiments reveals that FtsQ containing the TMS of either MalF or FtsL is capable of complementing an ftsQ temperature-sensitive mutant but that these mutants grow as filaments at the restrictive temperature (19). Also, green fluorescent protein (GFP) fusions to FtsQ swap constructs containing the MalF TMS show a twofold reduction in localization efficiency compared to wild-type FtsQ (7). In both studies, the capacity of swap constructs to support growth was used as the major criterion for functionality, but these results also support a role for the FtsQ TMS in FtsQ function, as the swap constructs are not fully efficient in complementation and localization. More evidence for a role for the FtsQ TMS in FtsQ function has come from two recent reports. First, a ΔftsK strain can be rescued by overexpression of ftsQ (13). Rescue is dependent on the N-terminal domain and TMS of FtsQ, as a construct that overexpressed these domains fused to the periplasmic domain of FtsL (QQL) also rescued ΔftsK, whereas a construct that contained the N-terminal domain and TMS of MalF fused to the periplasmic domain of FtsQ (FFQ) did not (13). Second, a single amino acid substitution in the FtsQ periplasmic domain (V92D) causes FtsQ to localize less efficiently, but in the presence of this mutant a swap of the cytoplasmic domain and TMS of FtsQ for those of MalF is no longer tolerated and localization is lost completely. This finding points to a role for the first part of FtsQ, comprising the TMS, in protein localization (17). Additional evidence for a potential role of a TMS in cell division protein localization comes from the finding that the TMS of FtsI (PBP3)—which cannot be swapped (19)—is capable of localization to the cell division site on its own (40).
In this paper we address the role of the FtsQ TMS in more detail. We have generated a set of TMS mutants that are either not affected, partially defective, or fully defective in function. The deficiencies were analyzed by testing the capabilities of the mutant proteins to efficiently incorporate into the membrane, to localize to the cell division site, and to interact with cell division proteins FtsB and FtsL. We show that the loss of function of our partially defective mutant is caused by a loss of localization to the division site, consistent with a role for the FtsQ TMS in division site localization.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
Strains and plasmids are listed in Table 1. Strain Top10F′ was used for routine plasmid handling and expression of ftsQ from pBAD24, strain MJC129 was used for complementation experiments, strain LMC500 was used for GFP-FtsQ localization, and strain MRE600 was used to obtain translation lysate for in vitro translation experiments.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli strains | ||
| MC4100 | F−araD139 ΔlacU169 relA1 rpsL150 thi mot flb5301 deoC7 ptsF25 rbsR | Laboratory collection |
| MJC129 | KS272 ftsQ(Ts) recA::cat | 19 |
| Top10F′ | F′[lacIq Tn10(Tetr)] mcrA (mrr-hsdRMS-mcrBC) φ80dlacZM15 lacX74 recA1 araD139 (ara-leu)7697 galU galK rpsL endA1 nupG Strr | Invitrogen |
| LMC500 | F−araD139 Δ(argF-lac)U169 deoC1 flbB5301 ptsF25 rbsR relA1 rpsIL150 lysA1 | 35 |
| MRE600 | RNase 1 gene | Laboratory collection |
| JOE309 | MC4100 ara+ | 7 |
| JOE417 | JOE309 ftsQE14::kan/pJC10 | 6 |
| CR306-313 | JOE309 ftsQE14::kan Δ(λattL-lom)::bla lacIq pCR45-pCR50/pJC10 | This work (each strain carries a different integration plasmid encoding a Flag-tagged ftsQ version) |
| SU202 | lexA71::Tn5 recA+sulA211 Δ(lacIPOZYA)169/F′lacIqlacZΔM15::Tn9 op408/op+lacZ | 10 |
| MM39 | araD lacΔU1269 malEΔ444 Strr | Laboratory collection |
| Plasmidsa | ||
| pBS-FtsQ | bla ftsQ | Laboratory collection |
| pBAD24 | araC PBADbla | 18 |
| pLD104 | araC PBADFFQ bla | 19 |
| pBAD24-FtsQ* | araC PBAD ftsQ* bla | This work |
| pTHV038 | lacIqPtrc-downgfp-mut2 bla | 8 |
| pTHV039 | lacIqPtrc-downgfp-mut2-ftsQ bla | 1 |
| pTHV-FtsQ* | lacIqPtrc-downgfp-mut2-ftsQ* bla | This work |
| pC4Meth108FtsQ | lacIqPT7ftsQ(5′1-108)-Met4-HindIII | 37 |
| pC4Meth108FtsQ* | lacIqPT7ftsQ*(5′1-108)-Met4-HindIII | This work |
| pC4Meth108FtsQ-TAG40 | lacIqPT7ftsQTAG40(5′1-108)-Met4-HindIII | 37 |
| pC4Meth-108FtsQ*-TAG40 | lacIqPT7ftsQ*TAG40(5′1-108)-Met4-HindIII | This work |
| pDSW204 | IPTG-regulated promoter, Ampr | 39 |
| pJC10 | pBAD33-ftsQ | 39 |
| pCR45-pCR50 | pDSW204-ftsQ*flag3 | This work |
| pBLM100 | bla lexA′-km-malE | 32 |
| pALM148 | bla lexA408′-km-malE | 32 |
Plasmids with asterisks contain coding sequences for ftsQ, ftsQL29R, ftsQL32R, ftsQV38P, ftsQL29/32R, and ftsQL29/32R;V38P. A superscript 1-108 signifies the ftsQ codons present.
Bacteria were grown in Luria-Bertani broth (LB; per liter, 10 g tryptone, 5 g yeast extract, 5 g NaCl) unless indicated otherwise. Solid medium was LB with agar (1.5%, wt/vol). Antibiotics were used at appropriate concentrations: ampicillin (100 μg/ml), chloramphenicol (30 μg/ml), and tetracycline (12.5 μg/ml). l-Arabinose and d-glucose were used at concentrations indicated to modulate expression of ftsQ constructs from the pBAD24 expression vector.
For co-IP assays, the strains were grown in NZY liquid broth or agar (as described elsewhere [6]) supplemented with chloramphenicol (10 μg/ml) and ampicillin at low concentration (25 μg/ml) and with l-arabinose or d-glucose added at a 0.2% final concentration, to induce or repress, respectively, the expression of FtsQ from pJC10.
Plasmid construction.
Mutations were introduced into FtsQ using nested PCR and plasmids pC4Meth108FtsQ-TAG40 or pC4Meth108FtsQ (37) as templates. Primers FtsQfw, FtsQ108Bamrev, and nested PCR primers were also used (sequences available at http://www.bio.vu.nl/vakgroepen/molmic/publications.html). The FtsQ L29/32R;V38P triple mutant was generated by a nested PCR on a construct containing the L29R mutation. Mutated FtsQ derived from the pC4Meth108FtsQ plasmids was used to clone the FtsQ mutants into pBAD24-FtsQ using the internal Asp718 restriction site in ftsQ. The ftsQ coding sequence was cloned into pBAD24 from pBS-FtsQ using EcoRI and HindIII, resulting in pBAD24-FtsQ. The 5′ parts of ftsQ containing the mutations were cut with EcoRI-Asp718 from mutated pC4Meth108FtsQ* and inserted into EcoRI-Asp718-digested pBAD24-FtsQ. The resulting pBAD24-FtsQ* constructs were used as template for PCR of full-length ftsQ containing the mutations to construct in-frame fusions of gfp to the 5′ end of ftsQ, using primers FtsQEcoRIsense and FtsQHindIIIrev. The PCR products were cut with EcoRI-HindIII and inserted into EcoRI-HindIII-digested pTHV038. All plasmid constructs were verified by sequencing. pCR45 to pCR50 were constructed by inserting EcoRI-XbaI-digested PCR products, generated using pBAD24-FtsQ* as a template and primers EcoRI-ftsQfor and XbaI-ftsQrev, into EcoRI-XbaI-digested pDSW204.
FtsQ complementation assay.
Overnight cultures of strain MJC129 [ftsQ1(Ts)] harboring the pBAD24 vector encoding FtsQ or FtsQ mutants, or pLD104 encoding the FFQ domain swap construct (19), were diluted to an optical density at 660 nm (OD660) of 0.01 in prewarmed LB containing 0.02% arabinose and grown at 30°C (control) and 42°C, and growth was monitored. Also, exponentially growing cells from cultures grown at 30°C without arabinose were diluted and spotted on LB agar with appropriate antibiotics and arabinose (0.01%, wt/vol) and incubated at 30°C or 42°C.
In vitro transcription, translation, and cross-linking.
Truncated mRNA was prepared as described previously (33) from HindIII-linearized pC4Meth-108FtsQ*TAG40 plasmids encoding FtsQ or FtsQ mutants. In vitro translation was performed in an E. coli cell- and membrane-free extract, and inner membrane vesicles prepared from the E. coli wild-type strain MC4100 (18.6 μg total protein) were added to allow targeting as described elsewhere (37), generating [35S]methionine-labeled FtsQ nascent chains of 108 amino acids in length, with the photoactivatable cross-linker (Tmd)Phe incorporated at position 40 through suppression of the UAG stop codon. Photo-cross-linking was performed as described previously, and carbonate extraction was used to separate soluble and peripheral membrane proteins from integral membrane proteins (33). The cross-linking adducts were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and phosphorimaging as described elsewhere (33).
Assay for in vivo membrane assembly.
Strain Top10F′ harboring the pBAD24 vector encoding FtsQ or FtsQ mutants was grown to early log phase. Cells were induced for 3 min with 0.2% l-arabinose, labeled with [35S]methionine (30 μCi/ml) for 5 min, and converted to spheroplasts as described previously (12). Aliquots of the spheroplast suspension were incubated with or without proteinase K (0.4 mg/ml) for 1 h on ice. Subsequently, phenylmethylsulfonyl fluoride (0.33 mg/ml) was added to inhibit proteinase K, and proteins were acid precipitated and immunoprecipitated with antisera against OmpA, trigger factor, or FtsQ, as described elsewhere (38). Samples were analyzed by SDS-PAGE, and proteins were visualized by phosphorimaging.
GFP-FtsQ fluorescence microscopy.
Strain LMC500 harboring the pTHV-FtsQ* vector, encoding FtsQ or FtsQ mutants, was grown in glucose minimal medium (GB1) (8). GFP-FtsQ* induction was constitutive from the weakened Ptrc promoter without induction. Exponentially growing cells were fixed and mounted on 1% agarose pads, and microscopy was performed as described elsewhere (8), using an Olympus BX-60 fluorescence microscope equipped with a UPLANFI 100×/1.3 numerical-aperture oil objective and a Photometrics Coolsnap-fx charge-coupled-device camera.
FtsQflag3 immunoprecipitation.
Cells grown overnight at 37°C in NZY medium supplemented with arabinose and antibiotics were diluted 1:100 in 5 ml of fresh NZY medium in the presence of arabinose and grown for 2 h at 37°C. Cells were then diluted 1:100 a second time into 50 ml of NZY medium supplemented with isopropyl-β-d-galactopyranoside, to induce ftsQ*flag3, and 0.2% glucose, to deplete pJC10-encoded ftsQ, and then grown for 3 h at 37°C (OD600 of around 0.4). As a control, JOE417 cells were grown in the same way with depletion of ftsQ. A total extract of each strain was prepared from 1 ml of culture, and 45 ml of each culture was harvested by centrifugation at 8,000 × g for 10 min at 4°C and resuspended in 300 μl of buffer I (10 mM Tris-HCl, 150 mM NaCl, pH 7.4). Cells were disrupted by sonication and centrifuged for 10 min at 8,000 × g to eliminate cells and debris. Membranes were collected by ultracentrifugation at 100,000 × g for 1 h at 4°C and resuspended in 300 μl of buffer I and 30 μl of 10% n-dodecyl-β-d-maltopyranoside (DDM; Anatrace). Membrane proteins were solubilized by 30 min of incubation at 4°C with gentle shaking, and solubilized membrane proteins were separated from insoluble material by ultracentrifugation at 100,000 × g for 30 min at 4°C. Insoluble fraction pellets were resuspended in 300 μl of buffer I and stored at −20°C. The protein concentration of solubilized membrane fractions was determined by the bicinchoninic acid assay (Pierce). In a final volume of 100 μl, solubilized membrane proteins (200 μg total protein) were incubated with 20 μl of anti-Flag M2 affinity beads (Sigma), previously washed with 500 μl of buffer I with 0.1% DDM, for 2 h at 4°C with gentle shaking. Beads were pelleted by low-speed centrifugation at 2,000 rpm for 3 min. The flowthrough (100 μl) was stored at −20°C, and the beads were washed four times with 500 μl of buffer I with 0.1% DDM and then resuspended in 80 μl of 1× SDS-PAGE sample buffer and incubated at 95°C for 5 min. Five μl of each sample was analyzed by SDS-PAGE and Western blotting. Blots were probed with anti-Flag (monoclonal; Sigma) or anti-FtsL or anti-FtsB (polyclonals; laboratory collection) antibody.
RESULTS AND DISCUSSION
Construction of FtsQ TMS mutants and their ability to complement a temperature-sensitive ftsQ1 mutant.
In order to study the role of the FtsQ TMS, we introduced several mutations that would alter the hydrophobicity and charge of the TMS (leucine replaced by arginine) or that are predicted to disrupt the structure of the α-helical TMS (valine replaced by proline), as proline is known to introduce a kink into an α-helix (Fig. 1). Single replacements L29R and L32R were made, and both were combined to further decrease the hydrophobicity and increase the charge of the TMS. Also, the single replacement V38P was made, and this mutation was combined with the L29/32R double mutation to generate the L29/32R;V38P triple mutant. Importantly, all mutants were predicted to still form a TMS when analyzed by the topology prediction program TMHMM2.0 (24).
First, the functionality of the mutants was assayed. Wild-type FtsQ and the FtsQ mutants were introduced into the pBAD24 plasmid, resulting in FtsQ expression under the control of arabinose (18). The plasmids were introduced into the temperature-sensitive ftsQ1 mutant strain MJC129 and tested for their capability to restore growth at the nonpermissive temperature in the presence of arabinose, on a plate as well as in liquid culture. A control experiment with the original domain swap construct FFQ (19) confirmed that FFQ complements ftsQ1(Ts) both on plates and in liquid under our experimental conditions (Fig. 2). All single-residue replacements supported good growth in liquid as well as on plates, indicating that these FtsQ mutants are functional. The triple mutant L29/32R;V38P failed to complement the ftsQ1(Ts) strain, both on plates and in liquid culture, indicating that this mutant is not functional. Finally, the L29/32R double mutant failed to complement in liquid culture but did show growth, albeit reduced, on plates. The partial complementation on plates is probably caused by elevated FtsQ L29/32R levels throughout the membrane (see below), which results in sufficient FtsQ at the cell division site to support division. When tested in the ftsQ depletion background used for co-IP studies (see below), FtsQ L29/32R gave rise to formation of wrinkled colonies, again indicative of defective FtsQ (17), whereas the single mutants fully complemented and the triple mutant did not complement at all (not shown). We conclude that the L29/32R mutant is partially defective.
FIG. 2.

Complementation of an ftsQ1(Ts) strain by plasmid-encoded FtsQ. A: Complementation experiment in liquid culture. MJC129 [ftsQ1(Ts)] with different plasmids was grown overnight at the permissive temperature (30°C), diluted to an OD660 of 0.01 in prewarmed LB medium supplemented with 0.02% arabinose, and grown at the nonpermissive temperature (42°C). Plasmids were as follows: pBAD24 (⧫), pBAD24-FtsQ (▪), pLD104 (FFQ) (⋄), pBAD24-FtsQL29R (▵), pBAD24-FtsQL32R (▴), pBAD-FtsQL29/32R (○), pBAD24-FtsQL29/32R;V38P (•), and pBAD24-FtsQV38P (□). B: Complementation experiment on plates. Exponentially growing cells of MJC129 [ftsQ1(Ts)] with different plasmids, grown at 30°C, were spotted on plates containing 0.01% arabinose and incubated overnight at the permissive (30°C) or nonpermissive (42°C) temperature. wt, wild type.
The L29/32R;V38P triple mutant is not correctly assembled into the membrane.
Defects in FtsQ function due to a change in the TMS can have several causes. FtsQ is a target for the signal recognition particle pathway (36), which ensures correct targeting of FtsQ to the Sec translocon, where it is cotranslationally translocated (reviewed in reference 26). Defective targeting could result in folding and aggregation of FtsQ in the cytoplasm, which would render the protein translocation incompetent. Also, a change in the TMS could affect the interaction of FtsQ with the Sec translocon and associated proteins such as YidC, resulting in incorrect membrane insertion and translocation. Furthermore, defects could be caused by the inability of FtsQ to localize to the cell division site or the inability to recruit downstream proteins.
To analyze whether the FtsQ TMS mutants were still targeted to the Sec translocon, we made use of a site-specific cross-linking assay developed in our laboratory. Previously, we established that FtsQ nascent chains with a length of 108 amino acid residues and a photoactivatable cross-linker coupled to a Phe residue incorporated at position 40 (108FtsQ) are efficiently cross-linked to both SecY and YidC when the nascent chains are synthesized in the presence of E. coli inverted membrane vesicles (IMVs) (33, 37). Nascent chains of all FtsQ TMS mutants were synthesized, incubated with IMVs, and photo-cross-linked (Fig. 3). Carbonate extraction was used to separate soluble and peripherally associated membrane proteins from IMPs, and the IMP fraction was analyzed for the presence of cross-linked adducts to FtsQ nascent chains. The amount of cross-linked adduct was corrected for the amount of labeled nascent chain present in the reaction and compared with wild-type FtsQ. All constructs except the triple L29/32R;V38P mutant were found to be efficiently cross-linked to both SecY and YidC (Fig. 3A), with a notable increase in cross-linking efficiency for the V38P single mutant and a slight decrease in efficiency for the L29/32R double mutant. This result indicates that the triple mutant is severely affected in targeting to the membrane but that in all other cases the FtsQ TMS (which contains the photoactivatable cross-linker) is targeted to the translocon and presumably inserted into the membrane.
FIG. 3.

Targeting of the FtsQ mutants to SecY/YidC and translocation of the mutants. (A) In vitro cross-linking of FtsQ nascent chains to YidC and SecY. In vitro translation of nascent 108FtsQ was performed in an E. coli cell- and membrane-free extract in the presence of [35S]methionine. After addition of IMVs, targeting of ribosome-nascent complexes to the membrane was assayed using a photoactivatable cross-linker incorporated into the FtsQ TMS. Inner membrane proteins were separated from soluble and membrane-associated proteins by carbonate extraction, and the inner membrane protein fraction was analyzed for cross-linked adducts to 108FtsQ. Cross-linked adducts to SecY and YidC were identified, and the ratio of radiolabeled cross-linked adduct to the total amount of labeled nascent chains present in the reaction mixture was determined. Subsequently, cross-linking efficiency was calculated from these ratios, with cross-linking to 108FtsQ set at 100%. (B) Proteinase mapping of FtsQ translocation. FtsQ and the FtsQ mutants were expressed and pulse-labeled in Top10F cells. The cells were converted to spheroplasts and treated with proteinase K to monitor translocation of FtsQ across the cytoplasmic membrane. The samples were immunoprecipitated using antibodies directed against FtsQ (top panel) or antibodies directed against OmpA and TF (bottom panel). OmpA (outer membrane protein A) and TF (cytoplasmic protein; asterisk) were analyzed as controls for proteinase K treatment and spheroplast formation, respectively. Immunoprecipitated material was analyzed by SDS-PAGE and visualized by phosphorimaging. The proteinase-sensitive band below TF is an unidentified protein that is routinely found in TF/OmpA immunoprecipitates (38). wt, wild type.
To study the correct translocation of the full-length FtsQ mutant constructs, we made use of a proteinase accessibility assay. When E. coli cells expressing wild-type FtsQ are converted to spheroplasts and subsequently incubated with proteinase K, the large periplasmic domain of FtsQ is degraded and FtsQ can no longer be recovered from the spheroplasts by IP (38). Proteinase accessibility can therefore be used to monitor the translocation of the periplasmic domain. All FtsQ TMS mutants were expressed from pBAD24 in the presence of [35S]methionine before converting the cells to spheroplasts and proteinase K treatment. Subsequently, FtsQ was recovered by IP, as well as the outer membrane protein OmpA and the cytoplasmic protein trigger factor (TF), which served as controls for spheroplast formation and integrity, respectively. The only FtsQ mutant that could be recovered from proteinase-treated spheroplasts was the triple mutant L29/32R;V38P, indicating that this mutant is not translocated across the membrane (Fig. 3B). This is in accordance with the defect in targeting of this mutant to the translocon (see above). All other mutants could be recovered from untreated spheroplasts but were accessible to proteinase K, indicating translocation of the proteins. Importantly, all controls showed efficient spheroplast formation (proteinase accessibility of OmpA) and good spheroplast integrity (proteinase inaccessibility of TF) (Fig. 3B). A slight difference in motility on SDS-PAGE was observed for all mutants except L29R, but it is not uncommon for TMS mutations to change protein mobility (33). Also, we observed that the L29R single mutant was expressed at lower levels in repeated experiments. The reason for the reduced expression level is not known, but as the plasmid used in this assay is the same that restored viability to the ftsQ1(Ts) mutant, we conclude that the L29R mutant is expressed to sufficient levels.
The L29/32R double mutant does not efficiently localize to the cell division site.
The functional deficiency of the L29/32R double mutant is not caused by a defect in membrane insertion and translocation, as shown above. Therefore, we tested the capability of the mutant proteins to localize to the cell division site, a prerequisite for FtsQ function. GFP fusions to FtsQ were constructed in exactly the same way as an already existing and tested GFP fusion to the wild-type protein. GFP-FtsQ is expressed from plasmid, from an attenuated Ptrc promoter that is not completely shut down in the absence of inducer (1). Strains carrying GFP fusions to either wild-type or mutant FtsQ were grown at steady state in the absence of inducer to allow expression of detectable but low-level amounts of GFP-FtsQ. As expected, all single mutants localized to the cell division site, corresponding with their observed functionality (Fig. 4). In contrast, the L29/32R double mutant localized to the membrane but in a diffuse pattern without clear enrichment at cell division sites (Fig. 4). This failure to efficiently localize to the division site explains why the double mutant is not fully functional, but it may contain sufficient FtsQ at the division site at high levels of protein expression. Finally, the triple mutant, which was found to be incapable of correct membrane insertion and translocation, localized in a diffuse cytosolic pattern, in accordance with its failure to integrate into the membrane.
FIG. 4.

Localization to the cell division site is abolished in the L29/32R double mutant. Phase-contrast and fluorescence images are of cells expressing GFP fusions to FtsQ (A), FtsQL29R (B), FtsQL32R (C), FtsQV38P (D), FtsQL29/32R (E), and FtsQL29/32R;V38P (F), grown to steady state at 28°C in GB1 medium. Some division sites are indicated with arrowheads. Bar (same for all), 2 μm.
The FtsQ TMS does not seem to interact with the TMS of other division proteins.
We wanted to test whether the FtsQ TMS can directly interact with the TMS of other bitopic membrane proteins involved in cell division, as the loss of such an interaction could explain the FtsQ L29/32R phenotype. The TMSs of FtsQ, FtsI, FtsB, FtsL, and FtsQ L29/32R were assayed for their capacities to form either homo- or hetero-oligomers using the GALLEX system, which allows the detection of transmembrane helix-helix interactions in the E. coli membrane (32). None of the TMSs was found to interact (data available at http://www.bio.vu.nl/vakgroepen/molmic/publications.html). So, the FtsQ TMS does not interact with TMSs of proteins with which it is known to interact, like FtsB and FtsL, at least not to the extent that the interaction can be detected by the GALLEX assay.
The L29/32R double mutant interacts with FtsB and FtsL.
Cell division proteins FtsB and FtsL are dependent on each other and on FtsQ for localization to the cell division site (4). FtsQ, FtsB, and FtsL form a protein complex in the membrane, but strikingly, this complex can be assembled independently of the localization of FtsQ to the cell division site (3). As our FtsQ L29/32R double mutant was defective for localization, we wanted to test whether this mutant still bound FtsB and FtsL. A C-terminal Flag3 tag was added to wild-type as well as the mutant FtsQ proteins to facilitate immunoprecipitation of FtsQ. A complementation experiment with the FtsQflag3 variants indicated that FtsQflag3 is fully functional (not shown). FtsQflag3 was immunoprecipitated from DDM-solubilized membranes, and the precipitates were screened for the presence of FtsB and FtsL. FtsB and FtsL were both efficiently precipitated with wild-type FtsQ or mutant FtsQ, but the FtsQ L29/32R;V38P triple mutant did not efficiently precipitate FtsB and FtsL (Fig. 5). FtsB and FtsL were not detected in a mock experiment using solubilized membranes from cells that did not express FtsQflag3 (lane 1), indicating that the precipitation is dependent on the presence of a Flag3-tagged variant of FtsQ. We conclude that the FtsQ L29/32R mutant can still interact with FtsB and FtsL, despite having lost the ability to localize to the division site. Surprisingly, the FtsQ L29/32R;V38P mutant was detected in solubilized membranes even though the protein is poorly targeted to and not functionally inserted in the membrane (see above). We attribute the detection of the triple mutant, and low amounts of precipitated FtsB and FtsL (lane 6), to the formation of insoluble aggregates by FtsQ L29/32R;V38P that precipitate with the membrane fraction. Subsequently, partial solubilization of the aggregates facilitates aspecific interaction of the triple mutant with FtsB and FtsL. Indeed, we found that of the FtsQ L29/32;V38P associated with the membrane fraction, only ∼50% could be solubilized with 1% DDM, whereas all other FtsQ variants were solubilized efficiently (not shown).
FIG. 5.
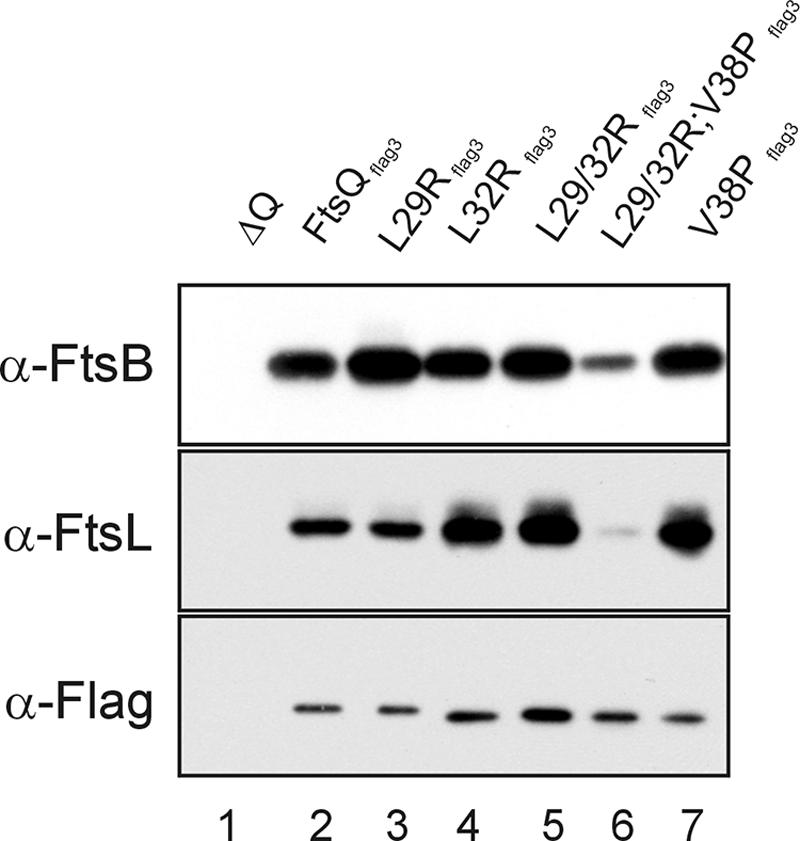
Coimmunoprecipitation of FtsQ mutants with FtsB and FtsL. Solubilized membranes from cells depleted of FtsQ (negative control, lane 1) and cells expressing FtsQflag3 (lane 2), FtsQL29Rflag3 (lane 3), FtsQL32Rflag3 (lane 4), FtsQL29/32Rflag3 (lane 5), FtsQL29/32R;V38Pflag3 (lane 6), and FtsQV38Pflag3 (lane 7) were immunoprecipitated with anti-Flag antibodies coupled to beads (see text). Immunoprecipitates were analyzed by Western blotting and probed with polyclonal antibodies against FtsB and FtsL and a monoclonal antibody against the Flag epitope.
Concluding remarks.
In this study we provide evidence for a contributory role of the FtsQ TMS in targeting to the site of cell division. Original domain swap experiments suggested that both the cytoplasmic domain and the TMS of FtsQ are not essential for FtsQ function, as they can be replaced by nonhomologous sequences (7, 19). However, a closer inspection of these results shows that they may, in fact, be consistent with a role for the FtsQ TMS in FtsQ function, as the swap constructs are not fully efficient in complementation and localization. More recent reports (13, 17) also have hinted at a role for the FtsQ TMS that extends beyond its functioning as an inert membrane anchor. Our results provide support for this role. It is of course striking that the replacement of the FtsQ TMS with the unrelated TMS of MalF still allows FtsQ localization and function, whereas the double mutant described in this paper has lost the capability to efficiently localize to the division site. The defects observed in the L29/32R double mutant and L29/32R;V38P triple mutant must be caused by an additive effect of the combined single mutations, as none of the single mutants was defective in growth and localization. The L29/32R double mutant was shown to be capable of membrane insertion and complex formation with FtsB and FtsL. Therefore, a likely explanation for the localization defect is that the interaction of FtsQ L29/32R with an upstream division protein, most likely FtsK, is lost or altered. The FtsQ TMS consists of 25 amino acid residues, which is rather long. An attractive hypothesis is that the introduction of two positive charges in the α-helix, close to the cytoplasmic face of the lipid bilayer, causes the helix to reorient in such a way that the charges are closer to the anionic lipid headgroups rather than embedded in the membrane. This reorientation could cause a change in the tilt of the TMS or pull FtsQ slightly more “into” the membrane. Both structural changes could affect the interaction with FtsK, if the FtsQ-FtsK interaction site is located in the cytoplasmic and/or TMS domain of FtsQ. A change in TMS tilt or pulling FtsQ into the membrane could also alter the structure or accessibility of the periplasmic domain. This could affect the interaction with FtsK (or some other protein) when the two proteins interact via the membrane-proximal regions in their periplasmic domains. As FtsB and FtsL still interact with the FtsQ double mutant, we expect that the more-membrane-distal regions of the FtsQ periplasmic domain are still correctly folded and positioned. Alternatively, the introduction of two positive charges in the FtsQ TMS could repel the TMS of an interacting protein (like FtsK), thereby disrupting other interactions between the two proteins. The MalF TMS, like the wild-type FtsQ TMS, is completely noncharged and would therefore not have the same repulsion effect. Our observations are consistent with the increasing evidence that cell division proteins contact each other through multiple interactions using multiple domains (17). Finally, the introduction of the positive charges could disturb the interaction of the helix with a specialized lipid domain at the division site. The presence of lipid domains enriched in negatively charged nonbilayer lipids at the site of cell division has been observed in E. coli and B. subtilis (27), and this specific lipid composition could play a role in the recruitment of membrane proteins. We will test these hypotheses in future studies. The set of experiments presented in this paper allows a detailed characterization of FtsQ biogenesis, from targeting to the Sec translocon to localization at the division site, and has great potential for the characterization of the biogenesis of other membrane proteins.
Acknowledgments
We thank Corinne ten Hagen-Jongman, Jorrit Boekel, Maija Tusa, Pieter Smit, and Wouter Jong for technical assistance. We thank Dirk Schneider (University of Freiburg, Germany) for the kind gift of GALLEX strains and plasmids.
D.J.S. is funded by a VENI fellowship from the Research Council for Earth and Life Sciences from The Netherlands Organization for Scientific Research. C.R. holds a Marie Curie Outgoing International Fellowship. G.J.H. was funded by EU grant QLRT-1999-30082. T.D.B. is funded by Vernieuwingsimpuls grant 016.001.024 of The Netherlands Organization for Scientific Research.
Footnotes
Published ahead of print on 10 August 2007.
REFERENCES
- 1.Aarsman, M. E., A. Piette, C. Fraipont, T. M. Vinkenvleugel, M. Nguyen-Disteche, and T. den Blaauwen. 2005. Maturation of the Escherichia coli divisome occurs in two steps. Mol. Microbiol. 55:1631-1645. [DOI] [PubMed] [Google Scholar]
- 2.Buddelmeijer, N., M. E. Aarsman, A. H. Kolk, M. Vicente, and N. Nanninga. 1998. Localization of cell division protein FtsQ by immunofluorescence microscopy in dividing and nondividing cells of Escherichia coli. J. Bacteriol. 180:6107-6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buddelmeijer, N., and J. Beckwith. 2004. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol. Microbiol. 52:1315-1327. [DOI] [PubMed] [Google Scholar]
- 4.Buddelmeijer, N., N. Judson, D. Boyd, J. J. Mekalanos, and J. Beckwith. 2002. YgbQ, a cell division protein in Escherichia coli and Vibrio cholerae, localizes in codependent fashion with FtsL to the division site. Proc. Natl. Acad. Sci. USA 99:6316-6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campo, N., H. Tjalsma, G. Buist, D. Stepniak, M. Meijer, M. Veenhuis, M. Westermann, J. P. Muller, S. Bron, J. Kok, O. P. Kuipers, and J. D. Jongbloed. 2004. Subcellular sites for bacterial protein export. Mol. Microbiol. 53:1583-1599. [DOI] [PubMed] [Google Scholar]
- 6.Chen, J. C., M. Minev, and J. Beckwith. 2002. Analysis of ftsQ mutant alleles in Escherichia coli: complementation, septal localization, and recruitment of downstream cell division proteins. J. Bacteriol. 184:695-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, J. C., D. S. Weiss, J.-M. Ghigo, and J. Beckwith. 1999. Septal localization of FtsQ, an essential cell division protein in Escherichia coli. J. Bacteriol. 181:521-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Den Blaauwen, T., M. E. Aarsman, N. O. Vischer, and N. Nanninga. 2003. Penicillin-binding protein PBP2 of Escherichia coli localizes preferentially in the lateral wall and at mid-cell in comparison with the old cell pole. Mol. Microbiol. 47:539-547. [DOI] [PubMed] [Google Scholar]
- 9.Di Lallo, G., M. Fagioli, D. Barionovi, P. Ghelardini, and L. Paolozzi. 2003. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology 149:3353-3359. [DOI] [PubMed] [Google Scholar]
- 10.Dmitrova, M., G. Younes-Cauet, P. Oertel-Buchheit, D. Porte, M. Schnarr, and M. Granger-Schnarr. 1998. A new LexA-based genetic system for monitoring and analyzing protein heterodimerization in Escherichia coli. Mol. Gen. Genet. 257:205-212. [DOI] [PubMed] [Google Scholar]
- 11.D'Ulisse, V., M. Fagioli, P. Ghelardini, and L. Paolozzi. 2007. Three functional subdomains of the Escherichia coli FtsQ protein are involved in its interaction with the other division proteins. Microbiology 153:124-138. [DOI] [PubMed] [Google Scholar]
- 12.Fröderberg, L., E. Houben, J. C. Samuelson, M. Chen, S. K. Park, G. J. Phillips, R. Dalbey, J. Luirink, and J. W. De Gier. 2003. Versatility of inner membrane protein biogenesis in Escherichia coli. Mol. Microbiol. 47:1015-1027. [DOI] [PubMed] [Google Scholar]
- 13.Geissler, B., and W. Margolin. 2005. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Mol. Microbiol. 58:596-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goehring, N. W., and J. Beckwith. 2005. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr. Biol. 15:R514-R526. [DOI] [PubMed] [Google Scholar]
- 15.Goehring, N. W., M. D. Gonzalez, and J. Beckwith. 2006. Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Mol. Microbiol. 61:33-45. [DOI] [PubMed] [Google Scholar]
- 16.Goehring, N. W., F. Gueiros-Filho, and J. Beckwith. 2005. Premature targeting of a cell division protein to midcell allows dissection of divisome assembly in Escherichia coli. Genes Dev. 19:127-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goehring, N. W., I. Petrovska, D. Boyd, and J. Beckwith. 2007. Mutants, suppressors, and wrinkled colonies: mutant alleles of the cell division gene ftsQ point to functional domains in FtsQ and a role for domain 1C of FtsA in divisome assembly. J. Bacteriol. 189:633-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzman, L. M., D. S. Weiss, and J. Beckwith. 1997. Domain-swapping analysis of FtsI, FtsL, and FtsQ, bitopic membrane proteins essential for cell division in Escherichia coli. J. Bacteriol. 179:5094-5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huitema, E., S. Pritchard, D. Matteson, S. K. Radhakrishnan, and P. H. Viollier. 2006. Bacterial birth scar proteins mark future flagellum assembly site. Cell 124:1025-1037. [DOI] [PubMed] [Google Scholar]
- 21.Janakiraman, A., and M. B. Goldberg. 2004. Recent advances on the development of bacterial poles. Trends Microbiol. 12:518-525. [DOI] [PubMed] [Google Scholar]
- 22.Karimova, G., N. Dautin, and D. Ladant. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187:2233-2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koppelman, C. M., T. Den Blaauwen, M. C. Duursma, R. M. Heeren, and N. Nanninga. 2001. Escherichia coli minicell membranes are enriched in cardiolipin. J. Bacteriol. 183:6144-6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krogh, A., B. Larsson, G. von Heijne, and E. L. Sonnhammer. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567-580. [DOI] [PubMed] [Google Scholar]
- 25.Lam, H., W. B. Schofield, and C. Jacobs-Wagner. 2006. A landmark protein essential for establishing and perpetuating the polarity of a bacterial cell. Cell 124:1011-1023. [DOI] [PubMed] [Google Scholar]
- 26.Luirink, J., and I. Sinning. 2004. SRP-mediated protein targeting: structure and function revisited. Biochim. Biophys. Acta 1694:17-35. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto, K., J. Kusaka, A. Nishibori, and H. Hara. 2006. Lipid domains in bacterial membranes. Mol. Microbiol. 61:1110-1117. [DOI] [PubMed] [Google Scholar]
- 28.Mileykovskaya, E., and W. Dowhan. 2000. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 182:1172-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robson, S. A., and G. F. King. 2006. Domain architecture and structure of the bacterial cell division protein DivIB. Proc. Natl. Acad. Sci. USA 103:6700-6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudner, D. Z., Q. Pan, and R. M. Losick. 2002. Evidence that subcellular localization of a bacterial membrane protein is achieved by diffusion and capture. Proc. Natl. Acad. Sci. USA 99:8701-8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheffers, D.-J., and M. G. Pinho. 2005. Bacterial cell wall synthesis: new insights from localization studies. Microbiol. Mol. Biol. Rev. 69:585-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider, D., and D. M. Engelman. 2003. GALLEX, a measurement of heterologous association of transmembrane helices in a biological membrane. J. Biol. Chem. 278:3105-3111. [DOI] [PubMed] [Google Scholar]
- 33.Scotti, P. A., M. L. Urbanus, J. Brunner, J. W. de Gier, G. von Heijne, C. van der Does, A. J. Driessen, B. Oudega, and J. Luirink. 2000. YidC, the Escherichia coli homologue of mitochondrial Oxa1p, is a component of the Sec translocase. EMBO J. 19:542-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiomi, D., M. Yoshimoto, M. Homma, and I. Kawagishi. 2006. Helical distribution of the bacterial chemoreceptor via colocalization with the Sec protein translocation machinery. Mol. Microbiol. 60:894-906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taschner, P. E. M., P. G. Huls, E. Pas, and C. L. Woldringh. 1988. Division behavior and shape changes in isogenic ftsZ, ftsQ, ftsA, pbpB, and ftsE cell division mutants of Escherichia coli during temperature shift experiments. J. Bacteriol. 170:1533-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian, H., D. Boyd, and J. Beckwith. 2000. A mutant hunt for defects in membrane protein assembly yields mutations affecting the bacterial signal recognition particle and Sec machinery. Proc. Natl. Acad. Sci. USA 97:4730-4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urbanus, M. L., P. A. Scotti, L. Fröderberg, A. Saaf, J. W. de Gier, J. Brunner, J. C. Samuelson, R. E. Dalbey, B. Oudega, and J. Luirink. 2001. Sec-dependent membrane protein insertion: sequential interaction of nascent FtsQ with SecY and YidC. EMBO Rep. 2:524-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Bloois, E., G. J. Haan, J. W. de Gier, B. Oudega, and J. Luirink. 2006. Distinct requirements for translocation of the N-tail and C-tail of the Escherichia coli inner membrane protein CyoA. J. Biol. Chem. 281:10002-10009. [DOI] [PubMed] [Google Scholar]
- 39.Weiss, D. S., J. C. Chen, J.-M. Ghigo, D. Boyd, and J. Beckwith. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181:508-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wissel, M. C., J. L. Wendt, C. J. Mitchell, and D. S. Weiss. 2005. The transmembrane helix of the Escherichia coli division protein FtsI localizes to the septal ring. J. Bacteriol. 187:320-328. [DOI] [PMC free article] [PubMed] [Google Scholar]