

Abstract
The genome sequence of the non-sugar-assimilating mesophile Methanococcus maripaludis contains three genes encoding enzymes: a nonphosphorylating NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase (GAPN), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and glyceraldehyde-3-phosphate ferredoxin oxidoreductase (GAPOR); all these enzymes are potentially capable of catalyzing glyceraldehyde-3-phosphate (G3P) metabolism. GAPOR, whose homologs have been found mainly in archaea, catalyzes the reduction of ferredoxin coupled with oxidation of G3P. GAPOR has previously been isolated and characterized only from a sugar-assimilating hyperthermophile, Pyrococcus furiosus (GAPORPf), and contains the rare metal tungsten as an irreplaceable cofactor. Active recombinant M. maripaludis GAPOR (GAPORMm) was purified from Escherichia coli grown in minimal medium containing 100 μM sodium molybdate. In contrast, GAPORMm obtained from cells grown in medium containing tungsten (W) and W and molybdenum (Mo) or in medium without added W and Mo did not display any activity. Activity and transcript analysis of putative G3P-metabolizing enzymes and corresponding genes were performed with M. maripaludis cultured under autotrophic conditions in chemically defined medium. The activity of GAPORMm was constitutive throughout the culture period and exceeded that of GAPDH at all time points. As GAPDH activity was detected in only the gluconeogenic direction and GAPN activity was completely absent, only GAPORMm catalyzes oxidation of G3P in M. maripaludis. Recombinant GAPORMm is posttranscriptionally regulated as it exhibits pronounced and irreversible substrate inhibition and is completely inhibited by 1 μM ATP. With support from flux balance analysis, it is concluded that the major physiological role of GAPORMm in M. maripaludis most likely involves only nonoptimal growth conditions.
Glyceraldehyde-3-phosphate ferredoxin oxidoreductase (GAPOR) catalyzes a dual substrate electron-transfer reaction in which oxidation of glyceraldehyde-3-phosphate (G3P) yields 3-phosphoglycerate (3PG) concomitant with reduction of oxidized ferredoxin. GAPOR and its homologs are found mainly in archaea including both euryarchaeota and crenarcheaota and especially in hyperthermophiles (26, 38, 42). Homologs can also be found in draft sequences of deltaproteobacterial species (M. A. Aklujkar, J. E. Butler, G. Reguera, E. S. Shelobolina, A. L. Lapidus, D. Bruce, and P. M. Richardson, presented at the 106th General Meeting American Society for Microbiology, Orlando, FL, 21 to 25 May 2006). It has been proposed that the role of GAPOR is to replace the conventional two-step reduction of G3P to 3PG catalyzed by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and 3-phosphoglyceric acid phosphokinase (PGK) in only the glycolytic direction of the Embden-Meyerhof-Parnas pathway (26). GAPOR uses ferredoxin as an electron acceptor while GAPDH uses NAD(P)+. A glycolytic pathway dependent on GAPOR is supposed to be a primordial pathway in early evolution (38) since at least some ferredoxins are more stable and more thermodynamically preferable for electron transport than pyridine nucleotides at high temperatures (36).
In Pyrococcus furiosus, a hyperthermophilic archaea, it has been suggested that the GAPOR reaction is coupled to synthesis of molecular hydrogen as the final electron acceptor (26). Nevertheless, GAPOR is found not only in hydrogen-producing species, such as Thermococcus celer as an active enzyme (38) and in Pyrococcus abyssi (10) and Thermococcus kodakarensis (13) as hypothetic homologs, but also in non-hydrogen-producing, i.e., hydrogen-consuming species, such as Desulfurococcus amylolyticus as an active enzyme (38), and Thermoproteus tenax (39), Methanocaldococcus jannaschii (8), and Methanococcus maripaludis (17) as hypothetic homologs. The rationale behind the evolution of a GAPOR-dependent glycolytic pathway in place of a GAPDH-PGK-dependent pathway, particularly in the latter group of organisms, remains unsolved.
M. maripaludis is the sole example so far of a mesophile that contains a gene annotated as GAPOR (GAPORMm) in its genome sequence (43). M. maripaludis is a facultative autotroph (18) which is capable of autotrophic growth in mineral medium in the presence of hydrogen and carbon dioxide. On the other hand, M. maripaludis readily incorporates acetate and a variety of amino acids when they are present (44, 45). The amino acids are incorporated into protein but are not subject to further catabolism (45). M. maripaludis has also been reported to be capable of synthesis of intracellular glycogen reserves (46). This was suggested from the result that activities of glycolytic enzymes were enhanced while the intracellular glycogen content increased (46). However, M. maripaludis has never been reported to be able to assimilate any extracellular sugars. It is therefore surprising that M. maripaludis possesses a homolog of P. furiosus GAPOR (GAPORPf) in its genome despite limited opportunities for glycolysis. Furthermore, M. maripaludis also possesses two homologs of putative NAD(P)-dependent glyceraldehyde-3-phosphate dehydrogenases (phosphorylating [GAPDH] and nonphosphorylating [GAPN]), which all in theory are capable of catalyzing the G3P-dependent step of the glycolytic pathway. Yu et al. (46) earlier performed GAPDH assays using crude extracts of M. maripaludis; however, no genome sequence was available, and GAPOR had not yet been discovered at that time.
The present study was initiated with an effort toward understanding the physiological role of a gene product which at first instance would not be expected to play an important role in an organism best known for its autotrophic growth capability. Toward this aim, we verified the catalytic function of GAPORMm through synthesis and purification of the recombinant protein in Escherichia coli and investigated its biochemical and biophysical characteristics with a particular focus on metal specificity. Subsequently, we investigated the relationship between the three enzymes (GAPDH, GAPN, and GAPOR) that are all theoretically capable of catalyzing G3P metabolism in M. maripaludis.
MATERIALS AND METHODS
Media and growth conditions of M. maripaludis.
Techniques used for growing methanogens were those of Miller et al. (25); a serum bottle capped with a butyl rubber inner cap and aluminum sealer was used for cultivation. M. maripaludis ATCC 43000, a strain which is not identical with the S2 strain used for genomic sequencing (17), was grown at 37°C in MPl (DSM medium 141 [http://www.dsmz.de/microorganisms/html/media/medium000141.html] with omission of acetate, yeast extract, and Trypticase) which was sparged (H2-CO2 [ratio of 80/20], liquid for 5 min; headspace for 30 s) prior to autoclaving. While the medium was hot, 17% cysteine · H2O · HCl (wt/vol) was added through a 0.2-μm-pore-size filter until the medium lost the color of the redox indicator. The inocula were 10% (vol/vol) of cells in the mid-exponential phase. After the cells were inoculated, the headspace was overpressurized to ∼2 atmospheres with the same gas filtered through a 0.2-μm-pore-size filter (Millex; Millipore). For GAPOR assays, cells were harvested on a 0.2-μm-pore-size filter from 5 ml of culture in an N2-CO2-H2 (90/5/5 ratio) atmosphere and lysed by the addition of BugBuster Reagent (Novagen) containing nonionic surfactants under the same atmosphere, with or without the addition of 4% (vol/vol) Triton X-100. The protein concentration in the lysate was determined using a detergent-compatible protein assay (DC Protein Assay; Bio-Rad). The cell lysate was stored at −80°C under the same atmosphere until use. For transcript assays, cells were harvested by centrifugation from 1 ml of culture under the same atmosphere. Three individual cultures were treated the same way.
Transcript analysis.
Total RNA was prepared using an EZNA Fungal RNA Miniprep Kit (Omega Bio-Tek, Inc.) according to the manufacturer's instructions. For real-time PCR assays, 30 ng of total RNA was used as a template. Primers GdF (5′-GTAGCTAAACAGGACGATATGAAAG) and GdR (5′-TCAACTACAATATCTGCATCTTCAA) for GAPDH (locus MMP0325), GnF (5′-GAATCAATATCTAAAAATGCGAAAA) and GnR (5′-ATGTTTCAACAACTTCTTCTTCAAC) for GAPN (locus MMP1487), and GpF (5′-GTAGCTAAACAGGACGATATGAAAG) and GpR (5′-TCAACTACAATATCTGCATCTTCAA) for GAPOR (locus MMP0945) were designed to yield fragments of approximately equal size (200 bp). Primers 16SF (5′-ATGCGAGTCTATGGTTTCGGCCATG) and 16SR (5′-GACGCTTTAGGCCCAATAAAAGTGG) were designed to amplify 16S rRNA as an internal control. Real-time PCR was performed with an AccessQuick reverse transcription-PCR System (Promega). Following the reverse transcription reaction (at 45°C for 45 min), PCR was performed according to the following conditions: 95°C for 2 min and 40 cycles of 95°C for 30 s, 58°C for 30 s, and 68°C for 1 min. The PCR products were analyzed by agarose gel electrophoresis. Reverse transcriptase was excluded from negative controls. Relative expression levels of target genes were quantitated by the comparative cycle threshold method (4) using 16S rRNA as an internal control.
Cloning of GAPOR.
Genomic DNA was extracted from actively growing M. maripaludis using a DNeasy Tissue Kit (QIAGEN) after repeated cycles of freezing and thawing (3). The entire open reading frame (accession number CAF30501; locus MMP0945) of the gene encoding GAPORMm was amplified by PCR using the following oligonucleotide primers: 5′-GACGACGACAAGATGAACATTTTGATTGATGG and 5′-GAGGAGAAGCCCGGTTTATTCTTTTAATTTCCAG in the presence of 8% (vol/vol) dimethyl sulfoxide and cloned into pET46 Ek-LIC vector (Novagen) using T4 DNA polymerase (Novagen). As the reverse primer included a stop codon, the pET46-Ek/LIC-GAPOR vector was expected to generate a fusion protein encoding only an N-terminal His tag. The DNA was used to transform E. coli strains BL21(DE3) or Rosetta-gami 2(DE3) (Novagen).
Preparation and purification of protein samples.
E. coli was cultured (37°C; aerobic) in M9 minimal medium (0.8 g/liter of NH4Cl, 0.5 g/liter of NaCl, 7.5 g/liter of Na2HPO4 · 2H2O, 3.0 g/liter of KH2PO4), to which the following separately sterilized components were added (per liter of medium): 2 ml of 1 M MgSO4 · 7H2O, 1 ml of 0.1 M CaCl2, 0.3 ml of 1 mM filter-sterilized thiamine-HCl, and 10 ml of a trace element solution containing (per liter) 1 g of FeCl3 · 6H2O, 0.18 g of ZnSO4 · 7H2O, 0.12 g of CuCl2 · 2H2O, 0.12 g of MnSO4 · H2O, and 0.18 g of CoCl2 · 6H2O. Sterilized glucose was added to a final concentration of 2 g per liter. Any of the following was added further (per liter of medium): 10 ml of 10 mM Na2WO4 · 2H2O, 10 ml of 10 mM Na2MoO4 · 2H2O, 5 ml of 10 mM Na2WO4 · 2H2O and 5 ml of 10 mM Na2MoO4 · 2H2O, or 10 ml of deionized ultrafiltered water. For M9 medium precultures, the last condition was used. A total of 40 mg/liter of l-leucine was added to all media for cultivation of Rosetta-gami 2(DE3) cells. Large-scale expression cultures were conducted using either a 10-liter fermentor (MBF-1000; Eyela, Japan) with constant aeration of 0.1 MPa (at 120 rpm and 37°C) or 1- to 5-liter shake flasks (at 200 rpm and 37°C). Expression was induced at an optical density at 600 nm of 0.5 to 0.6 or of 0.1 for the 1- or 8-liter scale culture, respectively, with isopropyl-β-d-thiogalactopyranoside (IPTG), and bacteria was grown for 12 h. Alternatively, bacteria was grown in the same culture sparged with N2 (4 ml/min) after the addition of IPTG for anaerobic expression using a Minifor system (Lambda). The bacteria were harvested by centrifugation (8,000 × g for 20 min at 4°C), washed, and resuspended in cold phosphate buffer (10 mM; pH 8.0), and then lysed with BugBuster Reagent (Novagen) containing benzonase nuclease (Novagen) and 1 mM phenylmethylsulfonyl fluoride. Anaerobically induced samples were harvested and lysed under an N2 or N2-CO2-H2 atmosphere. The lysate was centrifuged (20,000 × g for 20 min at 4°C), and the pellet was washed in buffer A (10 mM phosphate buffer [pH 8.0], 1 mM EDTA, 4% [wt/vol] Triton X-100), centrifuged (20,000 × g for 20 min at 4°C), and resuspended in buffer A containing 8 M urea. The resuspended pellet was dialyzed (for 3 h at 4°C) against 20 mM buffer B (buffer A with the addition of 10 mM urea and 100 mM NaCl) and subsequently, under oxygen-limited conditions, against buffer B with the addition of 1 mM reduced glutathione and 0.2 mM oxidized glutathione (for 3 h at 4°C). Finally, the protein preparation was centrifuged (25,000 × g for 1 h at 4°C), and the supernatant was recovered. Protein preparation using GAPORMm expressed in Rosetta-gami 2(DE3) differed from that of BL21(DE3), as the supernatant obtained after the first centrifugation following lysis contained the majority of the activity. In this case, the addition of Triton X-100 and urea was not required for solubilization of GAPORMm. GAPORMm was purified to >75% homogeneity through a HisTrap HP (GE healthcare) column and gel filtration using Superdex 200 10/300 (GE healthcare) calibrated with low-molecular-weight and high-molecular-weight gel filtration calibration kits (GE healthcare). Protein preparations were routinely monitored with an Agilent 2100 Bioanalyzer.
Cofactor analysis.
The purified protein was subjected to metal content analysis using inductively coupled plasma-mass spectrometry ([ICP-MS] SPQ-9000 with SII nanotechnology) after wet-sample digestion by 5 ml of HNO3 and 1 ml of HClO4 (11). Controls were conducted using [2Fe-2S]-ferredoxin from spinach (Sigma) and bovine serum albumin (Sigma).
Pterin content was measured using 0.5 mg of GAPOR by the previously described methods (24, 32) with fluorescence detection (lambda excitation at 363 nm; lambda emission at 442 nm). The pterin content was calculated by comparison with pterin-6-carboxylic acid. Xanthine oxidase from buttermilk (Sigma) was used as a positive control.
Acid-labile sulfide was measured by a method described elsewhere (5, 29). The positive control was Na2S · 9H2O, and 1 nmol of S2− gave an A670 of 0.0014.
Enzyme assay.
All assays were conducted in screw-cap cuvettes (Sigma) with butyl rubber caps (Voigt Global Distribution, LLC, Kansas City, KS) at room temperature. All assay mixtures (excluding protein) and headspace were sparged with N2 (99.9999%) for 2 min and 15 s, respectively; protein was added under a N2 atmosphere, and thereafter cuvettes were capped. GAPOR activity was routinely measured as previously reported by Mukund et al. (26) by following the reduction of benzyl viologen (BV) at A600 (ɛBV of 7,400 M−1 cm−1) (1). The reaction mixture contained 30 μM G3P, 3 mM BV, 56 μM Na2MoO4, 50 mM EPPS (N-[2-hydroxyethyl]piperazine-N′-[3-propanesulfonic acid]) buffer, pH 8.4. G3P-dependent reduction of ferredoxin was monitored by following the reduction of metronidazole (MNZ) at A320 (ɛMNZ of 9.3 mM−1 cm−1) at room temperature, as modified from the procedure of Soboh et al. (40). The reaction mixture contained 30 μM G3P, 2 mM Clostridium pasteurianum ferredoxin, 0.1 mM MNZ, 56 μM Na2MoO4, and 50 mM EPPS buffer, pH 8.4. C. pasteurianum ferredoxin was used instead of M. maripaludis ferredoxin, which is not available from commercial resources. The assays were started with the addition of 63 mM cysteine-HCl, which is added to ensure completely anoxic conditions, using syringes. Extracts prepared aerobically from aerobically grown cells yielded a reversibly inactivated protein preparation as GAPOR activities measured in anoxic buffer under N2 atmosphere could be recovered after the addition of l-cysteine. Without the reducing agent, GAPOR reacted very slowly (38-fold decrease with 100 μM G3P). The presence or absence of oxygen during expression and protein purification did not influence total yield of GAPOR activity or the requirement for l-cysteine-dependent activation.
Cofactor inhibition was tested using 0.1 mM G3P by the addition of either AMP, NADH, NADPH (all at 10 μM), or ATP (1 and 10 μM) in the reaction mixture using GAPORMm synthesized with Rosetta-gami 2(DE3). To maintain pH above 8.0 in reaction mixtures to which ATP was added, as confirmed using a pH indicator paper (Whatman), the concentration of EPPS buffer in each reaction mixture was increased to a 100 mM final concentration, including control reactions to which no cofactor was added.
Reverse GAPOR activity, i.e., catalysis of G3P formation, was measured by following the oxidation of prereduced BV at A600. The reaction mixture contained 100 μM 3PG, 100 μM BV, 56 μM Na2MoO4, and 50 mM EPPS buffer, pH 8.4. Assays were started by the addition of 100 μM sodium dithionite using syringes.
GAPDH activity was determined in both glycolytic and gluconeogenic directions as described previously (47) with minor modifications. The assay mixtures contained 2 mM NADP+, 0.3 mM G3P, 50 mM Tris-Cl (pH 8.0), with or without 5 mM K2HPO4 (glycolytic direction) or 0.2 mM NADPH, 30 nM 3-phospho-d-glyceroyl phosphate, 30 nM ATP, and 50 mM Tris-Cl (pH 8.0) (gluconeogenic direction). Each assay was started by the addition of protein. The reduction of NADP+ and oxidation of NADPH were monitored by fluorescence spectrometry. GAPN activity was determined as for GAPDH activity above, except with the omission of ATP.
Negative control reaction assays were conducted using reaction mixtures in which either protein, G3P, or electron acceptor was omitted. No activity was observed in any control reaction. Omission of IPTG during expression of recombinant GAPORMm resulted in complete loss of measurable GAPOR activity in crude lysates. One unit of activity is defined as 1 μmol of G3P oxidized per min.
Flux balance analysis.
Flux balance analysis was performed using the condensed stoichiometric model of M. maripaludis and Desulfovibrio vulgaris syntrophic coculture developed by Stolyar et al. (41). The input files of the published model were imported without modification and analyzed using the software CellNetAnalyzer (21), Matlab (version 7.0), and the Matlab Optimization module (Mathworks). The following constraints were fixed prior to flux optimization: lactate uptake (D86), 10; ethanol synthesis (D78), 0; glycerol synthesis (D83), 0; SO4 uptake (D01), 0; alanine uptake (M79), 0. If GAPDH (M32) is constrained to zero prior to flux optimization, all values reported in Fig. 2 of Stolyar et al. (41) are replicated with the exception of reaction M16. In a modified model, changes were incorporated in the reactions catalyzed by GAPDH (M32, 1 ZM_GAP + 1 ZM_ADP = 1 ZM_3PG + 1 ZM_ATP + 1 ZM_NADH; change to 1 ZM_GAP + 1 ZM_ADP = 1 ZM_3PG + 1 ZM_ATP + 1 ZM_NADPH) and GAPOR (M33, 1 ZM_GAP + 1 ZM_ADP = 1 ZM_3PG + 1 ZM_ATP + 1 ZM_Fdx(red); change to 1 ZM_GAP = 1 ZM_3PG + 1 ZM_Fdx(red), an irreversible reaction). To simulate the effect of variation in H2 availability, reaction M15 (uptake of H2 by M. maripaludis) was constrained in a range between 0 to 16.2 μmol/h prior to flux optimization.
FIG. 2.

Effect of variation (v) in concentrations of G3P (A) and BV (B) on the activities (v) of purified Mo-GAPOR synthesized using BL21(DE3). The activities were measured at a BV concentration fixed at 0.5 mM (inset) and 3 mM (A) or a concentration of G3P fixed at 0.1 mM (B). The concentration of the second substrate was varied from 10 to 2,000 μM. No activity was detected when substrates were omitted. The substrate inhibition pattern was confirmed with Mo-GAPOR synthesized using Rosetta-gami 2(DE3). As the inhibition was nearly identical, only data obtained with GAPORMm synthesized with BL21(DE3) are shown.
RESULTS
Amino acid sequence analysis of known and putative GAPOR-encoding genes.
The sequence of the gene encoding GAPORMm displays 44% similarity at the amino acid level to the gene encoding GAPORPf. Ligands to a [4Fe-4S] cluster (e.g., C298, C302, C306, and C514) and molybdopterin (e.g., R60 and D348), which were identified in the GAPORPf amino acid sequence (9, 42) and which define other aldehyde oxidoreductase (AOR) family protein sequences (22), are completely conserved in GAPORMm.
The GAPOR activity of cell extracts of E. coli expressing GAPORMm is dependent upon Mo and inhibited by the addition of W.
The gene encoding GAPORMm was subcloned into pET46 and first expressed in E. coli BL21(DE3). All of the measurable activity was precipitated after centrifugation of the cell extract even though prediction of the solubility of GAPORMm (http://sosui.proteome.bio.tuat.ac.jp) based on the amino acid sequence suggested that GAPORMm is a soluble protein. Attempts to enhance the solubility of GAPORMm by expression as a glutathione S-transferase or a maltose binding protein fusion in strain BL21(DE3) resulted in lower yields of total activity and no changes with respect to solubility compared to pET46 (data not shown).
To test the metal requirement of GAPORMm, expression was conducted in M9 minimal medium with four variations of metal additions: (i) 100 μM sodium tungstate (M9/W+), (ii) 100 μM sodium molybdate (M9/Mo+), (iii) 50 μM sodium tungstate-50 μM sodium molybdate (M9/Mo+/W+), or (iv) without the addition of either sodium tungstate or sodium molybdate (M9/−). All cultures were induced with 1 mM IPTG at the exponential phase of growth, and the growth rate after induction was comparable in all media (data not shown). Synthesis of a polypeptide of the expected molecular weight of GAPORMm was observed in cells grown in all media (Fig. 1). However, measurable GAPOR activity could only be detected in M9/Mo+-grown cells (Table 1).
FIG. 1.
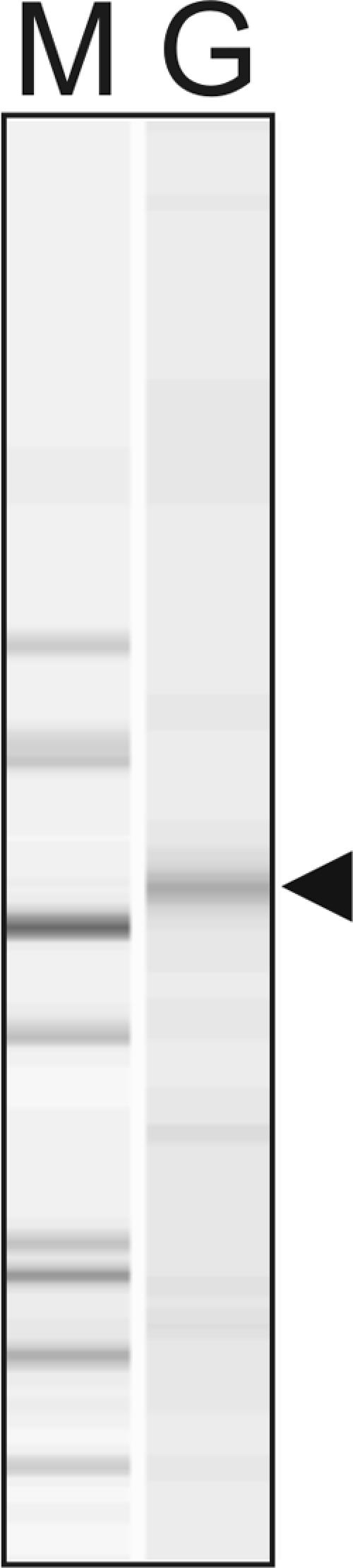
Electropherogram trace (Agilent 2100 Bioanalyzer) of purified GAPOR (Mo-GAPOR). M, molecular marker (Protein 200 Plus Ladder; Agilent); G, purified Mo-GAPOR (arrowhead indicates the expected size of GAPOR).
TABLE 1.
Characteristics of recombinant GAPOR purified from minimal medium containing different combinations of Mo and W
| Strain and culture condition | Mass (kDa) | GAPOR yield (mg/g of wet cells) | Specific activity (U/mg of protein)a | Amt detected (g-atoms/mol) of indicated substanceb
|
||||
|---|---|---|---|---|---|---|---|---|
| Mo | W | Fe | Pterin | Acid-labile sulfide | ||||
| BL21(DE3) | ||||||||
| W | 71 | 5.9 | UD | 0.013 | 0.020 | ND | ND | ND |
| Mo | 71 | 4.4 | 14 | 0.17 | 0.0007 | 1.3 | 0.34 | 6.1 |
| W+Mo | 71 | 5.8 | UD | 0.010 | 0.022 | ND | ND | ND |
| No Mo/no W | 71 | 3.5 | UD | 0.012 | 0.022 | ND | ND | ND |
| Rosetta-gami 2(DE3) | ||||||||
| W | 71 | 0.58 | UD | UD | UD | ND | ND | ND |
| Mo | 71 | 0.65 | 120 | 0.74 | UD | ND | ND | ND |
UD, undetectable.
ND, not determined; UD, undetectable.
GAPORMm catalyzes a BV- and G3P-dependent oxidoreductase reaction with pronounced substrate inhibition.
His6 affinity-purified GAPORMm eluted as a single major peak when subjected to size-exclusion gel chromatography with an estimated molecular mass of 70 kDa. The predicted molecular mass of the amino acid sequence encoding GAPORMm is 70.9 kDa (http://tw.expasy.org/tools/pi_tool.html), suggesting that recombinant GAPORMm was purified in a monomeric state. No loss of total activity was observed as a result of the His tag purification procedure (data not shown). GAPORMm purified from M9/Mo+ cultures (Mo-GAPOR) was reversibly inactivated by exposure of liquid preparations to air and reversibly activated by the addition of the reducing agent l-cysteine (data not shown). In contrast, the GAPOR activity of crude lysates of M. maripaludis was irreversibly inactivated by exposure to air. Most other previously characterized nonrecombinant AOR proteins (including GAPORPf) also appear to be irreversibly inactivated by oxygen exposure (16, 26, 28). The response of recombinant GAPORMm to l-cysteine reactivation may be related to the sulfide activation observed for a W-containing formaldehyde oxidoreductase purified from P. furiosus (34).
An estimation of kinetic constants for G3P was attempted in which the concentration of G3P was initially varied from 0.01 to 2 mM with the concentration of BV fixed at either 0.5 or 3 mM (Fig. 2). However, the catalytic rate was maximal at 0.1 mM G3P and significantly inhibited above 0.15 mM at both concentrations of BV. GAPORPf and AOR of P. furiosus also displayed substrate inhibition for each respective aldehyde (28), although less extreme than that of recombinant GAPORMm. To probe the nature of the inhibition, Mo-GAPOR was incubated with either 2 mM or 0.1 mM G3P for 10 min. Subsequently, the protein was desalted using Sephadex G-25 and then back-added to assay reaction mixtures containing 0.1 mM G3P and 3 mM BV. Reaction mixtures in which Mo-GAPOR had been preincubated with 2 mM G3P displayed 13-fold lower activity than Mo-GAPOR preincubated with 0.1 mM G3P, suggesting that the inhibition was both irreversible and independent of BV. Substrate inhibition was not observed for BV (Fig. 2B) as previously reported for other AORs. Kinetic constants were not estimated as a result of the severe substrate inhibition for G3P. The physiological electron acceptor of the general GAPOR reaction is proposed to be a ferredoxin, based on kinetic analysis with GAPORPf (26) in which a 70-fold greater affinity was observed with P. furiosus [4Fe-4S]-ferredoxin than with BV. As a ferredoxin from M. maripaludis was unavailable, a ferredoxin-dependent reaction was instead confirmed using a commercially available [4Fe-4S]-ferredoxin preparation from C. pasteurianum with MNZ as a reporter molecule. The reduction of MNZ was dependent upon the addition of both Mo-GAPOR and ferredoxin (data not shown).
A maximum specific activity of 1.8 U/mg of protein was observed for Mo-GAPOR expressed in strain BL21(DE3), almost 200-fold lower than that observed with purified GAPORPf (26). In order to enhance the activity, alternative host strains were tried for recombinant protein synthesis. Expression of GAPORMm in strain Rosetta-gami 2(DE3) in M9 medium with the addition of either Mo or W generated protein which, after purification to apparent homogeneity, yielded specific activities of 120 and 0 U/mg of protein, respectively (Table 1). Using the Rosetta-gami 2(DE3)-derived protein preparation, substrate inhibition was confirmed (data not shown), and the effect of physiological levels of key cofactors was also tested. The addition of a 10 μM concentration of either AMP, NADH, or NADPH to the enzyme assay reaction mixture had no effect on GAPOR activity although the addition of 1 to 10 μM ATP resulted in complete loss of GAPOR activity.
In vivo, it is assumed that GAPOR catalyzes only oxidation and not the formation of G3P, as the change in standard free energy of the reverse GAPOR reaction is far greater than the reverse PGK-GAPDH reaction due to a lack of ATP hydrolysis in the former case. As comparative estimates of actual free-energy changes in vivo are not easily calculated, particularly due to a lack of information regarding the relative ratio of reduced/oxidized ferredoxin in vivo, we instead tested whether Mo-GAPOR is enzymatically capable of catalyzing the reverse reaction in vitro. However, Mo-GAPOR was unable to catalyze 3PG-dependent oxidation of reduced BV (data not shown).
Metal and cofactor analysis of GAPORMm: the Mo-content of GAPORMm correlates with catalytic activity.
The metal content of acid hydrolysates of purified GAPORMm preparations was analyzed by ICP-MS (Table 1). Negative controls for ICP-MS using an equal amount (compared to GAPORMm) of bovine serum albumin or spinach [2Fe-2S]-ferredoxin did not contain detectable amounts of W and Mo (detection limit of <2.3 ppt for W and <0.3 ppt for Mo). The only catalytically active preparation (Mo-GAPOR) contained 10-fold greater levels of Mo than that derived from any other combination of metal additions and more than 100 times the amount of Mo than W.
Molybdopterin extracted from Mo-GAPOR emitted fluorescence with a peak at 442 nm when excited with light filtered at 363 nm with a bandwidth of 5 nm. The molybdopterin content of Mo-GAPOR was compared with a positive control consisting of pterin-6-carboxylic acid. The molybdopterin content of Mo-GAPOR was estimated to be 0.35 g-atoms per mol of protein, approximately twice the molar content of Mo. Mo-GAPOR was estimated to contain 6.1 g-atoms of acid-labile sulfide per mol of protein. Although the content of acid-labile sulfide was more than that of iron, disagreements in the relative content of Fe and S2− have been observed in other Fe-S-containing proteins (6, 19, 23).
Based on analysis of P. furiosus AOR and GAPORPf by Mukund and Adams (26, 28) and a comparison between the sequences coding for GAPORMm and GAPORPf, we expect ca. 1 g-atoms of W or Mo/mol and ca. 5 g-atoms of Fe/mol and S2−/mol if all recombinant GAPORMm molecules are fully intact and assembled correctly. Hence, both the ICP-MS and pterin analysis suggest that only one-fifth of Mo-GAPOR had acquired a molybdopterin cofactor and iron sulfur cluster.
Purified GAPORMm that had been expressed using Rosetta-gami 2(DE3) instead of BL21(DE3) in M9 medium with the addition of Mo contained 0.74 g-atoms of Mo and undetectable amounts of W (Table 1). No trace of the two metals was detected in GAPORMm expressed in Rosetta-gami 2(DE3) using M9 medium with the addition of W only. As the difference in specific activity between GAPORMm derived from BL21(DE3) and Rosetta-gami 2(DE3) was greater than the relative difference in Mo content, this suggests that the change in host affected more than just the Mo content of GAPORMm.
GAPOR catalyzes G3P oxidation and GAPDH catalyzes G3P synthesis in M. maripaludis grown under autotrophic conditions.
M. maripaludis was grown under autotrophic growth conditions in chemically defined medium containing added Mo and no added W (growth under these conditions is dependent on the presence of Mo). RNA and protein were extracted at several time points and analyzed for enzyme activity and transcript levels. As the yield of RNA was not sufficient to allow for Northern analysis (data not shown), real-time PCR was conducted instead.
GAPOR activity reached a maximum at day 12 and then decreased (Table 2). GAPDH activity was observed in the gluconeogenic direction only, and the activity was higher in early cultures and decreased thereafter. GAPOR-encoding transcript was observed at all time points, while only one cDNA preparation yielded detectable GAPDH transcript. No activity and no transcript encoding GAPN were detected at any stage of the culture period. Although there were discrepancies between the presence or absence of transcript and total enzyme activities, both data sets suggest that GAPOR is the dominant G3P-metabolizing enzyme and the only enzyme catalyzing oxidation of G3P under the tested conditions.
TABLE 2.
Changes in the gene expression profile and enzyme activity of G3P-related enzymes
| Day postinoculation | Relative gene expression ratioa
|
Enzyme activity (U/mg of protein)b
|
OD600c | |||||
|---|---|---|---|---|---|---|---|---|
| GAPDH | GAPN | GAPOR | GAPDH (G3P oxidation) | GAPDH (G-1,3-P reduction) | GAPN | GAPOR | ||
| 2 | UD | UD | 0.18 | UD | 1.9 | UD | 13 | 0.057 |
| 8 | UD | UD | 0.0031 | UD | 2.4 | UD | 23 | 0.041 |
| 12 | 0.00011 | UD | 0.027 | UD | 0 | UD | 72 | 0.065 |
| 15 | UD | UD | 0.019 | UD | 0.078 | UD | 25 | 0.037 |
The relative expression ratio was determined by the comparative cycle threshold method (5) using 16S rRNA as an internal control. UD, over the threshold (i.e., >45 cycles).
UD, undetectable.
Optical density at 600 nm. This value was 0.003 after inoculation (i.e., day 0).
Flux balance modeling predicts a zero GAPOR reaction rate under steady-state conditions.
Recently, a condensed in silico metabolic model of M. maripaludis in syntrophic coculture with D. vulgaris was published by Stolyar et al. (41). The published model was based on GAPDH and GAPOR reactions which differed (as summarized in Materials and Methods) from the reactions which in the present study had been verified using crude extracts and purified enzymes. If the GAPDH reaction was not constrained to zero, the original model predicted cyclic flux between GAP and 3PG at a rate close to 5 μmol/h that effectively would yield a net NADH:ferredoxin-oxidoreductase reaction without loss or gain of ATP and GAP. When the two GAPDH and GAPOR reactions were stoichiometrically corrected by changing the nucleotide pyridine cofactor specificity of GAPDH from NAD(H) to NADP(H) and by removing ADP/ATP from the GAPOR reaction, it was no longer necessary to eliminate the GAPDH reaction in order to achieve the published flux values (41). In the corrected model, flux through the irreversible reaction catalyzed by GAPOR was predicted to be zero, while predicted flux catalyzed by GAPDH (−0.6 μmol/h) was substantially reduced. Simulation of GAPOR deletion prior to flux optimization consequently resulted in no change. The effect of variation in H2 availability was tested by constraining M. maripaludis H2 uptake (reaction M15) between the value obtained under nonlimiting conditions (16.2 μmol/h) and zero (Fig. 3A). Such a condition may be found, for example, if other H2-consuming microorganisms also are present. When the available H2 content was reduced, initially the H2-dependent reduction of F420 was predicted to rapidly become zero (data not shown), and subsequently the H2-dependent reduction of ferredoxin also decreased (Fig. 3A). At this stage (between 5 to 10 μmol of H2/h), a cyclic GAPOR-GAPDH reaction catalyzing an ATP-dependent NADPH:ferredoxin-oxidoreductase reaction was predicted to commence, eventually (i.e., when H2 uptake was <5 μmol/h) reducing ferredoxin at a greater rate than the hydrogenase-catalyzed reaction.
FIG. 3.

(A) The predicted effect of H2 availability on biomass yield (MC_BIO) and relative reaction rate of H2-dependent reduction of ferredoxin (H2:Fdx), GAPDH in the direction of G3P-oxidation), and GAPOR in the direction of G3P-oxidation. For each fixed H2 value (constrained prior to flux optimization), the predicted reaction rate of each reaction was divided by the predicted biomass yield (left y axis) to obtain the reaction rate relative to biomass yield (right y axis). (B) Graphic summary of proposed pathways and regulation of glycogen metabolism in M. maripaludis. Only reactions related to the present study are shown; other reactions (amino acid metabolism and pentose phosphate pathway) utilizing the same metabolites have been omitted. Regulating molecules are indicated by a circle with a minus sign and, in the case of an unknown putative positive effector, by a plus sign. Abbreviations: G1P, glucose-1-phosphate; 1,3-DPG, 1,3-diphosphateglycerate; 3-PG, 3-phosphoglycerate; PYR, pyruvate. DCW, dry cell weight.
DISCUSSION
GAPORPf contains W (26), and it appears specific for W because even in medium containing excess Mo or vanadium (V) and trace amounts of W, no GAPORPf activity is detected (27). The factors which affect the choice of metals used by enzymes containing molybdenum cofactor (Mo-Co), a cofactor consisting of (i) organic molybdopterin and (ii) inorganic molybdenum or tungsten, remains a question of great interest (20). Kletzin and Adams suggested that W is obligatory in enzyme reactions which require very low redox potentials (≤−420 mV), such as the ferredoxin-dependent oxidoreductases AOR, GAPOR, and formaldehyde ferredoxin oxidoreductase (22). Analysis of recombinant GAPORMm expressed in minimal medium clearly indicated that GAPOR containing Mo and pterin is catalytically active and that the reaction catalyzed by GAPOR is not dependent on W. The accumulation of active GAPORMm in M. maripaludis cultured in chemically defined medium containing Mo but not W further supports the idea that GAPORMm containing Mo and not W is catalytically active. This verifies that GAPORMm encodes an enzyme with said activity and that the enzyme is actively synthesized under autotrophic growth conditions. The synthesis of active GAPOR containing only Mo suggests that the lack of GAPOR activity in extracts of P. furiosus cultured in the absence of W and presence of Mo or V (27) does not result from a chemical incompatibility related to the redox potential of the substrates and end products (22).
The addition of tungstate to the medium did not result in the formation of active Mo-Co containing recombinant GAPORMm, even in the absence of added Mo. This is unlikely to be due to a lack of capability by E. coli to synthesize W-containing Mo-Co. It has previously been demonstrated that E. coli can both incorporate tungstate using the same mechanism for molybdate uptake (33) and synthesize Mo-Co containing both W and Mo (7, 33). In addition, Mo-Co biosynthesis is an ancient, ubiquitous, and highly conserved pathway (31), and E. coli is no exception (35). Therefore, that active GAPORMm containing a tungsten cofactor was not synthesized is most likely explained, at least in this case, by the metal cofactor specificity of GAPORMm. An incompatibility between E. coli and GAPORMm with respect to maturation factor requirements or the presence or absence of a nucleotide, or a type of nucleotide, would be expected to influence equally the formation of both molybdenum- and tungsten-containing Mo-Co.
Mo-GAPOR is the first Mo-containing AOR member for which the corresponding gene sequence has been verified. Since the [4Fe-4S] cluster and molybdopterin ligands identified in sequences encoding other W-containing AORs (9) are conserved, the root of metal specificity must be determined by other as yet unidentified residues, if not dictated by the organism and/or environments as a whole.
The simultaneous addition of an equal concentration of Mo and W resulted in completely inactive MoW-GAPOR, suggesting that the presence of equimolar amounts of W inhibits the formation of Mo-containing Mo-Co, at least in the special case of GAPORMm. However, this should not pose limitations on the synthesis of Mo-dependent enzymes in natural environments since Mo is far more abundant than W in most geographical locations (22). GAPORMm therefore most likely evolved to utilize Mo in accordance with the metal abundance of the habitat of M. maripaludis, thereby differentiating from “ancestral” hyperthermophilic GAPOR at least with respect to metal specificity.
It has been suggested that GAPOR operates solely in the glycolytic direction (26) while GAPDH and PGK support a gluconeogenic pathway (26, 37) in sugar-utilizing P. furiosus. Mukund and Adams (26) also suggested that P. furiosus produces 2 mol less ATP per mol of glucose and disposes of excess electrons as molecular hydrogen as a result of exploiting GAPOR instead of GAPDH and PGK (26). This apparently inefficient mechanism may represent an ancestral form of glycolysis at the early stages of the evolutionary process of thermophilic organisms and can be explained by the fact that certain ferredoxins may be more stable than NAD(P)H at high temperatures (38). However, it is puzzling why GAPOR, which catalyzes the reaction in only the glycolytic direction, is found in a facultative autotroph that is not known to assimilate sugars. Furthermore, as the genome of M. maripaludis also contains two more candidates potentially capable of catalyzing G3P metabolism (GAPDH and GAPN), it is difficult to understand why ATP generation would be sacrificed if the organism is genetically capable of replacing the GAPOR reaction with ATP-generating GAPDH. We therefore performed a time course study to verify the presence of each of the three genes and encoded enzymes and to verify the directionality of the reactions that they catalyze (Table 2). Transcript and enzyme assays showed that only GAPORMm operates in the glycolytic direction, while only GAPDH operates in the gluconeogenic direction under autotrophic conditions, as the reactions catalyzed by GAPORMm and GAPDH were unidirectional with opposing directionality. This contrasts to earlier results (46) in which phosphorylating NADP+-dependent G3P oxidation was reported using crude extracts of M. maripaludis cultured under conditions similar to those used in the present study. Notably, the glycolytic GAPDH activity reported earlier was >90 and >2,000 times lower than the gluconeogenic GAPDH and glycolytic GAPOR activities, respectively, that were observed in the present study. While catalytically irreversible GAPDH isoenzymes that oxidize G3P (14) or catalytically reversible GAPDH isoenzymes with proposed physiological unidirectional roles (12) have previously been characterized, to our knowledge there are no previous reports of any phosphorylating GAPDH which catalyzes only formation, but not oxidation, of G3P. Further study is required to elucidate the cause of such apparent unidirectionality.
No sign of GAPN-encoding transcript or GAPN activity was observed at any time point of the time course study. It was recently demonstrated that a gene annotated as GAPN in Methanocaldococcus jannaschii that shares 56% amino acid identity with M. maripaludis GAPN (locus tag MMP1487) in fact encodes an aldehyde dehydrogenase with no specificity for G3P (15). The three residues proposed to be essential for binding of the phosphate moiety of G3P are also missing in the M. maripaludis GAPN sequence. Even if the M. maripaludis gene annotated as GAPN were expressed, it is likely that the encoded protein in any case would not catalyze nonphosphorylating NAD(P)+-dependent G3P oxidation.
Flux balance analysis, using a model corrected with respect to the stoichiometry and directionality of the reactions catalyzed by GAPOR and GAPDH, predicted that glycolytic flux would be strictly limited, at least under steady-state conditions of autotrophic growth, in order to achieve theoretically optimum biomass formation. As only GAPORMm is capable of catalyzing G3P catabolism and assuming that no other pathways of glycogen catabolism exist in M. maripaludis, this suggests that glycogen catabolism may also be limited under non-steady-state autotrophic growth conditions. Furthermore, although the GAPDH-GAPOR pair, together with other enzymes catalyzing reversible or a combination of irreversible steps of glycolysis/gluconeogenesis, would allow a cycle of glycogen storage and turnover as previously suggested (46), such a cycle of glycogen turnover, or a cycle between pyruvate and G3P alone, that is dependent upon both GAPDH and GAPOR in concert would constitute, in effect, an ATP-dependent NADPH:ferredoxin-oxidoreductase reaction. This raises the questions of how such a reaction cycle and/or glycogen turnover would be regulated and what would be the role of the metabolic reactions catalyzed by GAPDH-GAPOR in M. maripaludis.
Both the transcript and enzyme analysis suggested that GAPOR was constitutive throughout the growth period, while GAPDH activity was observed mainly in the early growth stages of the cultures. This contrasts with previous results with P. furiosus in which GAPORPf was suggested to be transcriptionally controlled and induced by the presence of extracellular sugar (41). As methanogens in general do not assimilate extracellular sugar, a similar regulation mechanism is not possible with M. maripaludis, which may explain why GAPOR activity was constitutive. Regulation may instead be posttranscriptional. Both GAPORPf (23) and GAPORMm (Fig. 2) are strongly inhibited by G3P; however, no other apparent allosteric interactions were previously noted with GAPORPf (23). It is difficult to imagine how the apparently irreversible substrate inhibition exhibited by GAPORMm alone would be able to exert any control over glycolytic flux. We therefore tested whether any cofactors would be able to affect the GAPOR-catalyzed reaction. Interestingly, ATP completely inhibited the reaction even at concentrations as low as 1 μM. As this concentration is lower than intracellular levels typically reported in other microorganisms (30), it is likely that an additional, as yet unknown, positive effector also influences GAPOR activity in vivo. Nevertheless, it is clear that, in contrast to GAPORPf, GAPORMm is posttranscriptionally regulated.
The results of flux balance analysis and assays determining the molecular regulation of GAPORMm and enzyme directionality of GAPOR and GAPDH together allow us to formulate a tentative hypothesis regarding the metabolic role of GAPORMm, which is summarized in Fig. 3B. Given that only GAPORMm is able to catalyze G3P catabolism and that the enzyme is subject to posttranscriptional regulation, it would appear that GAPORMm plays a central role in regulation of glycogen catabolism. The suggested roles of glycogen metabolism in microorganisms vary depending on the species, including temporary conditions of stress imposed by limitations in growth substrate (i.e., starvation) or chemical warfare or undefined continuous cycling (2). As flux balance analysis indicated no role for GAPORMm under steady-state autotrophic growth conditions, arguing against a role in continuous cycling, GAPORMm and glycogen catabolism are therefore more likely to play a metabolic role under non-steady-state growth conditions (for example, stationary phase or starvation) or under growth conditions dependent on alternative substrates. The fact that ATP is a potent negative regulator of GAPORMm further supports the notion that glycogen degradation in M. maripaludis may be limited to conditions of starvation, as the concentration of ATP would be expected to be lower under such conditions. Alternatively, the role of ATP as a negative regulator of GAPORMm may also be important in order to limit continuous ATP-driven and NADPH-dependent reduction of ferredoxin, a reaction that should be necessary only in the special case where both glycogen and H2, the major source of electrons for reduction of ferredoxin (41), are limited. In order to test this, we simulated the effect of reduced H2 availability on predicted reaction rates using flux balance analysis (Fig. 3A). At least stoichiometrically, a cyclic GAPOR-GAPDH reaction was predicted to maximize biomass formation in the event that the uptake of only H2 and no other external metabolites is limited. Whether such a putative reaction cycle takes place in vivo or not will require further investigation. Nevertheless, it can be concluded that both the proposed role of GAPOR and GAPDH in glycogen catabolism, as well as a putative ATP- and NADPH-dependent reduction of ferredoxin, most likely are important only for maintaining metabolic homeostasis under nonoptimal growth conditions.
Acknowledgments
We acknowledge and thank Peter McInerney for helpful discussions and ideas and Steve Van Dien (Genomatica) for helpful discussions and provision of stoichiometric models and network maps.
Footnotes
Published ahead of print on 17 August 2007.
REFERENCES
- 1.Ballantine, S. P., and D. H. Boxer. 1985. Nickel-containing hydrogenase isoenzymes from anaerobically grown Escherichia coli K-12. J. Bacteriol. 163:454-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballicora, M. A., A. A. Iglesias, and J. Preiss. 2003. ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol Rev. 67:213-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barns, S. M., R. E. Fundyga, M. W. Jeffries, and N. R. Pace. 1994. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc. Natl. Acad. Sci. USA 91:1609-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Shahar, Y., A. Robichon, M. B. Sokolowski, and G. E. Robinson. 2002. Influence of gene action across different time scales on behavior. Science 296:741-744. [DOI] [PubMed] [Google Scholar]
- 5.Brumby, P. E., R. W. Miller, and V. Massey. 1965. The content and possible catalytic significance of labile sulfide in some metalloflavoproteins. J. Biol. Chem. 240:2222-2228. [PubMed] [Google Scholar]
- 6.Bryant, F. O., and M. W. Adams. 1989. Characterization of hydrogenase from the hyperthermophilic archaebacterium, Pyrococcus furiosus. J. Biol. Chem. 264:5070-5079. [PubMed] [Google Scholar]
- 7.Buc, J., C. L. Santini, R. Giordani, M. Czjzek, L. F. Wu, and G. Giordano. 1999. Enzymatic and physiological properties of the tungsten-substituted molybdenum TMAO reductase from Escherichia coli. Mol. Microbiol. 32:159-168. [DOI] [PubMed] [Google Scholar]
- 8.Bult, C. J., O. White, G. J. Olsen, L. Zhou, R. D. Fleischmann, G. G. Sutton, J. A. Blake, L. M. FitzGerald, R. A. Clayton, J. D. Gocayne, A. R. Kerlavage, B. A. Dougherty, J. F. Tomb, M. D. Adams, C. I. Reich, R. Overbeek, E. F. Kirkness, K. G. Weinstock, J. M. Merrick, A. Glodek, J. L. Scott, N. S. Geoghagen, and J. C. Venter. 1996. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273:1058-1073. [DOI] [PubMed] [Google Scholar]
- 9.Chan, M. K., S. Mukund, A. Kletzin, M. W. Adams, and D. C. Rees. 1995. Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science 267:1463-1469. [DOI] [PubMed] [Google Scholar]
- 10.Cohen, G. N., V. Barbe, D. Flament, M. Galperin, R. Heilig, O. Lecompte, O. Poch, D. Prieur, J. Querellou, R. Ripp, J. C. Thierry, J. Van der Oost, J. Weissenbach, Y. Zivanovic, and P. Forterre. 2003. An integrated analysis of the genome of the hyperthermophilic archaeon Pyrococcus abyssi. Mol. Microbiol. 47:1495-1512. [DOI] [PubMed] [Google Scholar]
- 11.Evans, H. J., E. R. Purvis, and F. E. Bear. 1950. Molybdenum nutrition of alfalfa. Plant Physiol. 25:555-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fillinger, S., S. Boschi-Muller, S. Azza, E. Dervyn, G. Branlant, and S. Aymerich. 2000. Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J. Biol. Chem. 275:14031-14037. [DOI] [PubMed] [Google Scholar]
- 13.Fukui, T., H. Atomi, T. Kanai, R. Matsumi, S. Fujiwara, and T. Imanaka. 2005. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 15:352-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao, Z., and W. H. Loescher. 2000. NADPH supply and mannitol biosynthesis. Characterization, cloning, and regulation of the non-reversible glyceraldehyde-3-phosphate dehydrogenase in celery leaves. Plant Physiol. 124:321-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grochowski, L. L., H. Xu, and R. H. White. 2006. Identification of lactaldehyde dehydrogenase in Methanocaldococcus jannaschii and its involvement in production of lactate for F420 biosynthesis. J. Bacteriol. 188:2836-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heider, J., K. Ma, and M. W. Adams. 1995. Purification, characterization, and metabolic function of tungsten-containing aldehyde ferredoxin oxidoreductase from the hyperthermophilic and proteolytic archaeon Thermococcus strain ES-1. J. Bacteriol. 177:4757-4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrickson, E. L., R. Kaul, Y. Zhou, D. Bovee, P. Chapman, J. Chung, E. Conway de Macario, J. A. Dodsworth, W. Gillett, D. E. Graham, M. Hackett, A. K. Haydock, A. Kang, M. L. Land, R. Levy, T. J. Lie, T. A. Major, B. C. Moore, I. Porat, A. Palmeiri, G. Rouse, C. Saenphimmachak, D. Soll, S. Van Dien, T. Wang, W. B. Whitman, Q. Xia, Y. Zhang, F. W. Larimer, M. V. Olson, and J. A. Leigh. 2004. Complete genome sequence of the genetically tractable hydrogenotrophic methanogen Methanococcus maripaludis. J. Bacteriol. 186:6956-6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones, W. J., M. J. B. Paynter, and R. Gupta. 1983. Characterization of Methanococcus maripaludis sp. nov., a new methanogen isolated from salt marsh sediment. Arch. Microbiol. 135:91-97. [Google Scholar]
- 19.Kanai, T., S. Ito, and T. Imanaka. 2003. Characterization of a cytosolic NiFe-hydrogenase from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 185:1705-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisker, C., H. Schindelin, and D. C. Rees. 1997. Molybdenum-cofactor-containing enzymes: structure and mechanism. Annu. Rev. Biochem. 66:233-267. [DOI] [PubMed] [Google Scholar]
- 21.Klamt, S., J. Saez-Rodriguez, and E. D. Gilles. 2007. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol 1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kletzin, A., and M. W. Adams. 1996. Tungsten in biological systems. FEMS Microbiol. Rev. 18:5-63. [DOI] [PubMed] [Google Scholar]
- 23.Ma, K., and M. W. Adams. 2001. Hydrogenases I and II from Pyrococcus furiosus. Methods Enzymol. 331:208-216. [DOI] [PubMed] [Google Scholar]
- 24.Meckenstock, R. U., R. Krieger, S. Ensign, P. M. Kroneck, and B. Schink. 1999. Acetylene hydratase of Pelobacter acetylenicus. Molecular and spectroscopic properties of the tungsten iron-sulfur enzyme. Eur. J. Biochem. 264:176-182. [DOI] [PubMed] [Google Scholar]
- 25.Miller, T. L., and M. J. Wolin. 1974. A serum bottle modification of the Hungate technique for cultivating obligate anaerobes. Appl. Microbiol. 27:985-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukund, S., and M. W. Adams. 1995. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase, a novel tungsten-containing enzyme with a potential glycolytic role in the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 270:8389-8392. [DOI] [PubMed] [Google Scholar]
- 27.Mukund, S., and M. W. Adams. 1996. Molybdenum and vanadium do not replace tungsten in the catalytically active forms of the three tungstoenzymes in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 178:163-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukund, S., and M. W. Adams. 1991. The novel tungsten-iron-sulfur protein of the hyperthermophilic archaebacterium, Pyrococcus furiosus, is an aldehyde ferredoxin oxidoreductase. Evidence for its participation in a unique glycolytic pathway. J. Biol. Chem. 266:14208-14216. [PubMed] [Google Scholar]
- 29.Nielsen, F. S., P. S. Andersen, and K. F. Jensen. 1996. The B form of dihydroorotate dehydrogenase from Lactococcus lactis consists of two different subunits, encoded by the pyrDb and pyrK genes, and contains FMN, FAD, and [FeS] redox centers. J. Biol. Chem. 271:29359-29365. [DOI] [PubMed] [Google Scholar]
- 30.Peterkofsky, A., and C. Gazdar. 1973. Measurements of rates of adenosine 3′:5′-cyclic monophosphate synthesis in intact Escherichia coli B. Proc. Natl. Acad. Sci. USA 70:2149-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajagopalan, K. V., and J. L. Johnson. 1992. The pterin molybdenum cofactors. J. Biol. Chem. 267:10199-10202. [PubMed] [Google Scholar]
- 32.Rauh, D., A. Graentzdoerffer, K. Granderath, J. R. Andreesen, and A. Pich. 2004. Tungsten-containing aldehyde oxidoreductase of Eubacterium acidaminophilum. Eur. J. Biochem. 271:212-219. [DOI] [PubMed] [Google Scholar]
- 33.Rech, S., C. Wolin, and R. P. Gunsalus. 1996. Properties of the periplasmic ModA molybdate-binding protein of Escherichia coli. J. Biol. Chem. 271:2557-2562. [DOI] [PubMed] [Google Scholar]
- 34.Roy, R., S. Mukund, G. J. Schut, D. M. Dunn, R. Weiss, and M. W. Adams. 1999. Purification and molecular characterization of the tungsten-containing formaldehyde ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus: the third of a putative five-member tungstoenzyme family. J. Bacteriol. 181:1171-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sambasivarao, D., R. J. Turner, P. T. Bilous, R. A. Rothery, G. Shaw, and J. H. Weiner. 2002. Differential effects of a molybdopterin synthase sulfurylase (moeB) mutation on Escherichia coli molybdoenzyme maturation. Biochem. Cell Biol. 80:435-443. [DOI] [PubMed] [Google Scholar]
- 36.Schafer, G., M. Engelhard, and V. Muller. 1999. Bioenergetics of the Archaea. Microbiol. Mol. Biol Rev. 63:570-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schäfer, T., and P. Schönheit. 1993. Gluconeogenesis from pyruvate in the hyperthermophilic archaeon Pyrococcus furiosus: involvement of reactions of the Embden-Meyerhof pathway. Arch. Microbiol. 159:354-363. [Google Scholar]
- 38.Selig, M., K. B. Xavier, H. Santos, and P. Schonheit. 1997. Comparative analysis of Embden-Meyerhof and Entner-Doudoroff glycolytic pathways in hyperthermophilic archaea and the bacterium Thermotoga. Arch. Microbiol. 167:217-232. [DOI] [PubMed] [Google Scholar]
- 39.Siebers, B., B. Tjaden, K. Michalke, C. Dorr, H. Ahmed, M. Zaparty, P. Gordon, C. W. Sensen, A. Zibat, H. P. Klenk, S. C. Schuster, and R. Hensel. 2004. Reconstruction of the central carbohydrate metabolism of Thermoproteus tenax by use of genomic and biochemical data. J. Bacteriol. 186:2179-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soboh, B., D. Linder, and R. Hedderich. 2004. A multisubunit membrane-bound [NiFe] hydrogenase and an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis. Microbiology 150:2451-2463. [DOI] [PubMed] [Google Scholar]
- 41.Stolyar, S., S. Van Dien, K. L. Hillesland, N. Pinel, T. J. Lie, J. A. Leigh, and D. A. Stahl. 2007. Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 3:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Oost, J., G. Schut, S. W. Kengen, W. R. Hagen, M. Thomm, and W. M. de Vos. 1998. The ferredoxin-dependent conversion of glyceraldehyde-3-phosphate in the hyperthermophilic archaeon Pyrococcus furiosus represents a novel site of glycolytic regulation. J. Biol. Chem. 273:28149-28154. [DOI] [PubMed] [Google Scholar]
- 43.Verhees, C. H., S. W. Kengen, J. E. Tuininga, G. J. Schut, M. W. Adams, W. M. De Vos, and J. Van Der Oost. 2003. The unique features of glycolytic pathways in Archaea. Biochem. J 375:231-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitman, W. B., J. Shieh, S. Sohn, D. S. Caras, and U. Premachandran. 1986. Isolation and characterization of 22 mesophilic methanococci. Syst. Appl. Microbiol. 7:235-240. [Google Scholar]
- 45.Whitman, W. B., S. Sohn, S. Kuk, and R. Xing. 1987. Role of Amino Acids and Vitamins in Nutrition of Mesophilic Methanococcus spp. Appl. Environ. Microbiol. 53:2373-2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu, J. P., J. Ladapo, and W. B. Whitman. 1994. Pathway of glycogen metabolism in Methanococcus maripaludis. J. Bacteriol. 176:325-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeikus, J. G., G. Fuchs, W. Kenealy, and R. K. Thauer. 1977. Oxidoreductases involved in cell carbon synthesis of Methanobacterium thermoautotrophicum. J. Bacteriol. 132:604-613. [DOI] [PMC free article] [PubMed] [Google Scholar]