

Abstract
Horizontal gene transfer events followed by proper regulatory integration of a gene drive rapid evolution of bacterial pathogens. A key event in the evolution of the highly virulent plague bacterium Yersinia pestis was the acquisition of plasmid pPCP1, which carries the plasminogen activator gene, pla. This promoted the bubonic form of the disease by increasing bacterial dissemination from flea bite sites and incidentally enhanced replication in respiratory airways during pneumonic infection. We determined that expression of pla is controlled by the global regulator cyclic AMP (cAMP) receptor protein (Crp). This transcription factor is well conserved among distantly related bacteria, where it acts as a soluble receptor for the ubiquitous signaling molecule cAMP and controls a global network of metabolic and stress-protective genes. Crp has a similar physiological role in Y. pestis since loss of its function resulted in an inability to metabolize a variety of nonglucose substrates. Activation of pla expression requires a transcription activation element of the pla promoter that serves as a Crp binding site. Crp interaction with this site was demonstrated to occur only in the presence of cAMP. Alteration of the Crp binding site nucleotide sequence prevented in vitro formation of Crp-DNA complexes and inhibited in vivo expression of pla. The placement of pla under direct regulatory control of Crp highlights how highly adapted pathogens integrate laterally acquired genes to coordinate virulence factor expression with global gene networks to maintain homeostasis through the infectious life cycle.
The highly adapted pathogen Yersinia pestis coordinates the expression of a complex set of virulence factors to cause fulminant diseases in humans and other mammals (25). Normally, Y. pestis is vectored between hosts by fleas, which leads to initiation of bubonic plague episodes (30). The development of secondary pneumonic plague, however, presents an opportunity for person-to-person aerosol transmission, which can result in an epidemic outbreak (13). Bubonic plague and pneumonic plague are commonly fatal without rapid antibiotic intervention.
Y. pestis evolution has involved a variety of horizontal and lateral gene acquisition events, with extrachromosomal elements contributing numerous essential virulence factors and traits that promote association of this bacterium with fleas and pathogenesis in mammalian hosts (38). One virulence factor is the Ysc type III secretion (T3S) system of plasmid pCD1. Y. pestis and the enteropathogenic species Yersinia enterocolitica and Yersinia pseudotuberculosis, which cause self-limiting gastrointestinal illnesses, contain this specialized plasmid-encoded protein secretion system. Transfer of the genes appears to have been an early event in the evolution of Yersinia (38). Thus, the exceptional virulence of Y. pestis compared with the enteropathogenic Yersinia species involves additional traits, some of which reside in other laterally transferred species-specific genetic elements. Unique to Y. pestis is the 9.5-kb plasmid pPCP1 that encodes Pla, a surface-localized aspartyl protease which activates the plasminogen/plasmin cascade to dissolve fibrin clots. During bubonic plague, Pla has an important role in releasing the bacterium from the restricted subdermal site of delivery by a flea bite (30, 35, 37). Pla is more dramatically important for the progression of pneumonic plague as it promotes dissemination of Y. pestis from the lung (18). The genomic gain of pPCP1 and regulatory integration of its genetic content were an important adaptation of Y. pestis to its infectious life cycle.
Survival of Y. pestis in cold-blooded fleas and warm-blooded animal hosts selectively enforces the need to coordinately modulate gene expression patterns in response to environmental cues, including nutrient fluxes and environmental stresses. Consequently, colonization of each host niche requires the bacterium to balance metabolic resources with the need to produce products that circumvent host clearance mechanisms. In the Enterobacteriaceae family, which includes the genus Yersinia, physiological responses to internal and external stress cues are often mediated by modulating global networks of genes whose expression is controlled by transcription factors. One transcription factor is cyclic AMP (cAMP) receptor protein (Crp), a soluble receptor for the signaling molecule 3′,5′-cAMP found in diverse prokaryotic and eukaryotic organisms (6, 21). Crp responds to intracellular cAMP transported into the cell from the exterior milieu or synthesized by an endogenous adenylate cyclase (11). Allosteric changes in Crp occur when it is bound to cAMP, and these changes increase its affinity for specific cis-acting transcriptional elements to induce or repress gene transcription (24). Although it has not been previously examined for Y. pestis, control of gene expression by the cAMP-Crp regulatory system in other bacteria influences the metabolism of carbon and energy sources, respiration, osmotic and heat stress responses, and the elaboration of surface organelles, such as pili and flagella (6).
Investigation of Crp in Y. pestis revealed that this transcription factor was coopted to regulate synthesis of Pla. Considering the restrictive and highly adapted lifestyle of Y. pestis and the important role that Pla has in virulence, this observation provides insight into how a highly specialized and lethal pathogen evolved.
MATERIALS AND METHODS
Bacterial strains and media.
The strains and plasmids used in this study are listed in Table 1. Escherichia coli strains were routinely cultivated at 37°C in LB medium or on LB agar plates. Y. pestis strains were cultivated at 26°C in buffered L medium (1% trypsin, 0.5% yeast extract, 50 mM morpholineethanesulfonic acid [MOPS]; pH 7.0) supplemented with 0.1% glucose or 0.1% glycerol as indicated below. Bacterial samples used in the experiments were collected from exponentially growing cultures at an optical density at 600 nm (OD600) of 0.3 to 0.6. Antibiotics were used at the following final concentrations when they were needed: ampicillin, 100 μg/ml; chloramphenicol, 25 μg/ml for E. coli and 12.5 μg/ml for Y. pestis; kanamycin, 50 μg/ml for E. coli and 100 μg/ml for Y. pestis; streptomycin, 50 μg/ml; and tetracycline, 15 μg/ml for E. coli and 7.5 μg/ml for Y. pestis.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant properties | Source or reference |
|---|---|---|
| E. coli strains | ||
| BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm (DE3) | Novagen |
| DH5α | F− φ80lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ−thi-1 gyrA96 relA1 | Laboratory strain |
| One shot TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| S17-1 λpir | recA thi pro hsd(R− M+) RP4 2-Tc::Mu-Km::Tn7 λpir | 32 |
| Y. pestis strains | ||
| KIM5-3001 | pCD1+, pMT1+, pPCP1+, pgm | 7 |
| KIM8-3002 | pCD1+, pMT1+, pPCP1−, pgm | 7 |
| GY5475 | KIM5-3001, crp::pEP185.2 | This study |
| GY5476 | KIM5-3001, cyaA::pEP185.2 | This study |
| GY5477 | KIM8-3002, crp::pEP185.2 | This study |
| GY5478 | KIM8-3002, cyaA::pEP185.2 | This study |
| GY5523 | KIM5-3001, crp::pEP185.2, pWSK129 | This study |
| GY5524 | KIM5-3001, crp::pEP185.2, pGY776 | This study |
| GY5531 | KIM5-3001, pGY778 | This study |
| GY5533 | KIM5-3001, crp::pEP185.25, pGY778 | This study |
| GY5538 | KIM5-3001, crp::pEP185.2, pWSK129, pGY778 | This study |
| GY5539 | KIM5-3001, crp::pEP185.2, pGY776, pGY778 | This study |
| GY5564 | KIM5-3001, cyaA::pEP185.2, pGY778 | This study |
| GY5585 | KIM5-3001, cyaA::pEP185.2, pWSK129 | This study |
| GY5586 | KIM5-3001, cyaA::pEP185.2, pGY803 | This study |
| GY5595 | KIM5-3001, cyaA::pEP185.2, pWSK129, pGY778 | This study |
| GY5597 | KIM5-3001, cyaA::pEP185.2, pGY803, pGY778 | This study |
| GY5744 | KIM5-3001, pGY878 | This study |
| GY5745 | KIM5-3001, pGY879 | This study |
| GY5746 | KIM5-3001, pGY880 | This study |
| Plasmids | ||
| pCR-Blunt II-TOPO | pUC ori, Kanr | Invitrogen |
| pEP185.2 | mob+, oriR6K, Cmr | 15 |
| pET24b(+) | Kanr | Novagen |
| pRW50 | oriV, lacZYA, Tetr | 19 |
| pWSK129 | pSC101 ori, Kanr | 36 |
| pGY606 | pCR-Blunt II-TOPO with a 0.4-kb internal fragment of cyaA | This study |
| pGY609 | pEP185.2 with a 0.4-kb internal fragment of cyaA | This study |
| pGY725 | pCR-Blunt II-TOPO with a 0.3-kb internal fragment of crp | This study |
| pGY735 | pEP185.2 with a 0.3-kb internal fragment of crp | This study |
| pGY775 | pCR-Blunt II-TOPO with a 1.1-kb fragment containing crp | This study |
| pGY776 | pWSK129 with a 1.1-kb fragment containing crp | This study |
| pGY777 | pCR-Blunt II-TOPO with a 0.242-kb fragment containing the pla promoter | This study |
| pGY778 | pRW50 with a 242-bp fragment containing the pla promoter | This study |
| pGY802 | pCR-Blunt II-TOPO with a 3.0-kb fragment containing cyaA | This study |
| pGY803 | pWSK129 with a 3.0-kb fragment containing cyaA | This study |
| pGY809 | pET24b(+) with a 0.6-kb fragment containing crp | This study |
| pGY811 | pCR-Blunt II-TOPO with a 0.6-kb fragment containing crp | This study |
| pGY875 | pCR-Blunt II-TOPO with a 206-bp fragment containing the pla promoter | This study |
| pGY876 | pCR-Blunt II-TOPO with a 206-bp fragment containing the pla promoter with two point mutations altering the cAMP-Crp binding site | This study |
| pGY877 | pCR-Blunt II-TOPO with a 185-bp fragment containing the pla promoter missing the cAMP-Crp binding site | This study |
| pGY878 | pRW50 with a 206-bp fragment containing the pla promoter fused to lacZYA | This study |
| pGY879 | pRW50 with a 206-bp fragment containing the pla promoter with two point mutations altering the cAMP-Crp binding site that is fused to lacZYA | This study |
| pGY880 | pRW50 with a 185-bp fragment containing the pla promoter missing the cAMP-Crp binding site that is fused to lacZYA | This study |
Generation of bacterial strains and plasmid construction.
Mutant strains carrying an insertion mutation that inactivated crp or cyaA were generated using the suicide plasmid pEP185.2 as previously described (15). DNA fragments corresponding to an internal region of the target gene were generated by PCR with the following oligonucleotide primers: for crp, YP-CRP1 (5′-GTAAGCCACAAACAGACCCG-3′) and YP-CRP2 (5′-CCTGAATCAGTTGACGGAAC-3′); and for cyaA, Pestis cya 1 (5′-CAGATCTGAGTCTGCTACCGATCC-3′) and Pestis cya 2 (5′-CAGATCTGTCAACCAGGAAGAAGC-3′). The resulting fragments were cloned into plasmid pCR-Blunt II-TOPO according to the manufacturer's instructions (Invitrogen Corporation, Carlsbad, CA). DNA fragments were confirmed to not contain errors due to PCR amplification by DNA sequence analysis. The resulting plasmids were designated pGY725 (crp) and pGY606 (cyaA). The fragments were liberated by digestion with XhoI and SacI for the crp fragment and with BglII for the cyaA fragment. These fragments were then cloned into pEP185.2 at the corresponding restriction enzyme sites, resulting in plasmids pGY735 (crp) and pGY609 (cyaA). Plasmid derivatives were transferred to KIM5-3001 by conjugation, and mutants having the plasmid insertion in the target gene were screened on LB agar plates containing chloramphenicol and streptomycin. The mutants were confirmed by PCR using oligonucleotide primers that flanked the site of plasmid insertion.
Plasmids carrying the wild-type allele of either crp or cyaA were constructed by cloning PCR-amplified DNA fragments of the gene into low-copy-number plasmid pWSK129. Amplification of crp and cyaA DNA was done with the following oligonucleotide primers: for crp, CRP-start (5′-ACGCCGGTTTTTAGAGGGAA-3′) and CRP-end (5′-CCTGCTCCCGGTTAAATTTTC-3′); and for cyaA, CYAA-P-3 (5′-GACTCCGAGAAACTCATTGG-3′) and CYAA-P-4 (5′-ATAGGCAGAGGAGTAAAGCG-3′). Fragments were initially cloned into plasmid pCR-Blunt II-TOPO to obtain pGY775 (crp) and pGY802 (cyaA). The crp allele was then subcloned as an EcoRI fragment into pWSK129 to obtain pGY776. The cyaA allele was subcloned as a KpnI and XbaI fragment to obtain plasmid pGY803. These plasmids were transferred to Y. pestis by electroporation.
Transcriptional fusions between the pla promoter region and a promoterless E. coli lac operon were generated using pRW50 (19). Each of the fragments of interest was generated by PCR with primers, and these fragments correspond to DNA fragments B, C, and D described below for electrophoretic mobility shift assays (EMSA). DNA fragments were cloned into plasmid pCR-Blunt II-TOPO and then subcloned into pRW50.
Plasmid-encoded hexahistidine-tagged Crp was generated by cloning the crp gene into pET-24b(+). The gene was amplified by PCR with two primers, CRP-OE-S-P (5′-CAGCATATGGTTCTCGGTAAGCCAC-3′) and CRP-OE-E-P (5′-TATCTCGAGTTAACGGGTGCCGTAAACG-3′). The DNA fragment was initially cloned into plasmid pCR-Blunt II-TOPO to obtain plasmid pGY811. It was then subcloned as an NdeI- and XhoI-liberated fragment into plasmid pET-24b(+) (EMD Biosciences, Inc., San Diego, CA), resulting in plasmid pGY809. This plasmid was transformed into E. coli BL21(DE3) by electroporation.
Preparation of Yop proteins and SDS-PAGE analysis.
Secreted Yop proteins were prepared by using a modification of previously described methods (39). Y. pestis was grown overnight in buffered L medium supplemented with 0.1% glucose at 26°C and subcultured into calcium-chelated medium (1% tryptone, 0.5% yeast extract, 50 mM MOPS [pH 7.0], 16 mM sodium oxalate, 160 mM magnesium chloride). Cultures were incubated for 6 h at 37°C on a roller drum to provide mild aeration. The OD600 of cultures were measured, and bacterial cells were removed by centrifugation. Yop proteins from the culture supernatants were concentrated by precipitation with 10% (wt/vol) trichloroacetic acid at 4°C. After washing with ice-cold acetone, the precipitated proteins were suspended in protein sample buffer containing 2-mercaptoethanol. The volumes of the sample buffer were adjusted based on the OD600 of the cultures. Samples were heated at 95°C for 5 min and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with 10% polyacrylamide. Proteins were visualized by staining with Coomassie brilliant blue R (Sigma).
Fibrin matrix degradation assay.
Induction of fibrinolysis by Pla was qualitatively assayed by recording the dissolution of a fibrin matrix that was prepared as described previously, with minor modifications (3). The matrix contained 0.5 g of bovine fibrinogen (Sigma-Aldrich Co., St. Louis, MO) and 0.2% sodium azide (Sigma-Aldrich Co.) dissolved in 10 ml water. Sodium azide was included in the solution as a bacterial growth inhibitor. Separately, 25 NIH units of thrombin as defined by the manufacturer (Sigma-Aldrich Co.) was suspended in 100 mM sodium borate buffer (pH 7.75). The two solutions were gently mixed and poured into a petri dish. The fibrin matrix was stored for 30 min before the fibrinolysis assay was initiated. Exponentially growing cultures of bacteria were collected by centrifugation and resuspended in buffered L medium with 0.2% sodium azide. Cultures were diluted to obtain a concentration of 2 × 104 CFU/μl. Then 5 μl of each cell suspension was spotted onto the fibrin matrix and incubated for 20 h at 37°C. The appearance of a clear zone indicated that fibrinolysis occurred.
Quantitative measurement of activated plasmin.
Pla-mediated conversion of plasminogen to activated plasmin was monitored by measuring cleavage of the synthetic plasmin substrate t-butyloxycarbonyl-l-valyl-l-leucyl-l-lysine 4-methyl-coumaryl-7-amide (Boc-Val-Leu-Lys-MCA) (Peptide Institute Inc., Louisville, KY). The assays were completed in a 96-well microtiter plate (Nalge Nunc International, Rochester, NY). Release of the fluorescent product 7-amino-4-methylcoumarin was measured with an EL808-SynergyHT fluorimeter (Bio-Tek Instruments, Inc., Winooski, VT), and the data were analyzed using KC4 software. Fluorescence was measured at sensitivities of 50 and 75%. The excitation and emission filters were set at 360/40 and 460/40 nm, respectively. The 100-μl reaction mixture consisted of phosphate-buffered saline with 1 μg of human glu-plasminogen (American Diagnostic Inc., Stamford, CT), 0.5 μg of plasmin substrate, and approximately 2.5 × 107 CFU of the bacterial strain tested. A reaction mixture with no bacteria served as a negative control for this experiment. Each assay was conducted in triplicate.
Western blot analysis.
Steady-state levels of Pla were determined by Western blot analysis using rabbit polyclonal anti-Pla antibody. The antibody was raised against polyhistidine-tagged Pla purified by affinity chromatography (V. Motin, unpublished results). A New Zealand White rabbit was inoculated subcutaneously with 250 mg of antigen emulsified in TiterMax adjuvant (Sigma) and was boosted once at 4 weeks postimmunization. The animal handling procedures were approved by the UTMB Animal Care and Use Committee. Samples for Western blot analysis were normalized by adjusting the optical densities of logarithmically growing cultures to the same value. The OD600 of the culture was determined at the time of harvest. Bacterial cells were collected by centrifugation in a microcentrifuge at 16,000 × g for 5 min. The bacterial pellets were suspended in sample buffer containing 2-mercaptoethanol, and the volume was adjusted based on the OD600 of the culture. After heating at 95°C for 5 min, equivalent amounts of protein samples were separated by SDS-PAGE with 10% acrylamide and then transferred to nitrocellulose membranes. Detection of Pla was completed with anti-Pla antibodies (1:5,000), followed by incubation with goat anti-rabbit immunoglobulin G-horseradish peroxidase (1:10,000; Sigma Chemical). Western blot membranes were visualized by chemiluminescence (ECL Western blotting detection reagents; Amersham Biosciences, Piscataway, NJ).
Measurement of β-galactosidase activity.
Y. pestis was cultivated in buffered LB medium at 37°C or as indicated below. Bacterial samples were then harvested from exponentially growing cultures, and β-galactosidase activity was assayed as previously described (26). Each experiment was performed in duplicate and repeated on at least two separate occasions. The standard deviation of values for a given sample was less than 10%. Where indicated, the values for a sample were compared to the values obtained for the experimental control by a two-tailed t test.
Mapping the transcription start site for pla by 5′ RACE-PCR.
The procedure for mapping the 5′ end of an mRNA by rapid amplification of cDNA ends (RACE)-PCR was modified for use with Y. pestis (4). A sample of Y. pestis KIM5-3001 cultivated in buffered L medium was mixed with an equal volume of ice-cold RNAlater (Ambion Inc., Austin, TX). After the cells were harvested by centrifugation, total RNA was purified using an RNeasy mini kit according to the manufacturer's instructions (Qiagen Inc., Valencia, CA). pla cDNA was generated by reverse transcriptase-mediated synthesis of DNA in a reaction mixture containing 2 μg of total RNA, 5 μl of deoxynucleoside triphosphates, 1 μl of Moloney murine leukemia virus reverse transcriptase (New England Biolabs, Inc., Beverly, MA), 5 μl of 5× buffer, and oligonucleotide primer PLA-RACE-OUT (5′-GATCTTCCAGTCTAACTGGC-3′). cDNA was purified using a QIAquick PCR purification kit according to the manufacturer's instructions (Qiagen Inc.). Purified cDNA was mixed with 5 μl of 10× NEB buffer 4, 5 μl of 2.5 mM CaCl2, 10 μl of 10 mM dGTP, 0.5 μl of terminal transferase (New England Biolabs, Inc.), and 24.5 μl of water. A reaction mixture with no terminal transferase was used as a negative control. After incubation for 1 h at 37°C, the reaction was terminated by heating the sample to 75°C for 20 min. The 5′ poly(G)-tagged cDNA was then amplified by PCR using a poly(C) primer (5′-CCCCCCCCCCCCCCCCCC-3′) and primer PLA-RACE-IN (5′-GATCTTCCAGTCTAACTGGC-3′). The product of this reaction was separated from other reaction components by agarose gel electrophoresis and then purified using a Qiaex II gel extraction kit according to the manufacturer's instructions (Qiagen Inc.). The resulting cDNA was sequenced at the DNA sequencing facility of the University of California, Davis with an automated 3730 DNA analyzer (Applied Biosystems, Foster City, CA).
Purification of His6-tagged Crp.
E. coli strain BL21(DE3) was transformed with pGY809. Crp was then overexpressed and purified by affinity chromatography using a QIAexpressionist kit according to the manufacturer's instructions (Qiagen Inc.). Samples from each step of the expression and purification procedure were analyzed by SDS-PAGE with 10% acrylamide. The concentration of purified Crp was determined by the Bio-Rad protein assay (Bio-Rad Laboratories Inc.).
EMSA.
For the DNA binding studies, 1.8 mmol of defined DNA fragments carrying different portions of the pla regulatory region was incubated with increasing amounts of Crp in 20 μl of DNA binding buffer (10 mM Tris-HCl [pH 7.8], 50 mM KCl, 1 mM EDTA, 50 μg/ml bovine serum albumin, 1 mM dithiothreitol, 0.005% NP-40, 10% glycerol, 50 μM cAMP). The reaction mixture was incubated for 20 min at room temperature and then mixed with 3 μl of loading solution (50% glycerol and 0.1 mg/ml bromphenol blue). A 10-μl portion of the binding reaction mixture was loaded onto a 4% polyacrylamide gel buffered with Tris-acetate-EDTA containing 50 μM cAMP.
DNA fragments were generated by PCR and were purified using a QIAquick PCR purification kit (Qiagen Inc., Valencia, CA). The DNA concentration was determined by measuring the OD260. For amplification of DNA fragments by PCR, specific oligonucleotide primers were utilized. Fragment A (302 bp), extending from pla nucleotide position −169 to position 133, was amplified with PLA-P-S (5′-CTCCCGTTATCAGTACCATC-3′) and PLA-P-E (5′-AAGCTTAGCACTCCCGGACAGAAT-3′). Fragment B (210 bp), extending from position −77 to position 133, was generated with Ppla-CRP-W (5′-TCTTATGTGAGCAAAGTCACAT-3′) and PLA-P-E. Fragment C (210 bp), extending from position −77 to position 133 and containing two nucleotide changes (underlined) that altered the Crp binding site, was generated with Ppla-CRP-M (5′-TCTTATCTGAGCAAAGTCAGATAATTCTGT-3′) and PLA-P-E. Fragment D (189 bp), extending from position −56 to position 133, was generated with Ppla-CRP-D (5′-TAATTCTGTCAGACGACGAGA-3′) and PLA-P-E.
DNase I footprinting analysis.
To determine the location of the Crp binding site(s) within the pla regulatory region, DNase I footprinting analyses were carried out using the method of Galas and Schmitz (10), as modified by Huang and Igo (12). To label the DNA, pGY777 was digested with XhoI and treated with shrimp alkaline phosphatase (New England Biolabs). The digested fragments were labeled with [γ-32P]ATP (Amersham Biosciences) using T4 polynucleotide kinase and then digested with XbaI. The labeled fragments were purified by phenol-chloroform extraction, followed by use of a Qiagen nucleotide removal kit (Qiagen Inc.). The labeled fragments were incubated with purified Crp in a binding buffer as previously described (31), except that 2 μg of calf thymus DNA was included instead of salmon sperm DNA and 100 μM of cAMP was included in binding reaction mixtures where indicated. The DNA binding reaction mixtures were incubated at room temperature for 30 min, and DNase I (Worthington Biochemical Corporation) digestion was carried out as previously described (12). The reaction products were subjected to electrophoresis on an 8 M urea-8% polyacrylamide gel and analyzed by ImageQuant 5.0 using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). The location of the protected regions was determined by comparing the DNase I digestion patterns with a G+A sequencing ladder (20).
RESULTS
cAMP-Crp regulatory system modulates plasminogen activator-dependent proteolysis of secreted Yop proteins.
The Ysc T3S system is conserved among enteropathogenic Yersinia species and Y. pestis. Given that the cAMP-Crp regulatory system affects Ysc T3S-dependent secretion of the Yop effectors by Y. enterocolitica, we reasoned that loss of Crp function or loss of CyaA adenylate cyclase activity should similarly affect the Ysc T3S in Y. pestis (26). Examination of the Y. pestis KIM genome annotation resulted in the prediction that Crp and CyaA share 100 and 94% identity, respectively, with their counterparts in Y. enterocolitica and are encoded by open reading frames Y3956 and Y0382 (8). These loci, designated crp and cyaA, were inactivated by insertion mutagenesis in two strains derived from Y. pestis KIM differing in whether they carried plasmid pPCP1, which encodes the Pla protease. Commonly, studies of the Ysc T3S system have employed strains cured of pPCP1 because recovery of secreted Yop proteins is hampered by degradation due to Pla activity (34). This Pla-dependent Yop degradation phenomenon was confirmed (Fig. 1, compare lanes 2 to 5). Subsequent examination of the crp and cyaA mutants lacking pPCP1 showed that inactivation of the cAMP-Crp regulatory system caused a reduction in the amount of Yop proteins recovered from the culture medium (Fig. 1, lanes 5, 6, and 7). This result is consistent with results for Y. enterocolitica, where Crp is a positive regulator for expression and secretion of Yop proteins (26). It was expected that in the crp and cyaA mutants retaining pPCP1 the combination of less Yop production due to inactivation of the cAMP-Crp regulatory system and Pla-dependent Yop degradation would cause a severe reduction in the amount of Yop proteins recovered (Fig. 1). Surprisingly, this was not the case as significant amounts of secreted proteins were recovered from the growth medium of the cultures, suggesting that Pla activity was affected (Fig. 1, compare lane 2 to lanes 3 and 4). This observation led us to investigate how the cAMP-Crp regulatory system influences expression of Pla.
FIG. 1.

Accumulation of secreted Yop proteins is affected both by the cAMP-Crp regulatory system and by pPCP1. Selected strains of Y. pestis were cultivated to induce Yop secretion by the Ysc T3S system. Lane 1, molecular mass markers; lane 2, KIM5-3001 (pPCP1+); lane 3, GY5475 (crp, pPCP1+); lane 4, GY5476 (cyaA, pPCP1+); lane 5, KIM8-3002 (pPCP1−); lane 6, GY5477 (crp, pPCP1−); lane 7, GY5478 (cyaA, pPCP1−). Secreted Yop proteins were collected from the culture medium following the removal of bacterial cells. Proteins were separated by SDS-PAGE and stained with Coomassie brilliant blue. For each sample, an amount equivalent to 1 ml of culture at an OD600 of 1.0 was loaded.
Functional loss of the cAMP-Crp regulatory system reduces Pla activity.
The biological role of Pla in Y. pestis virulence is to promote dissemination of bacteria from the initial infection sites during the development of bubonic and pneumonic plague by inducing the plasminogen-plasmin proteolytic cascade to dissolve fibrin matrices (18, 30). Recapitulation of this cascade in vitro has demonstrated that fibrinolysis occurs through the Pla-dependent proteolytic processing of the plasminogen to plasmin (3). The activated plasmin cleaves fibrin at specific lysine residues, thereby degrading fibrin matrices. To evaluate whether the cAMP-Crp regulatory system affects Y. pestis in a way that alters this biologically relevant activity of Pla, whole cells were examined to determine their ability to induce fibrinolysis of a preformed fibrin matrix (Fig. 2A). When equal numbers of bacteria were spotted onto a fibrin matrix, the wild-type strain displayed a robust ability to induce fibrin degradation typified by a large zone of matrix dissolution, while a strain lacking pPCP1 was deficient. The crp and cyaA mutants were notably impaired, reflecting a reduced level of Pla activity (Fig. 2A). For both the crp and cyaA mutants, the ability to induce fibrinolysis was fully restored by in trans complementation of the mutant allele with a plasmid-located copy of the gene (Fig. 2A).
FIG. 2.

Y. pestis crp and cyaA mutants have reduced abilities to induce fibrinolysis and activate plasminogen. (A) Assay of fibrinolysis induction by Y. pestis. For each strain, approximately 1 × 105 CFU was spotted onto a preformed fibrin matrix containing 0.2% sodium azide. The sodium azide was included to prevent bacterial growth during the assay. Fibrinolysis was monitored by the appearance of a zone of clearing in the vicinity of bacteria spotted on the matrix surface. Position 1, KIM5-3001 (pPCP1+) (wild type) (WT); position 2, GY5475 (crp); position 3, GY5523 (crp, vector control); position 4, GY5524 (crp, vector + crp); position 5, GY5476 (cyaA); position 6, GY5585 (cyaA, vector control); position 7, GY5586 (cyaA, vector + cyaA); position 8, KIM5-3001 (wild type); position 9, KIM8-3002 (pPCP1−). (B) Quantitative measurement of Pla-induced plasmin activity. For all strains examined, equivalent numbers of bacteria were added to the plasminogen activation-plasmin activity assay mixture. Cleavage of the synthetic peptide substrate Boc-Val-Leu-Lys-MCA released the fluorescent compound 7-amino-4-methylcoumarin, which was spectrophotometrically detected; the results are expressed in arbitrary fluorescence units. A detailed description of assay conditions is provided in Materials and Methods. The following strains were assayed: KIM5-3001 (wild type), GY5475 (crp), GY5523 (crp, vector control), GY5524 (crp, vector plus crp), GY5476 (cyaA), GY5585 (cyaA, vector control), and GY5586 (cyaA, vector plus cyaA). A mock-inoculated control was also included.
As an additional measurement of Pla activity, the abilities of selected Y. pestis strains to activate purified plasminogen were determined using a fluorometry-based assay (Fig. 2B). Activated plasmin degrades the synthetic peptide substrate Boc-Val-Leu-Lys-MCA, releasing the fluorescent product 7-amino-4-methylcoumarin, which is spectrophotometrically quantified. The rate of product accumulation in this reaction is directly related to plasmin activation because plasminogen and the synthetic substrate are included at excessive concentrations in the reaction mixture. Equal numbers of wild-type Y. pestis and each of the genetically complemented mutants induced plasmin activity similarly (Fig. 2B). In contrast, less activity was detected for samples with the crp and cyaA mutants (Fig. 2B). Thus, two independent assessments of Pla function indicated that inactivation of the cAMP-Crp regulatory system results in a reduction in Pla activity.
Steady-state levels of Pla are decreased in crp and cyaA mutants.
One explanation for the reduction in Pla activity displayed by crp and cyaA mutants is that these strains produce less Pla. Alternatively, at least when heterologously expressed in other bacteria, Pla proteolytic activity is affected by changes to the bacterial cell envelope (27). This presents the possibility that production of Pla remains unchanged but its activity is altered by cAMP-Crp-mediated changes in gene expression that ultimately alter the cell envelope. In addition, localization of Pla to the outer membrane results in rapid processing of pre-Pla (36 kDa) to the predominant α-Pla form (35 kDa), but prolonged incubation of experimental samples results in autoproteolytic cleavage and accumulation of the β-Pla form (33 kDa) (17, 33). There is no experimental evidence demonstrating that α- and β-Pla differentially affect virulence or plasmin degradation (16), but an examination of Pla protein levels provided a general assessment of whether there was a gross change in Pla autoprocessing activity. To assess whether Pla synthesis is affected in crp and cyaA mutants, the steady-state level of whole cell-associated Pla protein was determined by Western blot analysis (Fig. 3). A comparison of wild-type bacteria to the crp and cyaA mutants revealed that either loss of adenylate cyclase function or inactivation of Crp results in a reduction in the amount of all forms of Pla. Complementation of the mutations with a functional gene restored Pla levels to levels equivalent to that observed for the wild-type strain (Fig. 3). This experimental outcome favors the hypothesis that functional loss of the cAMP-Crp regulatory system reduces Pla activity because expression of this protein is affected.
FIG. 3.

Steady-sate levels of Pla are reduced by mutations inactivating crp and cyaA. The following strains were assayed: KIM8-3002 (pPCP1−), KIM5-3001 (wild type) (WT), GY5475 (crp), GY5523 (crp, vector control), GY5524 (crp, vector + crp), GY5476 (cyaA), GY5585 (cyaA, vector control), and GY5586 (cyaA, vector + cyaA). A Western blot analysis of whole-cell lysates probed with polyclonal anti-Pla antibody was performed. The locations of the α and β forms of Pla are indicated on the right. Two cross-reactive proteins that appeared in the pPCP1− control are indicated by asterisks. For each sample, an amount equivalent to 1 ml of culture at an OD600 of 0.1 was loaded.
cAMP-Crp regulatory system affects transcription of pla.
An experiment to evaluate whether the low level of Pla produced by crp and cyaA mutants was due to changes in pla transcription was performed. A DNA fragment encompassing a portion of the pla open reading frame and extending 242 bp upstream was cloned upstream of a low-copy-number, plasmid-based, promoterless E. coli lacZYA operon to obtain pGY778 (Table 1). This Ppla-lacZYA transcriptional fusion was then introduced into Y. pestis strains and utilized to indirectly monitor expression of pla by quantifying the level of β-galactosidase activity (Fig. 4). Samples of bacteria were harvested from exponentially growing cultures. Consistent with the idea that the cAMP-Crp regulatory system affects transcription of pla, the crp and cyaA mutants exhibited a five- to sixfold reduction in β-galactosidase activity compared to the wild-type strain (Fig. 4). Complementation of the mutation with a functional copy of crp or cyaA restored expression to levels that were comparable to the values obtained for the wild-type strain (Fig. 4).
FIG. 4.

Transcription of pla is affected by the cAMP-Crp regulatory system. Levels of β-galactosidase expression were measured for selected strains transformed with pGY778 (Ppla-lacZYA). The strains examined were GY5531 (wild type) (WT), GY5533 (crp), GY5538 (crp, vector control), GY5539 (crp, vector + crp), GY5564 (cyaA), GY5595 (cyaA, vector), and GY5597 (cyaA, vector + cyaA). The results are averages and standard deviations from at least two independent experiments performed in duplicate. The mean value for each sample was compared the control (wild-type) value by the two-tailed t test. Values that were considered significantly different (P < 0.05) are indicated by an asterisk.
Exogenous cAMP restores pla expression in the cyaA mutant.
The role of cAMP as a second messenger is well established for both prokaryotes and eukaryotes. Bacteria actively export cAMP to the extracellular environment, and in E. coli and other bacteria, this signaling molecule is transported into the cell (2, 11). In animals, cAMP serves as a second messenger molecular that controls a variety of physiological activities, including modulation of the immune system (14). Y. pestis persists extracellularly in its eukaryotic hosts and survives inside macrophages. Both sites may expose Y. pestis to high localized concentrations of cAMP. We decided to investigate whether exogenous cAMP might serve as an inducer of pla expression, but we recognized that this analysis would be complicated due to endogenous cAMP synthesis by CyaA. Therefore, the analysis was performed with a cyaA mutant. In the absence of exogenously added cAMP, this strain exhibited low levels of expression from the pla-lacZYA fusion (Fig. 5A). Strikingly, exogenous cAMP, provided by addition of 1 mM cAMP to the culture medium, restored pla expression to wild-type levels (Fig. 5A). Importantly, a control experiment with the crp mutant demonstrated that the pla-lacZYA fusion was not induced in response to exogenous cAMP when the cell lacked Crp, which supported a regulatory model in which Crp plays a central role in perception of the cAMP signal and subsequent control of phenotypic responses (Fig. 5A). The concentration of extracellular cAMP needed to promote induction of pla was then estimated (Fig. 5B). The cyaA mutant was cultivated in a medium with cAMP concentrations ranging from 0 to 2 mM. This analysis showed that 50% maximal induction of pla occurred when the extracellular concentration of cAMP was 250 μM, and maximal expression required approximately 1 mM cAMP. Exogenous cAMP did not affect pla expression by the wild-type strain, indicating that under the current laboratory conditions the bacterial adenylate cyclase maintains intracellular cAMP at a concentration that fully induces pla. These results support the conclusion that cAMP is an important signaling molecule for pla activation.
FIG. 5.
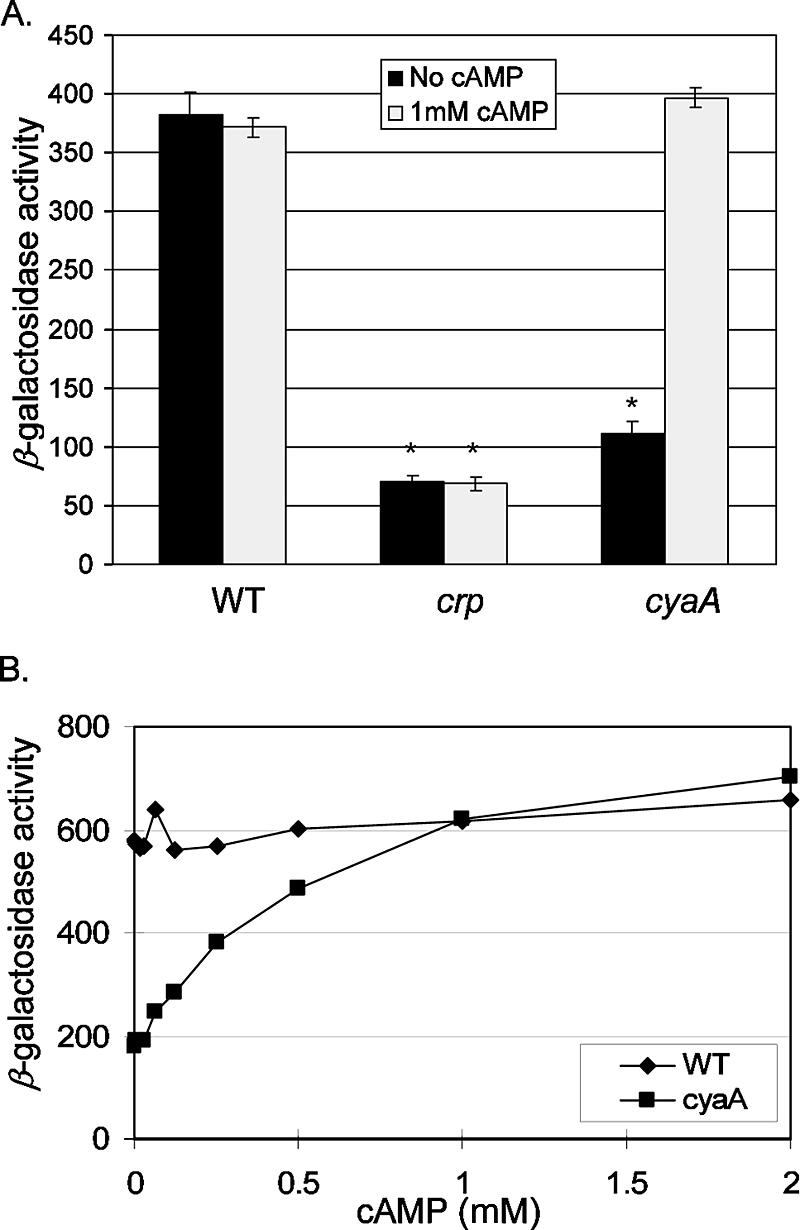
Regulation of pla expression in response to extracellular cAMP. (A) Levels of β-galactosidase expression were measured for selected strains cultivated for 4 h in buffered L medium without or with 1 mM cAMP. Each strain examined carried pGY778 (Ppla-lacZYA). The strains examined were GY5531 (wild type) (WT), GY5533 (crp), and GY5564 (cyaA). (B) Levels of β-galactosidase expression for GY5531 (wild type) (WT) and GY5564 (cyaA) cultivated for 4 h in buffered L medium supplemented with different amounts of cAMP, as indicated. The results are averages ± standard deviations from at least two independent experiments performed in duplicate. The mean value for each sample was compared to the control value (wild type, no cAMP) by the two-tailed t test. Values that were considered significantly different (P < 0.05) are indicated by an asterisk.
Effect of catabolite availability on Crp-mediated regulation of pla.
In other members of the family Enterobacteriaceae, the cAMP-Crp regulatory system controls the expression of a variety of genes that contribute to numerous physiological activities to maintain metabolic homeostasis. Among the repertoire of Crp-activated genes are the genes needed for utilization of nonglucose substrates that can serve as carbon and energy sources. We examined whether the cAMP-Crp regulatory system was needed for utilization of alternative carbon and energy sources. Like fermentation by other members of the family Enterobacteriaceae, Y. pestis fermentation of glucose was not affected by mutations in crp or cyaA, but these mutations abolished fermentation of alternative compounds, including maltose, galactose, mannitol, and glycerol (data not shown). In E. coli, uptake of glucose by the phosphotransferase transport system leads to inactivation of adenylate cyclase. Subsequent decreases in endogenous cAMP levels limit the DNA binding activity of Crp, and there are concomitant reductions in the transcription of Crp-activated genes, a phenomenon known as catabolite repression. To determine the extent to which pla expression is subject to catabolite repression, selected strains of Y. pestis were cultivated in L medium supplemented with either glucose or glycerol. Transcriptional analysis revealed that inclusion of glucose in the medium reduced expression of pla by approximately 40% compared to the expression in a medium supplemented with glycerol or with no supplement (Fig. 6). This reduction in pla expression was reflected by a modest decrease in the steady-state levels of Pla (data not shown). Thus, it appears that Crp in Y. pestis directly regulates pla transcription and retains control over catabolite utilization, which is a characteristic of members of the Enterobacteriaceae family.
FIG. 6.
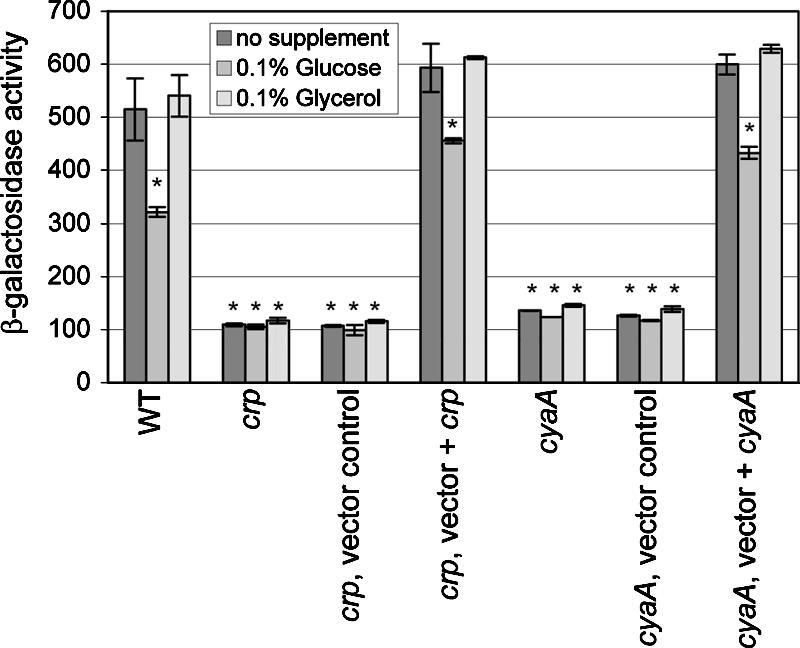
Catabolite modulation of pla transcription. (A) Selected strains of Y. pestis carrying pGY778 expressing Ppla-lacZ were cultivated in buffered L medium without or with glucose or glycerol. The strains examined were GY5531 (wild type) (WT), GY5533 (crp), GY5538 (crp, vector control), GY5539 (crp, vector + crp), GY5564 (cyaA), GY5595 (cyaA, vector control), and GY5597 (cyaA, vector + cyaA). The results are averages ± standard deviations from at least two independent experiments performed in duplicate. The mean value for each sample was compared to the control value (wild type, no supplement) by the two-tailed t test. Values that were considered significantly different (P < 0.05) are indicated by an asterisk.
Mapping the transcriptional start site of pla.
In other bacteria in which Crp-mediated gene activation has been characterized, there is universal involvement of a cis-acting nucleotide element mapping upstream of the site of transcription initiation. Assessment of this detail of pla activation required a better understanding of how pla transcription initiates. Therefore, RNA was isolated from Y. pestis and used to map the 5′ end of the pla mRNA by 5′ RACE-PCR (Fig. 7A). Nucleotide sequence analysis of the cDNA product generated by this procedure indicated that the initiating end of pla mRNA is 5′-ACAUUA. This places the transcription start site (position 1) at a location 77 bases upstream of the pla open reading frame (Fig. 7B). Efforts were then directed at evaluating potential cis-acting Crp-dependent regulatory elements, which would be indicative of a direct mechanism for pla activation by Crp.
FIG. 7.

Mapping promoter elements of pla. (A) cDNA of pla mRNA was generated by 5′ RACE-PCR. Lane 1, nucleotide size markers; lane 2, control sample in which reverse transcriptase was not included in the reaction mixture; lane 3, sample of the complete reaction mixture (pla cDNA). The sizes of nucleotide markers (in base pairs) are indicated on the left. The arrow on the right indicates the location of the amplified pla cDNA. (B) Coding strand of DNA for the promoter region of pla. The location of the transcription start site (+1) established by 5′ RACE-PCR is indicated. Other predicted features are underlined and labeled, including −10 and −35 sites, the cAMP-Crp binding site, and the start codon. The first 19 amino acids of Pla are shown above the corresponding codons. The E. coli consensus sequences for a cAMP-Crp binding site and the −10 and −35 sites are indicated in bold type below the matching elements of the pla DNA sequence.
Crp binds to a site centered 60.5 bases upstream of the pla transcription start site.
Examination of the nucleotide sequence upstream of the pla transcription start site identified a sequence which shares similarity to the E. coli consensus −10 and −35 promoter regions. Further upstream was an element centered at position −60.5 that is identical to an E. coli consensus binding site for Crp (Fig. 7B). Several experiments were then conducted to test the prediction that Crp interacted with this site and to test whether it was an element necessary for pla expression. To facilitate this analysis, Y. pestis Crp was purified by affinity chromatography as a hexahistidine-tagged fusion protein expressed in E. coli (data not shown). Different portions of the pla promoter were incubated with increasing concentrations of Crp, and the formation of protein-DNA complexes was evaluated by EMSA. A retarded band representing Crp-DNA complexes was detected in each case where the DNA fragment contained the predicted Crp binding site (Fig. 8, fragments A and B). A DNA fragment lacking this site failed to form Crp-DNA complexes (Fig. 8, fragment D). Likewise, alteration of the nucleotide sequence with substitutions at residues of the predicted Crp binding half-sites (C → G at nucleotide −54 and G → C at nucleotide −67) prevented the formation of stable Crp-DNA complexes (Fig. 8, fragment C). These results demonstrate that Crp directly binds to the pla promoter and that specific nucleotides are necessary for Crp-DNA complex formation. Experiments were then conducted to further delineate the Crp binding site location.
FIG. 8.
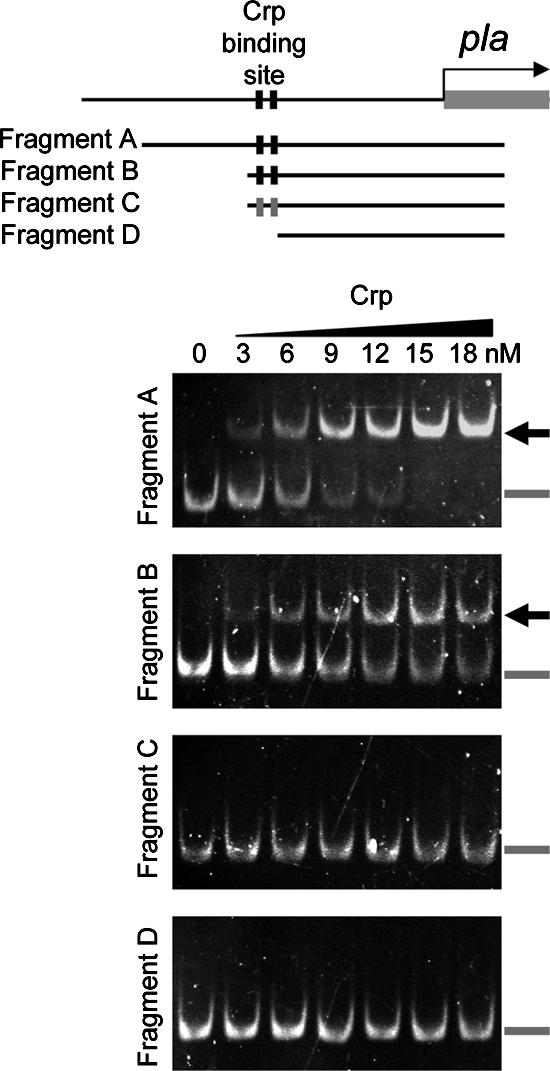
Crp complexes with DNA from the pla promoter region assessed by EMSA. The schematic diagram shows the location of the Crp binding site centered at nucleotide −60.5 (black boxes) relative to the pla open reading frame. Below this diagram the fragments of DNA evaluated for Crp-DNA complex formation are shown. Fragment A, nucleotides −169 to 133; fragment B, nucleotides −77 to 133; fragment C, nucleotides −77 to 133 with two nucleotide changes that alter each Crp binding half-site (gray boxes); fragment D, nucleotides −56 to 133 without the Crp binding site. Images for each EMSA are also shown; the samples used contained increasing amounts of purified Crp, as indicated. The location of Crp-DNA complexes is indicated by the arrows. The position of unbound DNA is indicated by the gray bars.
A DNase I footprinting approach was used to define the specific location where Crp was bound to the pla promoter. When the DNA binding reaction mixture included both Crp and cAMP, a region of protection was observed that overlapped with the predicted Crp binding site (Fig. 9, lane 4). As expected, omission of cAMP or Crp from the reaction mixture eliminated protection of this region (Fig. 9, lanes 2 and 3). Protection by Crp could be titrated by reducing the concentration of Crp (Fig. 9, lanes 4 to 9). These results indicate that Crp binds to a site upstream of pla between bp −70 and −52, which is consistent with the predicted binding site centered at position −60.5 of the pla promoter. The data clearly indicate that this transcription factor binds at a location where it can serve as an activator.
FIG. 9.

Interaction of Crp with the pla promoter region revealed by DNase I footprinting. The precise positions of the nucleotides protected by Crp were determined by comparison with the results of the Maxam-Gilbert G+A cleavage reaction shown. Purified Crp in the presence of 100 μM cAMP was used in all of the binding reactions except the control reaction shown in lane 2. Lane 1, G+A cleavage reaction; lane 2, control reaction with 0.9 μM Crp but no cAMP added; lane 3, control reaction with no Crp added; lane 4, 0.9 μM Crp; lane 5, 0.45 μM Crp; lane 6, 0.22 μM Crp; lane 7, 0.11 μM Crp; lane 8, 0.05 μM Crp; lane 9, 0.025 μM Crp. The DNA sequence of the pla promoter region is indicated on the left, and the protected nucleotide sites are indicated by bold type. The region protected from DNase I cleavage due to Crp binding of the DNA is indicated by the box.
Alteration or deletion of the Crp binding site abrogates transcription of pla.
To demonstrate that the in vitro binding analysis correlated with in vivo promoter activity, we turned to genetic analysis. Two Ppla-lacZYA transcriptional fusions were examined, each of which contained different mutations affecting the Crp binding site (Fig. 10). One transcriptional fusion differed by having a pla promoter devoid of nucleotides encompassing the Crp binding site. The other transcriptional fusion contained a pla promoter that carried the two point mutations altering the nucleotide sequence of each Crp binding half-site as described above for the EMSA experiments. The transcriptional fusions were each introduced into Y. pestis, and the level of β-galactosidase produced by each strain was quantified (Fig. 10). In both cases, there was a loss of lacZ expression compared to the expression with a fusion containing an intact pla promoter. The residual expression level for each of the fusions was not affected further by inactivation of crp or cyaA (data not shown). These results support the conclusion that expression of pla is subject to direct transcriptional control by the cAMP-Crp regulatory system.
FIG. 10.
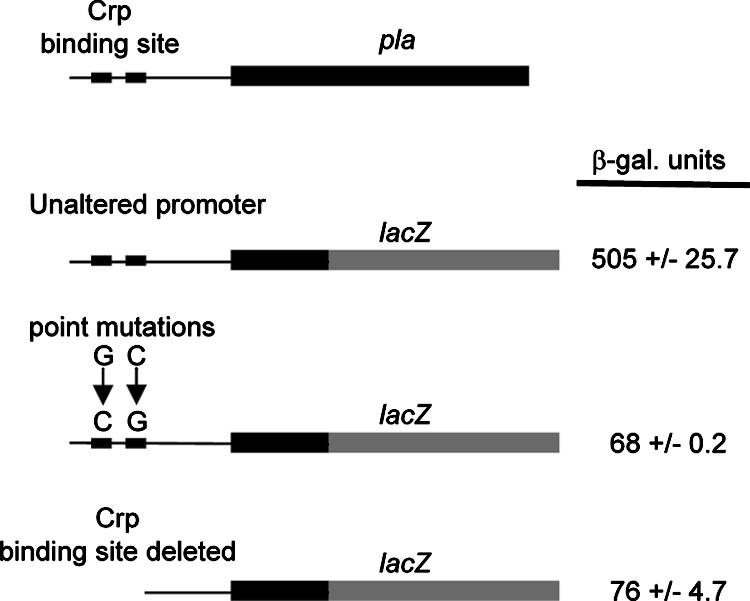
Upstream Crp binding site is essential for pla expression: schematic representation of the pla gene and different Ppla-lacZ transcriptional fusions (not to scale). The pla promoter region is indicated by a thin black line with two black boxes representing the half-sites of the Crp binding site. The pla open reading frame is indicated by a thick black line, and the lacZ open reading frame is indicated by a thick gray line. Each fusion, located on a plasmid (Table 1), was transformed into Y. pestis, and the level of β-galactosidase (β-gal.) expression was determined for samples collected following cultivation in buffered L medium. Each assay was completed at least three times, and the means ± standard deviations are shown.
DISCUSSION
The plague pathogen needs to monitor, respond to, and ultimately adapt to a spectrum of environmental stresses and host assaults in order to successfully establish an infection in an animal and to survive within a flea for vector-mediated transmission. One way to manage metabolism, environmental stress, and virulence factor expression is through transcriptional regulation. This ensures that virulence factor production is optimized and occurs during the correct stages of the Y. pestis life cycle. Pla has been shown previously to be important for the virulence of Y. pestis, but little was known about how its synthesis was controlled in response to environmental and metabolic stimuli. In this study, we present results that revealed a critical role for the cAMP-Crp regulatory system in controlling the transcription of pla. Our evidence supports a model in which Crp, in the presence of the signaling molecule cAMP, directly activates pla transcription by binding to a cis-acting regulatory element located within the promoter region.
The cAMP-Crp regulatory system has not been examined before for Y. pestis, but studies of other related bacteria serve as a foundation for comparison. E. coli has been the model organism for most studies that have defined how this signaling pathway operates. At least in the laboratory, endogenously synthesis appears to be the primary source of cAMP. This synthesis is dependent upon a class I adenylate cyclase homologous to Y. pestis CyaA. The E. coli protein consists of a catalytic domain and a regulatory domain that limits cAMP synthesis (9). Allosteric control by the regulatory domain is due to proteins of the phosphotransferase system responsible for the import of glucose and other carbon and energy sources. Upon glucose transport, cAMP synthesis by CyaA is limited, which decreases accumulation of intracellular cAMP. One effect of limitation of cAMP synthesis is a reduction in the expression of Crp-activated genes involved in catabolism of nonglucose substrates. Y. pestis ferments a number of different catabolites (8, 28). Among the compounds tested, fermentation of all but glucose required the cAMP-Crp regulatory system to be functional. This included the fermentation of glycerol, which is a defining metabolic characteristic of strains like strain KIM that belong to Y. pestis biovar Medievalis. The Crp-dependent control of nonglucose metabolism highlights a similarity between Y. pestis and E. coli. Regulation of pla was partially repressed when the growth medium was supplemented with glucose, but expression was not reduced to the level observed when cyaA was inactivated. We inferred from this comparison that CyaA retains some activity when Y. pestis is cultivated in a medium supplemented with glucose. This provides sufficient levels of cAMP for partial activation of pla by Crp.
Crp-mediated induction of pla could also occur in response to external cAMP. This mechanism of induction required cAMP to be present at a concentration equal to 250 μM to cause at least 50% activation of pla. This concentration of cAMP is greater than the reported levels of cAMP in biological samples. Nonetheless, we cannot overlook the potential for Y. pestis to occupy a microniche where it is exposed to quantities of cAMP sufficient to stimulate Crp. Our evaluation of the external concentration of cAMP needed for pla induction may have resulted in an overestimate. The Y. pestis genome was previously reported to contain open reading frames predicted to encode phosphodiesterases, which could have interfered with the analysis by rapidly degrading the cAMP signaling molecule as it entered the cell (8, 23). Situations where down-regulation of phosphodiesterase activity occurs would effectively sensitize the bacterium to external sources of cAMP. This idea is supported by studies with Salmonella enterica in which loss of phosphodiesterase activity resulted in a sharp increase in Crp activation of genes and phenotypes in response to external cAMP (1, 5). Clearly, this is an area where additional analysis is needed if this hypothesis is to be validated.
The mechanism by which Crp regulates pla was probed using biochemical approaches that revealed a site where the transcription factor binds the pla promoter. Crp-DNA complexes were detected by both EMSA and DNase I footprinting when the purified protein was coincubated with nucleotide fragments corresponding to the pla promoter region. Binding of DNA by Crp required cAMP as a cofactor, which provides confirmatory evidence for a model where this signaling molecule directly affects Y. pestis Crp affinity for DNA. The Crp binding site was mapped to a sequence centered at nucleotide −60.5 upstream of the transcription start site. These results and the location of the Crp binding site are consistent with features of class I Crp-dependent promoters of E. coli that enhance transcription activation from a site upstream by facilitating the binding of RNA polymerase to the promoter (29). Conservation of Crp residues known to form contact points with RNA polymerase further supports this model (40). Taken together, these data support the idea that Y. pestis Crp and E. coli Crp utilize similar mechanisms to recruit RNA polymerase for transcription initiation.
The complete repertoire of genes subject to regulatory control by Crp remains to be established, as does delineation of how the cAMP signal is integrated with other regulatory signals that affect virulence factor expression. Other studies have established that there are additional environmental cues, such as temperature, that additionally impact the global regulatory network of gene expression by Y. pestis during an infection (22). For the enteropathogen Y. enterocolitica, it was demonstrated that Crp is required for expression of the Ysc T3S system and other virulence factors (26). There is regulatory and functional conservation of genes encoding the Ysc T3S system among Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. It was therefore not surprising that functional loss of CyaA and Crp diminished Yop secretion by Y. pestis. It will be interesting to define the Crp-dependent regulatory checkpoint for the Ysc T3S system. Our current analysis of pla should serve as a firm foundation for these future studies.
The infectious cycle of Y. pestis involves stages in which the bacterium experiences a multitude of environmental changes. Survival within a mammal is distinctly different than survival within a blood-feeding arthropod. Y. pestis must establish an infection in both types of hosts and retain the capacity for efficient transmission to maintain its lifestyle. Colonization in each case requires complex coordination of physiological activities with specialized virulence factors that eliminate host barriers. Efficient transmission from the mammalian host by the flea is substantially dependent upon the bacterium reaching high titers in the bloodstream. Placement of virulence factors under control of the cAMP-Crp regulatory system links mammalian host colonization factors with activities that are essential for Y. pestis multiplication. Control of Pla production is a clear mechanistic example of how Crp mediates its regulatory effects by direct transcriptional control in Y. pestis. Moreover, defining the extended gene network affected by Crp should provide insight into how the cAMP-Crp regulon has been fine-tuned by this specialized pathogen.
Acknowledgments
We thank the anonymous reviewers for many insightful comments. Additionally, we are grateful to our colleagues in the Young lab for their generous contribution of reagents and for insightful feedback on this study.
This work was supported in part by grants AI156042 and AI067676 from the National Institute of Health National Institute of Allergy and Infectious Diseases to G.M.Y.
Footnotes
Published ahead of print on 12 October 2007.
REFERENCES
- 1.Alper, M. D., and B. N. Ames. 1975. Cyclic 3′,5′-adenosine monophosphate phosphodiesterase mutants of Salmonella typhimurium. J. Bacteriol. 122:1081-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ammerman, J. W., and F. Azam. 1982. Uptake of cyclic AMP by natural populations of marine bacteria. Appl. Environ. Microbiol. 43:869-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beesley, E. D., R. R. Brubake, W. A. Janssen, and M. J. Surgalla. 1967. Expression of coagulase and mechanisms of fibrinolysis. J. Bacteriol. 94:19-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bensing, B. A., B. J. Meyer, and G. M. Dunny. 1996. Sensitive detection of bacterial transcription initiation sites and differentiation from RNA processing sites in the pheromone-induced plasmid transfer system of Enterococcus faecalis. Proc. Natl. Acad. Sci. USA 93:7794-7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botsford, J. L. 1984. Cyclic AMP phosphodiesterase in Salmonella typhimurium: characteristics and physiological function. J. Bacteriol. 160:826-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Botsford, J. L., and J. G. Harman. 1992. Cyclic AMP in prokaryotes. Microbiol. Rev. 56:100-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowan, C., H. A. Jones, Y. H. Kaya, R. D. Perry, and S. C. Straley. 2000. Invasion of epithelial cells by Yersinia pestis: evidence for a Y. pestis-specific invasin. Infect. Immun. 68:4523-4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng, W., V. Burland, G. Plunkett III, A. Boutin, G. F. Mayhew, P. Liss, N. T. Perna, D. J. Rose, B. Mau, S. Zhou, D. C. Schwartz, J. D. Fetherston, L. E. Lindler, R. R. Brubaker, G. V. Plano, S. C. Straley, K. A. McDonough, M. L. Nilles, J. S. Matson, F. R. Blattner, and R. D. Perry. 2002. Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184:4601-4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deutscher, J., C. Francke, and P. W. Postma. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70:939-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galas, D., and A. Schmitz. 1978. DNase footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 5:3157-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldenbaum, P. E., and G. A. Hall. 1979. Transport of cyclic adenosine 3′,5′-monophosphate across Escherichia coli vesicle membranes. J. Bacteriol. 140:459-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang, K. J., and M. M. Igo. 1996. Identification of the bases in the ompF regulatory region, which interact with the transcription factor OmpR. J. Mol. Biol. 262:615-628. [DOI] [PubMed] [Google Scholar]
- 13.Inglesby, T. V., D. T. Dennis, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, A. M. Friedlander, J. Hauer, J. F. Koerner, M. Layton, J. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, M. Schoch-Spana, and K. Tonat. 2000. Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA 283:2281-2290. [DOI] [PubMed] [Google Scholar]
- 14.Kamenetsky, M., S. Middelhaufe, E. M. Bank, L. R. Levin, J. Buck, and C. Steegborn. 2006. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J. Mol. Biol. 362:623-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinder, S. A., J. L. Badger, G. O. Bryant, J. C. Pepe, and V. L. Miller. 1993. Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O:8 and construction of a transformable R−M+ mutant. Gene 136:271-275. [DOI] [PubMed] [Google Scholar]
- 16.Korhonen, T. K., M. Kukkonen, R. Vikola, H. Lang, M. Suomalainen, P. Kyllonen, and K. Lahteenmaki (ed.). 2004. The plasminogen activator Pla of Yersinia pestis: localized proteolysis and systemic spread. Horizon Bioscience, Norfolk, United Kingdom.
- 17.Kutyrev, V., R. J. Mehigh, V. L. Motin, M. S. Pokrovskaya, G. B. Smirnov, and R. R. Brubaker. 1999. Expression of the plague plasminogen activator in Yersinia pseudotuberculosis and Escherichia coli. Infect. Immun. 67:1359-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lathem, W. W., P. A. Price, V. L. Miller, and W. E. Goldman. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509-513. [DOI] [PubMed] [Google Scholar]
- 19.Lodge, J., J. Fear, S. Busby, P. Gunasekaran, and N. R. Kamini. 1992. Broad host range plasmids carrying the Escherichia coli lactose and galactose operons. FEMS Microbiol. Lett. 74:271-276. [DOI] [PubMed] [Google Scholar]
- 20.Maxam, A., and W. Gilbert. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 65:499-525. [DOI] [PubMed] [Google Scholar]
- 21.McKnight, G. S. 1991. Cyclic AMP second messenger systems. Curr. Opin. Cell Biol. 3:213-217. [DOI] [PubMed] [Google Scholar]
- 22.Motin, V. L., A. M. Georgescu, J. P. Fitch, P. P. Gu, D. O. Nelson, S. L. Mabery, J. B. Garnham, B. A. Sokhansanj, L. L. Ott, M. A. Coleman, J. M. Elliott, L. M. Kegelmeyer, A. J. Wyrobek, T. R. Slezak, R. R. Brubaker, and E. Garcia. 2004. Temporal global changes in gene expression during temperature transition in Yersinia pestis. J. Bacteriol. 186:6298-62305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkhill, J., B. W. Wren, N. R. Thomson, R. W. Titball, M. T. Holden, M. B. Prentice, M. Sebaihia, K. D. James, C. Churcher, K. L. Mungall, S. Baker, D. Basham, S. D. Bentley, K. Brooks, A. M. Cerdeno-Tarraga, T. Chillingworth, A. Cronin, R. M. Davies, P. Davis, G. Dougan, T. Feltwell, N. Hamlin, S. Holroyd, K. Jagels, A. V. Karlyshev, S. Leather, S. Moule, P. C. Oyston, M. Quail, K. Rutherford, M. Simmonds, J. Skelton, K. Stevens, S. Whitehead, and B. G. Barrell. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413:523-527. [DOI] [PubMed] [Google Scholar]
- 24.Passner, J. M., S. C. Schultz, and T. A. Steitz. 2000. Modeling the cAMP-induced allosteric transition using the crystal structure of CAP-cAMP at 2.1 A resolution. J. Mol. Biol. 304:847-859. [DOI] [PubMed] [Google Scholar]
- 25.Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis: etiologic agent of plague. Clin. Microbiol. Rev. 10:35-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen, S., and G. M. Young. 2002. Essential role for cAMP and its receptor protein in Yersinia enterocolitica virulence. Infect. Immun. 70:3665-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pouillot, F., A. Derbise, M. Kukkonen, J. Foulon, T. K. Korhonen, and E. Carniel. 2005. Evaluation of O-antigen inactivation on Pla activity and virulence of Yersinia pseudotuberculosis harbouring the pPla plasmid. Microbiology 151:3759-3768. [DOI] [PubMed] [Google Scholar]
- 28.Santer, M., and S. Ajl. 1955. Metabolic reaction of Pasteurella pestis. II. The fermentation of glucose. J. Bacteriol. 69:298-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savery, N. J., G. S. Lloyd, S. J. Busby, M. S. Thomas, R. H. Ebright, and R. L. Gourse. 2002. Determinants of the C-terminal domain of the Escherichia coli RNA polymerase alpha subunit important for transcription at class I cyclic AMP receptor protein-dependent promoters. J. Bacteriol. 184:2273-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebbane, F., C. O. Jarrett, D. Gardner, D. Long, and B. J. Hinnebusch. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. USA 103:5526-5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seoh, H. K., and P. C. Tai. 1999. Catabolic repression of secB expression is positively controlled by cyclic AMP (cAMP) receptor protein-cAMP complexes at the transcriptional level. J. Bacteriol. 181:1892-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 33.Sodeinde, O. A., and J. D. Goguen. 1988. Genetic analysis of the 9.5-kilobase virulence plasmid of Yersinia pestis. Infect. Immun. 56:2743-2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sodeinde, O. A., A. K. Sample, R. R. Brubaker, and J. D. Goguen. 1988. Plasminogen activator/coagulase gene of Yersinia pestis is responsible for degradation of plasmid-encoded outer membrane proteins. Infect. Immun. 56:2749-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sodeinde, O. A., Y. V. Subrahmanyam, K. Stark, T. Quan, Y. Bao, and J. D. Goguen. 1992. A surface protease and the invasive character of the plague. Science 258:1004-1007. [DOI] [PubMed] [Google Scholar]
- 36.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195-199. [PubMed] [Google Scholar]
- 37.Welkos, S. L., A. M. Friedlander, and K. J. Davis. 1997. Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain C092. Microb. Pathog. 23:211-223. [DOI] [PubMed] [Google Scholar]
- 38.Wren, B. W. 2003. The yersiniae—a model genus to study the rapid evolution of bacterial pathogens. Nat. Rev. Microbiol. 1:55-64. [DOI] [PubMed] [Google Scholar]
- 39.Young, B. M., and G. M. Young. 2002. YplA is exported by the Ysc, Ysa and flagellar type III secretion systems of Yersinia enterocolitica. J. Bacteriol. 184:1324-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou, Y., T. J. Merkel, and R. H. Ebright. 1994. Characterization of the activating region of Escherichia coli catabolite gene activator protein (CAP). II. Role at class I and class II CAP-dependent promoters. J. Mol. Biol. 243:603-610. [DOI] [PubMed] [Google Scholar]