

Abstract
The nonsegmented, negative-sense RNA genome of measles virus (MV) is encapsidated by the virus-encoded nucleocapsid protein (N). In this study, we searched for N-binding cellular proteins by using MV-N as bait and screening the human T-cell cDNA library by yeast two-hybrid assay and isolated the p40 subunit of eukaryotic initiation factor 3 (eIF3-p40) as a binding partner. The interaction between MV-N and eIF3-p40 in mammalian cells was confirmed by coimmunoprecipitation. Since eIF3-p40 is a translation initiation factor, we analyzed the potential inhibitory effect of MV-N on protein synthesis. Glutathione S-transferase (GST)-fused MV-N (GST-N) inhibited translation of reporter mRNAs in rabbit reticulocyte lysate translation system in a dose-dependent manner. Encephalomyocarditis virus internal ribosomal entry site-mediated translation, which requires canonical initiation factors to initiate translation, was also inhibited by GST-N. In contrast, a unique form of translation mediated by the intergenic region of Plautia stali intestine virus, which can assemble 80S ribosomes in the absence of canonical initiation factors, was scarcely affected by GST-N. In vivo expression of MV-N induced by the Cre/loxP switching system inhibited the synthesis of a transfected reporter protein, as well as overall protein synthesis. These results suggest that MV-N targets eIF3-p40 and may be involved in inhibiting MV-induced host translation.
Measles virus (MV) is a member of the genus Morbillivirus within the family Paramyxoviridae of the order Mononegavirales. This virus has a nonsegmented negative-sense single-stranded RNA genome that is encapsidated by multiple copies of the nucleocapsid protein (N) to form a helical ribonucleoprotein complex known as the nucleocapsid. Associated with the nucleocapsid are the two components of the viral RNA-dependent RNA polymerase: the phosphoprotein (P) and large protein (L). Together, these constitute the holonucleocapsid, which is packaged within a lipid envelope, bears the hemagglutinin and fusion glycoproteins (H and F), and is lined internally by matrix protein (M).
Recently, several reports showed that MV-N can induce systemic immunosuppression. The C-terminal part of MV-N binds to the Fc receptor on B cells and dendritic cells and induces immunosuppression by inhibiting antibody production (20), impairing dendritic cell function (14), preventing interleukin-12 production (15), and suppressing hypersensitivity responses (14, 15). Interestingly, a recent study showed that MV-N also activates signal cascades of innate immunity by phosphorylating IRF-3 (28). The C-terminal domain of MV-N associates with IRF-3, as well as the virus-activated kinase. These findings strongly imply that MV-N is involved in mediating the immune response, as well as inducing immunological abnormalities and pathogenicity during the virus life cycle.
In the present study, we searched for MV-N-binding cellular proteins by using a yeast two-hybrid screening system, and identified the p40 subunit of eukaryotic translation initiation factor 3 (eIF3-p40; eIF3γ) as a specific binding partner. We previously showed that MV shuts off host cell gene expression during virus infection (unpublished data). Therefore, we analyzed the implication of MV-N in the shutoff via binding with eIF3-p40 in the present study. We showed that MV-N inhibits in vitro translation of reporter mRNAs and in vivo protein synthesis in a dose-dependent manner. Based on these data, we proposed that MV-N shuts off host translation in MV-infected cells through protein-protein interactions with eIF3.
MATERIALS AND METHODS
Construction of yeast two-hybrid vectors.
To construct yeast two-hybrid bait vectors encoding the full-length MV-N gene of the wild-type HL strain (pGBKT-N) and two deletion clones (pGBKT-NΔ1 and pGBKT-NΔ6), cDNAs were amplified by PCR from the full-length MV-HL strain clone (unpublished data) using specific primer pairs (data not shown), and LA-Taq DNA polymerase (TaKaRa). PCR products were subcloned into pGEM-T Easy vector (Promega) and digested with NdeI and EcoRI. The resulting cDNA fragments were inserted separately into pGBKT7 bait vector (Clontech). To construct four yeast two-hybrid bait vectors encoding MV-N deletion clones (pGBKT-NΔ2, -NΔ3, -NΔ4, and -NΔ5), the pGEM-T Easy vector containing the full-length MV-N cDNA was PCR amplified using the specific primer pairs (data not shown) and Pfu Turbo DNA polymerase (Stratagene). The PCR fragments were phosphorylated with T4 polynucleotide kinase (TOYOBO) and self-ligated. The plasmids obtained were digested with NdeI and EcoRI, and the resulting cDNA fragments were inserted separately into pGBKT7 bait vector.
Two-hybrid screening and interaction assays.
Yeast AH109 cells were transformed with pGBKT7 bait plasmid containing full-length MV-N and the human T-cell cDNA library (107 clones) by using the lithium acetate method described in the Clontech manual. Transformed cells were plated on minimal selective synthetic dropout (SD) media (SD/−Ade/−His/−Leu/−Trp/X-α-Gal) containing 2.5 mM 3-aminotriazole (3-AT), and colonies were picked and replica plated after 5 to 7 days of incubation at 30°C. Plasmid DNA from positive clones was extracted by using the YeastMaker yeast plasmid isolation kit (Clontech) and electroporated into Escherichia coli ElectroMax DH10B competent cells (Invitrogen). The resulting plasmid recovered from E. coli, encoding eIF3-p40 cDNA, was retransformed into yeast with pGBKT7 vectors that encoded a series of MV-N deletion mutants (pGBKT-NΔ1 to -NΔ6). Positive clones were selected on SD medium in the absence of two nutrients (Leu and Trp), and the obtained colonies were spotted onto SD/−His/−Leu/−Trp plates in the presence of 0.5 mM 3-AT. Positive interactions were defined as the ability of transformed cells to grow on this medium.
Construction of eukaryotic expression plasmids.
To create a plasmid expressing hemagglutinin (HA)-tagged eIF3-p40 (pCMV-HA-eIF3-p40), total RNA isolated from human embryonic kidney 293 cells by ISOGEN (Nippon gene) was reverse transcribed by using a random primer (9-mer) and SuperScript II reverse transcriptase (Gibco-BRL), followed by PCR amplification with a specific primer pair corresponding to eIF3-p40 cDNA (5′-GAATTCGGATGGCGTCCCGCAAGGAAGG-3′ and 5′-CTCGAGATTAGTTGTTGTATTCTTGAAGAGCCTG-3′; restriction sites are underlined). This PCR product was inserted into the EcoRI/XhoI site of pCMV-HA (Clontech). To create plasmids expressing myc-tagged MV-N (pCMV-Myc-N) and two N deletions (pCMV-Myc-NΔ1 and NΔ2), cDNAs were PCR amplified from pGBKT7-N, pGBKT7-NΔ1, and pGBKT7-NΔ2, respectively, using specific primer pairs (data not shown), and the resulting cDNA fragments were inserted into the EcoRI/NotI site of pCMV-Myc (Clontech). To construct three expression vectors encoding myc-tagged deletions of MV-N (pCMV-Myc-NΔ2a, -NΔ2b, and -NΔ2c), pCMV-Myc-N was PCR amplified using specific primer pairs (data not shown) and Pfu Turbo DNA polymerase. PCR products were phosphorylated with T4 polynucleotide kinase and self-ligated.
Transfection and immunoprecipitation assay.
Cos-7 cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 100 U of penicillin G per ml, 100 μg of streptomycin per ml (Gibco-BRL), and 10% fetal bovine serum (Sigma). Cos-7 cells in 3.5-cm-diameter dishes were transfected with 1 μg of pCMV-HA-eIF3-p40 and pCMV-Myc-N or its deletion mutants using FuGENE6 transfection reagent (Roche). At 24 h posttransfection, the medium was replaced with 1.5 ml of DMEM containing one-tenth the normal amount of methionine, 10% fetal bovine serum, and 150 μCi of [35S] EasyTag Express protein labeling mix (Perkin-Elmer). At 16 h postlabeling, cells were lysed with lysis buffer (10 mM Tris-HCl [pH 7.5], 130 mM NaCl, 0.5% Triton X-100, 0.5 mM EDTA, 10 mM NaF, 1 mM Na3VO4) containing 2% (vol/vol) of a protease inhibitor cocktail (BD Bioscience) and clarified by centrifugation at 16,000 × g for 10 min. Cell lysates were incubated with a 1:200 dilution of anti-myc-tag monoclonal antibody (Clontech) or a 1:200 dilution of anti-HA-tag rabbit polyclonal antibody (Clontech), each containing 20 μl of protein A-Sepharose bead suspension, and rocked at 4°C overnight. The protein A-Sepharose beads were washed three times with phosphate-buffered saline (PBS), denatured at 100°C in sodium dodecyl sulfate (SDS) sample buffer, and subjected to SDS-10% polyacrylamide gel electrophoresis (PAGE). Immunoprecipitates were visualized by autoradiography.
MV infection and Western blotting.
COBL-a cells (a human lymphoid cell line) (13) were cultured in RPMI medium supplemented with 100 U of penicillin G per ml, 100 μg of streptomycin per ml, and 10% of fetal bovine serum. COBL-a cells in a 10-cm-diameter dish were infected with MV-HL (23) at a multiplicity of infection (MOI) of 0.001. After 48 h of infection, cells were harvested and then cross-linked with 1% formaldehyde in PBS for 10 min at room temperature. Cross-linking was stopped by the addition of glycine to a final concentration of 0.125 M. Cells were washed with PBS, lysed with the lysis buffer described above, and then subjected to a 30-s sonication with a Sonifier 450 (Branson). Cell lysate was clarified by centrifugation at 16,000 × g for 10 min and subjected to an immunoprecipitation assay using a 1:500 dilution of anti-N monoclonal antibody 8G (16), as described above. For cross-link reversal, the immunoprecipitates were boiled in SDS sample buffer for 10 min and then resolved on SDS-10% PAGE gels and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were incubated with a 1:1,000 dilution of anti-N rabbit polyclonal antibody or a 1:100 dilution of anti-eIF3-p40 goat polyclonal antibody (Santa Cruz Biotechnology) at 4°C overnight. The membranes were washed three times with PBS and then incubated with a 1:2,000 dilution of horseradish peroxidase-conjugated rabbit anti-goat, or goat anti-rabbit, immunoglobulin G (Dako) at room temperature for 1 h. Proteins that bound antibodies were detected by ECL Plus Western blotting detection reagents (Amersham).
Expression and purification of GST-fused proteins.
The cDNAs encoding MV-N and MV-NΔ2c were amplified by PCR from pCMV-Myc-N or pCMV-Myc-NΔ2c, respectively, using a specific primer pair (5′-CATATGCATGGCCACACTTTTGAGGAG-3′ and 5′-GAATTCCTAGTCTAGAAGATCTCTG-3′), as well as LA-Taq DNA polymerase. cDNA encoding MV-P was amplified from the full-length MV-HL strain clone with a specific primer pair (5′-CATATGCATGGCAGAAGAGCAGGCACG-3′ and 5′-GAATTCCTACTTCATTATTATCTTC-3′). These PCR fragments were cloned into the NdeI/EcoRI site of the baculovirus homologous recombination vector, pAcGHLT-A (Clontech), and recombinant baculoviruses were rescued according to the manufacturer's protocol. Briefly, the vector and BaculoGold linearized baculovirus DNA (BD Bioscience) were cotransfected into Sf9 insect cells by using Lipofectin (Gibco-BRL). After 3 days, the supernatant was screened for recombinant baculoviruses. Rescued recombinant baculoviruses were inoculated into Sf9 cells at an MOI of 5. After 4 days, the cells were harvested, washed with PBS, pelleted, lysed with lysis buffer described above on ice for 45 min, and centrifuged at 14,000 × g for 10 min at 4°C. Glutathione S-transferase (GST)-fused proteins were affinity purified by using prepacked glutathione-Sepharose 4B (Pharmacia Biotech) according to the manufacturer's protocol and dialyzed against 10 mM Tris-HCl (pH 7.5) and 20% glycerol. The dialyzed proteins were concentrated by using Vivaspin 500 (5,000 molecular weight cutoff; Vivascience) and stored at −70°C.
In vitro translation inhibition assay.
To synthesize capped luciferase mRNA (m7G-luc RNA), pRL-CMV vector (Promega) was linearized with BamHI and transcribed with the Ribomax large-scale RNA production system-T7 (Promega) in the presence of a cap analog, 7mGpppG (Promega). To synthesize encephalomyocarditis virus (EMCV)-internal ribosomal entry site (IRES)-luc RNA, pIRES (Clontech) was digested with XhoI and XbaI, and the fragment containing the EMCV-IRES sequence was inserted into pTNT (Promega) (pTNT-IRES). Firefly luciferase cDNA was amplified by PCR from pGL3-Basic (Promega) with the specific primer pair flanking the SalI and NotI sites (5′-GCGGTCGACGCCATGGAAGACGCCAAAAACATAAAG-3′ and 5′-GCGCGGCCGCTACACGGCGATCTTTCCGCC-3′) and inserted into pTNT-IRES. The obtained plasmid was transcribed with the Ribomax large-scale RNA production system-T7 in the absence of cap analog. For the synthesis of IGR-IRES-luc RNA, a pT7CAT-IRES-ΔaugRluc plasmid (25), containing the Plautia stali intestine virus (PSIV) intergenic region (IGR) sequence in front of the Renilla luciferase gene lacking the first ATG codon, was kindly provided by N. Nakashima (National Institute of Agrobiological Sciences of Japan). pT7CAT-IRES-ΔaugRluc was PCR amplified using the sense primer corresponding to nucleotides 5375 to 5401 in the PSIV genome (accession number AB006531) (5′-AGCTTTATTATTGGTCAAAATCTCTCC-3′) and the antisense primer corresponding to the end of the Renilla luciferase open reading frame (5′-TTATTGTTCATTTTTGAGAACTCGCTC-3′), as well as LA-Taq DNA polymerase, and the PCR product was ligated into pGEM-T Easy vector. After the direction of insertion was checked, the plasmid was digested with SpeI and transcribed with the Ribomax large-scale RNA production system-T7 in the absence of cap analog. Translation reactions were performed by using the Flexi rabbit reticulocyte lysate system (Promega) as recommended by the manufacturer. Briefly, 16.5 μl of rabbit reticulocyte lysate was preincubated with 2 μl of increasing concentrations of GST or GST-fused protein at 30°C for 90 min. Then, 250 ng of reporter RNA was added to 25-μl reactions containing preincubated rabbit reticulocyte lysate, 4 μCi of [35S]methionine (1,000 Ci/mmol; Amersham Bioscience), 20 μM amino acid solution minus methionine, 0.5 mM magnesium acetate, 100 mM KCl, and 40 U of RNase inhibitor (TOYOBO), followed by incubation for 90 min at 30°C. Alternatively, reporter RNA was preincubated with GST-fused MV-N at 30°C for 90 min and then added to rabbit reticulocyte lysate, followed by incubation for 90 min at 30°C. Translation products were resolved by SDS-12% PAGE and visualized by autoradiography. Luciferase bands were quantified by densitometric analysis. Translation inhibition experiments were performed a minimum of three times.
Conditional switching expression of MV-N in vivo.
A cDNA clone encoding the MV-N gene was amplified by PCR using a specific primer pair (5′-GATCGAATTCGATATCCGAGATGGC-3′ and 5′-GATCGAATTCGGTCCTAGTTTTT-3′) and was inserted into the EcoRI site of the pCALNL5 conditional expression vector which contains the expression-switching reporter unit CALNL consisting of a CAG promoter, a Cre/loxP cassette, and a neomycin resistance gene (9). The plasmid was linearized by ScaI and transfected into 293 cells by using FuGENE6 reagent. Cells were selected with 0.5 mg of Geneticin/ml, and neo-resistant cell clones were designated 293-MVN cells. A recombinant adenovirus expressing Cre recombinase, AxCANCre (9), was prepared by using a standard procedure (19) and purified by ultracentrifugation (10). To obtain MV-N switching expression, 293-MVN cells were infected with AxCANCre in 24-well plates at an MOI of 10, 20, or 50. After 24 h, cells were lysed with SDS sample buffer and sonicated for 6 s. Samples were boiled for 5 min and subjected to Western blotting with a 1:1,000 dilution of anti-N rabbit polyclonal antibody and a 1:2,000 dilution of goat anti-rabbit IgG as described above.
In vivo reporter synthesis inhibition assay.
Triplicate samples of 2 × 104 293-MVN and 293 cells were infected with AxCANCre in 24-well plates at an MOI of 10, 20, or 50. After 24 h, 50 ng of phRL-TK(Int-) vector encoding a Renilla luciferase reporter gene (Promega) was transfected into the cells using FuGENE6 reagent. After 24 h, the cells were harvested and washed with PBS. One aliquot of the cells was lysed with passive lysis buffer (Promega), and the luciferase activity was measured by using the Renilla luciferase assay system (Promega); another aliquot was treated with ISOGEN reagent, and the total RNA was subjected to real-time reverse transcription-PCR (RT-PCR) using the One-Step SYBR RT-PCR kit (TaKaRa) as recommended by the manufacturer's protocol. Briefly, total RNA was mixed with One-Step SYBR RT-PCR mixture containing Renilla luciferase sense primer (5′-CGTCCAGATTGTCCGCAACTA-3′) and antisense primer (5′-CAATAGCGTTGGAAAAGAACCC-3′) in a total volume of 50 μl. RT-PCR was carried out according to the manufacturer's instructions, and amplification data were analyzed by using the ABI Prism 7900HT sequence detection system.
In vivo protein synthesis inhibition assay.
Triplicate samples of 2 × 104 293-MVN and 293 cells were infected with AxCANCre in 24-well plates at an MOI of 10, 20, or 50. After 24 h, the culture medium was replaced with 0.5 ml of DMEM containing one-tenth the normal amount of methionine and 50 μCi of [35S]EasyTag Express protein labeling mix (Perkin-Elmer). At 24 h postlabeling, the cells were harvested and washed with PBS. An aliquot was lysed with lysis buffer (1% Triton X-100, 1% sodium deoxycholate, 5 mM Tris-HCl [pH 8.0], 30 mM NaCl) containing 2% (vol/vol) of a protease inhibitor cocktail and centrifuged at 16,000 × g for 10 min. Radioactivity incorporated into the cell lysate was measured by using a scintillation counter. An aliquot of the cells was treated with ISOGEN reagent, and total RNA was subjected to real-time RT-PCR with GAPDH (glyceraldehyde-3-phosphate dehydrogenase) sense primer (5′-GCCTCAAGATCATCAGCAATG-3′) and antisense primer (5′-GGTCATGAGTCCTTCCACGATA-3′) using the One-Step SYBR RT-PCR kit as described above. Alternatively, 293-MVN and 293 cells infected with AxCANCre were lysed with SDS sample buffer and sonicated for 6 s. Samples were boiled for 5 min and subjected to Western blotting with a 1:1,000 dilution of anti-GAPDH monoclonal antibody (Chemicon) and a 1:2,000 dilution of rabbit anti-mouse immunoglobulin (Dako) as described above.
RESULTS
eIF3-p40 interacts with MV-N in a yeast two-hybrid assay.
To identify proteins that bind to MV-N, a yeast two-hybrid screen was performed using the N protein of wild-type MV (the HL strain) as bait to screen a human B-cell library. Two-hybrid screening of 107 cDNAs identified several independent cDNA clones that had potential to interact with MV-N. Sequence analysis identified a clone that encoded the p40 subunit of eIF3 (eIF3-p40). The specificity of the interaction between MV-N and eIF3-p40 was confirmed by cotransforming the yeast, AH109, with the bait and the eIF3-p40 construct. Growth was only observed when yeast were transfected with MV-N and eIF3-p40 (Fig. 1A). To confirm the interaction between MV-N and eIF3-p40 in mammalian cells, the entire eIF3-p40 cDNA was inserted into an HA-tagged mammalian expression vector, and MV-N cDNA was cloned into a myc-tagged mammalian expression vector. These vectors were simultaneously transfected into Cos-7 cells and immunoprecipitated. Anti-myc antibody against myc-tagged MV-N coprecipitated eIF3-p40 (Fig. 1B). The anti-HA antibody, in contrast, failed to coprecipitate MV-N (data not shown), suggesting that this antibody may interfere with the protein-protein interaction. To further confirm whether MV-N binds to eIF3-p40 during MV infection, COBL-a cells, which have a high sensitivity to wild-type MV (13), were infected with MV-HL and then cross-linked with formaldehyde, followed by an immunoprecipitation assay with anti-N antibody. Endogenous eIF3-p40 was detected in the immunoprecipitate obtained from the MV-infected cells (Fig. 1C), suggesting that MV-N attaches, at least in part, to eIF3-p40 during MV infection.
FIG. 1.

Interaction between eIF3-p40 and MV-N. (A) Yeast strain AH109 was cotransfected with the following pairs of expression vectors and plated onto the selection medium without His, Leu, and Trp: (1) BD vector+AD-eIF3-p40, (2) BD-MV-N+AD-vector, (3) BD-MV-N+AD-eIF3-p40, (4) BD-lamin C+AD-eIF3-p40, (5) BD-MV-N+AD-SV40 large T antigen, and (6) BD-P53+AD-SV40 large T antigen. (B) Cos-7 cells were mock transfected (lane 1) or transfected with myc-tagged MV-N (lane 2) or myc-tagged MV-N and HA-tagged eIF3-p40 (lane 3). Immunoprecipitation was performed on total cell lysates using anti-myc antibody. Immunoprecipitates were detected by SDS-PAGE, followed by autoradiography. (C) COBL-a cells were infected with MV-HL at an MOI of 0.001. At 48 h postinfection, cells were cross-linked with formaldehyde, and the cell lysate was immunoprecipitated with anti-N monoclonal antibody. Immunoprecipitates were de-cross-linked by boiling and then detected by Western blotting with the indicated antibodies.
MV-N protein inhibits translation of a reporter RNA in rabbit reticulocyte lysates.
eIF3 is a 650-kDa complex formed by 11 or 12 different subunits and is one of the initiation factors required for protein synthesis in eukaryotes. Many viruses shut off host cell gene expression during virus infection using a variety of different strategies. Several RNA viruses inhibit the initiation of host mRNA translation by modulating various host translation initiation factors. Similarly, our preliminary data indicates that MV infection shuts off host protein synthesis without altering the level of cellular mRNAs (unpublished data), suggesting that inhibition occurs at the levels of host translation. To examine the functional consequences of the MV-N and eIF3-p40 interaction, we monitored the effect of MV-N on protein synthesis using an in vitro system. MV-N fused with GST (GST-N) was expressed by a recombinant baculovirus expression system and purified to homogeneity. GST and GST-fused MV-P protein (GST-P) were used as controls and prepared in a manner similar to that used for GST-N. Rabbit reticulocyte lysates were preincubated with increasing amounts of GST, GST-N, or GST-P, and the translation reaction was carried out using a capped reporter RNA. Translation products were detected by SDS-PAGE. As shown in Fig. 2, GST and GST-P did not interfere with the translation reaction, whereas GST-N markedly inhibited RNA translation in a dose-dependent manner (Fig. 2A), with up to 95% suppression observed at 150 nM GST-N. On the other hand, preincubation of reporter RNA with GST-N before addition to the rabbit reticulocyte lysate system had little effect on translation (Fig. 2B), indicating that the suppression by GST-N did not result from nonspecific binding between reporter RNA and GST-N. These results suggest that translation suppression is caused by interactions between MV-N and cap-dependent translation initiation factor(s) such as endogenous eIF3-p40.
FIG. 2.

Inhibition of in vitro translation by GST-fused MV-N. (A) Increasing amounts of recombinant GST, GST-N, or GST-P were incubated with rabbit reticulocyte lysates. A reporter RNA encoding the luciferase gene was added and translated in the presence of [35S]methionine. Translated products were separated by SDS-PAGE and detected by autoradiography. The amount of luciferase synthesized was quantified by densitometric analysis. The results shown represent the means of three experiments. (B) A reporter RNA was preincubated with increasing amount of GST-N and then added to rabbit reticulocyte lysates. Translated products were quantified in a similar manner as panel A.
Mapping of the eIF3-p40-interacting domain of MV-N.
To map the domain of MV-N that is required for its interaction with eIF3-p40, six two-hybrid bait vectors with distinct deletions in MV-N (Fig. 3A) were constructed and cotransformed into yeast cells with the eIF3-p40 construct. Positive interactions were defined as the ability of transformed cells to grow on selection medium (SD/−His/−Leu/−Trp/3-AT). Two deletion clones (NΔ1 and NΔ2) failed to induce growth of the yeast cells (Fig. 3A). To confirm the region involved in binding, a mammalian expression vector containing the deletion clones NΔ1 or NΔ2 was coimmunoprecipitated with eIF3-p40. NΔ1 still bound eIF3-p40, whereas NΔ2 did not (Fig. 3B). These results indicate that the N-terminal residues, 81 to 192, of MV-N are required for binding to eIF3-p40. To identify the eIF3-p40-binding domain of MV-N more precisely, we generated three deletion clones between positions 81 and 192 (NΔ2a, -b, and -c) and performed coimmunoprecipitation in a similar manner. All of the deletion clones failed to bind eIF3-p40 (Fig. 3C), indicating that the tertiary structure of the N-terminal region (residues 81 to 192) and/or residues 81 to 192 of MV-N is important for the interaction between MV-N and eIF3-p40.
FIG. 3.

Mapping the eIF3-p40-interacting domain of MV-N. (A) Parts of the N gene corresponding to amino acid residues 1 to 80, 81 to 192, 193 to 252, 253 to 336, 337 to 420, and 421 to 525 were deleted, and the resulting deletion clones were inserted into a BD vector for yeast two-hybrid analysis. These constructs were cotransformed with the AD-eIF3-p40 vector into yeast and plated onto selection medium lacking Leu and Trp (SD/−Leu/−Trp). The colonies obtained were spotted on SD/−His/−Leu/−Trp/3-AT plates and incubated at 30°C for 4 days. The apparent binding strength was assessed by the degree of growth and is scored as strong (+++), intermediate (++), weak (+), or absent (−). (B) Myc-tagged MV-N and its deletion clones, NΔ1 and NΔ2, were coexpressed with eIF3-p40 in Cos-7 cells and coimmunoprecipitated using anti-myc antibody. Asterisks indicate immunoprecipitated MV-N or its deletions. An arrowhead indicates the band of eIF3-p40 that was coprecipitated. (C) Parts of the NΔ2 deletion region corresponding to amino acid residues 81 to 120, 121 to 160, and 161 to 192 were further deleted, and the resulting deletion clones were coexpressed with eIF3-p40 in Cos-7 cells and coimmunoprecipitated. The asterisks indicate MV-N or its deletion clones precipitated using anti-myc antibody. Arrowhead indicates the band of eIF3-p40 coprecipitated.
Inhibition of in vitro translation by MV-N is mediated by its interaction with eIF3-p40.
To examine the effect of eIF3-p40 binding to the N protein on MV-N-induced suppression of in vitro translation, the deletion clone, NΔ2c, which failed to interact with eIF3-p40, was fused with GST and added to the in vitro translation system. As expected, an equimolar amount of GST-NΔ2c showed a little ability to inhibit translation (Fig. 4). This result indicates that the inhibition of translation by MV-N is mediated by its specific interaction with eIF3-p40.
FIG. 4.

MV-N lacking eIF3-p40-binding ability shows no inhibitory effect on translation. Increasing amounts of recombinant GST or GST-NΔ2c were incubated with rabbit reticulocyte lysates. A reporter RNA encoding the luciferase gene was added and translated in the presence of [35S]methionine. Translated products were separated by SDS-PAGE and detected by autoradiography. The amount of luciferase synthesized was quantified by densitometric analysis. The results shown represent means of three experiments.
MV-N exhibits distinct effects on two types of IRES-mediated translation.
To further evaluate the effect of the interaction between MV-N and eIF3 on translation, we investigated in vitro translation initiation mediated by two different IRESs. The mouse EMCV IRES utilizes canonical initiation factors, including eIF3, for translation initiation (18), and the RNA genome of PSIV possesses a unique IGR that functions as an IRES (22). The IGR-IRES can assemble 80S ribosomes in the absence of canonical initiation factors and can initiate translation in a methionine-independent manner (22, 29). We tested whether MV-N inhibited mRNA translation under the control of the EMCV-IRES or IGR-IRES. A reporter gene was added downstream of the EMCV-IRES or IGR-IRES, and these RNAs were translated in vitro with increasing amounts of GST-N. EMCV-IRES-mediated translation was inhibited by GST-N in a dose-dependent fashion (Fig. 5B). The inhibition rate was similar to that of cap-dependent translation, with up to 95% inhibition observed at 150 nM GST-N. On the other hand, inhibition of IGR-IRES-mediated translation was markedly reduced, with ca. 32% inhibition observed in the presence of equimolar amounts of GST-N (Fig. 5C). These results demonstrate that IGR-IRES-mediated translation is apparently insensitive to inhibition by GST-N compared to cap-dependent or EMCV-IRES-mediated translation and that MV-N inhibits host translation by directly interacting with eIF3.
FIG. 5.

MV-N selectively inhibits cap-dependent and EMCV-IRES-mediated translation but not IGR-IRES-mediated translation. Increasing amounts of GST-N were incubated with rabbit reticulocyte lysate. m7G-luc RNA (A), EMCV-IRES-luc RNA (B), and IGR-IRES-luc RNA (C) were added and translated in the presence of [35S]methionine. Translated products were separated by SDS-PAGE and detected by autoradiography. The amount of luciferase synthesized was quantified by densitometric analysis. The results shown represent the means of three experiments.
MV-N inhibits translation in vivo.
The physiological effect of MV-N-mediated translation inhibition was examined using whole cells. We examined the effects of MV-N on a cotransfected reporter gene by using a transient-transfection assay and observed that MV-N only slightly inhibited translation, probably because it was expressed in low levels (data not shown). As a result, we established a stable cell line that possessed a Cre/loxP expression-switching unit containing the MV-N gene (293-MVN cells). MV-N expression in 293-MVN cells was induced by infection with the recombinant adenovirus, AxCANCre, which expresses Cre recombinase. 293-MVN and parental 293 cells were infected with increasing amounts of AxCANCre, and MV-N expression was confirmed by Western blotting (Fig. 6A).
FIG. 6.

Inhibition of protein synthesis by MV-N in vivo. (A to D) 293-MVN cells or 293 cells were plated in 24-well plates and infected with a recombinant adenovirus, AxCANCre, expressing Cre recombinase at an MOI of 10, 20, or 50. (A) At 24 h postinfection, switching expression of MV-N in 293-MVN cells was detected by Western blotting. (B) At 24 h postinfection, a reporter plasmid encoding the luciferase gene was transfected and incubated for 24 h. Cells were harvested, an aliquot was lysed, and luciferase activity was measured by using a luminometer. Another aliquot was harvested, and the total RNA was analyzed for luciferase transcripts using one-step real-time RT-PCR. The luciferase activity and mRNA levels in 293-MVN cells are expressed relative to those in 293 cells. The results shown represent means of three experiments. (C) At 24 h postinfection, cells were labeled with [35S]methionine for 24 h. An aliquot of the cells was lysed, and the level of radioactivity incorporated in all proteins was measured by using a scintillation counter. An aliquot of the cells was harvested, and the total RNA was analyzed for transcripts of the housekeeping gene, GAPDH, using one-step real-time RT-PCR. The levels of radioactivity and GAPDH mRNA in 293-MVN cells are expressed relative to those in 293 cells. The results shown represent means of three experiments. (D) At 48 h postinfection, cells were lysed and endogenous GAPDH was detected by Western blotting.
To examine the ability of MV-N to inhibit translation in whole cells, 293-MVN and parental 293 cells were infected with increasing doses of AxCANCre and MV-N was allowed to accumulate. The cells were transfected with a reporter gene encoding Renilla luciferase, and luciferase mRNA levels and activity were measured simultaneously. The luciferase levels and activity were comparable in 293-MVN and 293 cells (Fig. 6B). However, luciferase synthesis was progressively suppressed in 293-MVN cells as MV-N began to accumulate, resulting in 59% inhibition in the presence of AxCANCre used at an MOI of 50. To further investigate the effect of MV-N on the overall rate of cellular protein synthesis, AxCANCre-infected 293 and 293-MVN cells were pulse-labeled with [35S]methionine in vivo, and the amount of incorporated radioactivity was determined. mRNA levels were also measured based on expression of the housekeeping gene, GAPDH (Fig. 6C). There was no difference in the rate of GAPDH mRNA transcription between 293 cells and 293-MVN cells, but overall protein synthesis in 293-MVN cells decreased up to 48% compared to 293 cells in the presence of AxCANCre used at an MOI of 50. In addition, Western blotting analysis demonstrated that the protein level of GAPDH was also decreased in 293-MVN cells (Fig. 6D). These results demonstrate that intracellular accumulation of MV-N results in a partial but substantial reduction in the overall rate of cellular protein synthesis.
DISCUSSION
In a previous study, it was demonstrated that although MV infection induces significant host shutoff, infected cells contain normal levels of host mRNA (unpublished data). This suggests that MV-induced shutoff occurs at the level of host translation. In most eukaryotic mRNAs, translation initiation commences with the recruitment of cap binding protein complex, eIF4F, composed of eIF4E (cap binding protein), eIF4A, and eIF4G, to the capped 5′ end (6). The 40S ribosomal subunit, which carries eIF3 and the ternary initiator tRNAiMet-eIF2-GTP complex, is then recruited to the 5′ end of the mRNA through interactions between eIF3 and eIF4G (6). The 40S subunits scan the mRNA in a 5′-to-3′ direction until an appropriate start codon is encountered. At this point, the anticodon in initiator tRNA (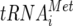 ), positioned in the ribosomal P site, engages in base pairing with the start codon in the mRNA. The large ribosomal 60A subunit joins and protein synthesis commences (6). It is known that several RNA viruses disrupt host translation initiation as part of their selective translation strategies. These viruses primarily target the eIF4F complex and related auxiliary factors in order to inhibit host translation. For example, during picornavirus infection, eIF4G is cleaved by virus-encoded proteases (5, 11, 17). In cells infected with EMCV and poliovirus, an eIF4E-binding protein (4E-BP) is dephosphorylated, resulting in sequestration of eIF4E by 4E-BP (3). During coxsackievirus and poliovirus infection, the poly(A)-binding protein, which forms a closed-loop translation complex in conjunction with eIF4G, is proteolyzed (8, 12). In influenza virus-infected cells, eIF4E is partially inactivated by dephosphorylation (2). Our preliminary studies indicate that no modifications of the eIF4F complex or any related auxiliary factors are observed during MV infection (unpublished data). In the present study, however, we show that MV-N has the ability to bind to eIF3-p40 and suppress mRNA translation in vitro (Fig. 1 and 2). Binding was necessary for suppression since the MV-N deletion clone, which does not interact with eIF3-p40, scarcely inhibited translation reactions (Fig. 4). These results indicate that MV-N uniquely suppresses translation reactions by binding to eIF3-p40. Our preliminary studies revealed that 293 and Cos-7 cells, which overexpressed SLAM (a receptor for wild-type MV) by plasmid transfection, permitted wild-type MV infection and replication (unpublished data). This implies that the results observed in Cos-7 cells (Fig. 1 and 3) and 293-MVN cells (Fig. 6) reflect the phenomenon during MV infection. To further confirm the effect of MV-N on translation in MV-infected cells, we attempted to generate a recombinant MV that possesses mutant N protein lacking the binding site to eIF3-p40 using reverse genetics. However, the variant virus could not be rescued by several trials using conditions with which other recombinant MVs were well rescued (data not shown). This result implied that the amino acid residues that participate in interaction with eIF3-p40 may also be important for the other function of N protein.
), positioned in the ribosomal P site, engages in base pairing with the start codon in the mRNA. The large ribosomal 60A subunit joins and protein synthesis commences (6). It is known that several RNA viruses disrupt host translation initiation as part of their selective translation strategies. These viruses primarily target the eIF4F complex and related auxiliary factors in order to inhibit host translation. For example, during picornavirus infection, eIF4G is cleaved by virus-encoded proteases (5, 11, 17). In cells infected with EMCV and poliovirus, an eIF4E-binding protein (4E-BP) is dephosphorylated, resulting in sequestration of eIF4E by 4E-BP (3). During coxsackievirus and poliovirus infection, the poly(A)-binding protein, which forms a closed-loop translation complex in conjunction with eIF4G, is proteolyzed (8, 12). In influenza virus-infected cells, eIF4E is partially inactivated by dephosphorylation (2). Our preliminary studies indicate that no modifications of the eIF4F complex or any related auxiliary factors are observed during MV infection (unpublished data). In the present study, however, we show that MV-N has the ability to bind to eIF3-p40 and suppress mRNA translation in vitro (Fig. 1 and 2). Binding was necessary for suppression since the MV-N deletion clone, which does not interact with eIF3-p40, scarcely inhibited translation reactions (Fig. 4). These results indicate that MV-N uniquely suppresses translation reactions by binding to eIF3-p40. Our preliminary studies revealed that 293 and Cos-7 cells, which overexpressed SLAM (a receptor for wild-type MV) by plasmid transfection, permitted wild-type MV infection and replication (unpublished data). This implies that the results observed in Cos-7 cells (Fig. 1 and 3) and 293-MVN cells (Fig. 6) reflect the phenomenon during MV infection. To further confirm the effect of MV-N on translation in MV-infected cells, we attempted to generate a recombinant MV that possesses mutant N protein lacking the binding site to eIF3-p40 using reverse genetics. However, the variant virus could not be rescued by several trials using conditions with which other recombinant MVs were well rescued (data not shown). This result implied that the amino acid residues that participate in interaction with eIF3-p40 may also be important for the other function of N protein.
Mammalian eIF3 is the largest (650 kDa) of all initiation factors and is composed of 11 or 12 different subunits whose precise patterns of interaction with other factors and stoichiometry remain poorly understood. To analyze the binding manner of MV-N on eIF3 complex, we attempted to immunoprecipitation assay using antibodies against several subunits of eIF3, but they did not pull down the whole eIF3 complex effectively, and the exact interaction between MV-N and eIF3 complex could not be defined (data not shown). Recent reports have revealed that a viral protein and several host factors regulate host protein synthesis by modulating eIF3 function. For example, the virus- and interferon (IFN)-inducible human protein, p56, binds to the p48 subunit of eIF3 and inhibits in vitro translation and cellular protein synthesis (4). Mouse p56 also inhibits in vitro translation by binding to the p110 subunit of eIF3 (4). Cyclin-dependent kinase 11 interacts with the p47 subunit of eIF3 and suppresses translation by phosphorylating a specific serine residue in the p47 subunit (24). In Norwalk virus, the RNA genome-linked protein VPg binds to the p66 subunit of eIF3 and inhibits in vitro translation reactions (1), which are thought to initiate protein synthesis from viral RNA. Our present study reveals a new mode of mammalian protein synthesis regulation that involves the modulation of eIF3 function via the p40 subunit.
GST-N inhibited both cap-dependent and EMCV-IRES-mediated translation in a dose-dependent manner (Fig. 5). The EMCV-IRES requires canonical eIFs, including eIF3, in order to initiate translation (18). Stable binding of eIF3 to the IRES is essential for the attachment of the 40S subunit to the IRES (26). This suggests that the suppression of EMCV-IRES-mediated translation by MV-N may occur through binding of MV-N to eIF3-p40, as seen in cap-dependent translation. In contrast, IGR-IRES-mediated translation is apparently insensitive to the inhibitory effects of GST-N (Fig. 5). The IGR-IRES can assemble 80S ribosomes from purified 40S and 60S ribosomal subunits in the absence of eIF2, 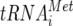 , or GTP hydrolysis and without any known canonical eIFs (29). Thus, eIF3 is not involved in IGR-IRES/40S subunit assembly. These unique characteristics may explain why IGR-IRES-mediated translation is only modestly inhibited by GST-N. At high concentrations of GST-N, slight suppression of IGR-IRES-mediated translation is observed; however, this may result because endogenous initiation factors are still be present in the lysates, and binding of MV-N to endogenous eIF3 may be interfering with 40S subunit binding to IGR-IRES.
, or GTP hydrolysis and without any known canonical eIFs (29). Thus, eIF3 is not involved in IGR-IRES/40S subunit assembly. These unique characteristics may explain why IGR-IRES-mediated translation is only modestly inhibited by GST-N. At high concentrations of GST-N, slight suppression of IGR-IRES-mediated translation is observed; however, this may result because endogenous initiation factors are still be present in the lysates, and binding of MV-N to endogenous eIF3 may be interfering with 40S subunit binding to IGR-IRES.
MV-N induced the suppression of protein synthesis at the level of translation both in vitro and in whole cells (Fig. 6). However, suppression in whole cells was partial, and suppression rates reached a plateau at ca. 50 to 60% inhibition. Our preliminary data indicate that MV-infected cells suppress more than 90% of host protein synthesis at 36 h postinfection (unpublished data), suggesting that one or more additional pathways are involved in MV-mediated host shutoff. Previous reports on other viral infections show that type I IFN induced by infection activates multiple pathways to shutoff host function, including phosphorylation of eIF2α by double-stranded RNA-activated protein kinase (21), activation of RNase L and subsequent degradation of mRNA by 2′,5′-oligoadenylate synthetase (27), and induction of virus-inducible protein P56 which binds to eIF3-p48 (4, 7). However, a previous report showed that the suppression of overall cellular protein synthesis by IFN is not complete but rather only ca. 50% inhibition (4). Indeed, we have shown that MV infection induces phosphorylation of eIF2α, but its participation for the inhibition of host protein synthesis in the overall host shutoff is partial and restricted at an early phase of infection (Inoue et al., unpublished). These data indicate that significant host shutoff induced by MV may occur through multiple cooperating pathways.
In addition to the significant suppression of host translation, we have confirmed that the selective translation of viral mRNA still occurs in MV-infected cells (unpublished data). Recently, our preliminary study identified a host factor that is implicated in the preferential translation of MV mRNAs in infected cells (unpublished data). From these data, it can be speculated that MV utilizes advanced strategies that control the host cells while evade MV itself from shutoff state in the infected cells.
Acknowledgments
We are grateful to Nobuhiko Nakashima of the National Institute of Agrobiological Sciences of Japan for the gift of pT7CAT-IRES-ΔaugRluc.
This study was supported by a grant from the Bio-oriented Technology Research Advancement Institution (BRAIN) and a grant-in-aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Footnotes
Published ahead of print on 8 August 2007.
REFERENCES
- 1.Daughenbaugh, K. F., C. S. Fraser, J. W. Hershey, and M. E. Hardy. 2003. The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 22:2852-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feigenblum, D., and R. J. Schneider. 1993. Modification of eukaryotic initiation factor 4F during infection by influenza virus. J. Virol. 67:3027-3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gingras, A. C., Y. Svitkin, G. J. Belsham, A. Pause, and N. Sonenberg. 1996. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc. Natl. Acad. Sci. USA 93:5578-5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo, J., D. J. Hui, W. C. Merrick, and G. C. Sen. 2000. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J. 19:6891-6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hentze, M. W. 1997. eIF4G: a multipurpose ribosome adapter? Science 275:500-501. (Erratum, 275:1553.) [DOI] [PubMed] [Google Scholar]
- 6.Hershey, J. W. B., and W. C. Merrick. 2000. The pathway and mechanism of initiation of protein synthesis, p. 33-88. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- 7.Hui, D. J., C. R. Bhasker, W. C. Merrick, and G. C. Sen. 2003. Viral stress-inducible protein p56 inhibits translation by blocking the interaction of eIF3 with the ternary complex eIF2.GTP.Met-tRNAi. J. Biol. Chem. 278:39477-39482. [DOI] [PubMed] [Google Scholar]
- 8.Joachims, M., P. C. Van Breugel, and R. E. Lloyd. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73:718-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanegae, Y., G. Lee, Y. Sato, M. Tanaka, M. Nakai, T. Sakaki, S. Sugano, and I. Saito. 1995. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site-specific Cre recombinase. Nucleic Acids Res. 23:3816-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanegae, Y., M. Makimura, and I. Saito. 1994. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn. J. Med. Sci. Biol. 47:157-166. [DOI] [PubMed] [Google Scholar]
- 11.Keiper, B. D., W. Gan, and R. E. Rhoads. 1999. Protein synthesis initiation factor 4G. Int. J. Biochem. Cell Biol. 31:37-41. [DOI] [PubMed] [Google Scholar]
- 12.Kerekatte, V., B. D. Keiper, C. Badorff, A. Cai, K. U. Knowlton, and R. E. Rhoads. 1999. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J. Virol. 73:709-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobune, F., Y. Ami, M. Katayama, M. Takahashi, R. Tuul, G. Korukluoglu, T. Kiyohara, R. Miura, H. Sato, M. Yoneda, and C. Kai. 2007. A novel monolayer cell line derived from human umbilical cord blood cells shows high sensitivity to measles virus. J. Gen. Virol. 88:1565-1567. [DOI] [PubMed] [Google Scholar]
- 14.Marie, J. C., J. Kehren, M. C. Trescol-Biemont, A. Evlashev, H. Valentin, T. Walzer, R. Tedone, B. Loveland, J. F. Nicolas, C. Rabourdin-Combe, and B. Horvat. 2001. Mechanism of measles virus-induced suppression of inflammatory immune responses. Immunity 14:69-79. [DOI] [PubMed] [Google Scholar]
- 15.Marie, J. C., F. Saltel, J. M. Escola, P. Jurdic, T. F. Wild, and B. Horvat. 2004. Cell surface delivery of the measles virus nucleoprotein: a viral strategy to induce immunosuppression. J. Virol. 78:11952-11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masuda, M., H. Sato, H. Kamata, T. Katsuo, A. Takenaka, R. Miura, M. Yoneda, K. Tsukiyama-Kohara, K. Mizumoto, and C. Kai. 2006. Characterization of monoclonal antibodies directed against the canine distemper virus nucleocapsid protein. Comp. Immunol. Microbiol. Infect. Dis. 29:157-165. [DOI] [PubMed] [Google Scholar]
- 17.Morley, S. J., P. S. Curtis, and V. M. Pain. 1997. eIF4G: translation's mystery factor begins to yield its secrets. RNA 3:1085-1104. [PMC free article] [PubMed] [Google Scholar]
- 18.Pestova, T. V., C. U. Hellen, and I. N. Shatsky. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 16:6859-6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Precious, B., and W. C. Russell. 1985. Virology: a practical approach. IRL Press, Oxford, England.
- 20.Ravanel, K., C. Castelle, T. Defrance, T. F. Wild, D. Charron, V. Lotteau, and C. Rabourdin-Combe. 1997. Measles virus nucleocapsid protein binds to FcγRII and inhibits human B-cell antibody production. J. Exp. Med. 186:269-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuel, C. E. 1993. The eIF-2α protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 268:7603-7606. [PubMed] [Google Scholar]
- 22.Sasaki, J., and N. Nakashima. 1999. Translation initiation at the CUU codon is mediated by the internal ribosome entry site of an insect picorna-like virus in vitro. J. Virol. 73:1219-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato, H., F. Kobune, Y. Ami, M. Yoneda, and C. Kai. Immune responses against measles virus in cynomolgus monkeys. Comp. Immunol. Microbiol. Infect. Dis., in press. [DOI] [PubMed]
- 24.Shi, J., Y. Feng, A. C. Goulet, R. R. Vaillancourt, N. A. Sachs, J. W. Hershey, and M. A. Nelson. 2003. The p34cdc2-related cyclin-dependent kinase 11 interacts with the p47 subunit of eukaryotic initiation factor 3 during apoptosis. J. Biol. Chem. 278:5062-5071. [DOI] [PubMed] [Google Scholar]
- 25.Shibuya, N., T. Nishiyama, Y. Kanamori, H. Saito, and N. Nakashima. 2003. Conditional rather than absolute requirements of the capsid coding sequence for initiation of methionine-independent translation in Plautia stali intestine virus. J. Virol. 77:12002-12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sizova, D. V., V. G. Kolupaeva, T. V. Pestova, I. N. Shatsky, and C. U. Hellen. 1998. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J. Virol. 72:4775-4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 28.tenOever, B. R., M. J. Servant, N. Grandvaux, R. Lin, and J. Hiscott. 2002. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J. Virol. 76:3659-3669. (Erratum, 76:6413.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson, J. E., T. V. Pestova, C. U. Hellen, and P. Sarnow. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell 102:511-520. [DOI] [PubMed] [Google Scholar]