

Abstract
The Epstein-Barr Virus (EBV) early protein EB2 (also called BMLF1, Mta, or SM) promotes the nuclear export of a subset of early and late viral mRNAs and is essential for the production of infectious virions. We show here that in vitro, protein kinase CK2α and -β subunits bind both individually and, more efficiently, as a complex to the EB2 N terminus and that the CK2β regulatory subunit also interacts with the EB2 C terminus. Immunoprecipitated EB2 has CK2 activity that phosphorylates several sites within the 80 N-terminal amino acids of EB2, including Ser-55, -56, and -57, which are localized next to the nuclear export signal. EB2S3E, the phosphorylation-mimicking mutant of EB2 at these three serines, but not the phosphorylation ablation mutant EB2S3A, efficiently rescued the production of infectious EBV particles by HEK293BMLF1-KO cells harboring an EB2-defective EBV genome. The defect of EB2S3A in transcomplementing 293BMLF1-KO cells was not due to impaired nucleocytoplasmic shuttling of the mutated protein but was associated with a decrease in the cytoplasmic accumulation of several late viral mRNAs. Thus, EB2-mediated production of infectious EBV virions is regulated by CK2 phosphorylation at one or more of the serine residues Ser-55, -56, and -57.
In eukaryotic cells, a continuous abundant bidirectional flow of macromolecules occurs between the cytoplasm and the nucleus via the nuclear pores. In metazoans, the nuclear export of most mRNAs generated from intronless and intron-containing genes is mediated by many conserved proteins, which coat the nascent RNA cotranscriptionally to form export-competent messenger RNP (mRNP) particles that are capped at their 5′ ends, spliced, and cleaved/polyadenylated at their 3′ ends (for a review, see reference 1). This multistep process involves the binding of RNA export adaptor proteins, including Aly/REF (28), the SR proteins 9G8 and SRp20 (20), and the U2AF protein (48), which by interacting with the heterodimeric export receptor TAP/p15 facilitates the transport of mature mRNPs to and through the nuclear pores. Recent studies strongly suggest that for some intron-containing RNAs, the export machinery is recruited to the 5′ ends of mRNAs and is composed of the cap-binding protein CBP80, which makes contact with REF/Aly bound to UAP56 in the TREX complex, a process that is splicing dependent (6). For mRNAs generated from intronless genes, two export pathways have been described: one implies binding of the SR proteins 9G8 and SRp20 to mRNAs (20, 21), and the other implies recruitment of REF at the 5′ end of the mRNA by the cap-binding protein CBP20 (33).
Most herpesvirus early and late genes are intronless, and the corresponding mRNAs are efficiently exported. However, and intriguingly, despite the profusion of cellular mRNA export factors, the export of most intronless viral mRNAs is strictly dependent on a viral gene product that shares properties with known mRNA export adaptors, i.e., the herpes simplex virus type 1 protein ICP27 (5, 24, 39), the cytomegalovirus protein UL69 (27), the Kaposi's sarcoma-associated herpesvirus protein ORF57 (29), the herpesvirus saimiri protein ORF57 (45), and the Epstein-Barr virus (EBV) protein EB2 (9, 41).
The EB2 protein shuttles between the nucleus and the cytoplasm (9), binds RNA in vitro and in vivo via an arginine-rich region (17), and induces the cytoplasmic accumulation of some, but not all, early and late viral mRNAs (2, 14, 16), likely by interacting with cellular adaptors of the TAP/p15 receptor pathway, such as REF (18) and human OTT1/RBM15 (19), a novel RNA-binding mRNA export factor that binds directly to TAP/p15 (26). Accordingly, the deletion of the EB2 gene abolishes the production of infectious viral particles, demonstrating that EB2 is an essential viral factor whose function cannot be transcomplemented by cellular factors (14).
It has been previously established that EB2 is a phosphoprotein (46) and that it is phosphorylated in vitro by CK2 (7), but the effect of phosphorylation on its function in viral-mRNA export and production of infectious virions could not been addressed due to the lack of relevant functional assays. Moreover, whether EB2 interacts stably with CK2 is also not known. Protein kinase CK2 is a heterotetrameric complex consisting of a stable association of two catalytic subunits (α or α′) and two regulatory β subunits that phosphorylate many different substrates localized in different cellular compartments (for a review, see reference 38). Recent studies have demonstrated that free populations of CK2 subunits can also exist within the same cell (12). Both CK2 subunits are independently targeted to the nucleus, where they exhibit differential mobility. CK2α, but not CK2β, shuttles between the nucleus and the cytoplasm. Thus, CK2 can act as a heterotetrameric holoenzyme but also as free populations of both subunits, either alone or in association with different partners and in different subcellular compartments.
In this study, using tandem affinity purification followed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry, we show that the CK2 subunits α and β copurify with EB2. We also show that CK2 subunits α and β interact in vitro both individually and as a complex within EB2 N- and C-terminal domains and that EB2 is a substrate for CK2. Furthermore, EB2 coimmunoprecipitates from living cells with CK2, which phosphorylates EB2 at several N-terminal sites, including Ser-55, -56, and -57. By functional studies, we demonstrate that replacement of these three serines by alanines (EB2S3A), but not their replacement by glutamic acids (EB2S3E), strongly impairs the capacity of EB2 to transcomplement 293BMLF1-KO cells harboring an EB2-defective EBV genome for the production of infectious EBV particles. This defect was associated with a decrease in EB2-mediated cytoplasmic accumulation of several late mRNAs. Taken together, these results strongly suggest that EB2 phosphorylation by CK2 is critical for the production of infectious virions.
MATERIALS AND METHODS
Plasmids.
All eukaryotic expression vectors used were derived from the cytomegalovirus immediate-early promoter-based plasmids pCI (Promega) and pNTAP (Stratagene). F precedes the name of the protein when it has been tagged at the N terminus with the Flag epitope, which can be detected with the monoclonal antibody (MAb) M2 (Sigma). pCI.F.EB2 expresses the EB2 protein initiated at the BSLF2 AUG, and pRcCMVZ41 expresses the EB1 protein (2). pCI.F.EB2Δ2 expresses EB2 that has amino acids 39 to 63 deleted (4). In pCI.F.EB2M1, the 50 N-terminal amino acids of EB2 have been deleted. From pCI.F.EB2 or pCI.F.M1, we generated pCI.F.EB2S3A, pCI.F.EB2M1S3A, and pCI.F.EB2S3E by using the QuickChange Site-Directed Mutagenesis kit (Stratagene). In pCI.F.EB2S3A and pCI.F.EB2M1S3A, Ser-55, -56, and -57 were each replaced by alanine using the oligonucleotide primer 5′-CACAGATGGAGAGATTGCAGCCGCAGAGGAGGAGGATG-3′ and its complement on the opposite strand. In pCI.F.EB2S3E, Ser-55, -56, and -57 were each replaced by glutamic acid using the oligonucleotide primer 5′-CACTCCACAGATGGAGAGATTGAAGAGGAAGAGGAGGAGTG-3′ and its complement on the opposite strand. pNTAP.EB2Nter expresses the EB2 N-terminal 184 amino acids fused at the N terminus to a calmodulin-binding peptide and to a streptavidin-binding peptide. The PCR amplification and cloning of partially overlapping EB2 cDNA fragments in pGex4T-2 (Amersham Biosciences) has been described previously (18).
Transfections and TAP-tagging experiments.
HeLa cells, HEK293 cells, and HEK293T cells were grown at 37°C in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal calf serum. Transfections were performed by the calcium phosphate precipitate method as described previously (9). HEK293T cells were transfected with 1 μg of pNTAP or pNTAP.EB2Nter. Cells were harvested 48 h later, and a nuclear extract was prepared from 4 × 107 cells as previously described (8). To purify TAP-tagged EB2Nter and associated proteins, nuclear extracts were processed following the manufacturer's instructions (Interplay Mammalian TAP system; Stratagene). Eluted proteins were analyzed by two-dimensional (2D) gel electrophoresis with silver staining and identified by MALDI-TOF mass spectrometry.
2D gels and mass spectrometry.
Dry pellets of eluted proteins were dissolved in 400 μl of 2D electrophoresis buffer (7 M urea, 2 mM thiourea, 2% [vol/vol] Triton X-100, 20 mM dithiothreitol, and 0.6% Pharmalytes, pH 3 to 10). The samples were sonicated. An immobilized pH gradient strip, pH 3 to 10 (Bio-Rad), was equilibrated with the sample for 15 h. Isoelectrofocusing was performed using the Protean IEF cell following the manufacturer's instructions (Bio-Rad). Polypeptides were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). They were either further analyzed by Western blotting or silver stained for mass spectrometry. Silver-stained polypeptide spots were destained and in-gel digested overnight with 50 ng trypsin (Promega). The peptides were purified and analyzed by MALDI-TOF mass spectrometry using a cyano-4-hydroxycinnaminic acid matrix on a Voyager-DE-STR instrument (Applied Biosystems). A database search with the peptide mass list was performed against the NCBInr database using the MS-FIT algorithm (http://prospector.ucsf.edu/prospector/4.0.8/html/msfit.htm) or ProFound (http://prowl.rockefeller.edu/prowl-cgi/profound.exe).
RNA extraction and RT-PCR analysis.
Following transfection, cytoplasmic RNAs were extracted as previously described (15). Reverse transcriptions were performed using Stratascript reverse transcriptase (Stratagene) and oligodT18 (Roche Diagnostics) for 1 h at 42°C. PCRs were performed using Taq DNA polymerase (Invitrogen) in the presence of [α-32P]dCTP (0. 1 μCi) with a set of specific primer pairs (Table 1). The PCR products were analyzed by electrophoresis in 6% acrylamide gels. The amount of 32P incorporated was quantified using a PhosphorImager (Amersham).
TABLE 1.
Primers used in this study
| Gene | Primer | Sequence (5′ to 3′) | B95-8 coordinatesa |
|---|---|---|---|
| β-Actin | Forward | GCTGCGTGTGGCTCCCA | |
| Reverse | ATCTTCATTGTGCTGGGG | ||
| BFRF3 | Forward | GGGAGGCTCAAAGAAGTTA | 61628-61650 |
| Reverse | ATGAAGAAACAGAGGGGG | 61882-61903 | |
| BdRF1 | Forward | CACTATCAGGTAACGCA | 148710-148728 |
| Reverse | TCAAGCCAGCGTTTATTT | 149723-149744 | |
| BDLF1 | Forward | CAGATTTGAAAGTGGTAG | 133283-133201 |
| Reverse | TTATCTTAACCAGCAAGTG | 132400-132422 | |
| BcLF1 | Forward | GCAATTATCAAAAAACC | 133313-133332 |
| Reverse | GGTGCAGTTTGTA | 133528-133543 | |
| BMRF1 | Forward | CCAGACATACGGTCAGTCCA | 80881-80904 |
| Reverse | TGCTTCACTTTCTTGGG | 81071-81091 |
Positions of the primers in the EBV B95-8 genomic sequence.
In vitro GST pull-down assays.
Glutathione S-transferase (GST) and GST fusion proteins were purified from Escherichia coli BL21 codons plus strain extracts with glutathione-Sepharose 4B beads (Amersham Biosciences). Beads carrying GST or the GST fusion proteins were equilibrated in TNTB interaction buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.05% Tween 20, and 0.1 mg/ml bovine serum albumin) and incubated with in vitro-radiolabeled proteins as follows. [35S]methionine-labeled CK2α or CK2β subunits were synthesized in vitro as previously described (42) using the TnT Coupled Transcription/Translation system (Promega). Equal amounts of in vitro-synthesized CK2 subunits, either alone or mixed together and preincubated for 15 min at 4°C, were added to 500 μl of TNTB and incubated with TNTB-equilibrated glutathione-Sepharose 4B beads loaded with GST or the GST fusion proteins for 2 h at 4°C. The beads were washed once in TNTB and then twice in TNT without bovine serum albumin, and the bound proteins were fractionated by SDS-PAGE and revealed by autoradiography.
In vitro phosphorylation assays.
For kinase assays using HEK293T cell extracts, beads carrying GST or GST fusion proteins were washed five times with kinase buffer (25 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl2, 5 mM MnCl2, and 1 mM CaCl2) and incubated for 30 min at 30°C with 50 μl of cell extracts prepared as described previously (36) and supplemented with 6 μl of 5× kinase buffer and 20 μCi of [γ-32P]ATP. The beads were washed three times in MTPBS (150 mM NaCl, 16 mM Na2HPO4, 100 mM EDTA, 1% Triton X-100, pH 7.3). The GST and GST fusion proteins were fractionated by SDS-PAGE. After Coomassie blue staining, the gel was analyzed by autoradiography.
For kinase assays using purified recombinant CK2, beads carrying GST or GST fusion proteins were washed five times with kinase buffer. Recombinant oligomeric CK2 was expressed in Sf9 cells and purified to near homogeneity (11). Equilibrated GST and GST fusion proteins were incubated at 25°C in 15 μl of kinase buffer containing 30 μM [γ-32P]ATP in the presence of 180 ng oligomeric CK2. After 10 to 30 min, the 32P-labeled proteins were analyzed by SDS-PAGE and autoradiography as described above.
Western blotting, immunofluorescence, and heterokaryon assays.
Western blotting was carried out as described previously (14). For indirect immunofluorescence, 106 HeLa cells were seeded onto glass coverslips in 100-mm petri dishes. Following transfection by the calcium-phosphate precipitate method, the cells on coverslips were processed for immunofluorescence as previously described (18). For the various indirect-immunofluorescence experiments, either the M2 anti-Flag MAb (Sigma) or a rabbit anti-CK2β polyclonal antibody was used as a primary antibody. A Fluorolink Cy3-labeled goat anti-mouse antibody (Amersham) or an Alexa Fluor 488 goat anti-rabbit immunoglobulin G (IgG) (heavy and light) antibody (Invitrogen) was used as a secondary antibody. Cell nuclei were stained by incubation with 5 μg/ml Hoescht 33258 (Sigma). For the heterokaryon assays, 24 h after transfection, 2 × 105 HeLa cells were seeded onto glass coverslips with an equal number of NIH 3T3 cells in six-well plates. The cells were allowed to grow overnight and were then treated for 2 h with 100 μg/ml of cycloheximide to inhibit protein synthesis. The cells were then washed with phosphate-buffered saline (PBS), and heterokaryon formation was performed by incubating the cells for 2 min in 50% polyethylene glycol (PEG) 8000 (Sigma) in PBS. Following PEG-induced fusion, the cells were washed extensively with PBS and returned to fresh medium containing 100 μg/ml of cycloheximide. After 2 h at 37°C, the cells were fixed with 4% paraformaldehyde. Indirect immunofluorescence was performed essentially as described above, except that Alexa Fluor 488 goat anti-mouse IgG (heavy and light) antibody was used as a secondary antibody and F-actin was labeled with Alexa Fluor 546 phalloidin (Molecular Probes, Invitrogen) to visualize the heterokaryons.
Immunoprecipitations and immunoprecipitation-kinase assays.
Transiently transfected HEK293T cells (2 × 106) expressing F.EB2 or F.EB2 mutants were lysed in 600 μl of RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium desoxycholate, and 0.1% SDS) supplemented with protease inhibitors (Complete; Roche). Fifty microliters of M2 anti-Flag affinity gel (Sigma) was added to the lysate. After incubation under rotation at 4°C, the beads were washed four times in RIPA and three times in kinase buffer and resuspended in 50 μl of kinase buffer containing 10 μCi of [γ-32P]ATP. Where indicated, 20 μM of the CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) was added to the in vitro kinase assay. After incubation for 30 min at 30°C, the beads were washed twice in RIPA. Bound proteins were eluted with 20 μg of 3× Flag peptide M2 (Sigma) in RIPA, fractionated by SDS-PAGE, and transferred to Hybond ECL membrane (Amersham). The same membrane was submitted to autoradiography and to Western blotting using an M2 anti-Flag antibody and the Alexa Fluor 680 goat anti-mouse IgG (Molecular Probes, Invitrogen) as a secondary antibody. The amount of 32P incorporated was quantified using a PhosphorImager (Amersham), and the amounts of proteins were quantified using the Odyssey infrared imaging system (LI-COR Biosciences, Nebraska).
Transcomplementation assays.
HEK293BMLF1-KO cells contain an EBV genome with the EB2 gene deleted and carry the gene for green fluorescent protein (GFP) under the control of the cytomegalovirus immediate-early promoter/enhancer (14). The 293BMLF1-KO cells were transiently transfected either with an empty vector, with an EB1-expressing vector, or with both EB1- and EB2-expressing vectors as previously described (14). After 3 days, the cell medium was collected and filtered through a 0.45-μm filter. Infection of Raji cells with the filtered medium was carried out as previously described (10, 23). Due to constitutive expression of the GFP from the EBV genome, infected Raji cells were bright green under UV light and were quantified by fluorescence-activated cell sorter (FACS) analysis (FACSCALIBUR).
RESULTS
The N terminus of EB2 interacts with CK2.
To identify cellular factors that interact with EB2, we used a TAP-tagging approach. We first inserted a cDNA fragment corresponding to the EB2 N-terminal 184 amino acids (EB2Nter) into plasmid pNTAP. HEK293T cells were transfected with pNTAP.EB2Nter, and the tagged protein was isolated from the cell lysate. TAP-tagged EB2Nter and copurifying proteins were separated by 2D gel electrophoresis and identified by MALDI-TOF mass spectrometry. As shown in Fig. 1, EB2Nter migrated in the 2D gel as several isoelectric variants, most likely representing different phosphorylated forms of the protein. This strongly suggests that several differentially phosphorylated forms of EB2 coexist in vivo and that some of the phosphoacceptor sites are located at the N terminus of the protein. Several proteins copurified with EB2Nter, including p32, a splicing inhibitor (37), the protein chaperone HSP70, and CK2α and -β subunits. Such proteins were not recovered when a lysate from HEK293T cells transfected with the empty vector was submitted to tandem affinity purification (data not shown).
FIG. 1.
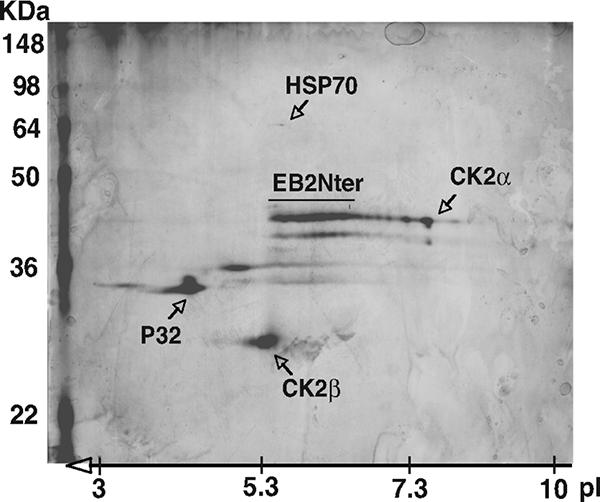
Both CK2α and -β subunits copurify with the 184 EB2 N-terminal amino acids. HEK293T cells were transfected with pNTAP.EB2Nter, expressing the EB2Nter protein fused at the N terminus to a calmodulin-binding peptide and to a streptavidin-binding peptide. TAP-tagged EB2 and associated proteins expressed in HEK293T cells were sequentially purified on streptavidin and calmodulin columns. The purified proteins were separated by 2D gel, silver stained, and identified by MALDI-TOF mass spectrometry. The unlabeled spots were not identified by mass spectrometry. The positions of the molecular mass markers are indicated on the left of the gel. pI, isoelectric point.
The CK2α and -β subunits interact with EB2.
To determine whether CK2 or CK2 subunits bind directly to EB2 and to which region, we performed in vitro binding assays using GST and the GST fusions GST-A, -Nter, and -Cter (Fig. 2A) and [35S]methionine-labeled CK2α and -β subunits produced by transcription-translation in vitro (Fig. 2B, lanes 1 to 3). GST-A was included in the assay because it contains the three serines that were shown previously to be phosphorylated by CK2 in vitro (7). As shown in Fig. 2B, top, CK2α bound to GST-Nter and -Cter (lanes 6 and 7). CK2β also bound to GST-Nter (lane 10), but more efficiently to GST-Cter (lane 11). Importantly, when the α and β subunits were mixed together prior to incubation with the GST fusion proteins (lane 3), a larger amount of both CK2α and CK2β bound to GST-Nter (lane 14) while only CK2β bound efficiently to GST-Cter (lane 15). To further determine where CK2 binds in EB2Nter, we performed GST pull-down experiments using GST and the GST fusion proteins GST-A, -B, -C, and -Nter (Fig. 2A) and purified CK2 (New England Biolabs) that was first 32P labeled in vitro by autophosphorylation of its β subunit. As shown in Fig. 2C, 32P-labeled CK2β (b, lane 1) bound detectably only to GST-B and GST-Nter (lanes 4 and 6). From the above-mentioned results, we concluded that in vitro, both CK2 subunits interact with EB2 individually, but more efficiently as a complex in a region that contains a CRM-1-independent nuclear export signal (NES) (18). CK2β also binds to the C terminus of EB2 with high affinity, but mostly not associated with the α subunit.
FIG. 2.

CK2 binding to EB2. (A) Schematic representation of EB2 and EB2 polypeptides used to generate GST fusion proteins. NLS, nuclear localization signal; RBD, RNA-binding domain. (B) Recombinant CK2α and CK2β in vitro labeled with [35S]methionine (lanes 1 to 3), either individually (lanes 4 to 11) or mixed together (lanes 12 to 15), for 15 min at 4°C were incubated with glutathione-Sepharose 4B beads loaded with GST or GST-A, -Nter, or -Cter. Proteins bound to the beads were eluted, resolved by SDS-PAGE, and visualized by Coomassie blue staining (bottom, lanes 4 to 15) and by autoradiography (top, lanes 4 to 15). *, nonspecific band. (C) Purified CK2, 32P labeled by autophosphorylation in vitro (b, lane 1), was incubated with beads loaded with GST or GST-A, -B, -C, and -Nter. Proteins bound to the beads were eluted, resolved by SDS-PAGE, and visualized by Coomassie blue staining (a) and by autoradiography (b).
CK2β colocalizes with EB2 in the nucleus.
Since CK2α and -β interact physically with EB2 in vitro, we assessed by indirect immunofluorescence and confocal microscopy whether they also interacted following transient expression of EB2 in HeLa cells. As shown in Fig. 3B, the endogenous CK2β subunit was localized mostly in the nucleoplasm with a granular staining pattern but could also be seen, although more sparsely, in the cytoplasm and in the nucleoli of the cells. The transfected EB2 protein was also nuclear but was totally excluded from the nucleoli (Fig. 3C). In HeLa cells in which EB2 was expressed, the confocal analysis revealed colocalization of EB2 and endogenous CK2β in well-defined nuclear speckles outside the nucleoli (Fig. 3D), which is compatible with an interaction in living cells.
FIG. 3.

EB2 and CK2β colocalize in the nuclei of living cells. HeLa cells expressing F.EB2 were immunostained using a rabbit anti-CK2β antibody and an Alexa Fluor 488-conjugated secondary antibody (B) and the M2 anti-Flag MAb and a Fluorolink Cy3-conjugated secondary antibody (C). (D) Colocalization of CK2β and EB2. The cell fluorescence was examined using confocal microscopy. Panel A shows the same cells in phase-contrast microscopy.
Only the 80 EB2 N-terminal amino acids are phosphorylated by a HEK293T cell extract.
EB2 has a docking site for CK2 between amino acids 61 and 140, and according to Scansite (http://scansite.mit.edu; 34), there are 10 potential CK2 phosphorylation sites in EB2 at S22, T35, S45, S49, T50, S55, S56, S57, T117, and T439 (Fig. 4A), when the search is done at low stringency. In order to locate all phosphorylation sites in EB2, overlapping peptides A, B, C, D, E, F, G, and H, spanning the entire EB2 protein (Fig. 4A) and fused to GST, were incubated with HEK293T cell extracts supplemented with kinase buffer and [γ-32P]ATP. GST-Nter was included in the assay, since it contains most of the phosphorylation sites. As shown in Fig. 4B and C, only GST-A and GST-Nter were phosphorylated, strongly suggesting that under our experimental conditions, all the phosphorylation sites are located within the 80 N-terminal amino acids of EB2. To evaluate if CK2 was the kinase phosphorylating GST-A and GST-Nter, these fusion proteins were incubated overnight with HEK293T cell extracts and then submitted to a kinase assay in the absence or presence of 20 μM of the CK2-specific inhibitor TBB (40) and analyzed as described above. As shown in Fig. 4D, GST-A (lane 9) and GST-Nter (lane 11) were phosphorylated. Addition of TBB reduced the phosphorylation of GST-A by 60% (lane 10) and of GST-Nter by 70% (lane 12). As CK2 interacted in vitro with GST-Nter but not with GST-A (Fig. 2), we then asked whether GST-Nter is phosphorylated by CK2 bound to it. To answer this question, GST and GST-A and -Nter were incubated overnight with HEK293T cell extracts, but the beads were extensively washed before the kinase assay was done in the absence or presence of 20 μM TBB. Under these conditions, only GST-Nter was phosphorylated (Fig. 4D, lane 11), and the phosphorylation was reduced by 60% by TBB. These results demonstrate that a kinase, likely CK2, stably interacts in vitro with and phosphorylates GST-Nter at sites located in the 80 N-terminal amino acids. To confirm this finding, GST-Nter, -A, -B, and -C were submitted to phosphorylation with [γ-32P]ATP by purified CK2 in vitro. Again, only GST-Nter and GST-A were phosphorylated, together with the β subunit of CK2 (Fig. 5).
FIG. 4.

CK2 in HEK293T cell extracts binds to and phosphorylates the EB2 N terminus. (A) Schematic representation of the EB2 peptides fused to GST to generate GST-A, -B, -C, -D, -E, -F, -G, -H, and -Nter. Putative CK2 sites shown over the sequences of peptides A, B, and H were determined using Scansite (http://scansite.mit.edu). (B and C) Glutathione-Sepharose 4B beads loaded with GST and the GST fusion proteins indicated above the lanes were incubated for 30 min at 30°C with a HEK293T cell extract supplemented with kinase buffer and [γ-32P]ATP. GST and the GST fusion proteins were eluted, resolved by SDS-PAGE, and visualized by Coomassie blue staining (gels a) and by autoradiography (gels b). (D and E) Glutathione-Sepharose 4B beads loaded with GST and the GST fusion proteins indicated above the lanes were incubated overnight with a HEK293T cell extract. The − wash assays were supplemented with kinase buffer and [γ-32P]ATP and further incubated for 30 min at 30°C. For the + wash assays, the beads were washed extensively with MTPBS and then submitted to a kinase assay. Where indicated, 20 μM TBB was added to the assay. GST and GST fusion proteins were eluted, resolved by SDS-PAGE, and visualized by Coomassie blue staining (gels a, lanes 1 to 6) and by autoradiography (gels b, lanes 7 to 12).
FIG. 5.

Purified CK2 holoenzyme phosphorylation sites are restricted to the N-terminal 80 amino acids of EB2. Glutathione-Sepharose 4B beads loaded with GST-Nter, -A, -B, and -C were incubated with purified CK2 holoenzyme in an in vitro kinase assay. The GST fusion proteins were eluted, resolved by SDS-PAGE, and analyzed by autoradiography.
EB2 immunoprecipitates contain catalytically active CK2.
Since GST-Nter interacts with and is phosphorylated by CK2 in vitro, we investigated if CK2 could be coimmunoprecipitated with full-length EB2 from living cells and if EB2 is phosphorylated by the coimmunoprecipitated CK2. EB2 proteins and interacting partners were captured from cell lysates prepared in RIPA buffer by incubation with the M2 anti-Flag affinity gel. The immunoselected proteins were subjected to a kinase assay. As shown in Fig. 6B, immunoselected EB2 was phosphorylated by a kinase (lane 3) whose activity was 88% inhibited by 20 μM TBB (lane 4), strongly suggesting that only CK2 interacted with and phosphorylated EB2 in living cells. There are several potential CK2 sites in the 80 N-terminal amino acids of EB2, but only Ser-55, -56, and -57 (Fig. 6A) have the highest probability to be phosphorylated by CK2 when a search using Scansite is conducted under high-stringency conditions. In order to know which sites are phosphorylated, the EB2 N-terminal mutants F.EB2Δ2, F-EB2S3A, F-EB2S3E, F.EB2M1, and F.EB2M1S3A, depicted in Fig. 6A, were also immunoprecipitated from HEK293T cells and submitted to a kinase assay. As shown in Fig. 6C, the EB2 mutants were all less phosphorylated than EB2 (Fig. 6C, a, lanes 2 to 7). Taken together, these results strongly suggest that EB2 immunoprecipitated from living cells stably interacts with CK2, which phosphorylates several EB2 N-terminal sites, including at least one of the three serine residues Ser-55, -56, and -57.
FIG. 6.

EB2 immunoprecipitates contains catalytically active CK2. (A) Amino acid sequences of the EB2 N-terminal 80 amino acids and of EB2 mutants Δ2, S3A, S3E, M1, and M1S3A. The Flag tag is in bold and italics. Amino acids added during the cloning procedure in Δ2 are in bold and italics. Putative CK2 sites are shown by a dot under the EB2 sequence. (B) Extracts from HEK293T cells, mock transfected or expressing F.EB2, were incubated with the M2 anti-Flag affinity gel. Immune complexes were submitted to an in vitro kinase assay in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of the CK2-specific inhibitor TBB (20 μM). Proteins in the immune complexes were resolved by SDS-PAGE and visualized by Western blotting (b) or autoradiography (a). IP, immunoprecipitation. (C) F.EB2, F.EB2M1, F.EB2Δ2, F.EB2S3A, F.EB2S3E, and F.EB2M1S3A expressed in HEK293T cells were analyzed as for panel B. In panels B and C, the relative amounts of EB2 immunoprecipitated were quantified using the Odyssey infrared imaging system. The relative intensities of the radioactive bands on the membrane were quantified using a PhosphorImager. The numbers under blots a correspond to the intensities of the radioactive bands (expressed in arbitrary units compared to EB2) normalized against the amounts of proteins immunoprecipitated (expressed in arbitrary units by comparison to EB2, shown by the numbers under blots b).
The EB2S3E mutant, but not the EB2S3A mutant, rescued infectious-virus production by 293 cells carrying an EB2-defective EBV genome.
In B cells latently infected with EBV, the ectopic expression of EB1, a virus-encoded transcription factor, is necessary and sufficient to induce the production of infectious virions. We recently reported the generation of 293BMLF1-KO cells containing an EBV mutant in which the early gene encoding the EB2 protein had been deleted (14). 293BMLF1-KO cells produce infectious EBV particles only when transfected with both an EB1 and an EB2 expression vector, demonstrating that EB2 is essential for the production of infectious virions (14). We evaluated the production of infectious EBV virions in such a transcomplementation assay, following transient expression of EB1 alone or expressed together with increasing amounts of F.EB2, F.EB2S3A, F.EB2S3E, F.EB2M1, or F.EBM1S3A. Three days following the transfection of 293BMLF1-KO cells, Raji cells were infected with the filtered medium. Due to the constitutive expression of GFP from the EBV recombinant genome, infected Raji cells are bright green under UV light and can be quantified by FACS analysis (FACSCALIBUR). As shown in Fig. 7A, expression of EB1 alone did not rescue the production of infectious virus (lane 1), whereas transient expression of both EB1 and EB2 did (compare lanes 1 and 2), and the amount of infectious virions produced increased with the amount of EB2 expressed (compare lanes 2 and 3). However, whereas the EB2 phosphomimetic mutant EB2S3E (lanes 6 and 7) and the EB2 deletion mutant EB2M1 (lanes 8 and 9) efficiently rescued the production of infectious viral particles when coexpressed with EB1, mutants EB2S3A (lanes 4 and 5) and EB2M1S3A (lanes 10 and 11) did not. As shown in Fig. 7A (bottom), F.EB2 and its mutant derivatives were expressed in comparable amounts. Taken together, these results strongly suggest that phosphorylation of EB2 at Ser-55, -56, and -57 is critical for the efficient production of EBV infectious virions.
FIG. 7.

The EB2S3A mutant does not rescue infectious-virus production by 293BMLF1-KO cells and inefficiently exports several late EBV mRNAs. (A) 293BMLF1-KO cells were transfected with 1 μg of EB1 expression vector alone (lane 1) or together with 0.25 μg (lanes 2, 4, 6, 8, and 10) or with 0.5 μg (lanes 3, 5, 7, 9, and 11) of F.EB2, F.EB2S3A, F.EB2S3E, F.EB2M1, and F.EB2M1S3A expression vectors. Three days after transfection, Raji cells were infected with the filtered medium, and after 48 h, infected cells were quantified by FACS analysis (FACSCALIBUR). Two representative Raji cell infections are presented. All errors bars represent standard deviations. WB anti-FLAG, levels of expression of F.EB2 and F.EB2 mutated proteins in transfected 293BMLF1-KO cells, as evaluated by Western blotting with the M2 anti-FLAG MAb. (B) Cytoplasmic RNAs were extracted from 293BMLF1-KO cells transfected as described for panel A, except that RNA was extracted only from cells transfected with 0.25 μg of F.EB2S3E (lane 6). They were reverse transcribed using oligo(dT)18 as a primer and PCR amplified in the presence of 0.1 μCi [α-32P]dCTP with the specific primers shown in Table 1. The DNA fragments generated by PCR were separated on a 6% polyacrylamide gel and revealed by autoradiography. (C) The relative amounts of 32P incorporated in the PCR products presented in panel B were quantified using a PhosphorImager (Amersham). The results are presented as 32P incorporation relative to that obtained at the highest EB2 concentration, arbitrarily expressed as 100 (lanes 3). All errors bars represent standard deviations.
The S3A mutation decreases the cytoplasmic accumulation of several viral late mRNAs.
EB2 facilitates the nuclear export of a subset of early and late viral mRNAs. Among them are the late mRNAs encoding the capsid proteins BcLF1 (major capsid protein [MCP]), BDLF1 (minor capsid protein [mCP]), and BFRF3 (smallest capsid protein [sCP]) and the protease/assemblin protein BdRF1 (Pr/AP) (2). However, EB2 has no effect on the nuclear export of the early mRNA encoding the polymerase processivity factor BMRF1 (EA-D) (14). Our results showing that EB2S3A, but not EB2S3E, inefficiently transcomplemented the production of infectious virions by 293BMLF1-KO cells could then be due, in part, to decreased export of viral mRNAs. We therefore analyzed, by reverse transcription-PCR, the cytoplasmic accumulation of BcLF1, BDLF1, BFRF3, BdRF1, and BMRF1 mRNAs in 293BMLF1-KO cells transiently expressing EB1 alone or EB1 together with EB2, EB2S3A, and EB2S3E. The 32P-labeled PCR products were separated by acrylamide gel electrophoresis and visualized by autoradiography (Fig. 7B), and the relative amount of 32P incorporated was quantified using a PhosphorImager (Fig. 7C). β-Actin mRNA was used to confirm that the same amount of RNA was used in each RT-PCR. In 293BMLF1-KO cells expressing EB1 alone, the PCR products corresponding to the EBV cytoplasmic mRNAs examined were almost undetectable except, and as expected, for BMRF1 mRNA (Fig. 7C, lanes 1). When both EB1 and EB2 were expressed, the amounts of cytoplasmic mRNAs increased with the amount of EB2-expressing vector transfected (Fig. 7C, compare lanes 2 and 3), except for BcLF1 mRNA. However, in 293BMLF1-KO cells expressing EB1 and EB2S3A, there was a clear decrease in the amount of PCR product obtained at the smaller amount of EB2S3A expression vector transfected (Fig. 7C, compare lanes 2 and 4), and the amounts of PCR products also increased with the amount of EB2S3A expression vector transfected (Fig. 7C, compare lanes 4 to lanes 5). It is noteworthy that the amounts of viral mRNAs present in the cytoplasm of 293BMLF1-KO cells expressing the smallest amount of EB2 or the phosphomimetic mutant EB2S3E (Fig. 7C, compare lanes 6 and 2) were comparable, except for BdRF1 cytoplasmic RNA, which was more abundant in cells expressing the EB2S3E mutant than in cells expressing EB2. As expected, the amount of cytoplasmic BMRF1 mRNA was not notably affected by expression of EB2 or the EB2 mutants. The above-mentioned results strongly suggest that CK2 phosphorylation of EB2 at one or several of the serine residues Ser-55, -56, and -57 is critical for the efficient cytoplasmic accumulation of several EBV late mRNAs.
The S3A mutation does not affect the nucleocytoplasmic shuttling of EB2.
Ser-55, -56, and -57 are located N terminal to the EB2 region that contains a CRM-1-independent NES (18). Since nuclear import (13, 22, 43), as well as nuclear export (35), has been reported to be regulated by CK2 phosphorylation, we studied the effect of the S3A mutation on nucleocytoplasmic shuttling of EB2 by performing human-mouse heterokaryon assays using HeLa cells transfected with a vector expressing either F.EB2 or F.EB2.S3A. Indirect immunofluorescence was performed to evaluate in how many heterokaryons the protein expressed in the HeLa cell nucleus had been transported to the mouse nucleus. As shown in Fig. 8, both EB2 (Fig. 8e) and EB2S3A (Fig. 8h) were localized in the nucleus and shuttled with the same efficiency. The endogenous nonshuttling hnRNPC protein was used as a control and, as expected, was not transported from human to mouse nuclei (Fig. 8b).
FIG. 8.

Nucleocytoplasmic shuttling of EB2 is not affected by the S3A mutation. F.EB2, and F.EB2S3A were expressed in HeLa cells. After 48 h, the HeLa cells were cocultivated overnight with NIH 3T3 cells. Prior to PEG-induced cell fusion, the cells were incubated for 2 h with cycloheximide. After fusion, the cells were further incubated for 2 h with cycloheximide. The cells were then immunostained using either the M2 anti-Flag MAb (panels e and h) or the anti-hnRNP-C (4F4) MAb (panel b) and an Alexa Fluor-conjugated secondary antibody. F-actin was labeled with Alexa Fluor 546 phalloidin to visualize the heterokaryons (panels c, f, and i). The cell nucleus was stained with Hoescht dye (panels a, d, and g). On the right sides of the panels the numbers indicate the numbers of heterokaryons with positive mouse cell nuclei immunostaining (indicating protein shuttling)/the total number of heterokaryons with positive HeLa nuclei immunostaining. IF, immunofluorescence.
DISCUSSION
The new findings presented in this study strongly suggest that CK2 bound to EB2 regulates the EB2-mediated production of infectious virions by phosphorylation of at least one of the serines 55, 56, and 57 of the protein. Interestingly, the phosphorylation of EB2 by CK2 at Ser-55, -56, and -57 was reported more than 10 years ago (7), but no function for this phosphorylation was demonstrated. ICP27 (44, 47) and Kaposi's sarcoma-associated herpesvirus ORF57 (30) also interact with and are phosphorylated by protein kinase CK2. However, whether protein kinase CK2 interaction with and/or phosphorylation of ICP27 and ORF57 affect their mRNA export functions is not known.
Our in vitro studies show that CK2α and -β subunits bind individually to the 184 N-terminal amino acids (EB2Nter) of EB2 and, more precisely, to amino acids 60 to 140 containing a CRM1-independent NES, but the amounts of both subunits bound are greater when they are mixed together before binding. These results suggest that CK2α and -β subunits bind in EB2Nter either individually or as a complex. Interestingly, we also showed that the β subunit binds individually and much more efficiently to the C-terminal 295 amino acids of EB2 (EB2Cter) than the α subunit, and this is unchanged even when the subunits are premixed before binding, although mixing the CK2α and -β subunits induces the formation of a stable heterotetrameric α2β2 complex (data not shown). These results suggest that binding of the β subunit to EB2Cter could destabilize the α2β2 complex. It is noteworthy that herpes simplex virus type 1 ICP27 also interacts in vitro with CK2β via its C-terminal domain, which is highly homologous to the EB2 C-terminal domain (44).
Our in vitro studies suggest that EB2 interacts in vivo with CK2 holoenzyme, but also with individual CK2 subunits in the region containing the NES. However, docking of CK2 or of CK2 subunits does not interfere with the nucleocytoplasmic shuttling of EB2. Moreover, it is unlikely that CK2α participates in the nucleocytoplasmic shuttling of EB2, since EB2 shuttling is resistant to the CRM-1 inhibitor LMB (leptomycin B) (18), whereas CK2α shuttling is inhibited by LMB (12). We are currently mapping the binding sites of CK2 holoenzyme and CK2 subunits on EB2 in order to evaluate how they participate in its function. Indeed, although EB2-bound CK2 phosphorylates several sites at the N terminus of EB2 in vitro, we do not know if it is the binding of CK2 or of its catalytic subunit to EB2 in vivo that is required for phosphorylation of EB2. Conversely, the identification of EB2 binding regions on both CK2α and CK2β subunits needs to be addressed in future studies to understand how EB2 recruits this cellular protein kinase.
Our results also demonstrate that CK2 not only binds to, but also phosphorylates, EB2 in vitro. EB2 expressed in living cells coimmunoprecipitated with and was phosphorylated by a kinase whose activity was reduced by close to 90% by TBB. Several mutations in the 80 N-terminal amino acids of EB2, including the replacement of the three serines at positions 55, 56, and 57 by alanines (EB2S3A) or glutamic acids (EB2S3E), strongly impaired the phosphorylation of the mutated EB2 proteins in the coimmunoprecipitation kinase assay. Interestingly, phosphorylation of the EB2M1 mutant, in which the 50 N-terminal amino acids of EB2 were deleted, was reduced by more than 90%, and phosphorylation of the EB2S3A mutant was reduced by 80%. These results suggest that several sites are differentially phosphorylated in vitro by CK2 bound to EB2, including Ser-55, -56, and -57. We have not mapped the CK2 sites that are phosphorylated in vivo in EB2 and, more specifically, which of the serine residues Ser-55, -56, and -57 are phosphorylated. However, EB2Nter expressed in HEK293T cells migrated in a 2D gel (Fig. 1) as several isoforms, which are probably differentially phosphorylated forms of the protein. We observed a similar migration pattern with immunoprecipitated full-length EB2 (data not shown). Nevertheless, only the phosphorylation of Ser-55, -56, and -57 appears to be important for the function of the protein in the production of infectious virions by 293BMLF1-KO cells, since the deletion mutant EB2M1 efficiently rescued virus production, whereas the phosphorylation ablation mutant EB2S3A and the EB2M1S3A mutant did not. However, it is noteworthy that phosphorylation per se is not essential, as the phosphomimetic mutant EB2S3E is functionally similar to EB2.
The defect of EB2S3A in efficiently inducing the production of virions correlates with a decrease in the cytoplasmic accumulation of several viral late mRNAs. We interpret these results in terms of a decrease in EB2-mediated nuclear export of viral mRNAs due to hypophosphorylation of EB2 at Ser-55, -56, and -57. However, viral-DNA replication is a prerequisite for the expression of the late genes. EB2 induces the replication of the viral genome, although indirectly, by inducing the export of some early mRNAs, including those encoding the primase, the single-stranded DNA-binding protein, and the DNA polymerase (14, 16). The decrease in the cytoplasmic accumulation of the late mRNAs might then be due to a decrease in viral replication due to mutation in EB2 of the three serines to alanines. However, it has been documented that the expression (export) of some late mRNAs was not significantly induced by replication while that of others was, but in both cases their expression was increased by EB2 (16). In our studies, when EB2S3A was expressed in 293BMLF1-KO cells, there was a decrease in the cytoplasmic accumulation of two of these replication-independent but EB2-dependent mRNAs, BdRF1 and BcLF1. This suggests that the effect of EB2S3A on the expression of some late genes is not only linked to replication. It has also been reported that EB2, by interacting with Sp110b, stabilizes some EBV mRNAs (32). It could be that EB2S3A does not interact with Sp110b, leading to decreased stability of some viral mRNAs and a decrease in their cytoplasmic accumulation. Alternatively, CK2 phosphorylation of EB2 may be critical for the recruitment of the RNA export adaptor OTT1 and/or the RNA export receptor TAP/p15. This hypothesis is under investigation. Another possible mechanism is that EB2S3A has a lower affinity for some viral mRNAs, leading to a reduction in their nuclear export, but this issue has not yet been addressed.
Our TAP-tagging experiment also suggests that the mitochondrial protein p32 (31), which was originally described as a protein interacting with the RNA-binding splicing regulator ASF/SF2 (25), is in a complex including EB2 and CK2. The HSV-1 RNA export factor ICP27 is also present in a complex containing CK2 and p32 (3). Since ASF/SF2 binds to TAP/p15 (20), it could also be that EB2 is recruited to viral mRNPs as a complex in which it is phosphorylated by CK2 and binds p32 associated with RNA-bound ASF/SF2. To explore this possibility, we are inactivating the p32-binding site in EB2.
In conclusion, our results strongly suggest that CK2 phosphorylation of EB2 at the level of at least one of the Ser-55, -56, and -57 residues, is critical for the efficient production of infectious virus, likely via cumulative effects on mRNA export and stability and on DNA replication. However, it is not yet known how phosphorylation of EB2 by CK2 regulates its function and how phosphorylation of EB2 is regulated.
Acknowledgments
We thank R. Buckland for reading the manuscript and F. Simian for expert assistance with confocal microscopy.
This work was supported by INSERM and grants from the Association pour la Recherche contre le Cancer (ARC 3420 to A.S.), the Agence Nationale de la Recherche (RPV06120CSA), and Lyon Biopôle. A.S. and E.M. are CNRS scientists. C.M.-P. is the recipient of an ARC fellowship.
Footnotes
Published ahead of print on 15 August 2007.
REFERENCES
- 1.Aguilera, A. 2005. Cotranscriptional mRNP assembly: from the DNA to the nuclear pore. Curr. Opin. Cell Biol. 17:242-250. [DOI] [PubMed] [Google Scholar]
- 2.Batisse, J., E. Manet, J. Middeldorp, A. Sergeant, and H. Gruffat. 2005. The Epstein-Barr virus mRNA export factor EB2 is essential for intranuclear capsid assembly and production of gp350. J. Virol. 79:14102-14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant, H. E., D. A. Matthews, S. Wadd, J. E. Scott, J. Kean, S. Graham, W. C. Russell, and B. Clements. 2000. Interaction between herpes simplex virus type 1 IE63 protein and cellular protein p32. J. Virol. 74:11322-11328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buisson, M., F. Hans, I. Kusters, N. Duran, and A. Sergeant. 1999. The C-terminal region but not the Arg-X-Pro repeat of Epstein-Barr virus protein EB2 is required for its effect on RNA splicing and transport. J. Virol. 73:4090-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, I. B., L. Li, L. Silva, and R. M. Sandri-Goldin. 2005. ICP27 recruits Aly/REF but not TAP/NXF1 to herpes simplex virus type 1 transcription sites although TAP/NXF1 is required for ICP27 export. J. Virol. 79:3949-3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, H., K. Dufu, C. Lee, J. L. Hsu, A. Dias, and R. Reed. 2006. Human mRNA export machinery recruited to the 5′ end of mRNA. Cell 127:1389-1400. [DOI] [PubMed] [Google Scholar]
- 7.Cook, I. D., F. Shanahan, and P. J. Farrell. 1994. Epstein Barr virus SM protein. Virology 205:217-227. [DOI] [PubMed] [Google Scholar]
- 8.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farjot, G., M. Buisson, M. Duc Dodon, L. Gazzolo, A. Sergeant, and I. Mikaelian. 2000. Epstein-Barr virus EB2 protein exports unspliced RNA via a crm-1 independent pathway. J. Virol. 74:6068-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feederle, R., M. Kost, M. Baumann, A. Janz, E. Drouet, W. Hammerschmidt, and H. J. Delecluse. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filhol, O., C. Cochet, G. N. Wedegaertner, G. N. Gill, and E. M. Chambaz. 1991. Coexpression of both alpha and beta subunits is required for assembly of regulated casein kinase II. Biochemistry 30:11133-11140. [DOI] [PubMed] [Google Scholar]
- 12.Filhol, O., A. Nueda, V. Martel, D. Gerber-Scokaert, M. J. Benitez, C. Souchiez, Y. Saoudi, and C. Cochet. 2003. Live-cell fluorescence imaging reveals the dynamic of protein kinase CK2 individual subunits. Mol. Cell. Biol. 23:975-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forwood, J. K., A. Brooks, L. D. Briggs, C. Xiao, D. A. Jans, and S. G. Vasudevan. 1999. The 37-amino-acid interdomain of dengue virus NS5 protein contains a functional NLS and inhibitory CK2 site. Biochem. Biophys. Res. Commun. 257:731-737. [DOI] [PubMed] [Google Scholar]
- 14.Gruffat, H., J. Batisse, D. Pich, B. Neuhierl, E. Manet, W. Hammerschmidt, and A. Sergeant. 2002. Epstein-Barr virus mRNA export factor EB2 is essential for production of infectious virus. J. Virol. 76:9635-9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gruffat, H., E. Manet, A. Rigolet, and A. Sergeant. 1990. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Res. 18:6835-6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han, Z., E. Marendy, Y. Wang, J. Yuan, J. T. Sample, and S. Swaminathan. 2007. Multiple roles of Epstein Barr virus SM protein in lytic replication. J. Virol. 81:4058-4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiriart, E., L. Bardouillet, E. Manet, H. Gruffat, F. Penin, R. Montserret, G. Farjot, and A. Sergeant. 2003. A region of the Epstein-Barr virus (EBV) mRNA export factor EB2 containing an arginine-rich motif mediates direct binding to RNA. J. Biol. Chem. 278:37790-37798. [DOI] [PubMed] [Google Scholar]
- 18.Hiriart, E., G. Farjot, H. Gruffat, M. V. Nguyen, A. P. Sergeant, and E. Manet. 2003. A novel NES and a REF interaction domain both promote mRNA export by the Epstein-Barr virus EB2 protein. J. Biol. Chem. 278:335-342. [DOI] [PubMed] [Google Scholar]
- 19.Hiriart, E., H. Gruffat, M. Buisson, I. Mikaelian, S. Keppler, P. Meresse, T. Mercher, O. A. Bernard, A. Sergeant, and E. Manet. 2005. Interaction of the Epstein-Barr virus mRNA export factor EB2 with human Spen proteins SHARP, OTT1 and a novel member of the family, OTT3, links Spen proteins with splicing regulation and mRNA export. J. Biol. Chem. 280:36935-36945. [DOI] [PubMed] [Google Scholar]
- 20.Huang, Y., R. Gattoni, J. Stevenin, and J. A. Steitz. 2003. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell 11:837-843. [DOI] [PubMed] [Google Scholar]
- 21.Huang, Y., and J. A. Steitz. 2001. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol. Cell 7:899-905. [DOI] [PubMed] [Google Scholar]
- 22.Hübner, S., C. Xiao, and D. A. Jans. 1997. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J. Biol. Chem. 272:17191-17195. [DOI] [PubMed] [Google Scholar]
- 23.Janz, A., M. Oezel, C. Kurzeder, J. Mautner, D. Pich, M. Kost, W. Hammerschmidt, and H. J. Delecluse. 2000. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrate the existence of additional viral ligands. J. Virol. 74:10142-10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koffa, M. D., J. B. Clements, E. Izaurralde, S. Wadd, S. A. Wilson, I. W. Mattaj, and S. Kuersten. 2001. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J. 20:5769-5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krainer, A. R., A. Mayeda, D. Kozak, and G. Binns. 1991. Functional expression of cloned human splicing factor SF2: homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell 66:383-394. [DOI] [PubMed] [Google Scholar]
- 26.Lindtner, S., A. S. Zolotukhtin, H. Uranishi, J. Bear, V. Kulkarni, S. Smulevitch, M. Samiotaki, G. Panayotou, B. K. Felber, and G. N. Pavlakis. 2006. RNA-binding motif protein 15 binds to the RNA transport element RTE and provides a direct link to the NXF1 export pathway. J. Biol. Chem. 281:36915-36928. [DOI] [PubMed] [Google Scholar]
- 27.Lischka, P., Z. Toth, M. Thomas, R. Mueller, and T. Stamminger. 2006. The UL69 transactivator protein of human cytomegalovirus interacts with DEXD/H-Box RNA helicase UAP56 to promote cytoplasmic accumulation of unspliced RNA. Mol. Cell. Biol. 26:1631-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo, M. L., Z. Zhou, K. Magni, C. Christoforides, J. Rappsilber, M. Mann, and R. Reed. 2001. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature 413:644-647. [DOI] [PubMed] [Google Scholar]
- 29.Malik, P., D. Blackbourn, and B. Clements. 2004. The evolutionary conserved Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with REF and acts as an RNA export factor. J. Biol. Chem. 279:33001-33011. [DOI] [PubMed] [Google Scholar]
- 30.Malik, P., and B. Clements. 2004. Protein kinase CK2 phosphorylation regulates the interaction of Kaposi's sarcoma-associated herpesvirus regulatory protein ORF57 with its multifunctional partner hnRNP K. Nucleic Acids Res. 32:5553-5569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Muta, T., D. Kang, S. Kitajima, T. Fujiwara, and N. Hamasaki. 1997. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 272:24363-24370. [DOI] [PubMed] [Google Scholar]
- 32.Nicewonger, J., G. Suck, D. Bloch, and S. Swaminathan. 2004. Epstein-Barr Virus (EBV) SM protein induces and recruits cellular Sp110b to stabilize mRNAs and enhance EBV lytic gene expression. J. Virol. 78:9412-9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nojima, T., T. Hirose, H. Kimura, and M. Hagiwara. 2007. The interaction between cap-binding complex and RNA export factor is required for intronless mRNA export. J. Biol. Chem. 282:15645-15651. [DOI] [PubMed] [Google Scholar]
- 34.Obenauer, J. C., L. C. Cantley, and M. B. Yaffe. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31:3635-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panasyuk, G., I. Nemazanyy, A. Zhyvoloup, M. Bretner, D. W. Litchfield, V. Filonenko, and I. T. Gout. 2006. Nuclear export of S6K II is regulated by protein kinase CK2 phosphorylation at Ser-17. J. Biol. Chem. 281:31188-31201. [DOI] [PubMed] [Google Scholar]
- 36.Paushkin, S. V., M. Patel, B. S. Furia, S. W. Peltz, and C. R. Trotta. 2004. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell 117:311-321. [DOI] [PubMed] [Google Scholar]
- 37.Petersen-Mahrt, S. K., C. Estmer, G. Ohrmalm, D. A. Matthews, W. C. Russell, and G. Akusjärv. 1999. The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J. 18:1014-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinna, L. A. 2002. Protein kinase CK2: a challenge to canons. J. Cell Sci. 115:3873-3878. [DOI] [PubMed] [Google Scholar]
- 39.Sandri-Goldin, R. M. 1998. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev. 12:868-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarno, S., H. Reddy, F. Meggio, M. Ruzzene, S. P. Davies, A. Donella-Deana, D. Shugar, and L. A. Pinna. 2001. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (′casein kinase-2′). FEBS Lett. 496:44-48. [DOI] [PubMed] [Google Scholar]
- 41.Semmes, O. J., L. Chen, R. Sarisky, Z. Gao, L. Zhong, and S. D. Hayward. 1998. Mta has properties of an RNA export protein and increases cytoplasmic accumulation of Epstein-Barr virus replication gene mRNA. J. Virol. 72:9526-9534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theis-Febvre, N., O. Filhol, C. Froment, M. Cazales, C. Cochet, B. Monsarrat, B. Ducommun, and V. Baldin. 2003. Protein kinase CK2 regulates CDC25B phosphatase activity. Oncogene 22:220-232. [DOI] [PubMed] [Google Scholar]
- 43.Vancurova, I., T. M. Paine, W. Lou, and P. L. Paine. 1995. Nucleoplasmin associates with and is phosphorylated by casein kinase II. J. Cell Sci. 108:779-787. [DOI] [PubMed] [Google Scholar]
- 44.Wadd, S., H. Bryant, O. Filhol, J. E. Scott, T.-Y. Hsieh, R. D. Everett, and J. B. Clements. 1999. The multifunctional herpes simplex virus IE63 protein interacts with heterogeneous ribonucleoprotein K and with casein kinase 2. J. Biol. Chem. 274:28991-28998. [DOI] [PubMed] [Google Scholar]
- 45.Williams, B. J. L., J. R. Boyne, D. J. Goodwin, L. Roaden, G. M. Hautbergue, S. A. Wison, and A. Whitehouse. 2005. The prototype g-2 herpesvirus nucleocytoplasmic shuttling protein, ORF57, transports viral RNA through the cellular mRNA export pathway. Biochem. J. 387:295-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong, K. M., and A. J. Levine. 1989. Characterization of proteins encoded by the Epstein-Barr virus transactivator gene BMLF1. Virology 168:101-111. [DOI] [PubMed] [Google Scholar]
- 47.Zhi, Y., and R. M. Sandri-Goldin. 1999. Analysis of the phosphorylation sites of herpes simplex virus type 1 regulatory protein ICP27. J. Virol. 73:3246-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zolotukhin, A. S., W. Tan, J. Bear, S. Smulevitch, and B. K. Felber. 2002. U2AF participates in the binding of TAP (NXF1) to mRNA. J. Biol. Chem. 277:3935-3942. [DOI] [PubMed] [Google Scholar]