

Abstract
The intracellular signaling molecule, cyclic-di-GMP (c-di-GMP), has been shown to influence bacterial behaviors, including motility and biofilm formation. We report the identification and characterization of PA4367, a gene involved in regulating surface-associated behaviors in Pseudomonas aeruginosa. The PA4367 gene encodes a protein with an EAL domain, associated with c-di-GMP phosphodiesterase activity, as well as a GGDEF domain, which is associated with a c-di-GMP-synthesizing diguanylate cyclase activity. Deletion of the PA4367 gene results in a severe defect in swarming motility and a hyperbiofilm phenotype; thus, we designate this gene bifA, for biofilm formation. We show that BifA localizes to the inner membrane and, in biochemical studies, that purified BifA protein exhibits phosphodiesterase activity in vitro but no detectable diguanylate cyclase activity. Furthermore, mutational analyses of the conserved EAL and GGDEF residues of BifA suggest that both domains are important for the observed phosphodiesterase activity. Consistent with these data, the ΔbifA mutant exhibits increased cellular pools of c-di-GMP relative to the wild type and increased synthesis of a polysaccharide produced by the pel locus. This increased polysaccharide production is required for the enhanced biofilm formed by the ΔbifA mutant but does not contribute to the observed swarming defect. The ΔbifA mutation also results in decreased flagellar reversals. Based on epistasis studies with the previously described sadB gene, we propose that BifA functions upstream of SadB in the control of biofilm formation and swarming.
The gram-negative bacterium Pseudomonas aeruginosa is an important model organism for the study of bacterial surface interactions, including biofilm formation and surface-mediated twitching and swarming motilities. However, the precise molecular mechanisms required for transition from a planktonic mode of existence to that of a surface-associated lifestyle are only beginning to come to light.
In the case of biofilm formation, microscopic studies, as well as genetic analyses, have shown that the initial surface attachment phase by P. aeruginosa proceeds in two distinct steps (8, 20). In the first step, known as reversible attachment, cells are loosely attached via a single cell pole and may readily detach and return to the planktonic phase. In the second step, cells that are tethered by a pole become attached via the long axis of the cell body. Such cells, deemed irreversibly attached, are more firmly attached to the surface. Genetic studies of initial attachment have led to the identification of SadB, a key component required for this transition from reversible to irreversible attachment in P. aeruginosa (8).
The ability to form a robust biofilm also requires the production of an exopolysaccharide (EPS) component of the biofilm matrix. Recent studies have identified genetic loci that are important for synthesis of an EPS component of biofilm matrix in several P. aeruginosa strains. In PA14, the pel genes are required for the production of a glucose-rich EPS (15, 16). In addition to the pel locus, the PAO1 strain also harbors a locus not found in PA14, the psl gene cluster, shown to produce a mannose-rich matrix (16, 23, 31). While these and other studies indicate that EPS production contributes to the overall structure of the mature biofilm (14), more recent data suggest that the pel and psl loci also play roles in initial attachment in strains PAK (54) and PAO1 (29), respectively.
Whereas biofilm formation is a surface-associated sessile behavior, swarming is a surface-associated motile behavior. In P. aeruginosa strain PA14, swarming motility is the flagellum-propelled movement of bacteria across surfaces, aided by the production of rhamnolipids as a surface-wetting agent (26, 53).
Given that biofilm formation and swarming motility are both surface-associated behaviors, it seems plausible that bacteria are able to transition from one behavior to another as dictated by changing surface conditions. One potential explanation for how cells might transition from one surface behavior to another is that these surface behaviors are coregulated. Caiazza et al. have provided evidence to establish that SadB coregulates both biofilm formation and swarming motility and is able to inversely control these two surface behaviors (7). Based on this evidence, these authors put forth a model wherein SadB controls biofilm formation and swarming motility by inversely regulating both the rate of flagellar reversals and the production of the pel-derived polysaccharide.
The inverse regulation of biofilm formation and swarming motility by SadB is reminiscent of the regulation of sessile and motile behaviors via a bacterial regulatory system involving the signaling molecule cyclic-di-GMP (c-di-GMP) (13, 24, 42, 43). In a growing number of bacterial systems, it has been shown that intracellular levels of this signaling molecule influence a myriad of bacterial behaviors, with the common theme being that the accumulation of c-di-GMP promotes sessile behaviors, such as biofilm formation, while the breakdown of c-di-GMP and subsequent decrease in cellular levels of this signal favors motile behaviors, such as swarming motility (5, 6, 19, 25, 40, 50).
Presumably, the steady-state level of c-di-GMP in the cell is controlled by the relative rates of synthesis and degradation of this molecule. The synthesis of c-di-GMP from two molecules of GTP is catalyzed by diguanylate cyclases (DGCs), enzymes characterized by the presence of a GGDEF domain (19, 40, 45, 51), whereas the breakdown of c-di-GMP is catalyzed by phosphodiesterases (PDEs), enzymes noted for possessing an EAL or an HD-GYP domain (5, 11, 44, 47, 51, 52).
In a companion manuscript, Merritt et al. establish that SadC, a novel GGDEF-containing protein that modulates cellular pools of c-di-GMP in P. aeruginosa PA14, functions genetically upstream of SadB in inversely controlling both biofilm formation and swarming motility (32). We postulated that a PDE should also be involved in degrading the c-di-GMP produced by SadC. Here we describe the identification of BifA, a c-di-GMP PDE that participates with SadC in the inverse regulation of biofilm formation and swarming motility and does so in a SadB-dependent manner.
MATERIALS AND METHODS
Strains and media.
Strains, plasmids and primers used in the present study are listed in Table 1. P. aeruginosa PA14 and Escherichia coli DH5α and JM109 were routinely cultured on lysogeny broth (LB) medium (4), solidified with 1.5% agar where appropriate. For P. aeruginosa, gentamicin (Gm) was used from 50 to 100 μg/ml. For E. coli, Gm was used at 10 μg/ml and ampicillin was used at 150 μg/ml. M63 minimal salts medium (39) supplemented with MgSO4 (1 mM), glucose (0.2%), and Casamino Acids (CAA; 0.5%) was used for all P. aeruginosa phenotypic assays unless otherwise noted. For arabinose-inducible plasmids, arabinose was added to cultures at a 0.5% final concentration.
TABLE 1.
Strains, plasmids, and primers
| Strain, plasmid, or primer | Relevant genotype or sequence | Source or reference |
|---|---|---|
| P. aeruginosa PA14 strains | ||
| SMC232 | WT | 41 |
| 12-3H6 (bifA)::Tn | 28 | |
| SMC3351 | ΔbifA | This study |
| SMC3417 | ΔbifA ΔsadC | This study |
| SMC3430 | ΔbifA sadB::Tn5 | This study |
| SMC3051 | pelA::pMQ89 | This study |
| SMC3381 | ΔbifA pelA::pMQ89 | This study |
| SMC3374 | WT::Tn7 | This study |
| SMC3403 | WT::Tn7-His-bifA+ | This study |
| SMC3369 | ΔbifA::Tn7 | This study |
| SMC3401 | ΔbifA::Tn7-His-bifA+ | This study |
| Plasmids | ||
| pEX-18-Gm | Knockout vector | 22 |
| pEX-ΔbifA | bifA deletion mutant construct | This study |
| pMQ89 | Suicide cloning vector | 48 |
| pMQ89-pelA-SCO | pelA SCO knockout vector | This study |
| pMQ80 | Cloning vector with arabinose-inducible promoter | 48 |
| pMQ80-His-bifA+ | Expresses WT His-tagged BifA under the control of an arabinose-inducible promoter | This study |
| pMQ80-His-bifA-AAAAA | Expresses His-tagged BifA-AAAAA under the control of an arabinose-inducible promoter | This study |
| pMQ80-His-bifA-AAL | Expresses His-tagged BifA-AAL under the control of an arabinose-inducible promoter | This study |
| pUX-BF13 | Helper plasmid for Tn7 transposition | 9 |
| pUC18-mini-Tn7T-Gm | Single-copy Tn7 insertion plasmid | 9 |
| pTn7-His-bifA+ | Single-copy Tn7 insertion plasmid containing the His-tagged bifA gene with its endogenous promoter region | This study |
| pQE-30 | Expression vector | Invitrogen, Inc. |
| pQE-30-bifA | Expresses His-BifA under an IPTG-inducible promoter | This study |
| pQE-30-bifA-AAAAA | Expresses His-BifA-AAAAA under an IPTG-inducible promoter | This study |
| pQE-30-bifA-AAL | Expresses His-BifA-AAL under an IPTG-inducible promoter | This study |
| Primers | ||
| KO-1 | CGACGAACAAGGAAGGCCCCGAGCAGGAGGCGTACATCATC | |
| KO-2 | GATGATGTACGCCTCCTGCTCGGGGCCTTCCTTGTTCGTCG | |
| KO-3 | tttctccatacccgtttttttgggctagcgaattcCTTGCAGAAGCCCGTCGCAG | |
| KO-4 | tcctttactcatatgtatatctccttccccgggtaccCCCGTGTCGCTGGCTTCAAG | |
| KO-3/HindIII | GGCGAAGCTTCTTGCAGAAGCCCGTCGCAG | |
| KO-4/XbaI | GGCGTCTAGACCCGTGTCGCTGGCTTCAAG | |
| pelA-HindIII | GGCGAAGCTTTGAAACATCCTTCGACCCATCGAG | |
| pelA-XbaI | GGCGTCTAGATCACCAACCGGCATGGATCGACTC | |
| pF-t1t2 | CAGACCGCTTCTGCGTTCTG | |
| pelA-3′end | ATCGGCAACTCGAACGTCCACAG | |
| bifA-5′Kpn | tttctccatacccgtttttttgggctagcgaattcaagcttTGGAACCTGGTCAACTGGGAC | |
| N-His-F | ACGAACAAGGAAGGCCCCTTGCACCACCACCATCACCACAAACTGGACTCCCGACACAG | |
| bifA-3′HIII | tcctttactcatatgtatatctccttccccgggtaccGAGGACGATCTCGCCGTGCGAC | |
| N-His-R | GTTTGTGGTGATGGTGGTGGTGCAAGGGGCCTTCCTTGTTCGTC | |
| glmS-up | CTCAAGTCGAACCTGCAGGAAGTC | |
| bifA-up | GCGAGACCACGGAGAAACTGC | |
| GG/Ala-F | CGGTTCGCTGGCGCGCCTGGCCGCCGCGGCCGCCGCCCTGGTCCAGGCCGAC | |
| GG/Ala-R | GTCGGCCTGGACCAGGGCGGCGGCCGCGGCGGCCAGGCGCGCCAGCGAACCG | |
| AAL-For | CACCGCGTGGTCGGCGTCGCCGCACTGCTGCGCTGGCAACATC | |
| AAL-Rev | GATGTTGCCAGCGCAGCAGTGCGGCGACGCCGACCACGCGGTG | |
| rplU1 | CGCAGTGATTGTTACCGGTG | |
| rplU2 | CAACCGCAATGGGCGCTATTGC | |
| pelA-F | TCAATCGCGGTTTCGAAGTGCTGC | |
| pelA-R | ATCCAGGTGACCCTTCAGCCAATC | |
| pelG-F | TATTGCTGGCGACCCTGTTCGATG | |
| pelG-R | ATGAAACGCAGCAGGTAGGCACAG | |
| EALHis-SacI | GGCGGAGCTCTTGAAACTGGACTCCCGACACAG | |
| EALHis-HindIII | GGCGAAGCTTTCAGGGCCGTTCGCTGCTGGTGG |
Saccharomyces cerevisiae strain InvSc1 (Invitrogen) was used for in vivo homologous recombination for plasmid constructions and was grown with yeast extract-peptone-dextrose (1% Bacto yeast extract, 2% Bacto peptone, and 2% dextrose) (48). Selections with InvSc1 were performed by using synthetic defined agar-uracil (Qbiogene, 4813-065) (48).
Construction of mutant strains.
A deletion of the bifA gene was constructed by first amplifying an ∼1-kb PCR fragment upstream of bifA using the primer pair KO-2 and KO-3 and an ∼1-kb PCR fragment downstream of bifA using primers KO-1 and KO-4 (primer sequences are listed in Table 1). PCR for strain constructions was performed with Phusion polymerase (New England Biolabs, Beverly, MA) according to the manufacturer's instructions, whereas PCR for screening and confirmation of mutations was performed with Taq polymerase (New England Biolabs) under standard conditions. Primers KO-1 and KO-2 are reverse complements of one another, with the underlined sequence located at the 5′ end of bifA and the italicized sequence located at the 3′ end of bifA (see Table 1). Primers KO-3 and KO-4 contain ∼35 bp of homology to the vector pMQ80 (Table 1, lowercase sequence). The two PCR fragments were cloned together into pMQ80 (linearized by SacI digestion) by in vivo homologous recombination in yeast as reported (48), resulting in a construct carrying a deletion of bifA from nucleotides 1 to 1941 of the 2,064-bp full-length gene surrounded by ∼1 kb of upstream and downstream flanking DNA. Plasmids were recovered from yeast as described, transformed via electroporation into E. coli DH5α, and screened by colony PCR, followed by plasmid isolation and sequencing to confirm the deletion. The bifA deletion construct was then reamplified by using primers KO-3/HindIII and KO-4/XbaI, digested with HindIII/XbaI, and cloned into the suicide vector pEX18-Gm (22). The resulting construct (pEX-ΔbifA) was transformed into P. aeruginosa by electroporation as described previously (10). Integrants were isolated on Gm and subjected to sucrose selection, and the resolved integrants were confirmed by PCR and sequencing. The pEX-ΔbifA construct was used in the same manner to generate a deletion of bifA in the sadB and sadC mutant backgrounds, resulting in the double ΔbifA sadB::Tn5 and ΔbifA ΔsadC mutants.
The pelA mutation was generated by a single-crossover (SCO) insertion using the pMQ89 suicide vector (48). The pMQ89-pelA-SCO construct consisted of a 620-bp 5′ fragment (nucleotides 52 to 672) of the pelA gene amplified using primers containing in-frame translational stop codons (pelA-HindIII and pelA-Xba) and cloned into HindIII/XbaI-digested pMQ89. This construct was transformed into P. aeruginosa, transformants were isolated, and insertions were confirmed by PCR using the vector primer pF-t1t2 and pelA-3′ end primer. The pMQ89-pelA-SCO construct was also introduced into the ΔbifA mutant to generate the ΔbifA pelA double mutant.
bifA complementation constructs. (i) Multicopy constructs for arabinose-inducible expression of the bifA gene.
An arabinose-inducible, N-terminal His6-tagged copy of bifA was generated by homologous recombination in yeast, using the vector pMQ80. Two overlapping PCR products spanning bifA and including the His6 tag were generated by using the primer pairs bifA-5′Kpn/N-His-R and bifA-3′HIII/N-His-F. These PCR products were joined with pMQ80 (linearized with SacI) by homologous recombination as described previously (48) to generate the full-length His-tagged bifA-containing plasmid designated pMQ80-His-bifA+.
N-terminal His6-tagged mutant versions of bifA were also generated by homologous recombination in pMQ80. Overlapping PCR fragments with mutations converting the GGDQF amino acid residues to alanine residues (GGDQF to AAAAA) were generated by using primer GG/Ala-F with bifA-3′HIII and primer GG/Ala-R with N-His-F (Table 1, the mutated residues are underlined). These fragments were combined with the bifA-5′Kpn/N-His-Rev fragment to generate the full-length His-tagged bifA-AAAAA-containing plasmid designated pMQ80-His-bifA-AAAAA.
Overlapping PCR fragments for mutating the conserved glutamic acid residue of the EAL domain (EAL to AAL) were generated using primers AAL-For with bifA-3′HIII and AAL-Rev with N-His-F. These fragments were combined with the bifA-5′Kpn/N-His-Rev fragment to generate the full-length His-tagged bifA-AAL construct designated pMQ80-His-bifA-AAL.
(ii) Tn7 constructs for single-copy chromosomal insertion of bifA and expression under the control of its native promoter.
The His-tagged bifA insert in the pMQ80-His-bifA+ construct was excised by using KpnI and HindIII and cloned into the pUC18-mini-Tn7T-Gm vector (9), yielding the plasmid pTn7-His-bifA+. Transposition of the Tn7 element was performed by coelectroporation of the pUC18-mini-Tn7T-Gm vector (or pTn7-His-bifA+) with the helper plasmid pUX-BF13 as described previously (9). Transposon insertions of the Tn7 alone or Tn7-His-bifA+ were generated in the wild type (WT) (yielding strains SMC3374 and SMC3403, respectively) and in the ΔbifA mutant (yielding strains SMC3369 and SMC3401, respectively). Transposon insertions were confirmed by PCR using the primers glmS-up and bifA-up.
Biofilm formation assays.
Biofilm formation in 96-well microtiter plates was assayed essentially as previously described (8, 37). For complementation of ΔbifA biofilm formation using pMQ80 constructs, arabinose was added to a 0.5% final concentration.
Microscopy. (i) ALI assay.
Air-liquid interface (ALI) analysis was performed as described previously (8) except that bacteria attached to the surface were visualized after 6 h.
(ii) Measurement of initial attachment.
Strains were grown and prepared for measurements of initial attachment as described previously (7), except that cultures were diluted 1:200, and cells were incubated for 30 min prior to enumeration. Surface-attached cells were counted over eight fields of view per strain, and the data are presented as the average number of cells per field.
(iii) Quantification of reversible and irreversible attachment.
The transition from reversible to irreversible attachment was monitored as described previously (8) except that cultures were diluted 1:200. Phase-contrast time-lapse images were captured at 1 frame per s for 60 s and converted to QuickTime movies for analysis. Irreversibly attached cells were scored as cells that did not move during the 1-min interval and were attached by the long axis of the cell. Reversibly attached cells were those that moved during the interval and were attached by a cell pole (8).
(iv) Measurement of swim reversal frequency.
Cells were grown and prepared for swim reversal frequency analysis as described previously (7), except that cultures were diluted 1:200 in M63 medium supplemented with glucose in the presence of 15% Ficoll (high-viscosity/swarming conditions) (53). Time-lapse images were captured every 0.3 s for 60 s, and images were converted to QuickTime movies for subsequent analysis. Approximately 15 cells per movie were counted in nine movies (∼135 cells total) for each strain to determine the reversal frequencies, which are expressed as the number of reversals per cell.
CR binding assays.
Congo red (CR) binding assays were performed as reported (7, 15, 16). Bacteria from LB-grown overnight cultures were spotted (2 μl) onto the plates and grown for 24 h at 37°C, followed by 48 h at room temperature.
qRT-PCR.
The WT and the ΔbifA mutant strain were grown under the conditions described for the CR assays, but without the dyes added (see above). Colonies were harvested and resuspended in M63 medium and normalized to an optical density at 600 nm (OD600) of 0.4. Cells were spun briefly at 16,000 × g to pellet, the supernatants were removed, and the pellets were flash-frozen in a dry ice-ethanol bath for 15 min. RNA was isolated, cDNA was prepared, and quantitative reverse transcription-PCR (qRT-PCR) was performed as reported (27). Expression levels were quantified in picograms of input cDNA using a standard curve method for absolute quantification, and these values were normalized to rplU expression. The primers used are listed in Table 1.
Motility assays.
Swim (0.3% agar) and twitch (1.5% agar) motility plates consisted of M63 medium supplemented with glucose, MgSO4, and CAA. Swim and twitch assays were carried out as reported previously (38, 55). The diameters of the swim zones were measured in millimeters and averaged over four replicate platings for each strain repeated twice. The diameters of twitch zones were measured in millimeters, and the data were averaged over three replicate platings repeated twice.
Swarm (0.5% agar) motility plates consisted of M8 minimal medium (26) supplemented with glucose, MgSO4, and CAA. Swarm plate preparation, inoculation, and incubation were performed as previously reported (53). Diameters of swarm zones were measured in millimeters for each strain, and the data are presented as averages over triplicate platings. To detect rhamnolipid production in the ΔbifA mutant, rhamnolipid assays were performed as reported earlier (49) using the same base medium as for the swarming assays but with 1.5% agar.
Assessing the stability of the WT and mutant BifA proteins for complementation of the ΔbifA mutant.
Strains carrying either pMQ80 alone, pMQ80-His-bifA+, pMQ80-His-bifA-AAL. or pMQ80-His-bifA-AAAAA were grown under the same conditions used for the biofilm complementation assays. LB-grown overnight cultures were normalized by the OD600; diluted (1:50) into M63 medium supplemented with glucose, MgSO4, CAA, and arabinose; and grown statically for 6 h at 37°C. Cells (20 ml) were harvested by centrifugation at 4,400 × g and resuspended in 1× sodium dodecyl sulfate (SDS) gel loading buffer with 100 mM dithiothreitol. Samples were boiled for 10 min and resolved by SDS-polyacrylamide gel electrophoresis (PAGE) in a 7.5% polyacrylamide gel (Bio-Rad, Hercules, CA). Proteins were transferred to a nitrocellulose membrane and probed with an anti-penta-His antibody (QIAGEN, Valencia, CA). Western blots were developed with the Western Lightning ECL detection kit (Perkin-Elmer, Boston, MA) according to the instructions of the manufacturer.
Cellular localization of BifA.
Cell fractionations were carried out as previously reported (34) with modifications. LB-grown overnight cultures were diluted (1:100) into LB (250 ml) containing Gm (25 μg/ml) and arabinose (0.5%) and grown for ∼4 h (OD600 ≈ 0.9) at 37°C with shaking. Bacterial cells were harvested by centrifugation at 5,520 × g for 15 min and washed once in phosphate-buffered saline. Cells were collected by centrifugation at 4,400 × g, the supernatants were removed, and the cell pellets were frozen at −80°C. Cell pellets were resuspended in buffer A (200 mM Tris-HCl [pH 7.5], 20 mM EDTA [pH 8.0], 1× Complete protease inhibitors [Roche Diagnostics Corp., Indianapolis, IN]) and lysed in a French pressure cell. Samples were centrifuged at 9,300 × g for 10 min at 4°C to remove unbroken cells, and supernatants were collected as whole-cell lysates. To separate the cytoplasmic fraction from the total membrane (TM) fraction, whole-cell lysates were centrifuged at 100,000 × g for 1 h at 4°C. The soluble fraction was collected as the cytoplasmic fraction. Membrane pellets were resuspended in buffer B (20 mM Tris-HCl [pH 7.6]) to yield the TM fraction. The TM was further fractionated by using Sarkosyl solubilization of the inner membrane. An equal volume of 4% Sarkosyl in buffer B was added to the TM fraction, and samples were incubated at room temperature for 20 min with shaking. Samples were then centrifuged at 100,000 × g for 1 h at 4°C. The soluble portion was collected as the inner-membrane fraction. Pellets were resuspended in buffer B to yield the outer-membrane fraction. Aliquots of all fractions were stored in 10% glycerol and protease inhibitors at −80°C.
For Western blots of cellular fractions, samples from each fraction were diluted to the same concentration (0.2 mg/ml) and 1 μg of total protein from each fraction was mixed with SDS loading buffer containing dithiothreitol (100 mM). Concentrations of total protein in each fraction were determined by using a Bio-Rad DC protein assay kit according to the manufacturer's instructions. Samples were resolved by SDS-PAGE using either 7.5% (BifA and SecY) or a 4 to 15% gradient (OprF) polyacrylamide gels (Bio-Rad). Proteins were transferred to a nitrocellulose membrane and probed with one of three antibodies: (i) anti-penta-His antibody (QIAGEN) to detect His-tagged BifA, (ii) SecY antisera (21) to detect the inner membrane protein SecY (1), and (iii) antisera to detect the outer-membrane protein OprF (3, 18, 30). Western blots were developed as described above.
Expression and purification of His-tagged BifA.
To express His-tagged BifA protein for purification in E. coli, the bifA gene was amplified by PCR using primers EALHis-SacI and EALHis-HindIII and cloned into the SacI/HindIII-digested pQE-30 vector (Invitrogen) harboring an N-terminal His6 tag. The bifA mutants (GGDQF to AAAAA and EAL to AAL) were generated by PCR amplification with the primers used above (EALHis-SacI and EALHis-HindIII) using the mutant bifA templates generated for the complementation constructs in pMQ80 (see above) followed by cloning into pQE-30.
E. coli JM109 strains carrying either pQE-30 alone, pQE-30-bifA+, pQE-30-bifA-AAL, or pQE-30-bifA-AAAAA constructs were grown overnight in LB-ampicillin, diluted 1:100, and grown for 4 h at 37°C. Expression of His-tagged BifA protein was induced with addition of 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside), followed by incubation at 12°C overnight. Cultures were harvested by centrifugation at 5,520 × g for 10 min and resuspended in wash buffer (20 mM imidazole, 20 mM Na2HPO4, 20 mM NaH2PO4, 500 mM NaCl, 1% Triton X-100, 4 mM MgCl2, and 10 mg of DNase I [Roche Diagnostics Corp., Indianapolis, IN]/ml). Cells were lysed in a French pressure cell, and the extract was clarified by centrifugation at 5,520 × g for 15 min. The soluble fraction was filtered through a 0.22-μm-pore-size filter. From this filtrate (crude extract), His-tagged BifA was purified by using a His-Trapp-FF NiSO4 column (GE Healthcare, Piscataway, NJ) and collected from the column in elution buffer (150 mM imidazole, 20 mM Na2HPO4, 20 mM NaH2PO4, 500 mM NaCl) by using a fraction collector (Bio-Rad). Eluate fractions containing BifA protein, as determined by SDS-PAGE separation and Coomassie blue staining, were treated with protease inhibitors (Roche Diagnostics), pooled, and dialyzed for 3 h using a Slide-A-Lyzer dialysis cassette (Pierce Biotechnology, Inc., Rockford, IL) against the dialysis buffer (75 mM Tris [pH 9.3], 30% glycerol, 200 mM NaCl, 10 mM MgCl2 25 mM KCl). Dialyzed samples were concentrated by using an Amicon Ultracel column (30,000 molecular weight cutoff) (Millipore, Billerica, MA). Protein concentrations were determined by using the RC DC protein assay kit. Protein purity was assessed by SDS-PAGE separation and Coomassie blue staining or Western blot analysis with a penta-His antibody (QIAGEN).
In vitro PDE activity assays. (i) bis-pNPP cleavage assays.
PDE activity of BifA was first assessed using the synthetic substrate, bis(p-nitrophenyl) phosphate (bis-pNPP), as previously described (5). Briefly, 20 μl of purified BifA (0.4 mg/ml) was incubated with bis-pNPP (5 mM) in buffer (5 mM MgCl2, 50 mM Tris-HCl [pH 9.3], 50 mM NaCl) for 60 to 90 min. The release of p-nitrophenol was quantified at OD410 in a spectrophotometer (Molecular Devices, Sunnyvale, CA). A dose-response curve was generated by assaying twofold serial dilutions of purified BifA in the reaction for 20 min. The PDE activity of crude extracts (20 μl of 1 mg of total protein/ml) containing WT BifA, BifA-AAL, BifA-AAAAA, or vector alone was also assayed as described above.
(ii) c-di-GMP cleavage assays.
Radiolabeled c-di-GMP substrate was synthesized by using purified PleD enzyme and [32P]GTP and processed as previously reported (33, 52). This substrate was then used in PDE activity assays which were performed as described previously (11, 33) using crude extracts. Reactions were incubated at room temperature (for 24 h) and terminated by the addition of 10 μl of 0.5 M EDTA (pH 8.0). Reaction products were separated by thin-layer chromatography (TLC) as described previously (33). TLC plates were exposed overnight on phosphorimaging screens (GE Healthcare) and analyzed on a Storm 860 (Molecular Devices).
In vivo quantification of c-di-GMP.
Whole-cell [32P]orthophosphate labeling, acid extraction, and two-dimensional TLC analysis of c-di-GMP for quantification were performed as reported elsewhere (19, 33). Quantification of labeled c-di-GMP was determined by using ImageQuant software v5.1 (Molecular Devices). The percentage of label incorporated into c-di-GMP was normalized to total 32P labeling and is expressed as the percent c-di-GMP.
RESULTS
Isolation and initial characterization of the PA4367::Tn mutant.
The PA4367::Tn mutant was initially isolated in a screen to identify novel genes that function together with the sadC gene in regulating biofilm formation in P. aeruginosa. The role of the sadC gene in biofilm formation is described in a companion manuscript (32). Merritt et al. report that sadC mutants exhibit severely reduced biofilm formation and also show reduced CR binding, indicating a decrease in polysaccharide production. The sadC mutant also shows enhanced swarming motility. The sadC gene encodes a protein with a GGDEF domain, which is associated with a c-di-GMP-synthesizing DGC activity. The phenotypes of sadC mutants are consistent with decreased levels of the signaling molecule, c-di-GMP—a decrease in c-di-GMP pools tends to promote a more motile lifestyle over a sessile mode of existence (13, 24, 42, 50).
Based on the studies of sadC described above, we predicted that genes functioning together with sadC in regulating biofilm formation would likely include those that impact the synthesis and/or stability of c-di-GMP. For example, proteins with EAL or HD-GYP domains are associated with c-di-GMP-degrading PDE activity (11, 17, 44, 47). Mutations in a c-di-GMP PDE would be expected to elevate intracellular levels of c-di-GMP and potentially lead to hyperbiofilm formation, increased CR binding, and decreased swarming motility (phenotypes opposite of those observed for the sadC mutant). Thus, we incorporated all three phenotypic criteria in a screen for such mutants.
Recently, Ausubel and coworkers reported the generation of a transposon insertion mutant library of P. aeruginosa strain PA14 (28). This PA14NR library is a nonredundant collection of 5,459 transposon mutants representing insertions in a majority of nonessential genes in P. aeruginosa PA14. To validate the utility of the PA14NR library, Liberati et al. screened for mutants with altered biofilm formation under the same minimal medium growth conditions used to isolate the sadC mutant (28). Approximately 420 mutants were isolated as biofilm defective, which included both reduced and enhanced biofilm formers relative to the WT (see Table S1 in the supplemental material). The resulting list of biofilm-defective mutants includes a number of genes previously shown to be important for biofilm formation, including mutations in genes required for flagellum and pilus synthesis, as well as the crc and sadB genes (8, 36, 37). The sadC gene (PA4332) was represented by a mutation in the sadC operon. A second GGDEF-containing gene, the previously characterized wspR gene (PA3702), was also identified. The wspR gene has been shown to be required for autoaggregation and biofilm formation in LB medium by P. aeruginosa strain PAO1 (12, 19). In addition to known biofilm factors, new genes with as-yet-uncharacterized roles in biofilm formation were also isolated. Together, these results represent the desired outcome for such a screen and, as such, confirm the utility of the PA14NR mutant library.
To screen for genes that might function in the same pathway as sadC, this subset of 420 transposon mutants with biofilm phenotypes was subjected to a second round of biofilm assays, in parallel with CR binding and swarm assays, to identify those mutants that formed hyper-biofilms, bound increased levels of CR, and showed decreased swarming motility. Of the 420 biofilm-altered mutants screened, we identified 55 mutants that were both hyper-biofilm formers and hyper-CR binders. Of those 55 mutants, 34 mutants were also completely defective for swarming motility (see Table S2 in the supplemental material).
From this list of 34 mutants, we chose to focus on the 12-3H6 mutant carrying a transposon insertion in the PA4367 open reading frame. The PA4367 gene is the only gene with an EAL or HD-GYP motif represented in this subset of mutants. While there are two other genes encoding EAL motifs represented in the larger collection of 420 biofilm-defective mutants, neither of these mutants satisfies all three of the phenotypic criteria used in the second round of screening. The PA4367 gene also contains a GGDEF-like motif, with the sequence GGDQF in the core motif. Therefore, the PA4367::Tn mutant satisfied our criteria of having biofilm, CR, and swarming phenotypes opposite of the sadC mutant, and motifs consistent with a role in c-di-GMP metabolism, indicating that PA4367 might antagonize the action of sadC with respect to these behaviors. Based on the biofilm phenotype of PA4367::Tn mutant, we propose to designate this gene bifA for biofilm formation.
The bifA gene negatively impacts biofilm formation at early stages.
To confirm and further analyze the biofilm phenotypes of the bifA::Tn mutant, we constructed a deletion mutation that removes nucleotides 1 to 1941 of the 2,046-nucleotide coding region of the bifA gene (See Materials and Methods for details). We then tested the ΔbifA deletion mutant in a 96-well microtiter assay compared to the bifA::Tn mutant. Quantification of the crystal violet-stained biofilms for this analysis is shown in Fig. 1A. After growth for 6 h, the bifA::Tn and the ΔbifA mutants both exhibited greatly enhanced biofilm formation (∼5.5-fold) over that of the WT. Both mutants grow at a rate indistinguishable from that of the WT (data not shown) under these same medium conditions, demonstrating that enhanced biofilm formation observed in the mutants is not due to increased growth rate.
FIG. 1.

Biofilm phenotypes of bifA mutants. (A) Quantification of biofilms formed by the WT, the bifA::Tn mutant and the ΔbifA mutant in the 96-well microtiter dish assay. Also shown are the single-copy complemented strains of the WT and the ΔbifA mutant carrying an insertion of either the Tn7 element alone (WT::Tn7 and ΔbifA::Tn7) or the Tn7 harboring a His-tagged version of bifA (WT::Tn7-His-bifA+ and ΔbifA::Tn7-His-bifA+). Cells were grown in M63 with glucose, MgSO4, and CAA for 6 h at 37°C prior to crystal violet staining. Crystal violet was solubilized in 30% glacial acetic acid and measured at OD550. (B) ALI assay. Top-down phase-contrast images of the WT and the ΔbifA mutant either alone or carrying insertions of the Tn7 (Tn7 vector) or the Tn7 with His-tagged bifA (Tn7-His-bifA+) are shown. Cells were grown in a 24-well plate for 6 h at 37°C, and images were recorded at a magnification of ×1,400. (C) Quantification of initial attachment of the WT and the ΔbifA mutant. Cells were incubated at 37°C for 30 min. Images were recorded at a magnification of ×1,400 over eight fields of view for each strain. The graph indicates the average number of cells attached to the substratum (n = 8) for each strain. (D) Quantification of reversible attachment of the WT, the sadB mutant and the ΔbifA mutant. Strains were incubated in 24-well plates for 5 min at 37°C. Time-lapse images were captured in 1-min intervals and converted to QuickTime movies for analysis. Irreversibly attached cells were scored as cells that did not move during the 1-min interval and were attached by the long axis of the cell. Reversibly attached cells were those that moved during the interval and were attached by a cell pole.
To show that the hyperbiofilm formed by the ΔbifA mutant is specifically due to mutation of the bifA gene, we generated a complementation construct (pTn7-His-bifA+) to express a His-tagged version of the bifA gene from its native promoter in single copy on the chromosome via Tn7 insertion at a previously characterized chromosomal site (9). The Tn7 element alone had no effect on biofilm formation in the WT or the ΔbifA mutant strain (WT::Tn7 and ΔbifA::Tn7) (Fig. 1A), indicating that neither the Tn7 element itself nor its physical insertion into the chromosome leads to any impact on biofilm formation. The Tn7-His-bifA+ element resulted in a ∼50% decrease in biofilm formed by the WT (WT::Tn7-His-bifA+) relative to the WT::Tn7 control strain. However, the Tn7-His-bifA+ complementation construct in the ΔbifA mutant (ΔbifA::Tn7-His-bifA+) reduced biofilm formation ∼7-fold relative to the ΔbifA::Tn7 control strain and resulted in a level of biofilm formation similar to that seen in the WT::Tn7 control strain. Thus, single-copy expression of the His-tagged BifA, expressed under its own promoter, is capable of restoring biofilm formation to WT levels in the ΔbifA mutant. These results support the conclusion that mutation of the bifA gene is solely responsible for the observed hyperbiofilm phenotype of the ΔbifA mutant.
Biofilms formed by the WT, the ΔbifA mutant, and the complemented strains were also examined microscopically (Fig. 1B) using the ALI assay (see Materials and Methods) under the same growth conditions used in Fig. 1A. The data show that cells of the WT alone or the WT::Tn7 control form distinct microcolonies of surface-attached bacteria surrounded by open (noncolonized) surface area at 6 h. Cells of the ΔbifA mutant alone or the ΔbifA::Tn7 control form much more densely packed microcolonies with greatly reduced noncolonized surface area compared to the WT cells. In contrast, cells of the ΔbifA mutant carrying the Tn7-His-bifA+ insertion are only sparsely attached and form far fewer microcolonies. WT cells carrying the Tn7-His-bifA+ insertion show slightly reduced attachment and microcolony formation compared to the WT or the WT::Tn7 control. These data are entirely consistent with the observations made with the 96-well plate assay (Fig. 1A).
The direct microscopic observations of biofilm formation described above indicate that there are more ΔbifA cells than WT cells attached to the surface after 6 h of biofilm formation. In order to assess whether this difference in attachment was apparent at an earlier time point, we repeated these assays, but instead of 6 h, cell attachment was observed after 30 min of exposure to the surface of the 24-well plate. These data show that after only 30 min there are almost 2.5-fold more ΔbifA cells (∼250 cells per field of view) than WT cells (∼100 cells per field of view) attached to the surface (Fig. 1C). These results confirm that ΔbifA mutant surface-attached cells outnumber WT cells. Inspection of these images and of planktonic cultures revealed no obvious difference in cell clustering of the ΔbifA mutant cells relative to the WT (data not shown), indicating that the increase in ΔbifA cell number occurs primarily via an increase in cell-to-surface interactions rather than cell-cell aggregation in the planktonic phase.
The initial attachment phase of biofilm formation has been described as occurring in two steps (8, 20). The first step, whereby cells attach to the surface by a single cell pole, is called reversible attachment. Cells at this step can easily detach from the surface and return to the planktonic phase. The second step, which occurs when reversibly attached cells become attached via the long axis of the cell, is designated irreversible attachment. Such irreversibly attached cells appear to interact more firmly with the surface. Because ΔbifA mutant cells show increased surface attachment at an early stage in biofilm formation, it seemed plausible that the ΔbifA mutant might also be altered in its ability to transition from reversible to irreversible attachment.
The reversible-to-irreversible transition was monitored by time-lapse microscopy after a 5-min encounter of cells with the surface. The sadB mutant was included as a control since this mutant has been shown to be defective at this step in biofilm formation (8). The results of this analysis (Fig. 1D) show that the ΔbifA mutant does not differ in the percentage of reversibly attached cells (∼40%) relative to the WT (∼37%), whereas the sadB mutant exhibits a much higher percentage of reversibly attached cells (∼89%), a finding for sadB that is consistent with previously published results (8). Thus, the increase in ΔbifA mutant cell attachment is likely not due to an increased transition from reversible to irreversible attachment.
The pel-derived polysaccharide is required for hyperbiofilm formation by the ΔbifA mutant.
Given that the original bifA::Tn mutant was isolated, in part, due to its increased CR binding relative to the WT, we hypothesized that bifA mutants produce an increased level of polysaccharide that directly impacts biofilm formation. In P. aeruginosa PA14, it has been shown that the primary polysaccharide component of biofilm matirix is synthesized by the pel locus and that this is the cellular component bound by CR (15, 16). Thus, we tested whether the pel locus was required for the enhanced CR binding observed in the ΔbifA mutant by introducing a pelA mutation into the ΔbifA genetic background. The results show that the pelA mutation completely eliminates both the CR binding and the wrinkled phenotype of the ΔbifA mutant (Fig. 2A). Furthermore, the ΔbifA pelA double mutant forms a biofilm that is nearly identical to that of the pelA single mutant after 24 h (Fig. 2B), indicating that increased pel-derived polysaccharide production in the ΔbifA mutant is required for the hyperbiofilm formation observed.
FIG. 2.

pelA is required for the enhanced CR binding and hyperbiofilm phenotypes of the ΔbifA mutant. (A) Representative images of CR binding. Shown are the WT::Tn7, the ΔbifA::Tn7 mutant, the complemented strains WT::Tn7-His-bifA+ and ΔbifA::Tn7-His-bifA+, the pelA mutant, and the ΔbifA pelA double mutant. Plates were incubated for 24 h at 37°C, followed by 48 h at room temperature. (B) Quantification of CV-stained biofilms. Strains were grown in M63 with glucose, MgSO4 and CAA for 24 h prior to CV staining. (C) qRT-PCR analysis of pelA and pelG expression in agar-grown colonies of the WT and the ΔbifA mutant. Expression is plotted as picograms of input cDNA for each strain.
We tested whether the overproduction of polysaccharide in the ΔbifA mutant was due to an increase in transcription of the pel locus. However, no statistically significant difference (P > 0.25) in pelA or pelG transcript levels was observed between the WT and the ΔbifA mutant when grown on an agar plate (Fig. 2C) or as a static planktonic culture (data not shown). These data suggest that increased pel EPS production in the ΔbifA mutant is controlled via a mechanism other than transcriptional control.
Increased pel-synthesized polysaccharide is not responsible for the defect in swarming motility observed in the ΔbifA mutant.
As indicated by the initial identification of the bifA::Tn mutant and confirmed in the ΔbifA deletion mutant, bifA mutants fail to swarm compared to the WT (Fig. 3). Given that P. aeruginosa PA14 swarming motility requires a functional flagellum and the production of rhamnolipids as a surface-wetting agent (26, 53), we tested whether bifA mutants synthesize a functional flagellum and produce rhamnolipids. In swimming assays, the bifA mutants swim nearly as well as the WT (swim zone measurements: ΔbifA = 22.9 ± 1.7 mm; bifA::Tn = 23 ± 1.6 mm; WT = 29 ± 2 mm), indicating that bifA mutants make a functional flagellum. The bifA mutants also produce rhamnolipids at a level similar to that for the WT (data not shown). We also determined whether bifA mutants were capable of type IV pilus-mediated twitching motility, a form of surface motility that has also been shown to be important for P. aeruginosa biofilm formation (37), and found that the bifA mutants twitch as well as the WT (twitch zone measurements: ΔbifA = 12.7 ± 1.6 mm; bifA::Tn = 12.2 ± 1.6 mm; WT = 13.5 ± 2.1 mm).
FIG. 3.
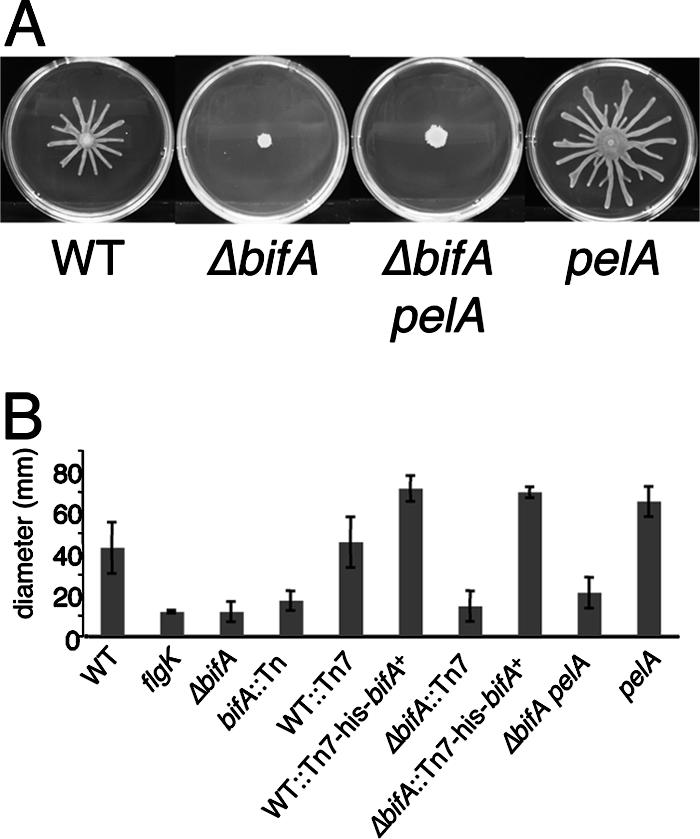
The pel-derived polysaccharide does not contribute to the swarming defects of the ΔbifA mutant. (A) Representative images of the WT, the ΔbifA mutant, the ΔbifA pelA double mutant, and the pelA mutant swarms after 16 h at 37°C. (B) Graph showing the average diameter (in millimeters) of swarms from triplicate platings for each strain.
On swarm plates, however, bifA mutants do not radiate outward from the point of inoculation but instead resemble the flgK mutant, a control strain that does not make a functional flagellum (Fig. 3B). The ΔbifA mutant forms a colony with the wrinkled morphology on the swarm plate similar to what was observed in the CR plate assays. This wrinkled phenotype, indicating overproduction of polysaccharide, prompted us to test whether enhanced polysaccharide production interferes with the ability of the ΔbifA mutant to swarm. Support for this notion comes from recent studies indicating that pelA mutants exhibit enhanced swarming relative to the WT, as reported by Caiazza et al. (7) and as shown in the present study (Fig. 3).
When tested on swarm plates, the ΔbifA pelA double mutant also shows no radiation of swarm tendrils out from the point of inoculation. Measurement of the swarm zone diameter shows there is no statistically significant difference between the ΔbifA pelA double mutant and the single ΔbifA mutant (Fig. 3B). As expected, however, the ΔbifA pelA mutant colony was no longer wrinkled due to the elimination of pel-mediated polysaccharide production. Thus, mutation of pelA in the ΔbifA mutant strain and subsequent loss of excess polysaccharide synthesis does not restore swarming motility in the ΔbifA mutant, indicating that overproduction of polysaccharide is not sufficient to inhibit swarming motility by the ΔbifA mutant.
The GGDQF and EAL domains of BifA are both required for complementation of the ΔbifA mutant.
The bifA gene encodes a protein of 688 amino acids with a predicted MW of ∼78 kDa. The two notable domains found in the BifA protein are a GGDEF-like domain and an EAL domain. GGDEF domains have been shown to be important for the activity of DGCs, which catalyze the synthesis of one molecule of c-di-GMP from two GTP molecules (40). The GGDEF-like domain of BifA maintains all but one of the conserved signature amino acid residues with the sequence GGDQF. EAL motifs, on the other hand, are required for the activity of c-di-GMP-degrading PDEs (11, 47), with the glutamic acid (E) residue previously shown to be critical for PDE activity in other systems (5, 52). The signature E, A, and L residues of this motif are conserved in BifA.
To test the importance of these motifs in the function of BifA, we generated mutations converting the key residues of each domain to alanine residues. We then assessed the ability of the WT His-BifA, His-BifA-AAAAA (GGDQF to AAAAA), and His-BifA-AAL (EAL to AAL) proteins, expressed from the multicopy pMQ80 plasmid, to complement the ΔbifA mutant in the 96-well plate assay. The results show that expression of His-tagged BifA protein from this plasmid in the ΔbifA mutant leads to a reduction in biofilm formation to approximately WT levels (Fig. 4A), a finding consistent with our observations in the single copy complementation experiments (see Fig. 1A). In contrast, expression of either His-BifA-AAAAA or His-BifA-AAL fails to suppress the hyperbiofilm phenotype of the ΔbifA mutant. Using Western blotting to detect His-BifA in these strains, we confirmed that the His-BifA-AAAAA and His-BifA-AAL mutant proteins are stable and expressed at levels similar to that for WT BifA (Fig. 4B). These results indicate that both the EAL domain and the GGDQF domain of BifA are required for proper function in vivo.
FIG. 4.

Assessment of function, stability, and cellular localization of BifA. (A) Assessment of the ability of the bifA gene, the bifA-AAAAA (GGDEF→AAAAA) mutant, and the bifA-AAL (EAL→AAL) mutant to complement the ΔbifA mutant when provided on an arabinose-inducible plasmid (pMQ80). The graph shows the quantification of biofilm formed by the WT and the ΔbifA mutant carrying either the pMQ80 vector alone or pMQ80 containing the His-tagged bifA gene (p-his-bifA+). Also shown is the quantification of biofilm formed by the ΔbifA mutant carrying pMQ80 with the bifA-AAAAA mutant (p-his-bifA-AAAAA) or the bifA-AAL mutant (p-his-bifA-AAL). Cells were grown for 6 h in the presence of 0.5% arabinose prior to CV staining. (B) Evaluation of expression and stability of WT BifA, BifA-AAAAA, and BifA-AAL proteins expressed from the pMQ80 constructs in panel A. Western blot showing the level of BifA expressed under the same conditions used in the biofilm assays in panel A. Equal amounts of cells were lysed and separated by SDS-PAGE. BifA was detected by using an anti-penta-His antibody. Purified His-BifA served as a control. (C) Cellular localization of BifA. Cellular fractions of the ΔbifA mutant carrying either vector alone (pMQ80) or vector containing the bifA gene (p-his-bifA+) were generated as described previously (see Materials and Methods). Approximately 1 μg of total protein from each fraction was separated by SDS-PAGE. Fractions are indicated as whole-cell (WC), soluble cytoplasmic (Cyt), total membrane (TM), inner-membrane (IM), and outer-membrane (OM) fractions. Western analysis was performed with either an anti-penta-His antibody, an anti-SecY antibody, or an anti-OprF antibody. The arrow indicates the BifA band. Purified His-BifA served as a control. SecY (∼50 kDa) served as a control for inner- membrane localization and OprF (∼35 kDa) served as an outer-membrane marker.
In the experiments described above, we utilized the multicopy pMQ80 vector for arabinose-inducible expression of His-BifA proteins in order to facilitate an assessment of protein levels and stability via Western blot analysis. It should be noted that we were unable to detect His-BifA when expressed in single copy, although this construct could complement a ΔbifA mutant. Although we could not assay protein levels of the single copy mutant constructs, we did observe that the His-BifA-AAAAA and His-BifA-AAL mutant proteins in single copy fail to complement the ΔbifA mutant (data not shown) in the same manner observed for the multicopy complementation experiments described above.
BifA localizes to the inner membrane.
Using the protein structure prediction algorithms TMHMM2 and Tmpred, the BifA protein is predicted to contain a N-terminal transmembrane domain, suggesting localization of BifA to the inner membrane, with the remaining C-terminal portion containing the GGDQF and EAL domains residing in the cytoplasm. To determine the subcellular localization of BifA, we performed cellular fractionations using a Sarkosyl extraction procedure to separate the inner and outer membranes (34), followed by Western blotting of these fractions to detect His-BifA. The results reveal that His-BifA is indeed highly enriched in the inner-membrane fraction (Fig. 4C). His-BifA could be weakly detected in whole-cell extracts and was enriched in the TM fraction and the inner-membrane fraction but was not detected in the outer-membrane fraction. The His-BifA-AAAAA and His-BifA-AAL mutant proteins are also enriched in the inner-membrane fraction, indicating that the GGDEF and EAL domains are not required for proper cellular localization (data not shown). Whether inner-membrane localization is required for the function of BifA is not yet clear. Deletion of the N-terminal region of BifA, including the predicted transmembrane (TRM) domain, leads to a loss of protein stability relative to the WT protein, making it difficult to draw any firm conclusions regarding the function of the BifA ΔTRM protein. However, when expressed in single copy, BifA ΔTRM shows partial complementation of the ΔbifA mutant biofilm defect, suggesting that the N-terminal region (and hence, membrane localization) is not absolutely required for BifA function in vivo (data not shown).
Western blotting with antibodies to SecY, an inner-membrane protein (1), and OprF, an outer-membrane-localized protein (3, 18), confirms that the inner and outer membranes were successfully fractionated.
BifA demonstrates PDE activity in vitro.
The complementation data described above indicate that the GGDQF and EAL domains of BifA are both important for its function (Fig. 4A). Thus, we set out to purify the WT His-BifA, His-BifA-AAAAA, and His-BifA-AAL proteins to test for both PDE and DGC activity. However, during the purification procedure, we found that His-BifA-AAAAA and His-BifA-AAL mutant proteins were stable in crude extracts (Fig. 5A) but that subsequent purification steps led to accelerated degradation of the mutant proteins relative to WT BifA. Therefore, we evaluated the activity of the WT His-BifA and the His-BifA mutant proteins in crude extracts compared to extracts prepared from the vector control strain.
FIG. 5.

Analysis of PDE activity of BifA in vitro. (A) Western blot showing the levels of WT His-BifA, His-BifA-AAL, and His-BifA-AAAAA protein in crude extracts of E. coli cells carrying either pQE-30 with the bifA gene, the bifA-AAL mutant, or the bifA-AAAAA mutant. Crude extracts were prepared as described previously (see Materials and Methods). Equal quantities (3 μg) of total protein were separated by SDS-PAGE and detected by Western blotting with an anti-penta-His antibody. (B) Assessment of PDE activity in the crude extracts (described above) compared to control extract (prepared from E. coli carrying the pQE-30 vector only) using the substrate bis-pNPP. The graph shows the dose-response curve generated by twofold serial dilutions of crude extract incubated in the presence of bis-pNPP for 20 min. The release of p-nitrophenol was quantified at OD410. The asterisk indicates statistical significance (P < 0.05). (C) Detection of PDE activity in crude extracts using the radiolabeled substrate, c-di-GMP. Crude extracts containing either WT BifA, BifA-AAL, and BifA-AAAAA mutant proteins in increasing concentrations (3 or 9 μg of protein per reaction) were incubated with c-di-GMP. A no-protein (NP) control lane indicates the amount of input substrate. CC3396 served as a positive control for PDE activity, converting c-di-GMP to the linear form, pGpG. (D) Purification of His-tagged BifA. His-tagged BifA was purified from crude extracts using a His-Trapp-FF NiSO4 column. The purity of BifA was evaluated by SDS-PAGE separation and Coomassie blue staining (lane 3) compared to crude extract (CE). The sizes of markers (M) in lane 1 are indicated in kilodaltons. (E) PDE activity of purified His-BifA. The graph shows a dose-response curve generated by twofold serial dilutions of BifA assayed as described in panel B.
We first assessed PDE activity in crude extracts by using a colorimetric assay involving PDE-mediated cleavage of the synthetic substrate, bis-pNPP (5). The data show that bis-pNPP is cleaved in crude extract containing the WT His-BifA protein and cleavage occurs in a concentration-dependent manner (Fig. 5B). Furthermore, the PDE activity observed in WT extracts is significantly higher at all concentrations shown than that of the vector control extract. In contrast, the activity in crude extract containing either the His-BifA-AAL or His-BifA-AAAAA mutant protein does not differ significantly from the control extract at any of the concentrations shown. These results suggest that mutational inactivation of either the GGDQF or EAL domain abolishes BifA PDE activity.
Next, we assessed the PDE activity in crude extracts using the native substrate, c-di-GMP. In WT extracts, the levels of c-di-GMP are reduced in a dose-dependent fashion compared to the input levels of c-di-GMP shown in the no-protein control, indicating the presence of c-di-GMP cleavage activity (Fig. 5C). CC3396, the purified protein serving as a positive control for PDE activity, cleaves c-di-GMP to form the linear product, pGpG. For the WT extracts, we observe the disappearance of c-di-GMP but do not see the accumulation of pGpG. This absence of pGpG product is likely due to subsequent cleavage and/or degradation of pGpG either by BifA itself or some other activity present in the extract. In fact, addition of crude extract prepared from the strain carrying the vector control to the CC3396 reaction leads to disappearance of the pGpG product (data not shown), indicating that degradation of pGpG occurs via a BifA-independent activity present in crude extracts.
In contrast to the WT extracts, crude extracts containing either His-BifA-AAAAA or His-BifA-AAL show no detectable reduction in c-di-GMP levels. The presence of the mutant proteins in these extracts at levels equivalent to the WT protein was confirmed by Western blotting (results not shown). These results confirm our observations in the colorimetric assay described above. Taken together, the data indicate that the WT BifA protein is able to cleave c-di-GMP and that cleavage requires both the GGDQF and the EAL domains of BifA.
In order to further confirm that the WT BifA protein has PDE activity, we continued with the purification of WT His-BifA from crude extracts using a nickel affinity resin (Fig. 5D) and tested its ability to cleave bis-pNPP in the colorimetric assay described above. As expected based on results with WT crude extracts, purified His-BifA also exhibits PDE activity in a concentration-dependent manner (Fig. 5E).
Given that BifA has a GGDEF-like motif with the sequence GGDQF, we also tested whether BifA possesses a DGC activity using established protocols (40). However, we could not detect synthesis of c-di-GMP using either purified His-BifA or crude extracts containing WT His-BifA, despite our ability in control experiments to synthesize c-di-GMP using purified PleD, a well-characterized DGC protein with known c-di-GMP cyclase activity (data not shown) (40). These data indicate that BifA either does not possess DGC activity or that such activity is below our level of detection.
The ΔbifA mutant exhibits increased cellular levels of c-di-GMP relative to WT.
Given that BifA possesses the ability to cleave c-di-GMP in vitro, we predicted that disruption of BifA activity might impact the cellular pools of c-di-GMP in vivo. Thus, we examined levels of c-di-GMP in ΔbifA mutant and WT cells when grown in the presence of radiolabeled inorganic phosphate and further analyzed as previously described (19, 33). Inspection of the two-dimensional TLC plate autoradiographs indicates that there is an increase in the levels of the c-di-GMP component in ΔbifA mutant cells relative to WT cells (Fig. 6A). Quantification of c-di-GMP and normalization to total 32Pi incorporation (for which there was no significant difference between the two strains [P = 0.42]) show that the ΔbifA mutant accumulates ∼2.5-fold more c-di-GMP than does the WT (Fig. 6B). This result is consistent with our in vitro observations and lends further support for the role of BifA as a c-di-GMP PDE.
FIG. 6.
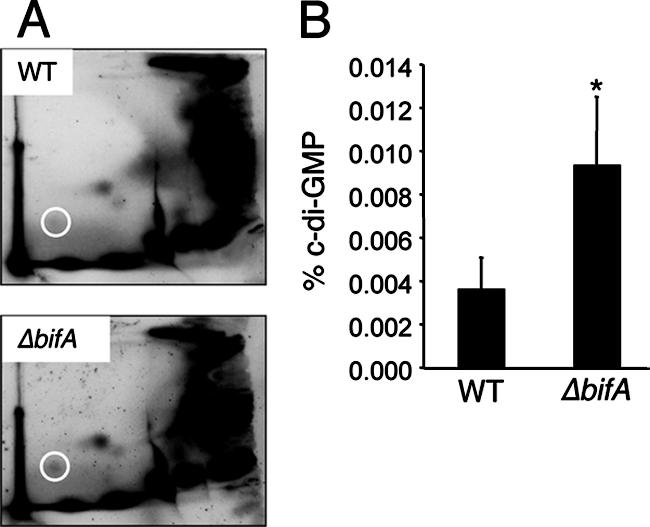
The bifA gene influences c-di-GMP levels in vivo. (A) Autoradiographs of representative two-dimensional TLC plates used to separate [32P]orthophosphate-labeled, acid-extracted whole-cell extracts prepared from both the WT and the ΔbifA mutant. The circle indicates the position of c-di-GMP. (B) Quantification of c-di-GMP levels. Autoradiographs were analyzed using the Storm 860 and ImageQuant software (v5.1). The percentage of label incorporated into c-di-GMP was normalized to total 32P labeling and expressed as the percentage of c-di-GMP. The asterisk indicates statistical significance (P < 0.02).
Genetic interactions between bifA and sadB in biofilm formation, pel-mediated polysaccharide synthesis, and swarming motility.
Recent findings from our laboratory have established the sadB gene as playing a key role in the inverse regulation of biofilm formation and swarming by P. aeruginosa (7). These studies show that sadB mutants are unable to form a biofilm but exhibit an enhanced ability to swarm. Thus, the sadB gene product promotes biofilm formation while simultaneously inhibiting swarming motility. This inverse regulation of surface behaviors mirrors the inverse regulation of motility and sessility by intracellular levels of c-di-GMP as has been shown in other systems (reviewed in reference 42). Given that the studies presented here establish a role for the bifA gene as a regulator of c-di-GMP levels, we wondered whether the sadB gene might function downstream of the bifA gene in response to c-di-GMP signaling in a pathway that inversely regulates biofilm formation and swarming motility in P. aeruginosa.
To determine the epistatic relationship between the sadB and bifA genes, we constructed a deletion of the bifA gene in the sadB mutant background. We then tested the resulting ΔbifA sadB double mutant for biofilm formation in the 96-well plate assay. In these assays, the biofilm formed by the ΔbifA sadB mutant is severely deficient relative to the WT and is nearly indistinguishable from the sadB single mutant (Fig. 7A). These results indicate that the sadB gene is required for the hyperbiofilm formed by the ΔbifA mutant and support a genetic pathway in which the sadB gene functions downstream of the bifA gene in regulating biofilm formation.
FIG. 7.

Genetic interactions between bifA and sadB in relation to biofilm formation, EPS production, and swarming. (A) Quantification of CV-stained biofilm formed by the WT, the ΔbifA mutant, the sadB mutant, and the ΔbifA sadB double mutant. Cells were grown for 8 h prior to CV staining. (B) CR binding by the indicated strains. Plates were incubated for 24 h at 37°C, followed by 48 h at room temperature. (C) Diameter (in millimeters) of swarm zones plotted for each of the indicated strains. Swarm plates were incubated for 16 h at 37°C. (D) Swim reversal frequencies of the indicated strains under high-viscosity (15% Ficoll) conditions are plotted. Approximately 135 total cells were counted per strain to determine the reversal frequency, expressed as the number of reversals per cell. The asterisks indicate statistical significance (P < 0.0005).
The recent report by Caiazza et al. also shows that sadB mutants are deficient in CR binding and produce colonies with a smooth morphology due to defects in the production of the pel-derived polysaccharide (7). In contrast, ΔbifA mutants exhibit hyper-CR binding and a highly wrinkled colony morphology due to increased production of pel polysaccharide (see Fig. 2A and 7B). To determine whether the bifA gene regulates polysaccharide production via SadB, we tested the CR-binding ability of the ΔbifA sadB mutant and found that CR binding is considerably reduced relative to the ΔbifA mutant, but CR binding is not eliminated, as is the case for the sadB mutant alone. Furthermore, the colony morphology of the ΔbifA sadB mutant is far less wrinkled than is the ΔbifA single mutant. These findings indicate that SadB contributes to increased pel polysaccharide production in the ΔbifA mutant. Taken together with the biofilm results described above, these data further support the notion that sadB functions downstream of bifA in regulating biofilm formation and that sadB does so by influencing pel-mediated polysaccharide synthesis.
The epistasis studies with the bifA and pelA genes described earlier indicated that the accumulation of polysaccharide in the ΔbifA mutant does not directly inhibit the swarming motility of the ΔbifA mutant (see Fig. 3A). Thus, some other factor(s) must be responsible for the swarming defect in the ΔbifA mutant. Given that sadB mutants are hyperswarmers and that overexpression of the sadB gene can repress the swarming motility of the WT strain (7), it seemed plausible that inhibition of the swarming motility in the ΔbifA mutant could be dependent on SadB. When tested on swarm agar plates, the ΔbifA sadB mutant is indeed a hyperswarmer relative to the WT, with a swarming zone comparable to that observed for the sadB mutant alone (Fig. 7C). This result indicates that SadB is required to inhibit the swarming motility of the ΔbifA mutant. The data also provide evidence that sadB is influenced by the bifA gene (and likely c-di-GMP signaling) in its role as a regulator of both biofilm formation and swarming motility.
Caiazza et al. also provided evidence that SadB controls swarming motility by modulating the rate of flagellar reversals (7). More specifically, they reported that sadB mutants show a higher rate of flagellar reversals in high viscosity medium (15% Ficoll), conditions that have previously been equated to conditions encountered while swarming (53). Because the ΔbifA mutant is inhibited for swarming motility, the opposite phenotype to that of the sadB mutant, we predicted that the ΔbifA mutant would show a reduced rate of flagellar reversals relative to the WT. To test this prediction, we monitored the swimming behavior of the ΔbifA mutant in 15% Ficoll and measured the frequency of swim reversals. Consistent with our hypothesis, the ΔbifA mutant shows a statistically significant ∼2-fold decrease in reversals compared to the WT. As a control, the sadB mutant showed the expected increase in reversals.
DISCUSSION
We present biochemical evidence that BifA, a dual EAL/GGDEF domain-containing protein, possesses PDE activity and is capable of cleaving both the synthetic substrate, bis-pNPP, and the native substrate, c-di-GMP, in in vitro cleavage assays. Mutation of the glutamic acid (E) residue of the core EAL motif shown to be critical for PDE function in other systems (5, 52) renders BifA nonfunctional in PDE activity assays. This mutation also abolishes the ability of BifA to complement any of the phenotypes associated with the ΔbifA mutant. A role for BifA as a PDE is further supported by our observations that the ΔbifA mutant has ∼2.5-fold-higher intracellular levels of c-di-GMP than does the WT in whole-cell labeling experiments.
Despite the presence of a GGDEF domain, BifA does not appear to possess DGC activity required for the synthesis of c-di-GMP. However, this is not entirely unexpected given that other dual EAL/GGDEF domain-containing proteins possess only PDE activity (11, 25, 33) and that the GGDEF-like domain of BifA differs from the consensus sequence in the core motif, with the sequence GGDQF. Recent data from Caulobacter cresentus indicates that the GGDEF domain of the PDE CC3396, which also differs from the consensus of the core motif with the sequence GEDEF, appears to act as a positive allosteric effector site for GTP (11). Consistent with a role for the BifA GGDQF domain in stimulating PDE activity, we observe that replacement of the GGDQF core residues with alanine residues greatly diminishes the PDE activity of BifA. Experiments to further address the role of this domain as an allosteric effector site are currently under way.
Mutating the bifA gene results in an increased pool of c-di-GMP and impacts both biofilm formation and swarming motility. How is this increased pool of c-di-GMP linked to the alterations in biofilm formation and swarming motility?
In the case of biofilm formation, mutating bifA results in increased production of the Pel polysaccharide, as indicated by increased binding of CR that is dependent on pelA. Like the pelA mutant, the ΔbifA pelA double mutant is biofilm defective, indicating that a functional pel locus is required for the increased biofilm formation observed in the ΔbifA mutant. According to our microscopy studies, enhanced biofilm formation by the ΔbifA mutant is triggered as early as the initial attachment phase, indicating that enhanced EPS production promotes initial attachment, a result consistent with recent findings from other groups (29, 54). Given that c-di-GMP is an allosteric activator of cellulose synthesis in Acetobacter xylinum (43), the increase in Pel polysaccharide could be due to the role of c-di-GMP as a positive allosteric effector of polysaccharide production, a result consistent with our observation that increased binding of CR is not correlated with increased expression of the pelA or pelG genes. However, no such allosteric regulatory role for c-di-GMP has yet been documented for Pel polysaccharide production in P. aeruginosa.
In addition to its hyperbiofilm phenotype, the ΔbifA mutant is completely defective for swarming and displays a wrinkled colony phenotype associated with EPS production when grown on swarming motility agar. Based on the observation that mutating the pel locus of the WT strain results in increased swarming motility, we predicted that the overproduction of the Pel polysaccharide might inhibit swarming motility. To our surprise, while mutating pelA in the ΔbifA mutant background eliminated the wrinkled colony phenotype, swarming motility was not restored. These data indicate that the defect in swarming due to the ΔbifA mutant was not due to excess polysaccharide production. As shown by genetic studies presented in the companion manuscript by Merritt et al., the accumulation of c-di-GMP mediated by SadC is required for loss of swarming in the ΔbifA mutant (32).
What then was causing the swarming defect in the ΔbifA mutant? We recently showed that mutating the sadB gene impacts flagellar function by modulating the rate of flagellar reversals specifically under high-viscosity conditions, an environment analogous to that encountered by cells while swarming (7). That is, the increased flagellar reversal rates of the sadB mutant were correlated with a hyperswarming phenotype. Given the swarm defect of the ΔbifA mutant, we predicted that this strain would show decreased flagellar reversals compared to the WT. Indeed, in the present study we showed the ΔbifA mutant has a decreased rate of flagellar reversals compared to the WT under high-viscosity conditions. Our data extend the correlation we observed previously between rates of flagellar reversals and the robustness of swarming motility.
Taken together with our recently published work (7, 32), phenotypic studies indicate that SadC, BifA, and SadB are all involved in the inverse regulation of biofilm formation and swarming motility. Mutations in the sadC and sadB genes result in strains defective for biofilm formation and a hyperswarming phenotype. In contrast, mutating bifA renders the strain a hyperbiofilm former and completely blocked for swarming motility. The similarity in phenotypes among these mutants suggested that they might function in the same genetic pathway, and our epistasis data published here and elsewhere (32) support this hypothesis.
Epistasis analysis presented in the companion study by Merritt et al. (32) indicates that BifA and SadC function in the same pathway in the inverse regulation of biofilm formation and swarming motility. Mutating sadC in the ΔbifA mutant background results in a significant reduction, but not elimination, of the hyperbiofilm and hyper-CR binding of the ΔbifA mutant and a partial restoration of swarming motility. These data indicate that at least one other DGC synthesizes c-di-GMP under the growth conditions used in our study (32).
The data presented above suggest that multiple DGCs generate c-di-GMP that is degraded by BifA, but BifA might be required to modulate c-di-GMP under a broad range of environmental conditions. Consistent with this hypothesis, the ΔbifA mutant is a hyperbiofilm former relative to the WT when tested for biofilm formation on at least 40 different carbon and energy sources, including when cells are grown on glucose, arginine, citrate, or LB (S. L. Kuchma and G. A. O'Toole, unpublished data). We also showed that the increased biofilm formation and the block in swarming motility observed for the ΔbifA mutant are dependent on the previously described sadB locus (7, 8). Similarly, the boost in biofilm formation and CR binding observed in strains carrying multiple copies of the sadC gene is largely eliminated when the sadB gene is mutated (32). Taken together, these genetic data suggest that SadC and BifA function upstream of SadB.
Previous studies of SadB led to the identification of a genetic pathway, including the chemotaxis-like CheIV locus, in the inverse regulation of biofilm formation and swarming motility (7). The identification and characterization of SadC by Merritt et al. (32), and of BifA, presented here, have expanded the SadB genetic pathway to include components that function upstream of SadB in regulating surface behaviors of P. aeruginosa. Furthermore, the characterization of SadC and BifA as contributing to c-di-GMP metabolism implicates this signaling molecule in the regulation of these surface behaviors. Based on these data, we propose that SadC and BifA function together to modulate the levels of c-di-GMP in the cell and that the resulting levels of this molecule impact both EPS production and flagellar function and thus biofilm formation and swarming motility. However, we cannot completely rule out the possibility that these genes function in similar but parallel pathways. For example, given that a sadB mutation does not eliminate CR binding by the ΔbifA mutant, it is plausible that BifA and SadB both contribute to Pel polysaccharide synthesis via distinct pathways. Alternatively, we would argue that c-di-GMP levels primarily influence Pel polysaccharide synthesis through a SadB-dependent pathway but that in a ΔbifA mutant the levels of c-di-GMP are sufficiently elevated to stimulate Pel activity via additional SadB-independent mechanisms.
Given that there are ∼40 proteins predicted to be involved in c-di-GMP metabolism in P. aeruginosa, a central question in the field now focuses on how this bacterium coordinates the activities of these proteins to effectively control cellular levels of c-di-GMP. We are interested in pursuing this question by exploring how SadC and BifA specifically impact surface behaviors under the conditions studied here. There are a number of mechanisms that might allow for coregulation of SadC and BifA. For example, the sadC and bifA genes might be cotranscriptionally regulated. However, based on a survey of published microarray studies, we find no evidence that the sadC and bifA genes are differentially expressed under conditions that impact biofilm formation (2, 35, 46, 56). Another mechanism to coordinate DGC and PDE activities may involve their spatial restriction within the cell. Recent work by Paul et al. illustrates the importance of cellular localization in the proper function of the PleD DGC (40). Together with Merritt et al., we have shown that both BifA and SadC localize to the cytoplasmic membrane. Colocalization of BifA and SadC to the same membrane hints at the potential for interaction between these proteins and may provide a mechanism for coregulation of their enzymatic activities. Finally, given that cellular GTP levels may reflect the energy status of the cell and the pool of substrate for DGC enzymes, allosteric regulation of BifA by GTP may provide an additional level of regulation for this system.
A more complete understanding of the role of SadC, BifA, and SadB, as well as the downstream chemotaxis-like components, should provide additional insight into how P. aeruginosa controls c-di-GMP-dependent biofilm formation and swarming motility as this microbe transitions from a planktonic to a surface-associated lifestyle.
Supplementary Material
Acknowledgments
We thank A. Malek for his assistance.
This study was supported by grants from the Cystic Fibrosis Foundation (AUSUBE04VO) and the Department of Energy (DE-FG02-ER63445) to F.M.A. and from the Cystic Fibrosis Foundation (OTOOLE06I0) and the National Institutes of Health (AI51360, 1-P20-RR01878) to G.A.O.
Footnotes
Published ahead of print on 22 June 2007.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Akiyama, Y., and K. Ito. 1985. The SecY membrane component of the bacterial protein export machinery: analysis by new electrophoretic methods for integral membrane proteins. EMBO J. 4:3351-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aspedon, A., K. Palmer, and M. Whiteley. 2006. Microarray analysis of the osmotic stress response in Pseudomonas aeruginosa. J. Bacteriol. 188:2721-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benz, R., and R. E. Hancock. 1981. Properties of the large ion-permeable pores formed from protein F of Pseudomonas aeruginosa in lipid bilayer membranes. Biochim. Biophys. Acta 646:298-308. [DOI] [PubMed] [Google Scholar]
- 4.Bertani, G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186:595-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobrov, A. G., O. Kirillina, and R. D. Perry. 2005. The phosphodiesterase activity of the HmsP EAL domain is required for negative regulation of biofilm formation in Yersinia pestis. FEMS Microbiol. Lett. 247:123-130. [DOI] [PubMed] [Google Scholar]
- 6.Boles, B. R., and L. L. McCarter. 2002. Vibrio parahaemolyticus scrABC, a novel operon affecting swarming and capsular polysaccharide regulation. J. Bacteriol. 184:5946-5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caiazza, N. C., J. H. Merritt, K. M. Brothers, and G. A. O'Toole. 2007. Inverse regulation of surface behaviors by Pseudomonas aeruginosa PA14. J. Bacteriol. 189:3603-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caiazza, N. C., and G. A. O'Toole. 2004. SadB is required for the transition from reversible to irreversible attachment during biofilm formation by Pseudomonas aeruginosa PA14. J. Bacteriol. 186:4476-4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi, K. H., J. B. Gaynor, K. G. White, C. Lopez, C. M. Bosio, R. R. Karkhoff-Schweizer, and H. P. Schweizer. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443-448. [DOI] [PubMed] [Google Scholar]
- 10.Choi, K. H., A. Kumar, and H. P. Schweizer. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391-397. [DOI] [PubMed] [Google Scholar]
- 11.Christen, M., B. Christen, M. Folcher, A. Schauerte, and U. Jenal. 2005. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J. Biol. Chem. 280:30829-30837. [DOI] [PubMed] [Google Scholar]
- 12.D'Argenio, D. A., M. W. Calfee, P. B. Rainey, and E. C. Pesci. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J. Bacteriol. 184:6481-6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Argenio, D. A., and S. I. Miller. 2004. Cyclic di-GMP as a bacterial second messenger. Microbiology 150:2497-2502. [DOI] [PubMed] [Google Scholar]
- 14.Davey, M. E., and G. A. O'Toole. 2000. Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64:847-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman, L., and R. Kolter. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675-690. [DOI] [PubMed] [Google Scholar]
- 16.Friedman, L., and R. Kolter. 2004. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J. Bacteriol. 186:4457-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galperin, M. Y., A. N. Nikolskaya, and E. V. Koonin. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203:11-21. [DOI] [PubMed] [Google Scholar]
- 18.Hancock, R. E., G. M. Decad, and H. Nikaido. 1979. Identification of the protein producing transmembrane diffusion pores in the outer membrane of Pseudomonas aeruginosa PA01. Biochim. Biophys. Acta 554:323-331. [DOI] [PubMed] [Google Scholar]
- 19.Hickman, J. W., D. F. Tifrea, and C. S. Harwood. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA 102:14422-14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinsa, S. M., M. Espinosa-Urgel, J. L. Ramos, and G. A. O'Toole. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol. Microbiol. 49:905-918. [DOI] [PubMed] [Google Scholar]
- 21.Hinsa, S. M., and G. A. O'Toole. 2006. Biofilm formation by Pseudomonas fluorescens WCS365: a role for LapD. Microbiology 152:1375-1383. [DOI] [PubMed] [Google Scholar]
- 22.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 23.Jackson, K. D., M. Starkey, S. Kremer, M. R. Parsek, and D. J. Wozniak. 2004. Identification of psl, a locus encoding a potential exopolysaccharide that Is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J. Bacteriol. 186:4466-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenal, U. 2004. Cyclic di-guanosine-monophosphate comes of age: a novel secondary messenger involved in modulating cell surface structures in bacteria? Curr. Opin. Microbiol. 7:185-191. [DOI] [PubMed] [Google Scholar]
- 25.Kazmierczak, B. I., M. B. Lebron, and T. S. Murray. 2006. Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol. Microbiol. 60:1026-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohler, T., L. K. Curty, F. Barja, C. van Delden, and J. C. Pechere. 2000. Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J. Bacteriol. 182:5990-5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuchma, S. L., J. P. Connolly, and G. A. O'Toole. 2005. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J. Bacteriol. 187:1441-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberati, N. T., J. M. Urbach, S. Miyata, D. G. Lee, E. Drenkard, G. Wu, J. Villanueva, T. Wei, and F. M. Ausubel. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA 103:2833-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma, L., K. D. Jackson, R. M. Landry, M. R. Parsek, and D. J. Wozniak. 2006. Analysis of Pseudomonas aeruginosa conditional psl variants reveals roles for the psl polysaccharide in adhesion and maintaining biofilm structure postattachment. J. Bacteriol. 188:8213-8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacEachran, D. P., S. Ye, J. Bomberger, D. A. Hogan, B. A. Stanton, and G. A. O'Toole. 2007. The Pseudomonas aeruginosa secreted protein, PA2934, decreases apical membrane expression of the cystic fibrosis transmembrane conductance regulator. Infect. Immun. 75:3902-3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsukawa, M., and E. P. Greenberg. 2004. Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J. Bacteriol. 186:4449-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merritt, J. H., K. M. Brothers, S. L. Kuchma, and G. A. O'Toole. 2007. SadC reciprocally influences biofilm formation and swarming motility via modulation of exopolysaccharide production and flagellar function. J. Bacteriol. 189:8154-8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monds, R. D., P. D. Newell, R. H. Gross, and G. A. O'Toole. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol. Microbiol. 63:656-679. [DOI] [PubMed] [Google Scholar]
- 34.Nunn, D., and S. Lory. 1993. Cleavage, methylation, and localization of the Pseudomonas aeruginosa export proteins XcpT, -U, -V, -W. J. Bacteriol. 175:4375-4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ochsner, U. A., P. J. Wilderman, A. I. Vasil, and M. L. Vasil. 2002. GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 45:1277-1287. [DOI] [PubMed] [Google Scholar]
- 36.O'Toole, G. A., K. A. Gibbs, P. W. Hager, P. V. Phibbs, Jr., and R. Kolter. 2000. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J. Bacteriol. 182:425-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Toole, G. A., and R. Kolter. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295-304. [DOI] [PubMed] [Google Scholar]
- 38.O'Toole, G. A., and R. Kolter. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449-461. [DOI] [PubMed] [Google Scholar]
- 39.Pardee, A. B., F. Jacob, and J. Monod. 1959. The genetic control and cytoplasmic expression of “inducibility” in the synthesis of β-galactosidase in Escherichia coli. J. Mol. Biol. 1:165-178. [Google Scholar]
- 40.Paul, R., S. Weiser, N. C. Amiot, C. Chan, T. Schirmer, B. Giese, and U. Jenal. 2004. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 18:715-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahme, L. G., E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins, and F. M. Ausubel. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899-1902. [DOI] [PubMed] [Google Scholar]
- 42.Römling, U., M. Gomelsky, and M. Y. Galperin. 2005. c-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57:629-639. [DOI] [PubMed] [Google Scholar]
- 43.Ross, P., H. Weinhouse, Y. Aloni, D. Michaeli, P. Weinberger-Ohana, R. Mayer, S. Braun, E. de Vroom, G. A. van der Marel, J. H. van boom, and M. Benziman. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 325:279-281. [DOI] [PubMed] [Google Scholar]
- 44.Ryan, R. P., Y. Fouhy, J. F. Lucey, L. C. Crossman, S. Spiro, Y. W. He, L. H. Zhang, S. Heeb, M. Camara, P. Williams, and J. M. Dow. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. USA 103:6712-6717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Ryjenkov, D. A., M. Tarutina, O. V. Moskvin, and M. Gomelsky. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 187:1792-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sauer, K., M. C. Cullen, A. H. Rickard, L. A. Zeef, D. G. Davies, and P. Gilbert. 2004. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 186:7312-7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt, A. J., D. A. Ryjenkov, and M. Gomelsky. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J. Bacteriol. 187:4774-4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shanks, R. M., N. C. Caiazza, S. M. Hinsa, C. M. Toutain, and G. A. O'Toole. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 72:5027-5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siegmund, I., and F. Wagner. 1991. New method for detecting rhamnolipids excreted by Pseudomonas aeruginosa species during growth on minimal agar. Biotechnol. Tech. 5:265-268. [Google Scholar]
- 50.Simm, R., M. Morr, A. Kader, M. Nimtz, and U. Romling. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 53:1123-1134. [DOI] [PubMed] [Google Scholar]
- 51.Tal, R., H. C. Wong, R. Calhoon, D. Gelfand, A. L. Fear, G. Volman, R. Mayer, P. Ross, D. Amikam, H. Weinhouse, A. Cohen, S. Sapir, P. Ohana, and M. Benziman. 1998. Three cdg operons control cellular turnover of cyclic di-GMP in Acetobacter xylinum: genetic organization and occurrence of conserved domains in isoenzymes. J. Bacteriol. 180:4416-4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamayo, R., A. D. Tischler, and A. Camilli. 2005. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J. Biol. Chem. 280:33324-33330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toutain, C. M., M. E. Zegans, and G. A. O'Toole. 2005. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J. Bacteriol. 187:771-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vasseur, P., I. Vallet-Gely, C. Soscia, S. Genin, and A. Filloux. 2005. The pel genes of the Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation. Microbiology 151:985-997. [DOI] [PubMed] [Google Scholar]
- 55.Whitchurch, C. B., M. Hobbs, S. P. Livingston, V. Krishnapillai, and J. S. Mattick. 1990. Characterization of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialized protein export system widespread in eubacteria. Gene 101:33-44. [DOI] [PubMed] [Google Scholar]
- 56.Whiteley, M., M. G. Bangera, R. E. Bumgarner, M. R. Parsek, G. M. Teitzel, S. Lory, and E. P. Greenberg. 2001. Gene expression in Pseudomonas aeruginosa biofilms. Nature 413:860-864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.