

Abstract
Model systems have previously been developed in which herpes simplex virus (HSV) is retained in human fibroblasts in a nonreplicating state known as quiescence. The HSV type 1 (HSV-1) immediate-early (IE) protein ICP0, an important activator of gene expression, reactivates the quiescent genome and promotes the resumption of virus replication. Previous studies reported that infection with ICP0-null HSV-1 mutants fails to reactivate quiescent HSV, even when the mutant itself undergoes productive replication, leading to the hypothesis that quiescent genomes exist in a silent configuration in which they are shielded from trans-acting factors. I reinvestigated these findings, using HSV-1 mutants with lesions in the transcription activators VP16, ICP0, and ICP4 to establish quiescent infection at high efficiency. Superinfection with ICP0-null HSV-1 mutants at a low multiplicity of infection (MOI), so that individual plaques were formed, reactivated expression from the quiescent genome, demonstrating that the requirement for ICP0 is not absolute. The previously reported failure to observe reactivation by ICP0-null mutants was shown to be a consequence of either a low initial MOI or a high superinfecting MOI. Competition between viral genomes at the level of gene expression and virus replication, especially when ICP0 was absent, was demonstrated during reactivation and also during normal infection of human fibroblasts. The results show that the multiplicity-dependent phenotype of ICP0-null mutants limits the efficiency of reactivation at low MOIs and that competition between genomes occurs at high MOIs. The conclusion that quiescent HSV genomes are extensively silenced and intrinsically insensitive to trans-acting factors must be reevaluated.
Infection with wild-type herpes simplex virus type 1 (HSV-1) usually results in productive virus replication and death of the host cell. Viral genes are expressed in a coordinated cascade characterized by three phases, immediate-early (IE), early, and late. IE genes are the first to be transcribed, and the IE proteins, particularly ICP4, ICP0, and ICP27, play crucial roles in the synthesis of early and late proteins. The ICP4 protein is essential for productive infection, stimulating early and late RNA synthesis through interactions with basal cellular transcription factors (7, 52). Protein ICP0 is not essential for virus replication, but infection in its absence is initiated inefficiently in many cell types (12, 42, 45, 46). Human fibroblasts are particularly restrictive for replication of ICP0-null mutants, but in contrast, the human osteosarcoma line U2-OS is fully permissive (51). ICP0 is a ubiquitin E3 ligase, suggesting that it acts by stimulating proteolysis of strategic cellular targets (4, 15). The tegument protein VP16 strongly stimulates IE transcription shortly after entry of the virion into the host cell, providing a further level of gene regulation (6, 35). Virus mutants that specify nonfunctional VP16 are phenotypically similar to ICP0-null mutants, exhibiting a cell type-dependent defect in initiating productive infection (2, 44).
After infection of humans or experimental animals, some neuronal cells survive infection and harbor HSV-1 in a latent state for the lifetime of the host (10, 25, 48). The mechanisms that silence the viral genome to prevent gene expression during latency are poorly understood, although recent studies have demonstrated that chromatin structure is probably an important factor, since lytic promoters are marked by histone modifications characteristic of nontranscribed cellular genes whereas the genome region that specifies the latency-associated transcript shows modifications associated with actively transcribed genes (3, 26, 27, 49).
To gain insight into the mechanisms of gene silencing, tissue culture systems that mimic aspects of latency have been developed. In the earliest of these, interferon-treated human fibroblasts were infected with HSV-1 and incubated for 7 days in the presence of an inhibitor of viral DNA synthesis to prevent lytic infection and cytopathology (50). The cultures were then transferred to 40.5°C and maintained without detectable virus replication, with reactivation achievable by downshift to 37°C or by superinfection with human cytomegalovirus (HCMV). This system was modified by increasing the initial incubation temperature to 42°C and avoiding the use of inhibitors (40). Human fibroblasts were infected with HSV-1 or HSV-2 at MOIs up to 0.03, and after incubation for 6 days at the supraoptimal temperature, the cultures could be transferred to the permissive temperature of 37°C and maintained without virus production. The quiescent virus resumed replication if the cultures were superinfected with HSV-1, HSV-2, or HCMV. Investigation of the functions required to reactivate quiescent HSV-2 demonstrated that superinfection with a range of HSV-1 mutants resulted in the resumption of HSV-2 replication, with the exception of the HSV-1 mutant dl1403 (41). This virus lacks the coding region for ICP0, suggesting a requirement for this IE protein in reactivation of quiescent HSV. The results were substantiated by the observation that ICP0 expressed by adenovirus recombinants was also able to reactivate quiescent HSV-2, whereas ICP0 mutant proteins that were inactive for stimulation of gene expression were nonfunctional for reactivation (19, 53). The nature of the block to virus replication imposed by incubation at 42°C is not known, and further developments of the quiescent system used HSV-1 mutants at the normal temperature of 37°C. It was found that dl1403, and the VP16 mutant in1814, could be retained in human fibroblasts for many days in a quiescent state and that superinfection of cultures with wild-type HSV-1 or HCMV resulted in reactivation (20, 45). In the studies with in1814, in which cultures were initially infected at an MOI of 0.03, superinfection with dl1403 again failed to reactivate expression from the quiescent genomes (20).
From these early investigations, it was concluded that ICP0 is required to reverse the quiescent state and reactivate viral-gene expression. Furthermore, reactivation was not detected even under conditions in which dl1403 itself replicated, suggesting that the quiescent genome was unaffected by the plethora of transcription and replication factors produced by the superinfecting dl1403. In turn, this observation led to the hypothesis that the quiescent genome is organized into a compact structure, possibly related to heterochromatin, and that reversal of this state uniquely requires ICP0.
Further manipuation of HSV-1 resulted in the isolation of mutants with greater impairment of IE gene expression and reduced cytotoxicity, which are suitable for the establishment of cultures containing quiescent genomes after initial infection at a high MOI. Mutants derived from in1814 and additionally impaired for the expression of ICP0 and ICP4 were used to obtain cultures in which most cells contained at least one quiescent genome (14, 37, 38). Even greater reduction in cytotoxicity was achieved by inactivating all IE genes, resulting in mutants that established quiescent infections in human fibroblasts and Vero cells (21, 23, 43). Mutant KM110 lacks the ICP0 coding region and the C-terminal activating domain of VP16, and although potentially capable of replication, this mutant is sufficiently impaired to establish quiescent infection at high efficiency in human fibroblasts without cytotoxicity (34). In each of these systems, provision of ICP0 reactivated the quiescent genome (21, 31, 37, 38, 43).
Recently, the inability of ICP0-null viruses to reactivate quiescent HSV-1 was reinvestigated by Minaker et al. (31). Based on the observation that the HSV-1 mutant KM110 can induce an antiviral response in human fibroblasts that is neutralized by ICP0 (11, 32, 33), these authors questioned whether cultures harboring quiescent HSV-1 were truly permissive for replication of ICP0-null mutants. By carrying out superinfection with viruses expressing enhanced green fluorescent protein as a marker, they demonstrated that, at an MOI of 30, ICP0-null viruses replicated as efficiently as wild-type HSV-1 at an MOI of 10 and that the mutant virus failed to reactivate expression from quiescent genomes (31). These experiments therefore supported the accepted view that quiescent genomes are strictly silenced and can be reactivated only by ICP0.
One disconcerting aspect of the studies described above concerns the complex phenotype of ICP0-null mutants, which exhibit cell type dependence in the ability to initiate productive replication. The human osteosarcoma line U2-OS permits normal virus replication in the absence of ICP0, whereas human fibroblasts represent the most impaired tissue culture examples, being more than 1,000-fold less permissive than U2-OS. Recent experiments suggest that U2-OS cells lack a repression mechanism that is active in human fibroblasts (18). Therefore, after infection at a low MOI, most ICP0-null genomes themselves become quiescent in human fibroblasts (13, 45). Interestingly, some genomes express distinct subsets of viral genes during the first few days of infection, suggesting that silencing is a random process (13). The crucial properties of the small number of cells in a human fibroblast culture that are permissive for replication are unknown at present. Most previous studies with ICP0-null mutants have reasonably emphasized that the defect in initiation of infection is greatest at low MOIs (less than 1), leading to the proposal that the absence of ICP0 is less important during infection at high MOIs (5, 8, 13, 46).
The experiments reported here were stimulated by a surprising observation that quiescent HSV-1 genomes could indeed be reactivated by superinfection with dl1403 under appropriate conditions. I therefore reinvestigated the requirements for reactivation of quiescent HSV-1.
MATERIALS AND METHODS
Plasmids.
Plasmid pCP58016 was constructed by excising the enhanced yellow fluorescent protein (YFP) coding region from pEYFP-C1 (Clontech) as a HindIII (end filled)/AgeI fragment and inserting it into pCP1802 (22) cleaved with NotI (end filled) and AgeI. This procedure results in a plasmid with YFP controlled by the HCMV major IE promoter (MIEP) and the simian virus 40 polyadenylation sequences, all embedded in the HSV-1 thymidine kinase (TK) coding region. Plasmid pCP376 has the Escherichia coli lacZ region, controlled by the HCMV MIEP, inserted into the coding sequences of the nonessential UL43 open reading frame, as described previously (37). Plasmid pMC17 consists of the HSV-1 VP16 coding region cloned into pUC9 (1). Plasmid pGX152 contains the HSV-1 EcoRI B fragment cloned into the EcoRI site of pBR328.
Cells and viruses.
Human fetal foreskin fibroblasts (HFFF2) and U2-OS cells were propagated in Dulbecco's modified Eagle medium (Invitrogen) supplemented with 5% (vol/vol) fetal calf serum and 5% newborn calf serum, with 100 units of penicillin and 100 μg of streptomycin per ml (D5 + 5). Syrian hamster fibroblasts (BHK-21) were propagated in Glasgow modified Eagle medium (Invitrogen) supplemented with 10% (vol/vol) newborn calf serum and 10% (vol/vol) tryptose phosphate broth plus 100 units of penicillin and 100 μg of streptomycin per ml. Wild-type HSV-1 was strain 17, and the derived mutants tsK (containing a temperature-sensitive mutation in the ICP4 coding region) and dl1403 (with most of the ICP0 coding region deleted) have been described previously (9, 36, 46). Mutant in1312 has mutations in the coding sequences for VP16, ICP0, and ICP4 (39). Mutant in1374 is in1312 with an insertion of pCP376 (37). Mutant in1330 was derived from in1312 by repair of the VP16 mutation, using methods described by Marshall et al. (28), and therefore, this virus lacks ICP0 and encodes a temperature-sensitive ICP4. Mutant dl1403Y was constructed by cotransfection of dl1403 DNA with ScaI-cleaved pCP58016 and subsequent plaque purification and propagation of TK-negative isolates. The genome structure and absence of parental virus were confirmed by Southern hybridization analysis of viral DNA, as described previously (24). Rescue of the ICP0 mutation of dl1403Y was achieved by cotransfection of dl1403Y DNA with EcoRI-cleaved pGX152, followed by purification and propagation of isolates that contained an intact ICP0 coding region, determined by Southern hybridization (46). Mutant dl1403/β-gal was constructed by cotransfection of dl1403 DNA with ScaI-cleaved pCP376, followed by selection and purification of a recombinant with the HCMV MIEP-lacZ insertion at UL43, using methodology described previously (37). Viruses were titrated on U2-OS cells at 31°C in the presence of 3 mM hexamethylene bisacetamide to overcome the effects of the VP16, ICP0, and ICP4 mutations (28, 29). Particle-to-PFU ratios were determined as described by Everett et al. (13); the values for the virus stocks used were 32 for dl1403, 11 for dl1403Y, and 9 for dl1403YR.
Infection of HFFF2 monolayers.
To establish cultures containing quiescent HSV-1, HFFF2 cells on 60-mm-diameter plates were infected; overlaid with Dulbecco's medium supplemented with 2% (vol/vol) fetal calf serum, 100 units of penicillin, and 100 μg of streptomycin per ml; and propagated at 38.5°C, as described previously (37). After incubation for 7 days, the monolayers were trypsinized and cells were seeded on 24-well plates. Superinfection was carried out after incubation at 38.5°C for a further 24 h. In many cases, cells were acid washed by incubation for 30 seconds at room temperature with pH 3 buffer, as described by Minaker et al. (31).
Analysis of infected cell DNA.
Total DNA was prepared from cultures, cleaved with BamHI, transferred to nitrocellulose membranes, and analyzed by Southern hybridization, using the VP16-coding fragment of pMC17 as a probe.
β-Gal assays.
Histochemical assay for β-galactosidase (β-Gal)-expressing cells, using 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside as a substrate and counterstaining, were carried out as described by Jamieson et al. (24). Quantification of β-Gal in cell extracts, using 4-methylumbelliferyl β-d-galactoside as a substrate, was carried out by fluorometric analysis as described previously (38).
YFP assays.
Cell extracts were prepared as for β-Gal assays. Extracts were placed in microtiter plates with blackened edges, and fluorescence was determined by use of a Hidex Chameleon plate reader with excitation at 485 nm and emission at 535 nm.
RESULTS
Reactivation by superinfection with dl1403.
Cultures of HFFF2 cells were infected with 3 PFU of the HSV-1 mutant in1374 per cell and incubated at 38.5°C for 8 days to enable full establishment of quiescent infection (37). After this treatment, no β-Gal-positive cells were detectable upon histochemical staining of cultures, as reported previously (37). Superinfection of cultures with 2 PFU of the ICP4 mutant tsK per cell resulted in approximately 50% of the cells expressing β-Gal, demonstrating that a substantial proportion of the cells harbored quiescent virus. Cultures were superinfected with the ICP0 deletion mutant dl1403 at various multiplicities and stained for the presence of β-Gal after incubation at 38.5°C for 2 days in the presence of 2% human serum to prevent secondary spread of virus. Surprisingly, and in contrast to the conclusions of previous studies, most plaques were β-Gal positive at the lowest multiplicities of dl1403 that resulted in plaque formation (Fig. 1A). The majority of β-Gal-positive plaques were uniformly stained, suggesting that reactivation occurred at the initial, or first few, rounds of replication. A minority, however, contained a mixture of β-Gal-positive and -negative rounded cells (not shown), presumably representing reactivation at a later stage in plaque formation. At an MOI of 0.1 (in terms of PFU on U2-OS cells, representing three dl1403 particles per cell), approximately 30 plaques formed on HFFF2 monolayers of 2 × 105 cells. As the MOI of dl1403 was increased, the total number of plaques rose sharply in a nonlinear fashion characteristic of infection of human fibroblasts with ICP0 mutants (12, 13, 45), and an increasing proportion did not express β-Gal (Fig. 1B). The numbers of β-Gal-positive plaques appeared to reach a maximum at a dl1403 MOI of 0.5, but it was not possible to obtain accurate data because discrete plaques could not generally be discerned within the extensive cytopathic effect (CPE) that developed during incubation at 38.5°C for 2 days (Fig. 1C). This experiment showed that β-Gal expression was readily activated in response to infection with dl1403 and that the effect was most obvious when superinfection was carried out at a low MOI. As expected, superinfection with wild-type HSV-1 yielded plaques that were uniformly β-Gal positive at all multiplicities tested (results not shown). Superinfection at 38.5°C with the UL26 mutant ts1201 resulted in cross-complementation with quiescent in1374, as described by Samaniego et al. (43), and the plaques obtained were also β-Gal positive (results not shown). In view of the findings shown in Fig. 1, the conclusion that quiescent genomes remain silent during productive infection with dl1403 clearly requires qualification.
FIG. 1.

Reactivation of β-Gal expression by superinfection with dl1403. HFFF2 cultures containing quiescent virus were established after initial infection with 3 PFU of in1374 per cell. At 8 days p.i., the cultures were superinfected with dl1403 at an MOI of 0.1 (A), 0.3 (B), or 0.5 (C) (based on U2-OS cell titers) and incubated for a further 2 days at 38.5°C in D5 + 5 with 2% (vol/vol) human serum added. Cultures superinfected with dl1403 at an MOI of 10 and incubated for 20 h in D5 + 5 with 50 μg/ml AraC added are shown in panel D.
The resumption of β-Gal expression could be due to a transcriptional activation of the HCMV MIEP that controls lacZ in in1374 or to recombination between the superinfecting and quiescent genomes. To determine whether DNA replication was required for stimulation of β-Gal expression, cultures containing quiescent in1374 were superinfected with dl1403 in the presence of cytosine arabinoside (AraC) and maintained at 38.5°C for 20 h. Upon histochemical staining, many β-Gal-positive cells were observed (Fig. 1D). Because CPE was much reduced during the shorter time when AraC was present compared with incubation for 2 days with 2% human serum, as in Fig. 1A to C, it was possible to detect positive cells at MOIs of dl1403 up to 10 (Table 1 and Fig. 1D). At an MOI of 10, approximately 0.5% of cells were β-Gal positive.
TABLE 1.
Reactivation by dl1403 and in1330a
| MOI | No. of β-Gal-positive cells after superinfection
|
|||
|---|---|---|---|---|
| dl1403 (31°C) | dl1403 (38.5°C) | in1330 (31°C) | in1330 (38.5°C) | |
| 0 | 5 | 0 | 4 | 0 |
| 0.1 | 5 | 7 | 7 | 0 |
| 0.15 | 9 | 7 | 10 | 1 |
| 0.3 | 24 | 75 | 27 | 2 |
| 0.5 | 38 | 125 | 37 | 2 |
| 1.0 | 79 | 155 | 77 | 1 |
| 1.5 | 93 | 171 | 90 | 0 |
| 2.5 | 106 | 243 | 100 | 1 |
HFFF2 cultures were infected with 3 PFU of in1374 per cell and incubated at 38.5°C for 8 days. After trypsinization and reseeding, the cells were superinfected with dl1403 or in1330 and incubated for a further 20 h at 38.5°C or 60 h at 31°C with 50 μg/ml AraC present. The numbers of β-Gal-positive cells per culture of 2 × 105 cells are expressed as the means of duplicate determinations.
To investigate further the functions of the superinfecting virus required to stimulate β-Gal production, mutant in1330, which has deletion of ICP0 plus the temperature-sensitive mutation from tsK in ICP4, was tested (Table 1). Cultures containing quiescent in1374 were superinfected with dl1403 or in1330 in the presence of AraC and maintained at 38.5°C for 20 h or 31°C for 60 h. Both viruses activated β-Gal expression at 31°C, but only dl1403 was effective at 38.5°C over a range of multiplicities. The expression of β-Gal upon superinfection with dl1403 is therefore dependent on functional ICP4 but not on DNA replication, strongly suggesting that initially the effect represents transcriptional activation of the quiescent genome rather than recombination with the superinfecting virus. After initial reactivation has occurred, reactivated and superinfecting viruses undergo recombination in the normal manner.
The above-mentioned findings apparently contradict previous studies, which showed that superinfection with ICP0-null mutants failed to reactivate quiescent HSV-2, in1814, or KM110 (20, 31, 41). There are differences in the experimental details of the previous studies and those represented in Fig. 1. Initial infections with HSV-2 or in1814 could not be performed at MOIs greater than 0.03, due to loss of cultures through escape virus replication or cytopathology (20, 31, 40). Mutant KM110 was sufficiently impaired to permit an initial MOI of 6, but superinfection was carried out only at high MOIs, 10 for wild-type virus and 30 for the ICP0-null mutant (31). The effects of altering initial and superinfecting MOIs on reactivation were therefore investigated to determine whether the differences in experimental protocols were responsible for the apparent discrepancies in results.
Construction of YFP-expressing dl1403 and rescuant.
Many of the studies described below involve quantifying reactivation by dl1403 and wild-type HSV-1. This is potentially problematic, because dl1403 preparations have a high particle-to-PFU ratio on HFFF2 cells, and thus comparison, even on the basis of titers on U2-OS cells, might result in the delivery of different functional genome loads to infected cells. To obtain appropriate virus stocks for superinfection studies, mutant dl1403Y, which is dl1403 with an insertion of YFP controlled by the HCMV MIEP at the TK locus, was constructed. A rescuant with the ICP0 deletion repaired, dl1403YR, was then produced. These viruses had similar particle-to-PFU ratios when titrated on U2-OS cells (11 for dl1403Y and 9 for dl1403YR). To ensure that infections with these preparations were comparable in human fibroblasts, two further tests were carried out. First, HFFF2 monolayers were infected at an MOI of 10 or 30, cells were acid washed to remove nonpenetrated virus, and viral-DNA levels were measured at 1.5 h postinfection (p.i.) by Southern blotting and hybridization. As shown in Fig. 2A, equivalent quantities of input DNA were delivered to cells at each MOI tested. Second, cultures of HFFF2 cells were infected with various amounts of dl1403Y or dl1403YR, coinfected with 10 PFU of wild-type HSV-1 per cell, and incubated overnight in the presence of AraC. YFP levels in the cultures were determined and shown to be very similar for the two viruses (Fig. 2B). Thus, infection at equal MOIs (as measured by titers on U2-OS cells) with dl1403Y or dl1403YR resulted in the delivery of equivalent numbers of potentially competent genomes to cells. Although this virus pair was used in most of the following experiments, the results were reproduced where possible (but are not presented) using dl1403 and wild-type HSV-1.
FIG. 2.

Comparison of dl1403Y and dl1403YR. (A) HFFF2 monolayers were infected with dl1403Y or dl1403YR at an MOI of 10 or 30. After a 1-h adsorption period, the cells were acid washed and incubated at 37°C for a further 30 min. Cellular DNA was prepared and cleaved with BamHI. A Southern blot was probed with radiolabeled VP16 coding region from pMC17. (B) HFFF2 monolayers were infected with dl1403Y or dl1403YR at various MOIs and coinfected with wild-type HSV-1 at an MOI of 10. The monolayers were incubated at 38.5°C for 20 h in D5 + 5 with 50 μg/ml AraC. YFP levels were determined in cell extracts.
Comparison with previously published results.
To investigate the effects of altering the initial MOI on the efficiency of reactivation of quiescent virus, HFFF2 monolayers were infected with in1374 at an MOI of 3, 0.3, or 0.03 and maintained at 38.5°C for 8 days. Superinfection was carried out with dl1403Y or dl1403YR at an MOI of 0.1, and cultures were maintained at 38.5°C until widespread CPE was observed. This reproduced the methodology used by Russell et al., (41) and additionally enabled analysis at a higher initial MOI through the use of the severely impaired mutant in1374. Intracellular DNA was extracted for analysis by Southern hybridization (Fig. 3). At all multiplicities, infection with dl1403YR resulted in the presence of DNA containing the VP16 mutation, identified by the existence of a novel BamHI site in the BamHI f fragment derived from replication of reactivated quiescent in1374 (Fig. 3, lanes 4 to 6) (2). As expected from the experiments shown in Fig. 1, cultures initially infected with 3 PFU of in1374 per cell and superinfected with dl1403Y also contained DNA with the VP16 insertion (Fig. 3, lane 1). At initial infection with 0.3 PFU of in1374 per cell, however, superinfection with dl1403Y did not yield replicated DNA containing the VP16 mutation (Fig. 3, lane 2), with one exception in five independent determinations (results not shown). At an initial MOI of 0.03, DNA containing the VP16 mutation was never detected. Therefore, the findings from the published experiments with quiescent HSV-2 or in1814 were reproduced, since reactivation by superinfection with the ICP0-null virus dl1403Y was detected reliably only in cultures initially infected with 3 PFU of in1374 per cell and never in cultures initially infected at an MOI of 0.03, the highest value used in the earlier studies.
FIG. 3.

Reactivation at low superinfecting MOIs. HFFF2 cultures containing quiescent virus were established after infection with in1374 at an MOI of 3 (lanes 1 and 4), 0.3 (lanes 2 and 5), or 0.03 (lanes 3 and 6). At 8 days p.i., the cultures were superinfected (si) with dl1403Y (lanes 1 to 3) or dl1403YR (lanes 4 to 6) at an MOI of 0.1 and incubated at 38.5°C until extensive CPE developed. Cellular DNA was prepared and cleaved with BamHI. A Southern blot was probed with radiolabeled VP16 coding region from pMC17, revealing a band of 8 kbp from wild-type virus (WT) and bands of 5 kbp and 3 kbp derived from genomes containing the BamHI site that gives rise to the VP16 mutation of in1374 (M).
The apparent discrepancy with the findings of Minaker et al. was also addressed. Cultures were infected with 3 PFU of in1374 per cell, maintained at 38.5°C for 8 days, and superinfected with dl1403Y or dl1403YR at different MOIs. After further incubation at 38.5°C for 22 h, the intracellular DNA was analyzed (Fig. 4). At all multiplicities tested, superinfection with dl1403YR resulted in the presence of replicated viral DNA containing the VP16 mutation resident in in1374 (Fig. 4, lanes 1 to 6). As the dl1403YR MOI increased above 0.3, however, the recovery of mutant DNA declined, indicating a degree of competition between genomes during replication. At an initial in1374 MOI of 0.3 or 0.03, superinfection with dl1403Y did not yield detectable amounts of replicated mutant DNA at any superinfecting MOI tested. At an initial in1374 MOI of 3 and multiplicities of dl1403Y greater than 0.1, DNA containing the VP16 mutation was present (Fig. 4, top, lanes 8 to 10). As the superinfecting multiplicity increased to 10 and 30, the amount of replicated mutant DNA decreased sharply (Fig. 4, top, lanes 11 and 12). At an MOI of 30 (lane 12), DNA containing the VP16 mutation was hardly detectable, in agreement with the results of Minaker et al. (31).
FIG. 4.

Reactivation by superinfection. HFFF2 cultures containing quiescent virus were established after infection with in1374 at an MOI of 3 (top), 0.3 (middle), or 0.03 (bottom). At 8 days p.i., the cultures were superinfected (si) with dl1403YR (lanes 1 to 6) or dl1403Y (lanes 7 to 12). After a 1-h adsorption period, the cells were acid washed and incubated at 38.5°C for 22 h. Cellular DNA was prepared, cleaved with BamHI, and probed with radiolabeled VP16 coding region from pMC17, as described in the legend to Fig. 3. The MOI of the superinfecting virus was 0.1 (lanes 1 and 7), 0.3 (lanes 2 and 8), 1 (lanes 3 and 9), 3 (lanes 4 and 10), 10 (lanes 5 and 11), or 30 (lanes 6 and 12).
Therefore, it appears that superinfection with ICP0-deficient mutants imposes two limitations on reactivation and replication of quiescent virus. At low MOIs, progression into lytic replication is restricted by the well-characterized defect in initiating productive infection. At high MOIs, competition for replication occurs between genomes, resulting in reduced levels of in1374-derived genomes even though the total amount of replicated DNA is high.
Competition between genomes during coinfection.
To investigate the prediction that competition for DNA replication occurs at high MOIs of dl1403, a reconstruction experiment was performed. HFFF2 cells, mock infected but incubated at 38.5°C for 8 days, were infected with 3 or 0.3 PFU of in1374 per cell, together with different amounts of dl1403Y or dl1403YR. The mock treatment was carried out for comparability with quiescent infection, although freshly seeded cells gave equivalent results (not shown). After incubation for 22 h, intracellular DNA was analyzed for the presence of the VP16 mutation (Fig. 5). For coinfection with dl1403YR, increasing the MOI from 0.1 to 1 resulted in a consistent proportion of replicated genomes containing the VP16 mutation (Fig. 5, lanes 1 to 3), as predicted, since replication of in1374 is possible only when a dl1403YR genome is also present. As the amount of dl1403YR increased further, the recovery of in1374-derived genomes declined, especially when in1374 was initially present at an MOI of 0.3 (Fig. 5, lanes 4 to 6). In the case of coinfection with dl1403Y, replication of both viruses increased up to an MOI of 1, as expected from the multiplicity-dependent phenotype of ICP0-null mutants at low MOIs (Fig. 5, lanes 7 to 9), but after this point the proportion of genomes containing the VP16 mutation declined, until at a dl1403Y MOI of 30, this value was very low, especially at an initial in1374 MOI of 0.3 (Fig. 5, lanes 10 to 12). Tellingly, in1374-derived genomes were always present at much lower levels when the coinfecting virus was dl1403Y than with dl1403YR, until the MOI was 30. Therefore, the reconstruction experiment demonstrated that competition for DNA replication of coinfecting viruses occurs at multiplicities of dl1403Y and dl1403YR greater than 1, in a manner similar to that observed during superinfection.
FIG. 5.

Coinfection of in1374 with dl1403Y or dl1403YR. HFFF2 monolayers were incubated at 38.5°C for 8 days, infected with in1374 at an MOI of 3 (top) or 0.3 (bottom), and coinfected with dl1403YR (lanes 1 to 6) or dl1403Y (lanes 7 to 12). After a 1-h adsorption period, the cells were acid washed and incubated at 38.5°C for 22 h. Cellular DNA was prepared, cleaved with BamHI, and probed with radiolabeled VP16 coding region from pMC17, as described in the legend to Fig. 3. The MOI of the coinfecting virus was 0.1 (lanes 1 and 7), 0.3 (lanes 2 and 8), 1 (lanes 3 and 9), 3 (lanes 4 and 10), 10 (lanes 5 and 11), or 30 (lanes 6 and 12).
A further demonstration that competition can result in some genomes failing to replicate at high MOIs of ICP0-null virus was obtained by carrying out an alternative coinfection experiment. The HSV-1 mutants dl1403 and dl1403/β-gal were mixed at a ratio of 1:3 in terms of PFU and used to infect HFFF2 monolayers. At a total MOI of 1, almost all cells contained a genome capable of expressing β-Gal, as shown by coinfection with wild-type HSV-1 in the presence of AraC and histochemical staining after 22 h (Fig. 6A). Significant CPE developed when cultures were incubated at 37°C for 40 h with 2% human serum present, but plaques were observed in certain areas of the monolayers and staining revealed that some did not express β-Gal (Fig. 6B). The plaques consisted of significant clusters of β-Gal-negative cells, virtually all of which (by extrapolation from the results of superinfection with wild-type HSV-1 [Fig. 6A]) contained a dl1403/β-gal genome. Thus, in these instances, the dl1403/β-gal genome failed to replicate, and furthermore, the HCMV MIEP (and presumably the entire dl1403/β-gal genome) was inactive even in cells that supported replication of dl1403. It is possible that the β-Gal-negative plaques arose from a cell initially infected with dl1403 alone, but cells subsequently infected during plaque formation must have contained dl1403/β-gal.
FIG. 6.
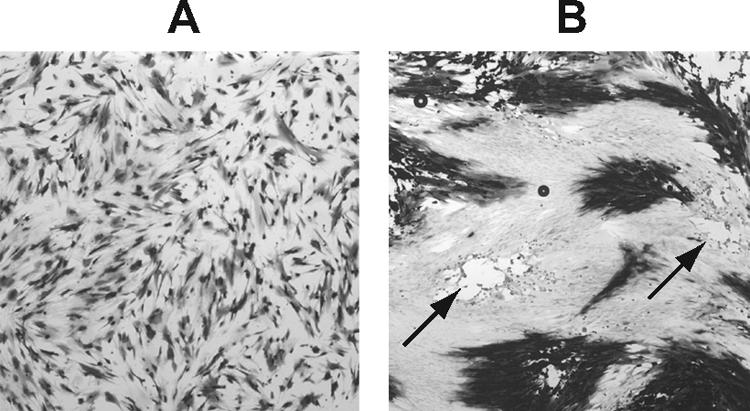
Competition between dl1403 genomes during coinfection. HFFF2 monolayers were infected with dl1403 and dl1403/β-gal, mixed in a 1:3 ratio on the basis of PFU, at a total MOI of 1. The cultures were coinfected with 3 PFU of wild-type HSV-1 per cell and incubated at 37°C for 22 h with 50 μg/ml AraC present (A) or incubated at 37°C for 40 h in D5 + 5 containing 2% human serum (B). Two plaques that did not contain β-Gal-expressing cells are marked with arrows.
Gene expression during coinfection and superinfection.
The experiments described above suggest that during ICP0-null infections, some viral genomes failed to replicate even in cells supporting active virus production. In other words, the interpretation of reactivation experiments also applies to coinfection. I pursued this finding to investigate whether similar effects could be observed at the level of gene expression. HFFF2 cells, incubated at 38.5°C for 8 days, were infected with a fixed amount (0.5 PFU per cell) of in1374, together with different amounts of dl1403Y or dl1403YR. As stated above, freshly seeded cells gave equivalent results (not shown). Cultures were maintained at 38.5°C for 22 h, with AraC present to ensure that gene expression represented the activities of input genomes without complications resulting from DNA replication. Cell extracts were analyzed for YFP and β-Gal content (Fig. 7A and C). The results showed that YFP production was much lower for dl1403Y than dl1403YR at a low total MOI, as expected from the known properties of ICP0-null mutants. At high MOIs, 30 PFU per cell of dl1403Y and 10 PFU per cell of dl1403YR produced similar amounts of YFP, confirming the results of Minaker et al. (31). The expression of β-Gal, however, was much higher during coinfection with dl1403YR, until at an MOI of 30, the values were comparable. At an MOI of 30, however, competition clearly occurred in dl1403YR-infected cells, since YFP levels were high but β-Gal expression was well below the maximum that was observed at MOIs of 0.3 to 3. Therefore, competition at the level of gene expression occurred in dl1403YR-infected cells at MOIs greater than 3. Maximal expression of dl1403Y (as judged by YFP production) occurred at MOIs of 30 or greater, but at this value, competition reduced the expression of β-Gal.
FIG. 7.

YFP and β-Gal expression during coinfection or superinfection. For coinfection (A and C), HFFF2 monolayers were incubated at 38.5°C for 8 days and then infected with in1374 (MOI, 0.5) and coinfected with dl1403YR (filled circles) or dl1403Y (open circles). After incubation at 38.5°C for 22 h with 50 μg/ml AraC, YFP (A) or β-Gal (C) levels were determined in cell extracts. For superinfection (B and D), HFFF2 monolayers were infected with in1374 (MOI, 3) and maintained at 38.5°C for 8 days. The cultures were then infected with dl1403YR (closed circles) or dl1403Y (open circles), and after incubation at 38.5°C for 22 h with 50 μg/ml AraC, YFP (B) or β-Gal (D) levels were determined in cell extracts.
The experiment shown in Fig. 7A and C demonstrated that expression of β-Gal from in1374 genomes is restricted during coinfection with dl1403Y, due to inefficient entry into lytic infection at low MOIs and competition at high MOIs. To make a quantitative assessment of whether the events observed during coinfection can account for the reduced reactivation of quiescent genomes upon superinfection with dl1403Y, an analogous experiment was carried out by superinfecting cultures containing quiescent in1374 (Fig. 7B and D). The production of YFP followed the pattern observed during coinfection, with equivalence between dl1403Y and dl1403YR almost reached at an MOI of 30 (Fig. 7A and B). Expression of β-Gal after superinfection with dl1403YR followed a pattern similar to that observed during coinfection, although competition was observed mainly at an MOI of 30 (Fig. 7C and D). Upon superinfection with dl1403Y, β-Gal expression was very low, detectable only at MOIs of 3 or greater. In relative terms, the maximum expression level during coinfection with dl1403Y was 22% of that reached during coinfection with dl1403YR, whereas for reactivation, this value was 2.5%. Therefore, this experiment suggests that gene expression during reactivation by dl1403Y is relatively less efficient than during coinfection.
DISCUSSION
Previous studies, using a variety of methodologies, have concurred that infection with ICP0-null HSV-1 mutants does not reactivate quiescent HSV. In the experiments described here, I demonstrated that by alteration of the experimental protocol it is possible to reach a different conclusion, namely, that reactivation does occur under conditions in which the superinfecting virus replicates from an initial low MOI. In view of these results, it is necessary to reassess the role of ICP0 in stimulating quiescent genomes to resume gene expression and, particularly, to review the hypothesis that quiescent genomes are shielded from trans-acting factors. The assertion that ICP0 alone is sufficient for reactivation in human fibroblasts, however, is not in doubt. Infection of cultures with adenovirus recombinants that express ICP0 results in reactivation of quiescent genomes, even when only small amounts of ICP0 are produced (19, 21, 53).
The results of previous investigations have essentially been reproduced here; thus, discrepancies with published conclusions are not due to experimental inconsistencies. After initial infection at a low MOI of 0.03, it was not possible to detect reactivation even when superinfection with dl1403 was at a low MOI and the virus was permitted to replicate and spread through the culture. The reasons for this observation are not entirely clear, since a significant proportion of cells (3%) should contain a quiescent genome, and the most likely explanation is that the rare initial infection that resulted in replication of dl1403 would be unlikely to occur in a cell containing a quiescent genome. Presumably, infection through secondary spread was effectively at high MOIs, resulting in competition that prevented either reactivation itself or detection of reactivated quiescent genomes. In addition, the dl1403 genomes that did not form plaques initially would become quiescent, competing with in1374 during superinfection. As a further consideration, the initiation of infection from incoming genomes may be more rapid than from quiescent genomes; if this is the case, competition from superinfecting virus would be exacerbated. The coinfection experiment shown in Fig. 6 confirmed that competition effects of this type can occur.
The failure to detect reactivation when both initial and superinfecting MOIs are high, reported by Minaker et al., was reproduced. At lower superinfecting MOIs, however, it was possible to detect reactivation by histochemical staining of plaques or analysis of DNA. Competition at high MOIs of dl1403Y accounted for the failure to detect reactivation, since this effect was also observed, albeit less dramatically, when superinfection was carried out with dl1403YR at high MOIs.
The interpretation of superinfection experiments was supported by reconstructions. Coinfection of cultures with in1374 and dl1403YR yielded results that were predictable and readily interpretable. Since dl1403YR provides the functions that are inactivated in in1374, the approach is tantamount to infecting cells with two wild-type viruses that can be differentiated by the presence of distinct marker genes. Although total replication (Fig. 5) and gene expression (Fig. 7) increased with the MOI, the relative contribution from the in1374 genome, kept at a fixed level, decreased at high total MOIs. There is a finite capacity within cells for transcription and replication (and for other processes, such as packaging) of viral genomes, and the reduction in contributions from in1374 genomes signifies that genomes compete for cellular and viral factors when the total MOI is high. Thus, although overall viral transcription and replication were higher at a dl1403YR MOI of 30 than at an MOI of 1, the degree of expression and replication of in1374 was much lower at the higher MOI. Indeed, Sacks and Schaffer previously noted competition between ICP0-null genomes and wild-type HSV-1 genomes, although the effect was not investigated further (42).
The situation is more complex during coinfection with in1374 and dl1403Y. Again, expression and replication of in1374 mirror those of dl1403Y, since the VP16 and ICP4 defects are complemented by dl1403Y. At low MOIs, however, ICP0-null genomes are transcribed and replicated poorly. To achieve efficient productive replication, it is necessary to increase the MOI to 30, at which point (from the observations of coinfection with dl1403YR) severe competition limits the transcription and expression of the in1374 genome. Indeed, I observed an analogous effect at the DNA synthesis level during coinfection of dl1403/β-gal (at a constant MOI of 0.1) with increasing amounts of dl1403: replication of dl1403/β-gal was severely reduced as the dl1403 MOI reached 30 (results not shown). A further demonstration of competition is shown in Fig. 6; the rare β-Gal-negative plaques must consist largely of cells infected with both dl1403 and dl1403/β-gal.
The findings reported here provide new information on the complexity of the ICP0-null mutant phenotype in restrictive cell types. Previous studies emphasized that the major impairment of gene expression is observed at low MOIs, with the defect overcome, either completely or partially, at high MOIs (5, 8, 12, 13, 42, 46). By including a distinguishable virus at a fixed low MOI, I demonstrated that although total gene expression and replication are similar at high MOIs (10 for dl1403YR and 30 for dl1403Y), the contributions from individual genomes at these multiplicities are well below maxima due to competition. It is not clear whether this signifies an overall reduction in expression and replication of all genomes or heterogeneity such that only selected genomes are transcribed and replicated, with the others possibly remaining quiescent.
The results presented here show that superinfection with dl1403 can indeed reactivate quiescent HSV, and it is important to consider whether the data completely refute the concept that the quiescent genome is shielded from trans-acting factors. Analysis of Fig. 4, 5, and 7 reveals that the relative amount of β-Gal expression or in1374 replication was approximately 10-fold lower during superinfection than during coinfection, when the response to dl1403Y was compared with the response to dl1403YR. The straightforward interpretation is that 90% of quiescent genomes are indeed refractory to superinfection with dl1403Y and that only 10% respond. This would imply heterogeneity among quiescent genomes, with some not repressed as tightly as the majority, in agreement with observations made at earlier times after infection (13). It should be noted, however, that all viral genomes appear to be altered in some way during conversion to a quiescent state. Neither the virion protein VP16 nor the chemical hexamethylene bisacetamide affects expression from quiescent genomes, whereas they stimulate transcription of in1374 or KM110 if added at the time of infection (18, 37, 38). While heterogeneity among quiescent genomes is the most likely interpretation of the data, some caveats must be recognized. In particular, the events during coinfection and superinfection may not be strictly comparable. Upon coinfection, all genomes are uncoated and transcribed with the same overall timing and through the same mechanisms. During superinfection, however, transcription of quiescent genomes may be delayed compared with that of the superinfecting ICP0-null virus, and if this is the case, the quiescent genome might suffer greater competition than was found for in1374 during coinfection. Therefore, although I favor the interpretation that there is heterogeneity in the extents of silencing of individual quiescent genomes, it remains possible that most genomes are potentially susceptible to trans-acting factors and that the complex phenotype of ICP0-null mutants precludes efficient reactivation.
A number of studies have addressed the role of ICP0 in reactivation of quiescent, or latent, HSV-1 in neuronal cells. In a system based on infection of rat pheochromocytoma (PC12) cells, quiescent HSV-1 was reactivated by superinfection of cultures with adenovirus recombinants that expressed either VP16, ICP0, or ICP4 (30). Latent HSV-1 in mouse ganglia was also reactivated by infection of dissociated ganglia with adenoviruses expressing VP16, ICP0, or ICP4 (16). Therefore, in these neuron-based systems, reactivation can be initiated by adenovirus-directed expression of VP16 or ICP4 in the absence of ICP0, either through a general effect on the viral genome or by stimulating ICP0 production from the quiescent or latent genome. The observation of ICP0-independent reactivation that I report here, therefore, highlights similarities between quiescence in fibroblasts and latency in neurons.
There is no evidence for the presence of ICP0 in neurons that harbor latent HSV, and thus, the initiation of reactivation must occur in the absence of this protein. Investigation of latency in the mouse has shown that, when careful attention is paid to equalizing the establishment of latency, ICP0-null mutants reactivate less efficiently than wild-type viruses upon explantation of ganglia (17, 47). These experiments indicate that ICP0 has an important role in reactivation in the mouse, but equally, they demonstrate that reactivation can occur in the complete absence of ICP0. Recent experiments have suggested that ICP0 is not required for the initiation of reactivation in vivo after hyperthermic treatment of mice but that the protein is important for later events in virus replication (47). Overall, reactivation in all neuron-based culture or animal model systems is inefficient when the number of viral genomes present is taken into account, with only a very small proportion responding to the stimuli, and it is therefore likely that latent genomes are heterogeneous in terms if their responsiveness to reactivation signals. The findings I report here also suggest heterogeneity in the responsiveness of quiescent genomes to reactivation in the absence of ICP0, signifying a convergence of the fibroblast model with the neuron-based systems discussed above. Studies on the structure and reactivation of quiescent HSV in fibroblasts may thus have direct relevance to latency in vivo.
Acknowledgments
I thank Mary Jane Nicholl for skilled assistance and Duncan McGeoch, Roger Everett, and Nigel Stow for helpful comments on the manuscript.
Footnotes
Published ahead of print on 22 August 2007.
REFERENCES
- 1.Ace, C. I., M. A. Dalrymple, F. H. Ramsay, V. G. Preston, and C. M. Preston. 1988. Mutational analysis of the herpes simplex virus type 1 trans-inducing factor Vmw65. J. Gen. Virol. 69:2595-2605. [DOI] [PubMed] [Google Scholar]
- 2.Ace, C. I., T. A. McKee, J. M. Ryan, J. M. Cameron, and C. M. Preston. 1989. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J. Virol. 63:2260-2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amelio, A. L., N. V. Giordani, N. J. Kubat, J. E. O'Neil, and D. C. Bloom. 2006. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol. 80:2063-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai, W., and P. A. Schaffer. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 66:2904-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell, M. E. M., J. W. Palfreyman, and C. M. Preston. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 180:1-19. [DOI] [PubMed] [Google Scholar]
- 7.Carrozza, M. J., and N. A. DeLuca. 1996. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol. Cell. Biol. 16:3085-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, J., and S. Silverstein. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 66:2916-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davison, M. J., V. G. Preston, and D. J. McGeoch. 1984. Determination of the sequence alteration in the DNA of the herpes simplex virus type 1 temperature-sensitive mutant ts K. J. Gen. Virol. 65:859-863. [DOI] [PubMed] [Google Scholar]
- 10.Efstathiou, S., and C. M. Preston. 2005. Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res. 111:108-119. [DOI] [PubMed] [Google Scholar]
- 11.Eidson, K. M., W. E. Hobbs, B. J. Manning, P. Carlson, and N. A. DeLuca. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 76:2180-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett, R. D. 1989. Construction and characterisation of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 70:1185-1202. [DOI] [PubMed] [Google Scholar]
- 13.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everett, R. D., J. Murray, A. Orr, and C. M. Preston. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81:10991-11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halford, W. P., C. D. Kemp, J. A. Isler, D. J. Davido, and P. A. Schaffer. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 75:6143-6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 75:3240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hancock, M. H., J. A. Corcoran, and J. R. Smiley. 2006. Herpes simplex virus regulatory proteins VP16 and ICP0 counteract an innate intranuclear barrier to viral gene expression. Virology 352:237-252. [DOI] [PubMed] [Google Scholar]
- 19.Harris, R. A., R. D. Everett, X. Zhu, S. Silverstein, and C. M. Preston. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2. J. Virol. 63:3513-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris, R. A., and C. M. Preston. 1991. Establishment of latency in vitro by the herpes simplex virus type 1 mutant in 1814. J. Gen. Virol. 72:907-913. [DOI] [PubMed] [Google Scholar]
- 21.Hobbs, W. E., D. E. Brough, I. Kovesdi, and N. A. DeLuca. 2001. Efficient activation of viral genomes by levels of herpes simplex virus ICP0 insufficient to affect cellular gene expression or cell survival. J. Virol. 75:3391-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Homer, E. G., A. Rinaldi, M. J. Nicholl, and C. M. Preston. 1999. Activation of herpesvirus gene expression by the human cytomegalovirus protein pp71. J. Virol. 73:8512-8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson, S. A., and N. A. DeLuca. 2003. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. USA 100:7871-7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jamieson, D. R. S., L. H. Robinson, J. I. Daksis, M. J. Nicholl, and C. M. Preston. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus Vmw65 mutants. J. Gen. Virol. 76:1417-1431. [DOI] [PubMed] [Google Scholar]
- 25.Jones, C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin. Microbiol. Rev. 16:79-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubat, N. J., A. L. Amelio, N. V. Giordani, and D. C. Bloom. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J. Virol. 78:12508-12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubat, N. J., R. K. Tran, P. K. McAnany, and D. C. Bloom. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 78:1139-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall, K. R., R. H. Lachmann, S. Efstathiou, A. Rinaldi, and C. M. Preston. 2000. Long-term transgene expression in mice infected with a herpes simplex virus type 1 mutant severely impaired for immediate-early gene expression. J. Virol. 74:956-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McFarlane, M., J. I. Daksis, and C. M. Preston. 1992. Hexamethylene bisacetamide stimulates herpes simplex virus immediate early gene expression in the absence of trans-induction by Vmw65. J. Gen. Virol. 73:285-292. [DOI] [PubMed] [Google Scholar]
- 30.Miller, C. S., R. J. Danaher, and R. J. Jacob. 2006. ICP0 is not required for efficient stress-induced reactivation of herpes simplex virus type 1 from cultured quiescently infected neuronal cells. J. Virol. 80:3360-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minaker, R. L., K. L. Mossman, and J. R. Smiley. 2005. Functional inaccessibility of quiescent herpes simplex virus genomes. Virol. J. 2:85-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mossman, K. L., P. F. MacGregor, J. J. Rozmus, A. B. Goryachev, A. M. Edwards, and J. R. Smiley. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mossman, K. L., H. A. Saffran, and J. R. Smiley. 2000. Herpes simplex viurs ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mossman, K. L., and J. R. Smiley. 1999. Truncation of the C-terminal acidic activation domain of herpes simplex virus VP16 renders expression of the immediate-early genes almost entirely dependent on ICP0. J. Virol. 73:9726-9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Hare, P. 1993. The virion transactivator of herpes simplex virus. Semin. Virol. 4:145-155. [Google Scholar]
- 36.Preston, C. M. 1979. Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature sensitive mutant tsK. J. Virol. 29:275-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Preston, C. M., and M. J. Nicholl. 2005. Human cytomegalovirus tegument protein pp71 directs long-term gene expression from quiescent herpes simplex virus genomes. J. Virol. 79:525-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Preston, C. M., and M. J. Nicholl. 1997. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 71:7807-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preston, C. M., A. Rinaldi, and M. J. Nicholl. 1998. Herpes simplex virus type 1 immediate early gene expression is stimulated by inhibition of protein synthesis. J. Gen. Virol. 79:117-124. [DOI] [PubMed] [Google Scholar]
- 40.Russell, J., and C. M. Preston. 1986. An in vitro latency system for herpes simplex virus type 2. J. Gen. Virol. 67:397-403. [DOI] [PubMed] [Google Scholar]
- 41.Russell, J., N. D. Stow, E. C. Stow, and C. M. Preston. 1987. Herpes simplex virus genes involved in latency in vitro. J. Gen. Virol. 68:3009-3018. [DOI] [PubMed] [Google Scholar]
- 42.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in culture. J. Virol. 61:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samaniego, L. A., L. Neiderhiser, and N. A. DeLuca. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smiley, J. R., and J. Duncan. 1997. Truncation of the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 produces a phenotype similar to that of the in1814 linker insertion mutation. J. Virol. 71:6191-6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stow, E. C., and N. D. Stow. 1989. Complementation of a herpes simplex virus type 1 Vmw110 deletion mutant by human cytomegalovirus. J. Gen. Virol. 70:695-704. [DOI] [PubMed] [Google Scholar]
- 46.Stow, N. D., and E. C. Stow. 1986. Isolation and characterisation of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 47.Thompson, R. L., and N. M. Sawtell. 2006. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J. Virol. 80:10919-10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner, E. K., and D. C. Bloom. 1997. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 10:419-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang, Q. Y., C. H. Zhou, K. E. Johnson, R. C. Colgrove, D. M. Coen, and D. M. Knipe. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 102:16055-16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wigdahl, B. L., A. C. Scheck, E. DeClercq, and F. Rapp. 1982. High efficiency latency and reactivation of herpes simplex virus in human cells. Science 217:1145-1146. [DOI] [PubMed] [Google Scholar]
- 51.Yao, F., and P. A. Schaffer. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zabierowski, S., and N. A. DeLuca. 2004. Differential cellular requirements for activation of herpes simplex virus type 1 early (tk) and late (gC) promoters by ICP4. J. Virol. 78:6162-6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu, X., J. Chen, C. S. H. Young, and S. Silverstein. 1990. Reactivation of latent herpes simplex virus by adenovirus recombinants encoding mutant IE-0 gene products. J. Virol. 64:4489-4498. [DOI] [PMC free article] [PubMed] [Google Scholar]