

Abstract
The flavoprotein WrbA, originally described as a tryptophan (W) repressor-binding protein in Escherichia coli, has recently been shown to exhibit the enzymatic activity of a NADH:quinone oxidoreductase. This finding points toward a possible role in stress response and in the maintenance of a supply of reduced quinone. We have determined the three-dimensional structure of the WrbA holoprotein from E. coli at high resolution (1.66 Å), and we observed a characteristic, tetrameric quaternary structure highly similar to the one found in the WrbA homologs of Deinococcus radiodurans and Pseudomonas aeruginosa. A similar tetramer was originally observed in an iron-sulfur flavoprotein involved in the reduction of reactive oxygen species. Together with other, recently characterized proteins such as YhdA or YLR011wp (Lot6p), these tetrameric flavoproteins may constitute a large family with diverse functions in redox catalysis. WrbA binds substrates at an active site that provides an ideal stacking environment for aromatic moieties, while providing a pocket that is structured to stabilize the ADP part of an NADH molecule in its immediate vicinity. Structures of WrbA in complex with benzoquinone and NADH suggest a sequential binding mechanism for both molecules in the catalytic cycle.
In 1993, a previously unknown protein was reported to copurify with the tryptophan repressor TrpR of Escherichia coli and was, due to this assumed interaction, termed tryptophan (W) repressor binding protein, WrbA (36). At closer inspection, WrbA was found to bind a flavin mononucleotide (FMN), making it a founding member of a novel family of flavodoxins (12). Based on sequence analyses, WrbA was predicted to show the typical β-α-β-fold of flavodoxins, with a twisted, five-stranded, parallel β-sheet and a binding site for the FMN cofactor at the carboxy terminal end of this β-sheet. Distinct from the basic flavodoxin fold (30), a conserved insertion was found in sheet β5 (residues 140 to 160) and was predicted to form an additional αβ-unit (11). Proteins with this type of insertion have been classified as long-chain flavodoxins (23). The functional role of WrbA remained controversial, as initial work indicated that E. coli WrbA promotes complex formation between the repressor TrpR and its operator DNA. WrbA alone failed to bind to DNA, such that it was consequently proposed to be an accessory element that could enhance TrpR-dependent repression of genes upon transition to stationary phase (36). Subsequent experiments addressed the influence of WrbA on the TrpR-DNA complex, and as these did not show any effect, the involvement of WrbA in transcription regulation was revoked (12).
While the function of WrbA was entirely enigmatic, sequence database searches hinted that the WrbA family of flavoproteins was homologous to a group of several biochemically characterized NAD(P)H:quinone oxidoreductases (NQO) from various species of fungi and green plants (2, 3, 5, 13, 17, 22, 35). Based on subsequent biochemical assays, NQO activity was demonstrated for the WrbA proteins of both E. coli and Archaeoglobus fulgidus, and this family of flavoproteins was therefore described as a new class of type IV NQOs (29). The E. coli protein bound NADH with a Km of 14 ± 0.43 μM and benzoquinone with a Km of 5.8 ± 0.12 μM. The affinities for WrbA from A. fulgidus were 19 ± 1.70 μM for NADH and 37 ± 3.60 μM for benzoquinone, and an analysis of apparent initial velocities indicated that the maximal activities of the enzymes were consistent with the optimal growth temperatures of the mesophilic E. coli and the hyperthermophilic A. fulgidus, respectively (29). Along with cyclic voltammetry measurements of WrbA (27), these results implicate WrbA proteins in the two-electron reduction of quinones as a measure of protection against oxidative stress.
We have crystallized WrbA from E. coli in its native state and with both the substrate benzoquinone and the cosubstrate NADH bound. Our structures point toward mutually exclusive binding of quinones and NADH to the FMN cofactor, indicating an ordered, sequential mechanism of catalysis. Furthermore, the structures underline the obvious homologies of WrbA with the iron-sulfur flavoproteins (ISF) from Methanosarcina thermophila and A. fulgidus that form the same characteristic homotetrameric structures but differ significantly in functionally relevant, structural details (1).
We note that at the time of submission of the manuscript, crystallization of E. coli WrbA and preliminary evaluation of data had been presented independently in two publications (33, 34).
MATERIALS AND METHODS
Crystallization and data collection.
Crystallization experiments for WrbA from E. coli were set up at 20°C for sitting-drop vapor diffusion using a 1:1 drop ratio composed of 10 to 50 mg/ml of protein in 20 mM Tris-HCl buffer at pH 7.8 and a suitable reservoir solution. The reservoir was composed solely of 15 to 35% (wt/vol) polyethylene glycol (PEG) 1500, and single crystals appeared after 1 to 2 days and continued to grow for 2 to 3 weeks (Fig. 1). For flash freezing prior to data collection, crystals of E. coli WrbA were washed for 1 to 5 min in solutions containing increasing amounts of PEG 1500 in steps of 5% until a final concentration of 50% (wt/vol) was reached. Diffraction data sets to a maximum resolution of 1.66 Å were collected using synchrotron radiation at EMBL/DESY, Hamburg, Germany. Crystals belonged to the tetragonal space group P43212, but occurred in two distinct crystal packing arrangements with different cell axes. The smaller cell (a = b = 62.80 Å; c = 201.53 Å) yielded a typical crystal packing with a Matthews parameter VM (21) of 2.26 Å3/Da, corresponding to a solvent content of 46%, but the larger cell (a = b = 94.36 Å; c = 175.36 Å) resulted in a very sparse crystal packing with a solvent content of 74% (VM of 4.39 Å3/Da), which is unusually high for soluble proteins. Nevertheless, crystals with both cells diffracted well, with the more frequently occurring, large cell giving the highest resolution data set. Data were indexed, integrated, and scaled using the HKL suite (28) and analyzed using XPREP (Bruker). Table 1 gives the data collection and refinement statistics.
FIG. 1.

Crystals of E. coli WrbA. The tetragonal bipyramidal shape of the crystals reflects their P422 symmetry.
TABLE 1.
Data collection statisticsa
| Data set | Value for the indicated crystal type
|
|||
|---|---|---|---|---|
| Native | NADH | Benzoquinone | Small cell | |
| Space group | P 43212 | P 43212 | P 43212 | P 43212 |
| Unit cell constants (Å) | ||||
| a | 94.36 | 94.20 | 94.24 | 62.80 |
| b | 94.36 | 94.20 | 94.24 | 62.80 |
| c | 175.36 | 173.64 | 175.55 | 201.53 |
| Resolution range (outer shell [Å]) | 50.00-1.66 (1.69-1.66) | 50.00-2.05 (2.15-2.05) | 50.00-1.95 (2.05-1.95) | 50.00-1.84 (1.94-1.84) |
| No. of unique reflections | 91,774 | 48,152 | 59,061 | 35,588 |
| Completeness (%) | 98.7 (97.3) | 96.9 (93.7) | 99.5 (97.3) | 99.4 (97.0) |
| Redundancy | 6.3 (5.3) | 4.1 (3.4) | 5.9 (5.2) | 6.7 (6.3) |
| Mean I/σ (I) | 12.1 (2.2) | 5.9 (1.9) | 6.2 (1.8) | 10.3 (2.6) |
| Rsym | 0.12 (0.68) | 0.12 (0.50) | 0.11 (0.58) | 0.06 (0.40) |
| Rpim | 0.06 (0.34) | 0.07 (0.36) | 0.03 (0.29) | |
Values in brackets represent the last resolution shells.
Substrate soaks and refinement.
For all soaking experiments, substrate and cosubstrate molecules were prepared as stock solutions of 200 mM NADH and 100 mM 1,4-benzoquinone. These were then added to the cryoprotectant buffer containing 50% (wt/vol) PEG 1500 to a final substrate concentration of 1 mM. Equivalent crystallization and soaking experiments were also carried out under anaerobic conditions, either with protein crystallized in a glove box containing less than 1 ppm of oxygen or by the addition of 2 mM sodium dithionite solution, pH 8, to all cryoprotectant solutions.
Structure solution and refinement.
The structure was solved by molecular replacement with programs from the CCP4 suite (6), using the homologous apo-WrbA structure of Deinococcus radiodurans (Protein Data Bank identifier [PDB ID] 1YDG) as a search model (10). For refinement, 5% of the total reflections were chosen at random and used as a test set for cross validation (4). In both unit cells, the replacement solution contained two monomers in the asymmetric unit. In the larger cell, crystal contacts were exclusively mediated between the protruding helix α3 from one monomer and the C-terminal parts of helices α2 and α7 of another monomer from a different WrbA tetramer. The replacement solutions were initially refined using the REFMAC program (25), which provided the first electron density maps. The electron density for the FMN cofactor that was absent in the search model was readily identified in the first Fo − Fc (the observed and calculated structure factor amplitudes, respectively) electron density maps. All model building was carried out in the programs O (14) and COOT (9). The model for E. coli WrbA comprises the entire polypeptide chain with the exception of the first residue (Met 1) that might have been naturally cleaved off. The initially planar conformation of the isoalloxazine ring of FMN did not fit the observed electron density. In order to allow the refinement programs to introduce a slight bend, the estimated standard deviations of the planarity restraints were increased in the library files.
With continuing improvement of the models, additional electron density that was at first modeled as water molecules became clearly continuous. It was then modeled with fragments of PEG of variable lengths. The final atomic model of native E. coli WrbA at a resolution of 1.66 Å comprises 198 amino acid residues plus one FMN molecule per monomer and a total of 463 water molecules and four PEG fragments of various lengths. Stereochemical analyses of the structures were carried out with PROCHECK (17). In a Ramachandran plot, all residues were found in the most favored or additionally allowed regions. Refinement statistics are given in Table 2. All figures were made with MOLSCRIPT (16) and Raster3D (24) or PyMOL (7).
TABLE 2.
Refinement statistics
| Data seta | Value for the indicated crystal type
|
|||
|---|---|---|---|---|
| Native | NADH | Benzoquinone | Small cell | |
| Resolution range (Å) | 50.00-1.66 | 50.00-2.05 | 50.00-1.95 | 50.00-1.84 |
| Rcryst | 0.169 | 0.168 | 0.172 | 0.187 |
| Rfree | 0.186 | 0.199 | 0.208 | 0.223 |
| RMSD in bond lengths (Å) | 0.012 | 0.018 | 0.017 | 0.019 |
| RMSD in bond angles (°) | 1.328 | 2.046 | 1.686 | 1.671 |
| Avg B-factor for protein (Å2) | 26.5 | 30.7 | 29.9 | 35.2 |
| Avg B-factor for waters (Å2) | 47.7 | 49.1 | 45.5 | 47.3 |
| Cruickshank's DPI (Å) | 0.047 | 0.087 | 0.087 | 0.096 |
DPI, diffraction-component precision index.
Protein Data Bank accession codes.
Structure factors and coordinates have been deposited with the Protein Data Bank at http://www.pdb.org. Accession codes are 3B61 for the native crystal, 3B6J for the NADH complex, 3B6K for the benzoquinone complex, and 3B6M for the second crystal form (small cell).
RESULTS
Crystal structure of E. coli WrbA.
WrbA from E. coli shows high homologies to the previously described WrbA proteins from D. radiodurans (PDB ID 1YRH; root mean square deviation [RMSD] for all Cα atoms, 1.53 Å) and P. aeruginosa (PDB ID 1ZWL; RMSD, 1.20 Å) (10). However, the E. coli protein was always crystallized in its holo-form, i.e., with the cofactor FMN tightly bound to the protein. The structural hallmarks of the fold of WrbA family proteins are five parallel β-strands forming a twisted β-sheet, with each strand, β1 to β5, followed by an α-helix, α1 to α5 (Fig. 2). Three insertions add to this topology: a loop inserted between strand β2 and helix α2b (residues 38 to 64; loop 1) that contains a further helical segment, α2a; the aforementioned insertion within strand β5 (loop 2); and a short insertion before helix α5 (loop 3). In all structures, the entire protein chain is clearly defined, and the absence of residue Met 1 is most likely due to posttranslational processing, as the amino terminus at Ala 2 and the terminal carboxy group at Gly 198 form a direct salt bridge of 2.6 Å. Disorder was observed, however, in the second crystal form with the smaller unit cell. Here, the loop 2 region showed significant disorder, and residues 146 to 156 were not visible in electron density maps. In this structure, residue Tyr 143, moderately conserved among the WrbA family (Fig. 3) was flipped outward, now facing away from the active site. This disorder cannot be explained by intermolecular interactions within the crystal lattice, as the protein surface surrounding the loop 2 region is not involved in crystal lattice contact formation in either of the two unit cells. A possible functional significance of this conformational change can therefore presently not be excluded. No significant differences were observed in data sets obtained from crystals reduced with dithionite.
FIG. 2.

Cartoon representation of WrbA from E. coli. The stereo image of the protein chain shows the N terminus in blue and the C terminus in red. Strands of the central, parallel β-sheet and the surrounding α-helices are numbered according to their occurrence in the protein sequence. The FMN cofactor is bound peripherally at the C-terminal end of the β-sheet.
FIG. 3.
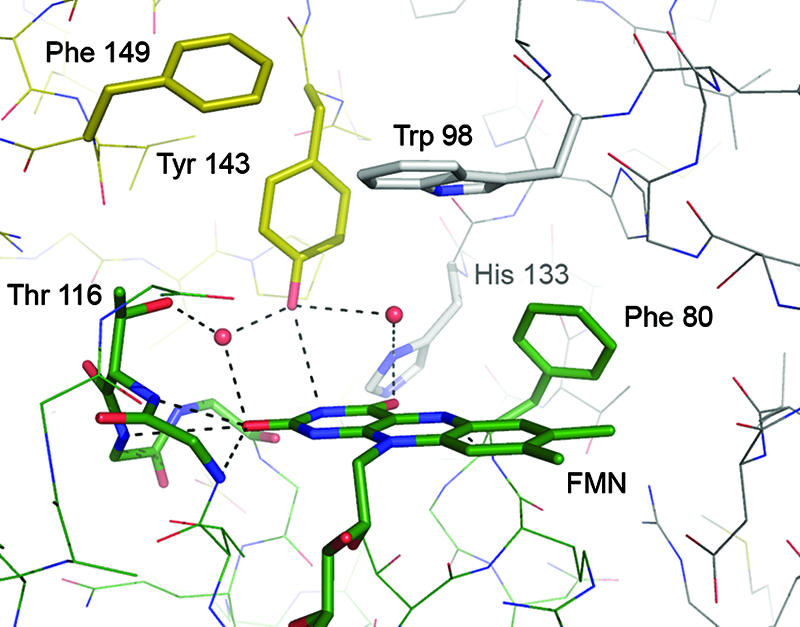
Close-up of the active site of E. coli WrbA. Residues from three monomers combine to create a specific, hydrophobic active-site pocket. Aromatic substrates can be stacked in between the FMN moiety and the side chain of Trp 98. All residues are colored by chain.
The FMN cofactor is bound on the periphery of the monomer, in a hydrogen-bonding network formed by the loop regions at the C termini of the β-sheets. It packs against the protein with its re face, whereby the catalytically relevant N5 atom of the isoalloxazine ring forms a long (3.2 Å) hydrogen bond to the backbone nitrogen of Phe 80. Below the FMN, an arginine residue, Arg 79, stabilizes the cofactor through a stacking interaction. While Arg 79 is conserved among WrbA proteins, it seems to be consistently replaced by a tyrosine in the ISF family of flavoproteins (see below) (29). Contacts between FMN and WrbA predominantly occur through the oxygen or nitrogen atoms of the backbone amide bonds, with the notable exception of His 14 that forms a hydrogen bond to an oxygen atom of the FMN phosphate.
WrbA has been found to behave like a dimer in size exclusion chromatography and like a tetramer in a dynamic light scattering experiment (29). The crystal structures of all WrbA proteins known today strongly support a homotetrameric quaternary structure with four independent active sites. The FMN cofactor becomes buried in a distinct active-site cavity upon multimerization, and residues from three different monomers participate in composing the structural features of this cavity. The environment of the active site itself is largely hydrophobic. Its rear wall is formed by the side chain of Phe 80, and its roof is formed by Trp 98 from a neighboring subunit, ideally suited to stack aromatic moieties—such as quinones or nicotinamide—between the indole side chain and the FMN. A second residue involved from the same subunit as Trp 98 is His 133, forming a hydrogen bond to oxygen O4 of FMN. From the third subunit involved in forming the active site, Tyr 143 forms a long (3.25 Å) hydrogen bond to N3 of the FMN, and Phe 149 shields this residue against the solvent (Fig. 3). Residues His 133 and Tyr 143, although close in protein sequence, in this way form parts of two different active sites within the tetramer (Fig. 4A).
FIG. 4.

Tetrameric arrangement of WrbA and binding of NADH. (A) Cartoon representation of the tetramer. A single monomer is shown, colored as described in the legend of Fig. 1. Note the extended loop 2 region (Fig. 2) interacting with a neighboring monomer. (B) Surface representation of the tetramer, centered on the active site of one monomer. The picture shows a PEG molecule bound at the active site, an NADH molecule in close proximity, and several further PEG molecules along a dimer interface. (C) Stereo representation of a close-up of the active site in the same orientation as in panel B. The left and right images are for the left and right eyes, respectively. The nicotinamide ring of NADH stacks onto the side chain of Tyr 12.
E. coli WrbA was crystallized from a precipitant solution containing exclusively PEG 1500, and in the electron density maps, several ordered, continuous features were detected that were modeled with PEG chains (Fig. 4B). Most of these are wrapped around one of the dimer interfaces, with the longest segment involving the extended side chain of Lys 195. Also, an undefined electron density feature was located on top of the FMN cofactor in a putative binding position for substrate and cosubstrate. This density was most prominent in the NADH soak, where it was modeled as a PEG chain. In all other structures, this electron density was weaker and was consequently modeled with only two or three water molecules, but we cannot exclude at least partial presence of a PEG molecule.
NADH binding to WrbA.
In soaks of E. coli WrbA with NADH, the dinucleotide was found to bind in close proximity to the FMN cofactor to a specific binding pocket that is predominantly composed of the extended loop 1 region but is situated at the interface of two monomers in the tetramer (Fig. 4B and C). In this binding mode, excellent electron density and low B-factors were observed for the ADP-part of NADH, while the remaining nicotinamide-ribityl moiety was less ordered. Strikingly, contacts between NADH and protein are almost exclusively mediated by water molecules, with only a single hydrogen bond being observed between Oη of Tyr 12 and a phosphate oxygen of NADH. In the complex structure, the putative substrate binding pocket between the FMN cofactor and the side chain of Trp 98 was occupied by what was interpreted to be the end of a PEG chain, with its terminal hydroxo group hydrogen bonded to His 133. At the pH of 7.5 used in the crystallization setup, this histidine is likely to be deprotonated. Since crystallization and stabilization of crystals were critically dependent on the presence of PEG 1500, we did not succeed in replacing the putative PEG molecule in soaks with NADH. As a consequence, the nicotinamide-ribityl part of NADH was not modeled on the flavin ring but, rather, in a pocket next to it, with the nicotinamide ring in a π-stacking interaction with the side chain of Tyr 12, a residue that is not conserved among either WrbA or ISF family members. This observed position of the nicotinamide can be considered nonfunctional (Fig. 4C). However, manual removal of the PEG molecule and rotation of the less-well-ordered part of the NADH molecule show that the specific binding of the ADP moiety places the remainder of the dinucleotide in a perfect position to interact with the flavin and form an electron transfer-competent complex.
Complex of WrbA with benzoquinone.
Binding of the known substrate benzoquinone to the FMN site of WrbA was observed in a complex structure at a resolution of 1.95 Å. The aromatic ring system of the substrate was found to stack between the isoalloxazine ring of FMN and the indole side chain of Trp 98, placing it in an ideal position for electron transfer (Fig. 5). In the benzoquinone soaks, the quality of the observed electron density map varied consistently between the two monomers of WrbA present in the asymmetric unit of the crystal. Consequently, after refinement higher B-factors were observed for the benzoquinone molecule on the less ordered side. It can be assumed that full occupancy of the binding sites with the substrate was not achieved, possibly due to the partial binding of PEG molecules at this location, as observed in the NADH soak. However, comparisons of different electron density features with native crystals clearly confirmed the presence of the planar benzoquinone molecule (Fig. 5).
FIG. 5.
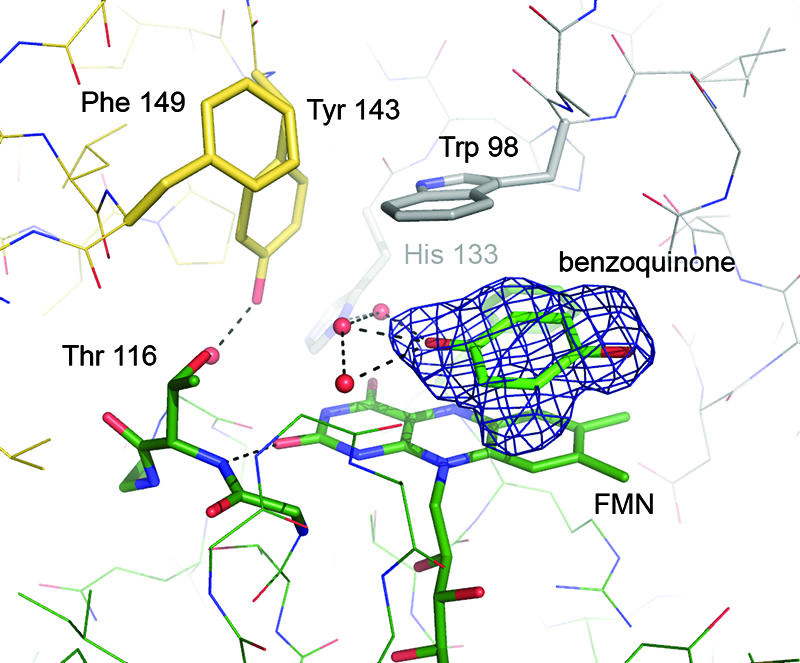
Binding of benzoquinone to the FMN site of WrbA. The picture shows the amino acid residues surrounding the active site and an Fo − Fc density map, contoured at 3.5 σ, from which the benzoquinone molecule was omitted.
Structural comparison with the ISF of M. thermophila.
The tetrameric quaternary structure of WrbA of E. coli, as well as of its homologs from D. radiodurans and P. aeruginosa, was first observed in a different family of flavoproteins, the ISFs from M. thermophila and A. fulgidus (1, 37). These proteins show the same, typical flavodoxin fold with a five-stranded β-α-β-topology, but they are characterized by an additional loop region between strand β2 and helix α2 that contains a compact Cys-X2-Cys-X2-Cys-X5-Cys motif. A cubane-type [4Fe:4S] cluster is bound to this motif, and the loop attains a conformation that brings this cluster in close proximity to an FMN cofactor not of the same, but of a neighboring, subunit in the tetramer (1). A structural comparison between WrbA and ISF underlines the high homology of both classes of proteins, particularly in the region of the central β-sheet (residues 70 to 150), where the structures of M. thermophila ISF and E. coli WrbA align with an RMSD of their Cα atoms of 0.74 Å (Fig. 6). The three loop regions inserted into the WrbA sequence with respect to regular flavodoxins are also found in ISF family proteins. The first of them, loop 1, holds the [4Fe:4S] cluster in ISF, but its conformation varies drastically between ISF and WrbA, where it plays a crucial role in forming the binding cleft for the cosubstrate NADH (Fig. 4C and 6). In contrast to the position of the cluster in ISF, this binding cleft allows for electron transfer from NADH bound predominantly by residues of the same subunit that holds the corresponding FMN cofactor.
FIG. 6.

Superposition of the monomers of E. coli WrbA (red) and M. thermophila ISF (black). Based on a common flavodoxin-like fold, the structures differ in three characteristic loop regions. Strongest variations are visible in the loop region 1, where ISF binds a [4Fe:4S] cluster while WrbA forms a specific binding cleft for NADH at a very different position in the monomer. However, multimer formation then places both putative electron donor sites at very similar positions in respect to the FMN cofactor.
The two remaining insertions, loop 2 within β-strand β5 from residues 140 to 160 and loop 3 from residues 165 to 174, have been found to be more extensive in the structure of ISF-3 from A. fulgidus than in ISF of M. thermophila (1). They have been hypothesized to increase the stability of the tetrameric arrangement of subunits, and indeed loop 2 of WrbA forms extensive interactions with neighboring subunits (Fig. 4A). As highlighted by the architecture of the active site and by the location of the NADH binding site at the interface of two subunits, the tetramer seems to be essential for the functionality of WrbA, and its additional stabilization by the extended loop 2 region supports this hypothesis.
At the FMN site, the N5 atom is shielded by a phenylalanine residue, Phe 80, and a similar situation is found in ISF-3 of A. fulgidus, where Phe 84 is found at this position; in the ISF of M. thermophila a methionine, Met 86 provides a potentially more reactive environment (1). In all cases, the roof of the active site cavity is set to interact with aromatics in a stacking interaction between the FMN moiety and Trp 98 in E. coli WrbA, Arg 104 in M. thermophila ISF, and Arg 102 in A. fulgidus ISF-3. However, the exact environment of this site, the proximity of putative donor and acceptor sites for hydrogen-bonding interaction with a substrate, and the electrostatic potential are very different. Similar to A. fulgidus ISF-3, E. coli WrbA shows a strongly negative electrostatic surface potential (data not shown), while that of M. thermophila ISF is strongly positive (1).
DISCUSSION
Reaction mechanism of WrbA.
The structure of E. coli WrbA in complex with an NADH molecule outlines a clear reaction pathway. The two-electron carrier NADH binds to its high-affinity binding site in immediate proximity to the oxidized FMN cofactor. With the nicotinamide ring stacking on the isoalloxazine, two electrons and a proton can be transferred, and the remaining NAD+ then leaves the active site, making room for the binding of a substrate molecule such as benzoquinone. Subsequent two-electron reduction of the substrate returns the FMN cofactor back to its oxidized state and closes the reaction cycle. A ping-pong mechanism of this kind, with overlapping sites for the hydride donor and acceptor, has been proposed for other oxidoreductases such as NADPH:quinone reductase from rat liver (19). The significance of the conserved tetrameric arrangement of this class of proteins lies in providing residues required to construct an intricate active-site cavity. As the WrbA monomer is evolutionarily derived from flavodoxins, it binds the FMN cofactor at the very periphery of the protein, with no possibility or only very limited possibilities to arrange residues around the exposed si face of the flavin. In WrbA, multimer formation brings two additional regions in close proximity to a neighboring FMN: the C termini of helices α3 (residues Gly 95 and Trp 98) and α4 (His 133) as well as the characteristic insertion in strand β5, the loop 2 region between residues 140 and 150 (Tyr 143 and Phe 149) (Fig. 2 and 4A).
Moreover, the arrangement of the loop 1 region in WrbA creates a specific binding cleft for NADH, and although our structure shows a flexible binding of the nicotinamide-ribityl part of the NADH molecule, the positioning of the binding sites clearly allows for an interaction in which the nicotinamide ring stacks right above the N5 position of FMN. Very similar arrangements are commonly observed in structures of nicotinamide dinucleotides bound to flavoproteins, as for example in ferredoxin:NADP+ reductase at the end of the photosynthetic electron transport chain (15, 31).
WrbA and ISF as reductases under oxidative stress.
Similar to WrbA, ISF proteins were not discovered in conjunction with a defined enzymatic function, and their exact role in the bacterial cell remains to be elucidated. They are widespread in bacteria and archaea and are able to accept electrons from ferredoxins, such that they were originally proposed to function as one-electron/two-electron switches in electron transfer chains (18). However, the discovery of a large family of these proteins soon suggested a broader functionality (37). Recently, a reducing activity toward O2 and H2O2 was shown for M. thermophila ISF, suggesting a protective role toward oxidative stress, similar to the one assumed for WrbA (29).
In this light, the obvious, evolutionary kinship of ISF and WrbA proteins outlines a class of modular, tetrameric proteins that act as putative reductases of aromatic substrates. Their active sites show substantial differences, but a π-stacking interaction involving both the FMN isoalloxazine and an amino acid residue from a neighboring subunit seems to be a common theme. At the same time, the proteins show considerable flexibility in their choice of electron donor: the ISF family exemplifies a system optimized to interact with soluble electron donors such as ferredoxins. To this end they possess a [4Fe:4S] cluster that serves as the initial electron acceptor. In ISF, a flavin semiquinone was not detected during redox titrations (18), meaning that the first electron delivered may be stored in the cluster while the second electron to arrive then triggers a two-electron reduction of the flavin to its hydroquinone form. In WrbA the electron donor system has been changed to a specific binding cleft for the two-electron donor NADH, thus removing the need for a separate one-electron storage site while retaining a possibly similar mechanism at the FMN.
Further support for the hypothesis of a widespread family of FMN reductases comes from another candidate member, the NADPH:FMN reductase YhdA from Bacillus subtilis (8, 32). This structure has been independently deposited in the PDB by the Midwest Structural Genomics Consortium in 2003 (PDB ID 1NNI) and by the Northeast Structural Genomics Consortium in 2006 (PDB ID 2GSW). However, no publication further describing the structure is available to date. YhdA shows a fold with high homology to ISF and WrbA but with a very open and accessible FMN site. The region equivalent to loop 1 does not harbor a [4Fe:4S] cluster as in ISF (Fig. 6), but it adopts a conformation close to that of ISF and distinct from the one observed in WrbA. This implies that the NADH binding groove of WrbA is absent, and thus (5) YhdA must employ a very different binding mode for NADPH. YhdA also fully retains the tetrameric quaternary structure typical for this protein family, which has been suggested to contribute to the significant thermostability of both WrbA (26) and YhdA (8). However, although its FMN and the putative substrate azobenzene are aromatic, the structure of YhdA does not show any residues placed to engage in a π-stacking such as Trp 98 in E. coli WrbA or in cation-π-stacking such as Arg 104 in M. thermophila ISF.
In contrast, one of the few structures of flavodoxin-like proteins from eukaryotic organisms, the NADPH:FMN reductase YLR011wp (Lot6p) from Saccharomyces cerevisiae, was purified and crystallized as a dimer, and although a similar tetramer is generated by crystal packing, the interaction of its two monomers is loose, putting them at a much larger distance than in any WrbA or ISF structure (20). Accordingly, YLR011wp lacks the extended loop 2 region that we presume to act in stabilizing the tetramer and is therefore to be classified as a short-chain flavodoxin (23).
As an increasing number of primary sequences and three-dimensional structures become available through genomics and structural genomics efforts, the tetrameric oxidoreductases of the WrbA and the ISF families turn out to be a widespread class of redox proteins with a wide range of possible substrates. In spite of the wealth of information gained from this broadening base of data, however, the tools of protein biochemistry and enzymology will remain crucial for achieving a comprehensive understanding of the physiological function of the individual enzymatic machineries in question.
ADDENDUM IN PROOF
While this paper was under review, a description of the structure was published (J. Carey, J. Brynda, J. Wolfová, R. Grandori, T. Gustavsson, R. Ettrich, and I. K. Smatanová, Protein Sci. 16:2301-2305, 2007). It is in accordance with our data but does not show any ligand binding to the FMN.
Acknowledgments
This work was supported by a Marie Curie Intra-European Fellowship within the 6th Framework Programme (S.L.A.A.), by the EMBO Young Investigator Programme (O.E.), by Deutsche Forschungsgemeinschaft (S.L.A.A. and O.E.), and by grant DE-FG02-95ER20198 from the U.S. Department of Energy (J.G.F.).
Diffraction data were collected at EMBL/DESY, Hamburg, Germany. We thank Ralf Ficner for continuous support.
Footnotes
Published ahead of print on 19 October 2007.
REFERENCES
- 1.Andrade, S. L. A., F. Cruz, C. L. Drennan, V. Ramakrishnan, D. C. Rees, J. G. Ferry, and O. Einsle. 2005. Structures of the iron-sulfur flavoproteins from Methanosarcina thermophila and Archaeoglobus fulgidus. J. Bacteriol. 187:3848-3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brock, B. J., and M. H. Gold. 1996. 1,4-benzoquinone reductase from the basidiomycete Phanerochaete chrysosporium: spectral and kinetic analysis. Arch. Biochem. Biophys. 331:31-40. [DOI] [PubMed] [Google Scholar]
- 3.Brock, B. J., S. Rieble, and M. H. Gold. 1995. Purification and characterization of a 1,4-benzoquinone reductase from the basidiomycete Phanerochaete chrysosporium. Appl. Environ. Microbiol. 61:3076-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brünger, A. T. 1993. Assessment of phase accuracy by cross validation: the free R-value. Methods and applications. Acta Crystallogr. D 49:24-36. [DOI] [PubMed] [Google Scholar]
- 5.Cohen, R., M. R. Suzuki, and K. E. Hammel. 2004. Differential stress-induced regulation of two quinone reductases in the brown rot basidiomycete Gloeophyllum trabeum. Appl. Environ. Microbiol. 70:324-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collaborative Computational Project No. 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 7.DeLano, W. L. 2002. The PyMOL molecular graphic system. DeLano Scientific, Palo Alto, CA.
- 8.Deller, S., S. Sollner, R. Trenker-El-Toukhy, I. Jelesarov, G. M. Gubitz, and P. Macheroux. 2006. Characterization of a thermostable NADPH:FMN oxidoreductase from the mesophilic bacterium Bacillus subtilis. Biochemistry 45:7083-7091. [DOI] [PubMed] [Google Scholar]
- 9.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 10.Gorman, J., and L. Shapiro. 2005. Crystal structures of the tryptophan repressor binding protein WrbA and complexes with flavin mononucleotide. Protein Sci. 14:3004-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandori, R., and J. Carey. 1994. 6 New candidate members of the alpha/beta twisted open-sheet family detected by sequence similarity to flavodoxin. Protein Sci. 3:2185-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grandori, R., P. Khalifah, J. A. Boice, R. Fairman, K. Giovanielli, and J. Carey. 1998. Biochemical characterization of WrbA, founding member of a new family of multimeric flavodoxin-like proteins. J. Biol. Chem. 273:20960-20966. [DOI] [PubMed] [Google Scholar]
- 13.Jensen, K. A., Z. C. Ryan, A. V. Wymelenberg, D. Cullen, and K. E. Hammel. 2002. An NADH:quinone oxidoreductase active during biodegradation by the brown-rot basidiomycete Gloeophyllum trabeum. Appl. Environ. Microbiol. 68:2699-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones, T. A., J.-Y. Zou, S. W. Cowan, and M. Kjelgaard. 1991. Improved methods for building proteins in electron density maps and location of errors in these models. Acta Crystallogr. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 15.Karplus, P. A., M. J. Daniels, and J. R. Herriott. 1991. Atomic-structure of ferredoxin-NADP+ reductase: prototype for a structurally novel flavoenzyme family. Science 251:60-66. [PubMed] [Google Scholar]
- 16.Kraulis, P. 1991. MOLSCRIPT: a program to produce both detailed and schematic plots of proteins. J. Appl. Crystallogr. 24:946-950. [Google Scholar]
- 17.Laskowski, M. J., K. A. Dreher, M. A. Gehring, S. Abel, A. L. Gensler, and I. M. Sussex. 2002. FQR1, a novel primary auxin-response gene, encodes a flavin mononucleotide-binding quinone reductase. Plant Physiol. 128:578-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latimer, M. T., M. H. Painter, and J. G. Ferry. 1996. Characterization of an iron-sulfur flavoprotein from Methanosarcina thermophila. J. Biol. Chem. 271:24023-24028. [DOI] [PubMed] [Google Scholar]
- 19.Li, R., M. A. Bianchet, P. Talalay, and L. M. Amzel. 1994. Structural studies of quinone reductase by X-ray crystallography. Biophys. J. 66:A265. [Google Scholar]
- 20.Liger, D., M. Graille, C. Z. Zhou, N. Leulliot, S. Quevillon-Cheruel, K. Blondeau, J. Janin, and H. van Tilbeurgh. 2004. Crystal structure and functional characterization of yeast YLR011wp, an enzyme with NAD(P)H-FMN and ferric iron reductase activities. J. Biol. Chem. 279:34890-34897. [DOI] [PubMed] [Google Scholar]
- 21.Matthews, B. W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33:491-497. [DOI] [PubMed] [Google Scholar]
- 22.Matvienko, M., A. Wojtowicz, R. Wrobel, D. Jamison, Y. Goldwasser, and J. I. Yoder. 2001. Quinone oxidoreductase message levels are differentially regulated in parasitic and non-parasitic plants exposed to allelopathic quinones. Plant J. 25:375-387. [DOI] [PubMed] [Google Scholar]
- 23.Mayhew, S. G., and M. L. Ludwig. 1975. Flavodoxins and electron-transferring flavoproteins. Academic Press, New York, NY.
- 24.Merritt, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505-524. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53:240-255. [DOI] [PubMed] [Google Scholar]
- 26.Natalello, A., S. M. Doglia, J. Carey, and R. Grandori. 2007. Role of flavin mononucleotide in the thermostability and oligomerization of Escherichia coli stress-defense protein WrbA. Biochemistry 46:543-553. [DOI] [PubMed] [Google Scholar]
- 27.Nöll, G., E. Kozma, R. Grandori, J. Carey, T. Schodl, G. Hauska, and J. Daub. 2006. Spectroelectrochemical investigation of a flavoprotein with a flavin-modified gold electrode. Langmuir 22:2378-2383. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski, Z., and W. Minor. 1996. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 29.Patridge, E. V., and J. G. Ferry. 2006. WrbA from Escherichia coli and Archaeoglobus fulgidus is an NAD(P)H:quinone oxidoreductase. J. Bacteriol. 188:3498-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sancho, J. 2006. Flavodoxins: sequence, folding, binding, function and beyond. Cell Mol. Life Sci. 63:855-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serre, L., F. Vellieux, M. Medina, C. Gomez Moreno, J. C. Fontecilla-Camps, and M. Frey. 1996. Crystal structures of a ferredoxin:NADP+ reductase and of a complex with NADP+. Biochem. Soc. Trans. 24:10S. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki, Y., T. Yoda, A. Ruhul, and W. Sugiura. 2001. Molecular cloning and characterization of the gene coding for azoreductase from Bacillus sp OY1-2 isolated from soil. J. Biol. Chem. 276:9059-9065. [DOI] [PubMed] [Google Scholar]
- 33.Wolfova, J., R. Grandori, E. Kozma, N. Chatterjee, J. Carey, and I. K. Smatanova. 2005. Crystallization of the flavoprotein WrbA optimized by using additives and gels. J. Cryst. Growth 284:502-505. [Google Scholar]
- 34.Wolfova, J., J. R. Mesters, J. Brynda, R. Grandori, A. Natalello, J. Carey, and I. Kuta Smatanova. 2007. Crystallization and preliminary diffraction analysis of Escherichia coli WrbA in complex with its cofactor flavin mononucleotide. Acta Crystallogr. F 63:571-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wrobel, R. L., M. Matvienko, and J. I. Yoder. 2002. Heterologous expression and biochemical characterization of an NAD(P)H: quinone oxidoreductase from the hemiparasitic plant Triphysaria versicolor. Plant Physiol. Biochem. 40:265-272. [Google Scholar]
- 36.Yang, W. P., L. Y. Ni, and R. L. Somerville. 1993. A stationary-phase protein of Escherichia coli that affects the mode of association between the Trp repressor protein and operator-bearing DNA. Proc. Natl. Acad. Sci. USA 90:5796-5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao, T., F. Cruz, and J. G. Ferry. 2001. Iron-sulfur flavoprotein (Isf) from Methanosarcina thermophila is the prototype of a widely distributed family. J. Bacteriol. 183:6225-6233. [DOI] [PMC free article] [PubMed] [Google Scholar]