

Abstract
The goal of this project was to better define the relationship between the endoribonuclease activity of murine hepatitis virus (MHV) Nsp15 (mNsp15) and its role in virus infection. Molecular modeling demonstrated that the catalytic residues of mNsp15 are superimposable with its severe acute respiratory syndrome coronavirus ortholog. Alanine substitutions at three key residues in the mNsp15 catalytic pocket (H262, H277, and G275) and a double-mutant version (H262P and H277A) generated proteins with greatly reduced but detectable endoribonuclease activities. Furthermore, these mutant proteins demonstrated lower cleavage specificities for uridylate than wild-type (WT) mNsp15. These mutations were successfully incorporated into viruses named vH262A, vH277A, vG275A, and vH262P+H277A. All four mutant viruses formed plaques with diameters similar to that of MHV-A59 1000 (WT virus) on several different cell lines. Interestingly, viruses with a mutation at a noncatalytic residue, D324A, could not be recovered despite repeated attempts, and expression of mNsp15 containing the D324A mutation in Escherichia coli resulted in an insoluble protein. Plaques derived from vH262A produced approximately 6- to 13-fold fewer PFU than those from WT virus. Cells infected with mNsp15 mutant viruses accumulated lesser amounts of plus- and minus-sense subgenomic RNAs and spike protein than WT virus. The expression of mNsp15 in trans by transient transfection partially restored RNA synthesis by vH262A. These results demonstrate that mNsp15 is required for optimal infection by MHV.
Coronaviruses are significant pathogens of humans and animals. They are also interesting because they use replication mechanisms that are unusual among the positive-stranded RNA viruses (18). For example, coronaviruses produce minus-strand subgenomic RNAs (sgRNAs) by a discontinuous transcription mechanism and utilize these as templates for subgenomic mRNA (sgmRNA) synthesis (18, 22). In addition, the viral genome encodes a number of proteins that are not typically found in the positive-strand RNA viruses, including a potential RNA primase and several RNA-processing enzymes such as ExoN, XendoU, and cap methyltransferases (20, 24-26, 32).
One of these novel RNA-processing enzymes is nonstructural protein 15 (Nsp15), a hexameric endoribonuclease that preferentially cleaves 3′ of uridylates (12, 21, 28). Severe acute respiratory syndrome coronavirus (SARS-CoV) recombinant Nsp15 (sNsp15) produced in Escherichia coli has endoribonuclease activity that is stimulated by Mn2+, generating a 2′,3′ cyclophosphodiester cleavage product (2, 3). The structure of sNsp15 was first determined by electron microscopy and then by X-ray crystallography (3, 21). The structure of murine hepatitis virus (MHV) Nsp15 (mNsp15) was also recently solved (28). A mutation (D6408A) in the Nsp15 coding sequence of human coronavirus (HCoV) 229E prevented viral RNA accumulation, suggesting that HCoV-229E Nsp15 is required for viral replication (12).
Both the arterivirus and coronavirus families are members of the order Nidovirales and encode orthologs of Nsp15 (NendoU) endoribonuclease (9, 19). A thorough analysis of mutations in the ortholog of NendoU in the arterivirus equine arteritis virus (EAV) revealed that the endoribonuclease exerts significant and pleiotropic effects on EAV replication (19). Several mutations, including mutations in the putative active site (H2693 and H2978), reduced viral plaque formation significantly but did not abolish EAV infection in cultured cells. Other mutations, including D3014A, which corresponds to the D6408A mutation in HCoV-229E Nsp15, were lethal to EAV replication and abolished EAV infection. The complex phenotypes conferred by the mutant viruses suggest that Nsp15 may have more than one role in the replicative cycles of coronaviruses and their relatives.
Murine coronaviruses have provided good model systems for studies of multiple sclerosis, virus-induced demyelination, acute and chronic hepatitis, gastroenteritis, and pneumonitis (4-6, 11, 15, 27). Recent phylogenetic studies, including a comparison of an extensive set of amino acid sequences, revealed that SARS-CoV is best placed as an early branch of the group 2 coronaviruses, which includes MHV (9, 26). MHV strain 1-infected A/J mice develop a severe pulmonary infection characterized by congestion, pulmonary infiltrates, formation of a hyaline membrane, and hemorrhage, symptoms similar to those caused by SARS-CoV in humans (4, 5). Moreover, the RNA cis-acting elements, such as the 3′ untranslated region (3′ UTR) and stem-loops in the 5′ UTR, of SARS-CoV can functionally substitute for their counterparts in MHV-A59, confirming a close phylogenetic relationship between SARS-CoV and MHV (8, 13, 14). Taken together, these results suggest that MHV is an excellent model system for in vivo studies of SARS-CoV pathogenesis.
The goal of this study was to determine whether mNsp15 endoribonuclease activity is required for the MHV infection process. We produced wild-type (WT) and mutant mNsp15 and examined the requirements for its active-site residues in RNA cleavage in vitro. We also generated MHVs containing mutations in mNsp15 and found that the mNsp15 mutant viruses were defective for viral infection in permissive cells, especially in viral RNA accumulation. Lastly, we demonstrated that WT mNsp15 can partially complement both the RNA accumulation and the infectivity of mutant MHV in trans.
MATERIALS AND METHODS
Bacteria, viruses, and cells.
E. coli strain Top10 (Gibco-BRL), which was used for DNA manipulations, was cultured at 37°C in Luria-Bertani and 2× yeast extract-tryptone media. DBT and 17CL-1 cells were maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% calf serum (HyClone, Logan, UT). L2 cells were maintained at 37°C under 3% CO2 in DMEM supplemented with 10% calf serum. BHK-21 baby hamster kidney cells expressing the MHV receptor (BHK-R) were grown in minimal essential medium supplemented with 10% calf serum and G418 (800 μg/ml) to select for cells expressing the MHV receptor.
Recombinant Nsp15.
Recombinant Nsp15 containing an N-terminal His6 tag was expressed in E. coli and purified using immobilized metal affinity chromatography followed by Mono Q ion-exchange chromatography as previously described (2). Site-directed mutations in Nsp15 cDNA were made with the QuikChange II site-directed mutagenesis kit (Stratagene, San Diego, CA). The entire cDNA was sequenced to ensure that only the intended mutation was introduced.
Endoribonuclease assays.
RNAs used for endoribonuclease assays were chemically synthesized by Dharmacon, Inc. (Boulder, CO) and contain the SARS-CoV transcription regulatory sequence (TRS) (5′-CAACU/AAACGAAC [the slash indicates the expected cleavage site]) for sgmRNA3, encoding the spike (S) protein. RNAs used in the gel-based endoribonuclease assays were labeled at their 5′ termini with [γ-32P]ATP and T4 polynucleotide kinase according to the protocol described previously (2).
The standard RNA cleavage assay mixture contained 1 μM RNA (1 × 104 cpm) in buffer A (50 mM Tris [pH 7.5], 50 mM KCl, 1 mM dithiothreitol, and 5 mM MnCl2) at 30°C. The endoribonuclease reactions were terminated by the addition of a gel-loading buffer containing 7.5 M urea. Products were separated by electrophoresis in gels composed of 7.5 M urea and 20% polyacrylamide. Gels were wrapped in plastic and exposed to a PhosphorImager screen for quantification using Molecular Dynamics software. Each result shown was reproduced in at least two independent experiments.
The real-time endoribonuclease assay used a substrate from Integrated DNA Technologies, Inc. (Coralville, IA). The substrate has a carboxyfluorescein (FAM) fluorophore at the 5′ end and tetramethylrhodamine (TAMRA), which quenches FAM fluorescence, at the 3′ end. The substrate contains only 4 nucleotides (nt), of which 3 are deoxyribonucleotides while 1 is a ribonucleotide that is a potential cleavage site for Nsp15. The excitation wavelength was 492 nm, and the maximum emission of the substrate at 518 nm was determined in buffer A. Changes in fluorescence were measured over time using a BMG plate reader.
Molecular modeling.
The molecular docking program Dock 6.0 was used to perform flexible docking of 3′-UMP (from Protein Data Bank [PDB] identification number [ID] 4RSK; RNase A:3′-UMP complex structure) into the mNsp15 crystal structure (PDB ID 2GTH), which was kept rigid. A set of spheres that represent the negative image of the binding site were defined within the 10-Å radius of the catalytic-site residues H262, H277, K317, and T321 of mNsp15 protein to adopt the sphere-matching algorithm of Dock 6.0. Incremental construction (anchor-and-grow method) was used to allow the flexibility of the ligand. The automatic matching mode was used with 20 configurations per ligand building cycle. Interaction between the ligand and the receptor was evaluated by the grid score (a combination of van der Waals and electrostatic components) and visual inspection. The interhydrogen bonds between the highest-ranked 3′-UMP and the catalytic-site residues of mNsp15 were further refined by minor manual model building using Xfit (16).
The atomic coordinates of the WT mNsp15 crystal structure (28) (PDB ID 2GTH) were used to construct a model of the mNsp15 G275A mutant with Modeler (Insight II; Accelrys). Residues within a 10-Å radius of A275 were subjected to a simulated annealing optimization with respect to the variable target function of Modeler. The figures were prepared using USCF Chimera (http://www.cgl.ucsf.edu/chimera/).
Construction of plasmids used in an MHV reverse-genetic system.
The primers and plasmids used in this study are listed in Tables 1 and 2, respectively. The F plasmid of the MHV-A59 1000 reverse-genetic system containing the mNsp15 cDNA sequence (30) was used to introduce mutations into mNsp15. Briefly, the 3.38-kb BamHI-HpaI fragment of the F plasmid was amplified and cloned into the pGEM-T vector (Promega, Madison, WI) to produce pWtNsp15-PG. This plasmid harbors sequences of open reading frames for MHV Nsp15, Nsp16, and 2a protein. Using the QuikChange II site-directed mutagenesis kit (Stratagene, Inc., La Jolla, CA) according to the manufacturer's instructions, mutations H262A, G275A, H277A, and D324A were introduced into mNsp15 of pWtNsp15-PG to yield p1F-PG, p2F-PG, p3F-PG, and p4F-PG, respectively. The sequences of BamHI-HpaI fragments carrying single mutations in mNsp15 were determined in order to confirm that the desired sequences were obtained. The BamHI-HpaI fragments containing the mutations were excised and religated into BamHI-HpaI-digested plasmid F, and the ligation products were then transformed into the Top10 strain of E. coli. The region of the recovered clones between the BamHI and HpaI sites was sequenced again to verify that the desired mutation was recovered.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′→3′)a |
|---|---|
| H262A_SD_F | GGACTACGCGTTTGAAgcCGTTGTTTATGGTAGTTTTAACC |
| H262A_SD_R | GGTTAAAACTACCATAAACAACGGCTTCAAACGCGTAGTCC |
| G275A_SD_F | CCAGAAGATTATTGGAGcTTTGCATTTGCTTATTGGCTTAGCCC |
| G275A_SD_R | GGGCTAAGCCAATAAGCAAATGCAAAGCTCCAATAATCTTCTGG |
| H277A_SD_F | CCAGAAGATTATTGGAGGTTTGgcTTTGCTTATTGGCTTAGCCC |
| H277A_SD_R | GGGCTAAGCCAATAAGCAAAGCCAAACCTCCAATAATCTTCTGG |
| D324A_SD_F | GTAAGAGTGTGTGCACTGTTATTGCTTTATTGTTAGATGATTTTGTGG |
| D324A_SD_R | CCACAAAATCATCTAACAATAAAGCAATAACAGTGCACACACTCTTAC |
| Oligonucleotide 35-5_R | GCT CGG ATC CAC TAG TAA CGG CCG CCA GTG |
| Oligonucleotide 28-1_F | GTA TGT TAA CAA ACA TGC ATT CCA CAC C |
| MHVpcdna-ab | AAAAGGTACCATGGGTTTAGAAAATGTAGTG |
| MHVpcdna-bc | CGAAGGATCCTACAGATCCTCTTCAGAGATGAGTTTCTGCTCCTGCAAACGAGGATAGAAAGTCATGACCTTCTC |
| MHV 5′ 1-20 (+) | TATAAGAGTGATTGGCGTCC |
| 7059 (−) oligonucleotide 8d | CTGGTGTTACACAGGCAG |
| S_seq_R1 primere | ATTGCTGAGCGTAAACAACG |
| N (−) primerf | GGC ACT CCT TGT CCT TCT |
| B35For | TAG TAC TCT ACC TGG TTT T |
| 17TG | TTT TTT TTT TTT TTT TTG |
| GAPDH-F2 primer | ACCCAGAAGACTGTGGATGG |
| GAPDH-R2 primer | CACATTGGGGGTAGGAACAC |
Lowercase letters indicate mutations.
Sense primer containing a KpnI site, A at −3, a start codon (ATG), and G at +4 to replace S with G.
Antisense primer with a Myc tag, a stop codon (TGA), and BamHI site.
Corresponding to nt 611 to 628 in the MHV-A59 genome (GenBank accession number NC001846); used to specifically prime minus-strand sgRNA1.
Corresponding to nt 25320 to 25339 in the MHV-A59 genome; used to specifically prime minus-strand sgRNA3.
Corresponding to nt 29920 to 29937 in the MHV-A59 genome; used to specifically prime minus-strand sgRNA7.
TABLE 2.
Plasmids used in this study
| Plasmid | Description |
|---|---|
| Plasmid Fa | PCR-XL-Topo vector containing a 6.9-kb DNA fragment of MHV cDNA (bp 15755-22739 on the MHV-A59 genome) |
| pWtNsp15-PG | pGEM-T (Promega, Madison, WI) containing a PCR product corresponding to a 3.4-kb BamHI-HpaI fragment in plasmid F of MHV cDNA |
| p1F-PG | pWtNsp15-PG containing an H262A mutation in mNsp15 |
| pHK1Fb | Plasmid F containing the mNsp15 H262A mutation |
| p2F-PG | pWtNsp15-PG containing a G275A mutation in mNsp15 |
| pHK2F | Plasmid F containing the mNsp15 G275A mutation |
| p3F-PG | pWtNsp15-PG containing an H277A mutation in mNsp15 |
| pHK3F | Plasmid F containing the mNsp15 H277A mutation |
| p4F-PG | pWtNsp15-PG containing a D324A mutation in mNsp15 |
| pHK4F | Plasmid F containing the mNsp15 D324A mutation |
| pcD-mNsp15 | pcDNA1 (Invitrogen, Carlsbad, CA) containing mNsp15 coding sequence |
| pcD-sNsp15 | pcDNA1 containing sNsp15 coding sequence |
| pcD-GFP-sNsp15 | pcDNA1 containing GFP conjugated to the N terminus of the mNsp15 coding sequence |
One of seven plasmids used in the MHV-A59 reverse-genetic system (30).
The BamHI-HpaI fragment containing the H262A mutation in p1F-PG was excised and religated into BamHI-HpaI-digested plasmid F to construct pHK1F. Plasmids pHK2F, pHK3F, and pHK4F were constructed using p2F-PG, p3F-PG, and p4F-PG, respectively, with the same protocol as that for the construction of pHK1F.
Recovery of mNsp15 mutant viruses.
The reverse genetic system for MHV-A59 1000 used in this study was initially described by Yount et al. (30). cDNAs representing the entire MHV-A59 genome containing either the mNsp15 WT sequence or a mutant sequence were constructed by ligation of A and G cloned cDNAs as described previously (30). WT genomes containing mNsp15 WT and mutant sequences were transcribed in vitro and electroporated into BHK-R cells as previously described (30). Cultures were observed for as long as 72 h postinfection (hpi) for the development of cytopathic effects (CPE), which consist mainly of syncytium formation. Virus-infected cultures were then harvested and frozen at −70°C. Mutant viruses were subjected to one round of plaque purification and were amplified once in DBT cells to generate stocks for additional characterizations. The sequences of recovered viruses corresponding to the 5′ and 3′ UTRs, as well as the portions encoding mNsp15 mutant proteins, were amplified by reverse transcription-PCR (RT-PCR), followed by direct sequencing of the amplified products.
Plaque assays.
In order to determine growth curves for mNsp15 mutant viruses, DBT cells were grown in 96-well plates, and replicate wells were infected at a multiplicity of infection (MOI) of either 0.5 or 1.5 with mNsp15 mutant viruses or MHV-A59 1000 (WT virus). After the inocula were washed away, cultures were incubated until 0, 4, 8, 12, 16, or 24 hpi, at which time they were frozen at −70°C. Triplicate samples were obtained for all time points. Virus production was quantified by plaque assays on monolayers of L2 cells. The two growth curves were done with independent isolates. Isolated plaques were resuspended and titered on L2, 17CL-1, and DBT cells. The diameters of the resultant plaques were determined and compared with those formed by the WT virus.
Analysis of viral RNA accumulation.
DBT cells (2.25 × 105 per well) were seeded into 12-well plates and incubated at 37°C for 15 h to reach approximately 2.5 × 105 cells per well. Replicate cultures were then infected with either WT virus or viruses carrying mutations in mNsp15 at an MOI of 1, further incubated for 6 h, washed twice with phosphate-free DMEM, fed with DMEM supplemented with 2% dialyzed calf serum and 10 μg/ml of actinomycin D, and incubated at 37°C. Following a 15-min incubation, the medium was replaced with a phosphate-free medium containing 10 μg/ml actinomycin D, 2% dialyzed serum, and 200 μCi/ml 32PO4 and was incubated at 37°C for 5.5 h, by which time 90% of the cells infected with WT virus had formed syncytial giant cells. The labeled cultures were washed twice with cold phosphate-buffered saline, and RNA was extracted using an RNeasy minikit (Qiagen). The amount of RNA in each sample was measured using the RediPlate 96 RiboGreen RNA quantitation kit (Invitrogen Inc.). Equal amounts of radiolabeled viral RNA were denatured in formaldehyde gel-loading buffer containing ethidium bromide (20 μg/ml) at 65°C for 15 min and then electrophoresed through a 1% formaldehyde-agarose gel at 100 V for 6 h. Following electrophoresis, the gel was illuminated with UV light, the image was captured with a FluorChem 8900 (Alpha Innotech, San Leandro, CA) imaging system, and the relative amount of 28S rRNA bands was determined by densitometry. The gel was soaked in 70% methanol for 30 min, dried over a vacuum, and exposed to X-ray film. The amounts of radiolabel in individual sgRNAs and the genomic RNA (gRNA) were determined by exposing the dried gel to a Molecular Dynamics PhosphorImager equipped with Storm 8.2 software. The 28S rRNA in each sample was used to normalize the PhosphorImager signals to account for small differences in the amount of RNA loaded per sample.
Quantitative RT-PCR (RT-qPCR) was used to determine the effect of the H262A mutation on MHV minus-strand RNA accumulation. DBT cells (4 × 105) were seeded in parallel T25 flasks, incubated at 37°C for 48 h under 5% CO2, and then infected with either the H262A mutant or WT virus at an MOI of 0.5. Total RNAs were extracted at 8 and 12 hpi, and 2 μg of the RNA was subjected to Superscript II RNase H-reverse transcriptase (Invitrogen Inc.) in the presence of a primer specific for MHV minus-strand RNAs (primer MHV 5′ 1-20 [Table 1]). The resultant reverse transcription products were purified using a QIAquick PCR purification kit (Qiagen Inc., Valencia, CA), and 5% of the product was subjected to amplification with antisense primers specific for MHV RNA1, sgRNA3, and sgRNA7. The levels of the products were quantified with iQ SYBR Green Supermix (Bio-Rad, Philadelphia, PA). Primers used to specifically prime MHV minus-strand RNA1 and sgRNA3 and -7 are listed in Table 1. The level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in each sample was used as the internal control in each qPCR. A negative control in the absence of primers was performed for each reaction.
Accumulation of S protein during MHV infection.
DBT cells (2.5 × 105 per well) were seeded onto 12-well plates, incubated at 37°C for 12 h under 5% CO2, and infected with either an mNsp15 mutant or WT virus at an MOI of 1. At 8 and 10 hpi, total proteins were harvested as described previously (31). Total protein (2 μg per lane) was electrophoresed on sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes for Western blotting as described previously (31). Protein was detected with goat anti-MHV S protein antibodies (kindly provided by K. Holmes), horseradish peroxidase (HRP)-conjugated anti-goat immunoglobulin G (IgG) antibodies, and the Immobilon Western Detection reagents (Millipore, Billerica, MA) according to the manufacturer's recommendations. As an internal control, GAPDH was detected using a mouse primary antibody (Alpha Diagnostics, San Antonio, TX) and HRP-conjugated anti-mouse IgG as a secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
trans-Complementation assay.
DNA fragments encoding mNsp15 and sNsp15 were amplified using sense and antisense primers that harbored BamHI and KpnI restriction sites, respectively (Table 1). The fragments were first cloned into pGEM-T vectors and sequenced to confirm that there were no additional mutations in Nsp15. The BamHI-KpnI fragments were then excised from pGEM-T and subcloned into BamHI-KpnI-digested pcDNA3.1(+) (Invitrogen, Carlsbad, CA) to generate plasmids pcD-mNsp15 and pcD-sNsp15, respectively (Table 2). This placed mNsp15 and sNsp15 under the control of the cytomegalovirus immediate-early promoter in pcDNA3.1. The SARS-CoV Nsp15 protein was also tagged with an N-terminal Myc epitope by adding the sequence to the oligonucleotide primer (Table 1). One million DBT cells seeded in each well of a six-well plate were transfected with either pCDNA3.1(+), pcD-mNsp15, or pcD-sNsp15 by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as recommended by the manufacturer. Thirty hours later, the cells were infected with the H262A mutant virus (vH262A) or WT virus at an MOI of 1. Metabolic labeling of the MHV plus-strand RNAs was conducted at 6 hpi using the protocol described above. As a control, a pcDNA3.1 plasmid encoding green fluorescent protein (GFP) conjugated to the N terminus of sNsp15 was constructed and named pcD-GFP-sNsp15. GFP expression in DBT cells transfected with pcD-GFP-sNsp15 was used as an indication of transfection efficiency and sNsp15 expression in DBT cells.
RESULTS
Modeling of the active site of mNsp15.
sNsp15 cleaves RNA by a mechanism identical to that of RNase A (3, 21). To gain insight into the mechanism of RNA cleavage used by the MHV protein (mNsp15), we modeled the active site of mNsp15 complexed with 3′-UMP by using the coordinates from the crystal structure of mNsp15 (PDB ID 2H85) and the molecular docking program Dock 6.0 (17). The best model has UMP oriented so that the hydrogen-bonding face of uracil is contacted by T321 of mNsp15 (Fig. 1A). The putative catalytic histidine, H262 in mNsp15, is positioned to interact with the phosphate of the UMP (corresponding to the cognate phosphodiester in the substrate RNA), and H277 would interact with the ribose 2′ hydroxyl moiety (Fig. 1A, left). These results are consistent with the recognition of uridine by the active-site residues of sNsp15 (Fig. 1A, center) as proposed by Ricagno et al. (21), as well as with the proposed cleavage mechanism that results in the formation of a 2′-3′ cyclic phosphodiester on the RNA product (2). In fact, despite some differences between the amino acid residues in the two catalytic pockets, the two active sites are virtually superimposable (Fig. 1A, right).
FIG. 1.

Biochemical analysis of mNsp15. (A) Molecular modeling of the active sites for mNsp15 and sNsp15. The UMP is indicated in yellow, red, and blue, for carbon, oxygen, and nitrogen, respectively. (Left) The location of the UMP in mNsp15 was determined using the molecular docking program Dock 6.0. (Center) The comparable residues in sNsp15 are shown with UMP. (Right) Superposition of sNsp15 catalytic-site residues on mNsp15. (B) Recombinant mNsp15 WT and mutant proteins used in the biochemical analysis. The purified proteins were separated on a 4-to-12% acrylamide-sodium dodecyl sulfate gel and stained with Coomassie brilliant blue. (C) Gel-based analysis of the endoribonuclease activities of sNsp15 and mNsp15. Lanes C, uncleaved 13-nt TRS RNA substrate containing a single uridylate. The amount of protein added to the reaction mixture, in nanograms, is given above each lane, as well as the identity of the WT or mutant protein. Arrows identify the Nsp15 cleavage products.
Our modeling studies also predicted that G275 in mNsp15 would contribute to the active sites in two ways. First, the G275 backbone nitrogen should add to the anionic charge of that face of the active site. Second, the small hydrogen R group of G275 is needed to sterically accommodate UMP; substitution of the bulkier alanine should cause the Cβ atom to disrupt interactions between the catalytic histidine residues and UMP. Modeling of the mNsp15 active site with an alanine in place of the glycine at position 275 revealed that UMP could no longer be docked (data not shown).
Biochemical characterization of mNsp15.
To allow functional comparison of sNsp15 and mNsp15, mutants with alterations of residues H262, H277, and G275 in mNsp15 were constructed (Fig. 1B). WT mNsp15 and the three mutant proteins with single amino acid substitutions in the catalytic pocket (H262A, G275A, H277A) were tested for their abilities to cleave an RNA substrate containing the SARS-CoV TRS for sgmRNA3 by a gel-based endoribonuclease assay. The 13-nt TRS substrate contains uridylate at the fifth position from the 5′ terminus. Consistent with our previous report (10), both sNsp15 and mNsp15 were able to generate the 5-nt product expected from preferential cleavage at this uridylate, indicating specificity for uridylates (Fig. 1C). Quantification of cleared products from several gel-based assays showed that the G275A and H277A proteins retained endoribonuclease activity at 2% and 10% (Fig. 1C), respectively, of WT mNsp15 enzymatic activity, while cleavage by the H262A protein was at background for this assay (Fig. 1C and data not shown). Mutations of the comparable catalytic histidines (H249A and H234A) in sNsp15 significantly reduced endoribonuclease activity (10). These results indicated that, in agreement with the modeling studies, the catalytic residues comparable to those in sNsp15 are also required for efficient endoribonuclease activity of mNsp15.
To extend the comparisons of mNsp15 and sNsp15, we examined preferential cleavage of uridylates by using a fluorescent real-time endoribonuclease assay previously described by Bhardwaj et al. (2) (Fig. 2A). This assay uses one of four nucleic acid substrates, each of which contains a 5′ fluorophore paired with a 3′ quencher and one ribonucleotide; the remaining three nucleotides have deoxyriboses and cannot be cleaved by RNases. When cleavage takes place, the dissociation of the quenching group from the fluorophore results in fluorescence intensity that can be measured in real time. For brevity, each substrate is named by the cleavable ribonucleotide (e.g., rU denotes the substrate with a uridylate). In our standard assay performed with 0.04 μM Nsp15, neither sNsp15, WT mNsp15, nor any of the mNsp15 mutant proteins could cleave rA or rG substrates (data not shown). As expected, however, both sNsp15 and mNsp15 actively cleaved rU, at rates of 44.5 and 29.6 μM s−1, respectively. With the rC substrate, the rates of hydrolysis were lower for both sNsp15 and mNsp15, although the rate was higher for mNsp15 than for sNsp15 (5.8 versus 1.8 μM s−1, respectively). The ratio of rU to rC cleavage was calculated to determine the specificity factor (SF). sNsp15 had an SF of ∼25, while mNsp15 had an SF of ∼3.7 (mean from six trials; range, 5 to 3). Therefore, while both enzymes preferentially cleaved uridylates, sNsp15 had higher specificity for uridylate than did mNsp15.
FIG. 2.

Results of a real-time fluorescent endoribonuclease assay with sNsp15 and mNsp15. (A) Specificity for cleavage of rU or rC substrates by sNsp15 and mNsp15. The cleavable uridylate or cytidylate in the substrate is identified by the letters rU or rC, respectively. AFU, arbitrary fluorescence units. (B) Examination of the endoribonuclease activities of the H262A and H262P H277A mutant proteins. Endoribonuclease activity was tested at 0.08 μM WT or mutant mNsp15. The substrate used contains a cognate rU or rC, as indicted on the right. (C) Purity of WT and H262P+H277A mutant proteins detected by silver staining (Amersham, Piscataway, NJ).
Mutations in the RNase A catalytic triad are known to affect RNase activity by degrees rather than to abolish it altogether (7). While the H277A and G275A mutants retained detectable activity in the gel-based assay, the H262A mutant did not. The lack of cleavage by the H262A mutant in this assay could reflect an issue with the background level in the gel-based assay. To determine whether the mutations in the catalytic pocket completely abolished mNsp15 activity, we incubated twice the normal concentration of WT mNsp15 and the H262A mutant (each at 0.08 μM) for the fluorescence-based assay and monitored their activities over a 400-s period. We note that Nsp15 is enzymatically active only as a hexamer (2, 10), and a change in protein concentration will affect hexamer formation. As a control, the hepatitis C virus Ns5B protein was added at 0.24 μM with the rU substrate, and no increase in fluorescence was observed (data not shown). The H262A mutant did indeed release fluorescence from the rU substrate (Fig. 2B). Further increases in the concentration of the H262A mutant resulted in even more obvious rU cleavage (data not shown). Along with the results for the G275A and H277A mutants from the gel-based assay, our data suggest that none of the three mutations in the mNsp15 catalytic pocket completely abolished the endoribonuclease activity of mNsp15.
To examine the effects of changes in the catalytic pocket further, we expressed a protein that contains two mutations, H262P and H277A. This mutant corresponds to a virus we fortuitously obtained and characterized (see below). The H262P+H277A protein was expressed well and was purified over three columns to apparent homogeneity, as determined by silver-stained denaturing gel electrophoresis (Fig. 2C). When tested in the fluorescent assay, the H262P+H277A mutant retained detectable endoribonuclease activity, although at a slightly lower level than that of the H262A mutant (Fig. 2B). These results confirm that mutations in the catalytic pocket of mNsp15 significantly reduce, but do not completely abolish, endoribonuclease activity.
In the real time assay, we used rC as a control and noticed that the H262A mutant cleaved rC about as well as rU, while the WT protein preferentially cleaved rU (Fig. 2A). Therefore, we determined the SFs for the H262A and H262P+H277A mutants and found them to be 1.35 and 1.17, respectively. These results demonstrate that the active-site mutations decreased the specificity of RNA cleavage relative to that for WT mNsp15.
MHV with mutations in Nsp15.
To examine the biological effects of mutations in mNsp15, we assembled full-length MHV-A59 1000 genomes containing H262A, G275A, and H277A substitutions in mNsp15 by in vitro transcription from ligated cDNAs. We also attempted to assemble a virus with the D324A mutation, since the corresponding substitutions in HCoV and EAV have been reported to be lethal for virus production (12, 20). BHK-R cells electroporated with the WT genome or with genomes with the H262A, G275A, or H277A mutation all developed CPE after 24 h. The same manipulation with MHV gRNA carrying the D324A substitution did not produce CPE, and no mutant virus was recovered in three independent trials (Fig. 3A). In one of the experiments, we incubated the electroporated cells at 34°C and 40°C in the event that the mutation rendered the virus temperature sensitive, but we still failed to obtain viral plaques (data not shown). Thus, the MHV genome carrying the D324A substitution appeared to be nonviable, consistent with the results of previous studies of HCoV-229E and EAV (12, 19).
FIG. 3.

Plaque morphology and growth kinetics for mNsp15 mutant and WT viruses. (A) Cultures of BHK-R cells were electroporated with full-length WT or mutant mNsp15 transcripts and seeded on DBT cells in 75-cm2 flasks. The progeny was plaque purified and amplified once in DBT cells. Plaques from mNsp15 single- and double-mutant viruses and WT virus were produced in six-well plates seeded with L2 cells. Nonviable D324A mutant genomes (gD324A) did not form any visible plaques. (B and C) Triplicate DBT cell cultures in a 96-well plate were infected at the indicated MOI and harvested at the indicated times postinfection, and viral titers were determined by a plaque assay. Note that independent viral isolates were used in panels B and C. (B) Growth curves of vH262A, vG275A, vH277A, and WT virus at an MOI of 0.5. (C) Growth curves of vH262A, vG275A, vH277A, vH277A+H262P, and WT virus at an MOI of 1.5.
For clarity, the viruses are named with the letter “v” in front of the mutation. Mutants vH262A, vG275A, and vH277A produced plaques similar to those of the WT virus (Fig. 3A). The resultant viruses were recovered from the medium, plaque purified, and expanded in DBT cells. Sequencing of RT-PCR fragments containing mNsp15 confirmed that vH262A and vG275A possessed only their expected single mutations in mNsp15. From the electroporation to recover vH277A, we obtained plaques of the intended mutant without other changes in Nsp15. However, three of the independent plaques had a second spontaneous mutation in mNsp15 (L291Q, I250Q, or H262P). The virus with both the H277A and the H262P mutation, named vH262P+H277A, was characterized further. The fact that we recovered single-mutant viruses for all three intended mutations as well as several double mutants indicated that a reduction in mNsp15 endoribonuclease activity is not lethal to MHV.
To quantify the effects of the mutations, we examined the morphologies of the plaques from WT and mutant viruses on monolayers of L2 cells. Plaque diameters for the single and double substitution mutants were modestly reduced from those for WT virus (Fig. 3A and Table 3; also data not shown). Mutants vH262A, vG275A, vH277A, and vH262P+H277A averaged a 10 to 17% reduction in plaque diameter in comparison to WT virus. Similar results were obtained with the permissive cell types 17CL-1 and DBT. In no case was the difference in plaque diameter between the mutant and the WT virus statistically significant (P > 0.5).
TABLE 3.
Titration and diameters of virus particles within single plaques isolated from either H262A mutant virus or MHV-A59 1000 in L2 cellsa
| Virus | Mean titer ± SEb (%) | Plaque diam (mm)c |
|---|---|---|
| MHV-A59 1000 | 5.64 × 105 ± 0.80 × 105 (100) | 2.22 ± 0.06 |
| vH262A | 6.76 × 104 ± 1.77 × 104 (12) | 2.26 ± 0.06 |
WT virus or vH262A was isolated from 12 individual plaques formed on L2 cells by using Pasteur pipettes, resuspended in 1 ml of DMEM, sonicated three times for 20 s at 35% power in a cup sonicator, and titered.
From 12 plaques.
Calculated as an average for at least 25 plaques. α is 0.05, and Tukey's W procedure was performed.
The infectivities of the mutant viruses were analyzed further by determining their titers in several independent plaque-forming assays. Viruses were eluted from single plaques and titered on L2 cells. The mutant vH262A produced only 12% of the PFU of WT virus (Table 3). These results indicate that, despite the similarity in plaque morphology, the mutant viruses do not grow to the same level as the WT virus.
To confirm that the mutant viruses are defective in replication, the growth kinetics of WT and mutant viruses were determined using an MOI of 0.5 to infect DBT cells. All three mutants grew to lower titers (by as much as 1.5 log units) than WT virus (Fig. 3B). The maximum titer reached by vH277A was three- to fourfold higher than those of vH262A and vG275A (Fig. 3B). Interestingly, the reduction in viral titer was more pronounced after 12 hpi than at earlier times postinfection for all of the mutants. When the same experiment was performed at an initial MOI of 1.5, the more-pronounced reductions in the titers of mutant viruses after 12 h were again observed, but the differences from the WT were smaller than those observed in the experiments performed with an MOI of 0.5 (Fig. 3C). These results prompted us to examine the double mutant vH262P+H277A. Again, it had a slightly more pronounced defect 12 h after the initiation of infection than the viruses with single amino acid substitutions (Fig. 3C). Furthermore, vH262P+H277A produced less infectious virus than the single mutants.
RNA accumulation in cells infected with mNsp15 mutant viruses.
We examined whether one mode of viral RNA replication and/or transcription was differentially affected. MHV gRNA and sgmRNAs were metabolically radiolabeled in the presence of actinomycin D to suppress cellular transcription and were then analyzed by denaturing gel electrophoresis (Fig. 4A). All mNsp15 single-mutant viruses synthesized reduced amounts of gRNA and of all six sgmRNAs, but the relative ratios for the RNAs were approximately the same as those seen in a WT infection (Table 4). Mutant vH277A produced only 56% of the total viral RNAs detected in WT-infected cells. Mutants vH262A and vG275A had more-severe defects, producing total virus-specific RNAs at 31 and 22% of the level in WT-infected cells, respectively.
FIG. 4.

Effects of the mutations in mNsp15 on MHV-specific RNA and S protein accumulation. (A) RNA synthesis in cells infected with mNsp15 mutant viruses. Cells were infected with either an mNsp15 mutant virus or WT virus, and viral RNAs were metabolically labeled as described in Materials and Methods. The labeled viral RNAs were separated by formaldehyde agarose gel electrophoresis and visualized by autoradiography. The positions of bands of gRNA and sgmRNAs 1 through 7 are indicated on the right. (B) Effects of the H262A substitution in mNsp15 on MHV minus-strand RNA accumulation. Levels of minus-strand RNAs produced by vH262A and WT virus were determined by RT-qPCR. The relative levels of minus-strand RNA were determined from the threshold values normalized for GAPDH mRNA expression (endogenous control), and then the WT virus value was defined as 1.0. Error bars, standard errors of the means. The symbol + stands for WT virus. The relative levels of expression of the MHV minus-strand RNAs by vH262A (dotted bars) were compared with the levels of expression by WT virus (shaded bars) (α = 0.05). The values shown represent data from three independent experiments. A melting curve showed that there was a single product in each RT-qPCR, and agarose gel electrophoresis of RT-qPCR products confirmed the single RT-qPCR products (data not shown). (C) Expression of MHV S protein by mNsp15 mutant viruses. DBT cells were seeded in a T25 flask, incubated for 24 h, and infected with an mNsp15 mutant or WT virus at an MOI of 1. Total proteins were extracted from the DBT cells at 8 and 10 hpi and were then analyzed in order to detect S protein synthesized by mNsp15 mutant viruses and WT virus by using with a goat polyclonal antibody against MHV S protein (31). As a control, expression levels of GAPDH in DBT cells were determined using mouse anti-mouse GAPDH antibodies and HRP-conjugated anti-mouse IgG antibodies.
TABLE 4.
Relative molar amounts of virus-specific RNAs synthesized in DBT cells infected with mNsp15 mutant viruses
| RNA species | Relative molar amta (mean ± SE)
|
|||
|---|---|---|---|---|
| vH262A | vG275A | vH277A | MHV-A59 1000 | |
| RNA1 | 1 | 1 | 1 | 1 |
| sgmRNA2 | 2.1 ± 0.2 | 2.2 ± 0.4 | 2.0 ± 0.3 | 2.5 ± 0.4 |
| sgmRNA3 | 2.3 ± 0.3 | 2.4 ± 0.5 | 2.5 ± 0.5 | 3.1 ± 0.4 |
| sgmRNA4 | 8.8 ± 0.7 | 9.6 ± 2.0 | 9.6 ± 1.5 | 10.3 ± 0.8 |
| sgmRNA5 | 7.3 ± 0.3 | 7.6 ± 1.3 | 7.0 ± 1.2 | 8.2 ± 0.2 |
| sgmRNA6 | 14.0 ± 1.4 | 15.0 ± 2.6 | 13.5 ± 1.7 | 13.7 ± 1.4 |
| sgmRNA7 | 30.9 ± 3.5 | 33.6 ± 7.0 | 32.1 ± 8.0 | 27.4 ± 5.4 |
Relative to the molar amount of RNA1.
RT-qPCR was used to determine whether the altered growth phenotype of vH262A could be attributed to a defect in MHV minus-strand RNA synthesis. Expression levels of minus-strand gRNA1, sgRNA3, and sgRNA7 were evaluated in DBT cells infected with vH262A or WT virus (Fig. 4B). At 8 hpi, cells infected with vH262A contained minus-strand gRNA1, sgRNA3, and sgRNA7 at 62%, 52%, and 32%, respectively, of levels in cells infected with the WT virus. Average cycle threshold (CT) values for expression levels of the WT viral minus-strand RNAs were 19.6 for gRNA1 (CT for GADPH, 18.80), 20.1 for sgRNA3 (CT for GADPH, 19.40), and 15.2 for sgRNA7 (CT for GADPH, 19.10). In parallel, average CT values for expression levels of the vH262A viral minus-strand RNAs were 20.4 for gRNA1 (CT for GADPH, 18.96), 20.1 for sgRNA3 (CT for GADPH, 19.7), and 15.2 for sgRNA7 (CT for GADPH, 19.4). At 12 hpi, the expression levels of vH262A minus-strand RNAs were less than 38% of those in cells infected with WT virus. The more severe decrease in MHV minus-strand RNA synthesis at 12 hpi correlates with the defect in viral titers observed for the mutant viruses at this time (Fig. 3B and C).
S protein expression by mNsp15 mutant viruses.
Based on the reduced viral RNA levels, we expected that the mutant viruses would express viral proteins at reduced levels. The S protein was examined by Western blot analyses of lysates of DBT cells infected with either a mutant virus or WT virus (Fig. 4C). Relative to that in WT virus-infected cells, S protein accumulation by three single-substitution mutant viruses was less than 27% at 8 hpi. At 10 hpi, similar defects in S protein levels were also observed for vH262A and vG275A, but vH277A-infected cells produced ∼70% of the S protein level found in cells infected with WT virus. We did not pursue the basis for this change in vH277A.
trans-Complementation of vH262A with Nsp15 proteins derived from MHV-A59 and SARS-CoV.
We wanted to determine whether Nsp15 could function in trans to complement the observed defects. For this analysis, the MHV or SARS-CoV Nsp15 WT coding sequence was cloned into pCDNA3.1 under the control of the strong immediate-early cytomegalovirus promoter. We also expressed an N-terminal fusion of GFP and sNsp15 cloned into pCDNA3.1. The GFP-sNsp15 plasmid was transfected into DBT cells, and expression was detected by fluorescence microscopy (Fig. 5A). DBT cells were transfected with either pCDNA3.1 or one of the two plasmids that express MHV or SARS-CoV Nsp15. At 30 h posttransfection, the cells were infected with either WT virus or vH262A. MHV-specific RNAs were then detected by metabolic radiolabeling of cells with 32Pi in the presence of actinomycin D (Fig. 5B). Consistent with previous observations, vH262A exhibited a defect in the accumulation of all MHV plus-strand RNAs relative to that for the WT virus in the presence of the empty vector (Fig. 5B, first and second lanes). However, coexpression of the mNsp15 protein in cells infected with vH262A increased the levels of all of the plus-strand RNAs 2.3-fold (from 18% to 41%) (Fig. 5B, third lane). This result was confirmed by two other independent experiments, both of which showed a 1.5-fold or better increase in the abundance of the MHV plus-strand RNAs. Interestingly, expression of the sNsp15 protein in trans did not affect the levels of RNAs produced by vH262A, indicating that the SARS-CoV ortholog could not complement MHV plus-strand RNA levels in trans. Expression of the sNsp15 or mNsp15 WT protein had no obvious effects on the plus-strand RNAs produced by WT virus in an independent experiment (data not shown), demonstrating that expression of mNsp15 in trans did not affect MHV infection.
FIG. 5.
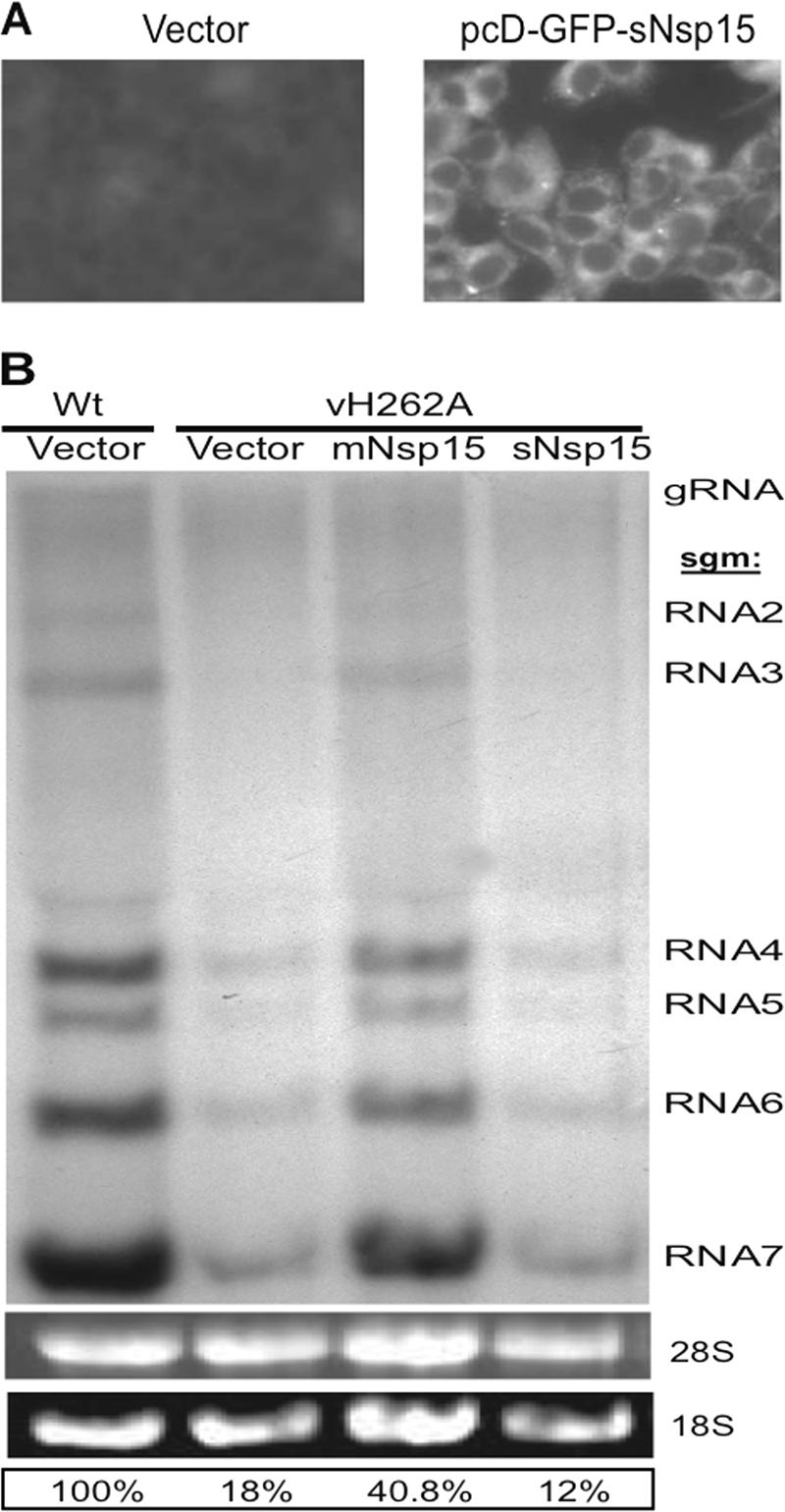
RNA accumulation in Nsp15-trans-complemented DBT cells infected with vH262A. (A) Detection of sNsp15 expression. DBT cells were transfected either with vector only or with pcD-GFP-sNsp15 and were subjected to GFP detection by fluorescent microscopy of cells at 30 h posttransfection. (B) DBT cells were first transfected either with vector only or with a plasmid driving the expression of Nsp15 derived from MHV-A59 or SARS-CoV (pcD-mNsp15 or pcD-sNsp15) and then infected with vH262A or WT virus 30 h later. At 6 hpi, viral RNAs were metabolically labeled as described in Materials and Methods. The labeled viral RNAs were resolved by formaldehyde agarose gel electrophoresis and visualized by autoradiography. The identities of the MHV RNAs are given on the right.
To confirm the complementation of vH262A with the mNsp15 protein in trans, titers of vH262A and WT virus were determined in DBT cells that had been transfected with vector only or with vector expressing mNsp15 or sNsp15 protein and then infected with vH262A or WT virus (Table 5). At 24 hpi, the cells were frozen at −80°C, thawed, sonicated, and clarified; then the resultant supernatants containing either vH262A or WT virus were titered. Mutant vH262A grown in DBT cells trans-complemented with the mNsp15 protein had yields of infectious virus >3.6 times higher than those in cells transfected with vector only or with sNsp15 protein. Along with the results for MHV plus-strand RNA production, these results demonstrate that MHV Nsp15 could act in trans in a step necessary for MHV replication.
TABLE 5.
Titration of vH262A in trans-complemented DBT cells
| Virus | Transfection | Titer (log10) (mean ± SE) |
|---|---|---|
| MHV-A59 1000 | Vector only | 7.36 ± 0.03 |
| vH262A | Vector only | 6.31 ± 0.06 |
| mNsp15 | 6.98 ± 0.02 | |
| sNsp15 | 6.41 ± 0.01 |
DISCUSSION
The Nsp15 proteins of coronaviruses such as HCoV-229E, SARS-CoV, and infectious bronchitis virus have been characterized as uridylate-specific endoribonucleases (2, 12). In this work, we examined the effects of single and double amino acid substitutions in the catalytic pocket of mNsp15 and determined that endoribonuclease activity was significantly affected by the mutations, consistent with the report of Xu et al. (28). However, residual endoribonuclease activity could still be detected even with the double amino acid substitution. Mutant viruses containing the same single or double amino acid substitutions were apparently unaffected in plaque morphology, but the infectious virus titer was reduced as much as 13-fold. There were also corresponding decreases in the levels of MHV plus- and minus-strand RNAs and the MHV S protein. Lastly, we demonstrated that the mNsp15 protein could partially complement the defect in mNsp15 mutant viruses.
Biochemical properties of MHV Nsp15.
The catalytic pocket of the MHV Nsp15 protein differed in several residues from that of SARS-CoV. However, when a UMP substrate was docked into the mNsp15 active site, the similarities between mNsp15 and sNsp15 were obvious. For mNsp15, residues H262, H277, and K317 form a catalytic triad that is superimposable with the comparable residues in sNsp15. The modeling generated results that are consistent both with the substrate interaction model proposed by Ricagno et al. (21) for sNsp15, based on the well-studied RNase A catalytic triad, and with the mechanism of RNA cleavage by Nsp15 (2). In addition, modeling with UMP shows that G275, H277, K317, T321, and T368 of mNsp15 are in close proximity.
Some notable differences exist in the other side of the catalytic cleft of mNsp15 and sNsp15. First, in sNsp15, residue S293 is likely responsible for specific recognition of the uracil. In mNsp15, residue T321 would serve as the specificity-determining residue (Fig. 1A). Second, the side chain oxygen atom of T368 in mNsp15 forms a hydrogen bond with the C-4 keto group of the UMP, while the comparable residue in sNsp15 is Leu345. Third, a ribose ring in UMP can make two additional hydrogen bond interactions with the side chain oxygen atoms of E362 in mNsp15 (at 2.5 and 2.6 Å) compared to T340 in sNsp15. The close positioning of the E362 side chain to the ribose in UMP should impose some restriction on the binding orientation. Fourth, the role of the C-terminal tail appears to differ between sNsp15 and mNsp15. Residues Y342 and P343 of sNsp15 appear to form hydrophobic stacks with the uracil and should stabilize UMP binding, while K365 and V366 would take their place in mNsp15. K365 can engage in hydrophobic interactions with the substrate through its side chain carbon atoms.
The mNsp15 protein was found to preferentially cleave uridylates, but the specific cleavage of uridylates was less stringent than that of sNsp15. A change of residue H262 to alanine resulted in a protein that can cleave uridylates and cytidylates equally well (Fig. 2B). This observation was not made with the orthologs from other nidoviruses (19, 21). At this point, we do not understand the molecular basis for this difference. However, we speculate that the difference in the interaction between E362 and the C-terminal residues of mNsp15 with the substrate could account for the slightly relaxed cleavage specificity of mNsp15. This model needs to be confirmed by further biochemical analyses.
Recombinant mNsp15 was previously produced in E. coli by Bhardwaj et al. (2) and was demonstrated to possess significantly lower endoribonuclease activity than the corresponding enzymes from SARS-CoV and infectious bronchitis virus. The mNsp15 protein used in this study is more biochemically active than the previous preparations (2). A basis for this difference is that the mNsp15 made previously contained His6 tags at both the N and C termini, while the version in this work contained only an N-terminal His6 tag. In the fluorescent real-time assay, we observed that the doubly tagged protein cleaved substrates at uridylates and cytidylates approximately equally well, with a specificity ratio of ∼1 rather than the ratio of 3 found for the N-terminally tagged mNsp15 (data not shown). Since the C termini of sNsp15 and mNsp15 contribute to the formation of the active sites, we now appreciate that additional residues at the C terminus of Nsp15 could significantly affect Nsp15 endoribonuclease activity and specificity.
Nsp15 and MHV infection.
Recombinant MHVs harboring either the single substitution H262A, G275A, or H277A or two mutations (H277A and H262P) in mNsp15 were infectious and produced plaques in L2, DBT, and 17CL-1 cells with a mean diameter of approximately 80% of that for MHV-A59. We had expected a greater effect of mutations in mNsp15 on MHV infectivity. However, it should be noted that reduced enzymatic activity was retained with all the mNsp15 mutant proteins (Fig. 2C and data not shown). Furthermore, mNsp15 with two mutations in the catalytic pocket had the least endoribonuclease activity, and vH262P+H277A produced less infectious virus than the mutants with single amino acid substitutions. We posit that the reduced endoribonuclease activity of mutant versions of Nsp15 is responsible for the reduced virus production. The observation that genomes containing the D324A substitution were nonviable further supports a claim for an essential role of Nsp15 in MHV infection; molecular modeling predicts that D324 is critical for maintaining a network of ionic bonds, and the change to alanine resulted in an insoluble protein (data not shown). In any case, the fact that mutations in Nsp15 reduced the level of infection by as much as 2 log units clearly shows that the Nsp15 endoribonuclease is needed for optimal MHV replication.
MHV Nsp15 mutant viruses have distinguishable features in plaque formation and viral RNA synthesis compared to EAV mutants with similar mutations. For example, the EAV mutant virus containing a mutation (H2963A) comparable to H262A in mNsp15 produced plaques with only 20% of the diameter of WT virus plaques (19). We note that in our experiments with MHV, similar results were observed with all of the cells tested (L2, DBT, and 17CL-1). In particular, 17CL-1 cells have been reported to produce interferon (1, 29). Thus, mutations in mNsp15 cannot be easily distinguished by plaque morphology alone, and the reduction of mNsp15 activity did not abolish plaque formation in interferon-positive cells. Nonetheless, the widespread defect in MHV gene expression caused by mutations in mNsp15 did not allow us to determine whether mNsp15 acts on one particular process required for viral infection.
Lastly, this study showed that two mNsp15 mutant viruses were partially complemented in trans for MHV plus-strand RNA synthesis and infectious particle production by expression of mNsp15 in DBT cells. Thus, mNsp15 forms another complementation group in MHV-A59 open reading frame 1b (23). Based on these results, future assessments of the correlation between activities of Nsp15 could be performed by expression of Nsp15 in trans rather than by the more laborious method of making recombinant viruses. It is also of interest that sNsp15 could not complement the mNsp15 mutation in trans. We cannot completely rule out the possibility that the failure of sNsp15 to complement mNsp15 mutations is due to lower levels of expression of sNsp15 than of mNsp15. However, the higher enzymatic activity of sNsp15 relative to mNsp15 makes it unlikely that our trans-complementation results are due to lower levels of enzymatic activity in cells transfected with the sNsp15 expression plasmid. Rather, we suggest that mNsp15 may need to specifically interact with other MHV-specific molecules.
Acknowledgments
We gratefully acknowledge support from the NIH (1R56AI06164 to C.C.K. and AI51493 to J.L.L.).
We thank Jeffrey D. Cirillo of the Department of Microbial and Molecular Pathogenesis for kindly providing a mouse anti-GAPDH primary antibody. We also thank Nick Makkinje, Shawn T. Williamson, and Pinghua Liu of the Leibowitz laboratory for help and encouragement.
Footnotes
Published ahead of print on 26 September 2007.
REFERENCES
- 1.An, S., C. J. Chen, X. Yu, J. L. Leibowitz, and S. Makino. 1999. Induction of apoptosis in murine coronavirus-infected cultured cells and demonstration of E protein as an apoptosis inducer. J. Virol. 73:7853-7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhardwaj, K., L. Guarino, and C. C. Kao. 2004. The severe acute respiratory syndrome coronavirus Nsp15 protein is an endoribonuclease that prefers manganese as a cofactor. J. Virol. 78:12218-12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhardwaj, K., J. Sun, A. Holzenburg, L. A. Guarino, and C. C. Kao. 2006. RNA recognition and cleavage by the SARS coronavirus endoribonuclease. J. Mol. Biol. 361:243-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Albuquerque, N., E. Baig, X. Ma, J. Zhang, W. He, A. Rowe, M. Habal, M. Liu, I. Shalev, G. P. Downey, R. Gorczynski, J. Butany, J. Leibowitz, S. R. Weiss, I. D. McGilvray, M. J. Phillips, E. N. Fish, and G. A. Levy. 2006. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 80:10382-10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Albuquerque, N., E. Baig, M. Xuezhong, I. Shalev, M. J. Phillips, M. Habal, J. Leibowitz, I. McGilvray, J. Butany, E. Fish, and G. Levy. 2006. Murine hepatitis virus strain 1 as a model for severe acute respiratory distress syndrome (SARS). Adv. Exp. Med. Biol. 581:373-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dick, G. W., J. S. Niven, and A. W. Gledhill. 1956. A virus related to that causing hepatitis in mice (MHV). Br. J. Exp. Pathol. 37:90-98. [PMC free article] [PubMed] [Google Scholar]
- 7.Gaur, D., and J. K. Batra. 2005. Role of aspartic acid 121 in human pancreatic ribonuclease catalysis. Mol. Cell. Biochem. 275:95-101. [DOI] [PubMed] [Google Scholar]
- 8.Goebel, S. J., J. Taylor, and P. S. Masters. 2004. The 3′ cis-acting genomic replication element of the severe acute respiratory syndrome coronavirus can function in the murine coronavirus genome. J. Virol. 78:7846-7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorbalenya, A. E., L. Enjuanes, J. Ziebuhr, and E. J. Snijder. 2006. Nidovirales: evolving the largest RNA virus genome. Virus Res. 117:17-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guarino, L. A., K. Bhardwaj, W. Dong, J. Sun, A. Holzenburg, and C. Kao. 2005. Mutational analysis of the SARS virus Nsp15 endoribonuclease: identification of residues affecting hexamer formation. J. Mol. Biol. 353:1106-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Homberger, F. R., L. Zhang, and S. W. Barthold. 1998. Prevalence of enterotropic and polytropic mouse hepatitis virus in enzootically infected mouse colonies. Lab. Anim. Sci. 48:50-54. [PubMed] [Google Scholar]
- 12.Ivanov, K. A., T. Hertzig, M. Rozanov, S. Bayer, V. Thiel, A. E. Gorbalenya, and J. Ziebuhr. 2004. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. USA 101:12694-12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang, H., M. Feng, M. E. Schroeder, D. P. Giedroc, and J. L. Leibowitz. 2006. Putative cis-acting stem-loops in the 5′ untranslated region of the severe acute respiratory syndrome coronavirus can substitute for their mouse hepatitis virus counterparts. J. Virol. 80:10600-10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang, H., M. Feng, M. E. Schroeder, D. P. Giedroc, and J. L. Leibowitz. 2006. Stem-loop 1 in the 5′ UTR of the SARS coronavirus can substitute for its counterpart in mouse hepatitis virus. Adv. Exp. Med. Biol. 581:105-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavi, E., D. H. Gilden, Z. Wroblewska, L. B. Rorke, and S. R. Weiss. 1984. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597-603. [DOI] [PubMed] [Google Scholar]
- 16.McRee, D. E. 1999. XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125:156-165. [DOI] [PubMed] [Google Scholar]
- 17.Moustakas, D. T., P. T. Lang, S. Pegg, E. Pettersen, I. D. Kuntz, N. Brooijmans, and R. C. Rizzo. 2006. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 20:601-619. [DOI] [PubMed] [Google Scholar]
- 18.Pasternak, A. O., W. J. Spaan, and E. J. Snijder. 2006. Nidovirus transcription: how to make sense? J. Gen. Virol. 87:1403-1421. [DOI] [PubMed] [Google Scholar]
- 19.Posthuma, C. C., D. D. Nedialkova, J. C. Zevenhoven-Dobbe, J. H. Blokhuis, A. E. Gorbalenya, and E. J. Snijder. 2006. Site-directed mutagenesis of the nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 80:1653-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Putics, A., W. Filipowicz, J. Hall, A. E. Gorbalenya, and J. Ziebuhr. 2005. ADP-ribose-1"-monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J. Virol. 79:12721-12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricagno, S., M. P. Egloff, R. Ulferts, B. Coutard, D. Nurizzo, V. Campanacci, C. Cambillau, J. Ziebuhr, and B. Canard. 2006. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. USA 103:11892-11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawicki, S. G., and D. L. Sawicki. 2005. Coronavirus transcription: a perspective. Curr. Top. Microbiol. Immunol. 287:31-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawicki, S. G., D. L. Sawicki, D. Younker, Y. Meyer, V. Thiel, H. Stokes, and S. G. Siddell. 2005. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PLoS Pathog. 1:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seybert, A., C. C. Posthuma, L. C. van Dinten, E. J. Snijder, A. E. Gorbalenya, and J. Ziebuhr. 2005. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J. Virol. 79:696-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi, S. T., and M. M. Lai. 2005. Viral and cellular proteins involved in coronavirus replication. Curr. Top. Microbiol. Immunol. 287:95-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snijder, E. J., P. J. Bredenbeek, J. C. Dobbe, V. Thiel, J. Ziebuhr, L. L. Poon, Y. Guan, M. Rozanov, W. J. Spaan, and A. E. Gorbalenya. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiss, S., and J. Leibowitz. Pathogenesis of murine coronavirus infections. In S. Perlman, T. Gallagher, and E. J. Snijder (ed.), The nidoviruses, in press. ASM Press, Washington, DC.
- 28.Xu, X., Y. Zhai, F. Sun, Z. Lou, D. Su, Y. Xu, R. Zhang, A. Joachimiak, X. C. Zhang, M. Bartlam, and Z. Rao. 2006. New antiviral target revealed by the hexameric structure of mouse hepatitis virus nonstructural protein nsp15. J. Virol. 80:7909-7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye, Y., K. Hauns, J. O. Langland, B. L. Jacobs, and B. G. Hogue. 2007. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 81:2554-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yount, B., M. R. Denison, S. R. Weiss, and R. S. Baric. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 76:11065-11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu, W., and J. L. Leibowitz. 1995. Specific binding of host cellular proteins to multiple sites within the 3′ end of mouse hepatitis virus genomic RNA. J. Virol. 69:2016-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziebuhr, J. 2005. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 287:57-94. [DOI] [PMC free article] [PubMed] [Google Scholar]