

Abstract
The use of endocytic pathways by viral glycoproteins is thought to play various functions during viral infection. We previously showed in transfection assays that herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is transported from the cell surface back to the trans-Golgi network (TGN) and that two motifs of gB cytoplasmic tail, YTQV and LL, function distinctly in this process. To investigate the role of each of these gB trafficking signals in HSV-1 infection, we constructed recombinant viruses in which each motif was rendered nonfunctional by alanine mutagenesis. In infected cells, wild-type gB was internalized from the cell surface and concentrated in the TGN. Disruption of YTQV abolished internalization of gB during infection, whereas disruption of LL induced accumulation of internalized gB in early recycling endosomes and impaired its return to the TGN. The growth of both recombinants was moderately diminished. Moreover, the fusion phenotype of cells infected with the gB recombinants differed from that of cells infected with the wild-type virus. Cells infected with the YTQV-mutated virus displayed reduced cell-cell fusion, whereas giant syncytia were observed in cells infected with the LL-mutated virus. Furthermore, blocking gB internalization or impairing gB recycling to the cell surface, using drugs or a transdominant negative form of Rab11, significantly reduced cell-cell fusion. These results favor a role for endocytosis in virus replication and suggest that gB intracellular trafficking is involved in the regulation of cell-cell fusion.
Herpes simplex virus type 1 (HSV-1), a member of the herpesviruses family, is a ubiquitous human pathogen mainly responsible for infections of mucocutaneous epithelia that may recur due to the virus latency-reactivation cycle. HSV-1 occasionally spreads to the central nervous system, causing severe encephalitis. HSV-1 envelope glycoprotein B (gB), one of the most conserved glycoproteins among herpesviruses, is a major determinant of virus infectivity, in vitro and in vivo. The essential functions of this type 1 transmembrane protein at various steps of viral replication such as entry, fusion, and cell-cell spread, have been extensively studied and in part assigned to specific domains of the protein (11-13, 16, 24, 27, 32, 33, 43, 53, 59, 79), although a precise picture of the mechanisms by which gB fulfills these functions is still missing. Various studies have also illustrated the role of gB in experimental pathogenesis, in particular in HSV-1 neuroinvasion (27, 41, 78, 83). How glycoproteins might be related to neuroinvasion has not been completely elucidated. However, transneuronal spread might be functionally related to the mechanisms of intracellular assembly and the formation of viral mature particles (25, 50, 69).
It is well established that endocytosis pathways are used by numerous viral glycoproteins in infected cells, in a strategy supposedly aimed to help successful replication and to promote pathogenesis (18, 45). Endocytosis of herpesviruses glycoproteins has, for instance, been involved in cell-cell spread and evasion from the immune response (9, 19, 20). Mostly, endocytosis is thought to favor the concentration of glycoproteins at the intracellular site where viral assembly would take place. Several herpesvirus envelope proteins, including gB homologues, follow an internalization process after having reached the surface of cells and accumulate in the trans-Golgi network (TGN) or in vesicles derived from it (7, 17, 19, 31, 63, 71). These observations support a model of herpesvirus assembly that states that after newly synthesized intranuclear capsids bud through the inner nuclear membrane, they are first de-enveloped through the outer nuclear membrane and then acquire their final envelope at the TGN (48, 49). Furthermore, endocytosis of fusogenic viral glycoproteins has been implicated in the regulation of virus-induced cell-cell fusion (58, 64, 76). Interestingly, although a relation between cell-cell fusion and gB intracellular trafficking has been suggested, a strict correlation between cell fusion activity and levels of gB cell surface expression has not been found (17, 64).
Previous studies reported that pseudorabies virus (PRV), cytomegalovirus (CMV), varicella-zoster virus (VZV), and HSV-2 gB are internalized from the cell surface (17, 20, 31, 71). We showed earlier in transfection assays that endocytosis and concentration in the TGN occurs for HSV-1 gB (7). Two motifs of the HSV-1 gB cytoplasmic domain, YTQV (amino acids [aa] 889 to 892) and LL (aa 871 to 872) are involved in two distinct steps of gB retrograde trafficking to the TGN. Disruption of the YTQV motif prevented internalization of gB, whereas disruption of the LL motif impaired its return to the TGN while enhancing its recycling to the plasma membrane. To further understand the role of gB endocytosis during virus replication, we constructed HSV-1 recombinants in which each of these gB tail endocytic motifs has been inactivated. We then investigated in infected cells the consequences of each mutation on gB trafficking, virus growth, and virus-induced cell-cell fusion. Our results suggested that the presence of gB at the surface of infected cells is regulated differentially by each of the investigated endocytosis motifs and that gB trafficking is involved in the production of infectious particles and in cell-cell fusion.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney Vero cells were grown in minimum essential medium supplemented with 10% fetal calf serum (FCS). Cos-7 cells and Vero-derived D6 cells (kindly provided by P. Desai and S. Person, Johns Hopkins University, Baltimore, MD) were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FCS. For D6, 0.5 mg of G418 (Geneticin; Gibco-BRL)/ml was added to the culture medium. The 143B human osteosarcoma TK- cell line (European Collection of Cell Cultures, Porton Down, United Kingdom) was cultured in DMEM-10% FCS containing 15 μg of 5-bromo-2′-deoxyuridine/ml as previously described (73). Cell culture media and reagents were all from Invitrogen (Cergy Pontoise, France), except for 5-bromo-2′-deoxyuridine, which was obtained from Sigma-Aldrich (Lyon, France). The reference KOS and the KgBct-GFP (61) viruses were propagated in Vero and D6 cells, respectively. For preparation of viral stocks, approximately 2 × 108 cells were infected at a multiplicity of infection (MOI) of 0.001 PFU/cell. At 5 to 7 days postinfection, culture supernatants were cleared by low-speed centrifugation, and extracellular virions were concentrated by centrifugation for 2 h in a Sorvall GSA rotor at 10,000 rpm.
Antibodies.
Immunocytochemical staining was performed using antibodies to gB (rabbit polyclonal serum R69; generously provided by R. J. Eisenberg and G. H. Cohen, University of Pennsylvania), ICP5 (mouse monoclonal antibody; Virusys Corp., Sykesville, MD), TGN 46 (sheep polyclonal antibody; Serotec, Oxford, United Kingdom), and CD71 (mouse monoclonal anti-human antibody; Sigma). Cy2-coupled anti-mouse immunoglobulin G (IgG), Cy3-coupled anti-rabbit IgG, Cy5-coupled anti-sheep IgG, and biotin-coupled anti-mouse IgG antibodies were all purchased from Jackson Immunoresearch Laboratories (West Grove, PA). Alexa 488-coupled anti-mouse IgG was from Invitrogen (Cergy-Pontoise, France). Horseradish peroxidase (HRP)-coupled anti-mouse IgG, mouse monoclonal anti-gB antibody (Abcam), and anti-rabbit IgG antibodies (Dako, Trappes, France) were used in Western blot analyses.
Plasmids and constructs.
Plasmids pgB-Y889A and pgB-LL871AA were constructed by eliminating the green fluorescent protein (GFP) coding sequence inserted at the NotI site of the UL27-gB gene from the previously described pGFP-gBY889A and pGFP-gBLL871AA plasmids (7). Plasmid pECFP-Rab11S25N, encoding a transdominant-negative form of Rab11 (80) fused to enhanced cyan fluorescent protein (ECFP) was kindly provided by J. Salamero (Institut Curie, Paris, France). Plasmid pECFP-N1 was obtained from Clontech BD Biosciences (Le Pont de Claix, France).
Construction of HSV-1 gB mutants.
Recombinant viruses were essentially constructed as previously described (62). Briefly, subconfluent monolayers of Vero cells were cotransfected with 1 μg of KgBct-GFP genomic DNA and with linearized plasmids pgB-Y889A or pgB-LL871AA in a fivefold molar excess using 9 μl of FuGENE-6 according to the manufacturer's recommendations (Roche, Meylan, France). At 4 to 5 days after transfection, cell cultures were screened for the presence of nonfluorescent plaques. Progeny viruses were plated and plaque purified three times on Vero cells. Stocks for the newly obtained KgBY889A and KgBLL871AA viruses were prepared as described for the KOS virus. The presence of the desired mutation in the gB gene of each recombinant virus was verified by BamHI restriction fragment length polymorphism of total viral DNA (62) and by PCR and sequencing using previously described primers (7).
Biotinylation of cell surface gB in HSV-1-infected cells.
The surface expression of gB was investigated by biotinylation according to a previously described procedure (44) with minor modifications. 143B cells cultured in 2.5-cm-diameter dishes were inoculated with KOS at an MOI of 5 for 1 h and then rinsed with an acid glycine solution to remove any residual nonpenetrated inoculum. At 8 h postinfection, the cells were biotinylated with NHS-LC biotin (1 mg/ml; Pierce, Rockford, IL). The labeled cells were then harvested in immunoprecipitation buffer (0.05 M Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA, and 1% NP-40, with protease inhibitors). Samples were precleared overnight with protein A-agarose beads (Roche) to minimize background and then reacted with the polyclonal R69 anti-gB antibody (1/800) for 3 h. Immunoprecipitation was then performed overnight at 4°C. Samples were washed extensively, before loading them on a 7% polyacrylamide gel for sodium dodecyl sulfate-polyacrylamide gel electrophoresis under nonreducing conditions. Immunoprecipitated biotinylated glycoproteins were detected with streptavidin-HRP by Western blotting. After a stripping step, the membranes were hybridized with a monoclonal mouse anti-gB antibody (Abcam).
Endocytosis of HSV-1 gB assayed by biotinylation.
143B cells cultured in 10-cm-diameter dishes were infected with KOS at an MOI of 5 for 1 h and then washed with acid-glycine. Eight hours after infection, the cells were biotinylated twice for 20 and 15 min by using cleavable Sulfo-NHS-SS biotin (2 mg/ml; Pierce) instead of NHS-LC biotin. After extensive washes with phosphate-buffered saline (PBS), cells were incubated at 37°C for 3 to 4 h to allow endocytosis of glycoproteins. Biotinylated infected cells were treated three times with freshly prepared glutathione (GSH) at 60 mg/ml for 20 min at 4°C to remove any biotin remaining at the cell surface. After extensive washes, cells were harvested in immunoprecipitation buffer and reacted with the anti-gB antibody. Biotinylated proteins were detected by streptavidin-HRP Western blotting and then visualized after stripping by an anti-gB antibody as described above. As controls, infected biotinylated cells were treated with GSH immediately after biotinylation and then immunoprecipitated.
Imaging analysis of gB internalization.
143B cells were infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 0.1. At 1 h after infection, the cells were rinsed extensively with PBS and fresh medium (DMEM with 2% FCS and without 5-bromo-2-deoxyuridine) was added. Seven hours after infection, cells were incubated in DMEM containing 10% (vol/vol) donkey serum for 30 min at 37°C, followed by incubation at 4°C for 30 min. Cells were incubated with the anti-gB R69 polyclonal serum (1/500 in PBS plus 10% donkey serum) for 45 min at 4°C and then washed twice with PBS containing 0.2% (wt/vol) bovine serum albumin (BSA) at room temperature and once with DMEM-10% FCS at 37°C. The culture medium was added again, and cells were placed at 37°C for several time intervals. After incubation at 37°C, cells were fixed with methanol for 10 min. Controls that were not allowed to internalize were washed and fixed immediately after incubation with the primary antibody. After fixation, cells were washed five times with PBS-0.2% BSA for 5 min and then incubated for 1 h with a PBS-0.2% BSA-10% donkey serum solution containing the anti-TGN 46 (1/200) and anti-ICP5 antibodies (1/300). After three washes with PBS-0.2% BSA, cells were stained with Cy2-coupled anti-mouse IgG antibody (1/200), Cy3-coupled anti-rabbit IgG antibody (1/750), and Cy5-coupled anti-sheep IgG antibody (1/500). Coverslips were washed five times with PBS-0.2% BSA and once with water and then mounted onto glass slides.
To better characterize the compartment where gB was endocytosed, internalization of the transferrin receptor, which cycles between the cell surface and early and recycling endosomes, was assayed together with gB, using a monoclonal anti-CD 71 (1/500 in PBS plus 10% donkey serum) in addition to the anti-gB antibody. The transferrin receptor-specific staining was revealed with an Alexa 488-coupled anti-mouse IgG. In this series of experiments, cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature and then washed with PBS-0.2% BSA-0.05% saponin. Fluorescence on 0.1-μm-thick optical sections was analyzed with a Leica TCS SP2 AOBS confocal microscope.
Subcellular distribution of gB in infected cells.
143B cells were infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 0.1. One hour after infection, cells were rinsed extensively with PBS, and fresh medium (DMEM with 2% FCS and without 5-bromo-2-deoxyuridine) was added. The cells were then incubated at 37°C for 6 h before fixation. For subcellular analysis, cells were fixed with paraformaldehyde; permeabilized; incubated with the anti-gB, anti-CD71, and anti-TGN antibodies; and revealed with the same secondary antibodies as described above.
Image processing and quantification.
The vertical optical resolution was 0.1 μm. Confocal imaging quantification was performed by using the ImageJ processing and analysis software. All images were corrected for background fluorescence by subtracting the local median background intensity observed in an area which did not contain cells. The background-corrected images were analyzed for quantification by using the colocalization plug-in of the software, which highlights the colocalized points of two 8-bit images. Two points were considered colocalized if their respective intensities were strictly higher than the threshold of their channels (= 30) and if the ratio of their intensities was strictly higher than a ratio of 60%.
Virus growth.
Confluent Cos-7 and 143B cells in 35-mm culture dishes were infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 5 for single-step or of 0.001 for multistep growth curves. One hour after infection, extracellular remaining virions were washed off by treatment with an acid-glycine-saline solution (13). After a thorough wash with PBS, fresh medium (DMEM-2% FCS) was added. Infected cell culture supernatants were harvested at different times postinfection and, after low-speed centrifugation to clear the supernatants, extracellular virions were titrated on Vero cells. Cell-associated virions were titrated without or with a low pH wash before harvesting to inactivate the virions on the cell surface.
Incorporation of gB in recombinant viruses.
The incorporation of mutated gB molecules into virions was verified by Western blotting. Extracellular virion particles were run on a 4 to 12% NuPage polyacrylamide gel under denaturing conditions before transfer onto a nitrocellulose membrane. The membrane was incubated with the R69 anti-gB and the anti-ICP5 antibodies and then with the secondary goat anti-mouse and goat anti-rabbit IgGs coupled to HRP. The immunoblot was revealed by enhanced chemiluminescence (Amersham). Signals were acquired by using a LAS-3000 apparatus (Fujifilm) for further quantification, using the provided Fujifilm Multi-Gauge software.
Cell fusion and inhibition by drugs.
To compare the characteristics of infected cell cultures in the absence or presence of drugs affecting the intracellular transport of proteins, confluent Cos-7 cell cultures were infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 1 and then extensively rinsed 1 h after infection and observed 20 h postinfection with a phase-contrast microscope. For chlorpromazine treatment, medium containing 5 μg of chlorpromazine hydrochloride (Largactil, Sanofi-Aventis, France)/ml was added to the cultures 2 h postinfection and maintained for 18 h, until fixation with 4% (wt/vol) paraformaldehyde in PBS for 30 min at room temperature. For bafilomycin A1 (Tocris, Bristol, United Kingdom) treatment analyses, DMEM containing the drug at a final concentration of 250 nM (1/1,000 dilution of a 250 μM bafilomycin A1 stock solution in dimethyl sulfoxide [DMSO]) or DMSO as a control (1 μl/ml) was added 2 h postinfection. Cells were observed with a phase-contrast microscope 20 h postinfection before fixation and permeabilization with methanol for 10 min at −20°C. After extensive rinses with PBS, cells were incubated with an anti-ICP5 antibody for 1 h, rinsed with PBS, and incubated for 1 h with a biotin-coupled anti-mouse IgG antibody. After additional washes with PBS, the cells were incubated with β-galactosidase-coupled streptavidine (Roche) at 1/1,000 in PBS for 1 h. The immunostaining was revealed by incubating the cells for 2 h at 37°C with a solution containing 4 mM potassium ferrocyanide, 4 mM potassium ferricyanide, 1 mM magnesium chloride. and 0.4 mg of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside)/ml in PBS and then stopped by removing the revealing solution and washing the cells thoroughly with PBS. To verify the effect of chlorpromazine and bafilomycin on gB endocytosis, an internalization assay was performed as described above except that the cells were treated with chlorpromazine or bafilomycin 2 h after infection.
Rab11S25N transdominant-negative assay.
Approximately 8 × 106 Cos-7 cells were transfected with 20 μg of plasmid pECFP-Rab11S25N or plasmid pECFP-N1 by electroporation using standard procedures and plated on 35-mm-diameter dishes. At 24 h after transfection, cells were mock infected or infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 2 PFU/cell. Cells were fixed with paraformaldehyde (4% in PBS) at 20 h postinfection and observed with a conventional Leica DMB fluorescence and phase-contrast microscope. To verify the effect of Rab11S25N on gB subcellular localization, transfected cells were fixed 7 h postinfection and stained with the anti-gB antibody as described above.
RESULTS
Construction of gB mutant viruses.
The cytoplasmic tail of HSV-1 gB contains two trafficking signals involved in the retrograde transport of the glycoprotein from the cell surface to the TGN (7). The tyrosine motif YTQV (aa 889 to 892) was shown to act as an endocytosis signal promoting retrieval of the glycoprotein from the cell surface. Indeed, mutation of the tyrosine residue (gBY889A) totally impaired internalization, and truncation of the tyrosine motif was associated with both accumulation at the plasma membrane and impaired internalization of the protein. The LL signal at position 871 to 872 was involved in a second transport step that directs gB to the TGN. When this motif was mutated (gBLL871AA), gB was still internalized, but its return to the TGN was impaired and recycling to the cell membrane was increased. To address the contribution of these signals to virus infection, we constructed recombinant viruses containing either gBY889A or gBLL871AA in place of gB, using classical cotransfection procedures. To obtain mutant viruses and screen for them easily, we used a previously published defective HSV-1 recombinant, KgBct-GFP. In this virus derived from the KOS strain, the cytoplasmic tail of gB has been disrupted by an in-frame incorporation of GFP. Due to a defect in maturation of the fusion gB-GFP protein, this virus is defective and grows only on gB complementing cells, where it produces fluorescent plaques (61). We assumed that the replacement of gB-GFP with either gBY889A or gBLL871AA would restore the infectivity of the virus. Vero cells were cotransfected with the KgBct-GFP infectious viral DNA and the linearized plasmids pgBY889A or pgBLL871AA. These plasmids encode a gB containing ATQV and AA in place of the YTQV and LL motifs, respectively. Progeny viruses isolated 4 to 5 days after transfection were plated on Vero cells. Recombinants were then easily screened for their ability to form plaques and for the loss of GFP fluorescence. The resulting viruses were plaque purified three times, and viral stocks were constituted and titrated on Vero cells. Each of the KgBY889A and KgBLL871AA genomes had lost the GFP encoding sequence and recovered the electrophoresis pattern of the KOS strain, as verified by BamHI restriction length fragment polymorphism analysis. In addition, insertion of the expected mutation in the gB gene was verified by PCR and sequencing.
Biotinylation and endocytosis of HSV-1 gB in infected cells.
To investigate whether internalization of gB from the cell surface occurred during infection, an endocytosis assay that did not rely on antibodies was performed using biotinylation of cell surface proteins. We first ensured that gB was detected on the surface of infected cells by biotinylation. 143B cells were infected with KOS and washed with acid-glycine to remove any remaining nonpenetrated virion particles, and cell surface proteins were biotinylated at 8 h postinfection at 4°C using noncleavable biotin. Whole-cell extracted protein samples were immunoprecipitated with an anti-gB-antibody, and biotinylated gB was revealed by streptavidin-HRP Western blotting under nonreducing conditions and then further identified by gB Western blotting (Fig. 1A). To investigate whether HSV-1 gB was internalized at early times of infection, the biotinylation experiment was repeated with Sulfo-NHS-SS cleavable biotin. Cells were infected and biotinylated at 8 h postinfection at 4°C with cleavable biotin. After a washing step, the infected cells were left 4 more hours at 37°C before the cells were treated with GSH to remove any noninternalized biotin from the cell surface. After immunoprecipitation and streptavidin-HRP Western blotting, biotinylated gB was detected in cells that were incubated for 4 h after biotinylation, whereas no signal was observed in cells that were treated with GSH immediately after biotinylation (compare Fig. 1B, lanes 2 and 3). This proved that HSV-1 gB present on the surface of infected cells had been internalized.
FIG. 1.

Endocytosis of gB during infection assayed by biotinylation. (A) Biotinylation of gB was assayed in 143B cells with noncleavable biotin 8 h after infection with KOS. Samples were immunoprecipitated with a polyclonal anti-gB antibody and then separated by sodium dodecyl sulfate-7% polyacrylamide gel electrophoresis under nonreducing conditions, and biotinylated glycoproteins were revealed with streptavidin-HRP by Western blotting (lanes 1 to 4). After stripping, the membrane was revealed with a monoclonal anti-gB antibody (lanes 5 to 8). Lanes: 1, 2, 5, and 6, uninfected control cells; lanes 3, 4, 7, and 8, infected cells; lanes 1, 3, 5, and 7, nonbiotinylated; lanes 2, 4, 6, and 8, biotinylated. (B) 143B cells were biotinylated with Sulfo-NHS-SS-cleavable biotin 8 h after infection with KOS and left for 4 h for infection to proceed. After treatment of the cells with GSH, lysed samples were gB immunoprecipitated and visualized with streptavidin-HRP (lane 3). As controls, cells were gB immunoprecipitated immediately after the biotinylation step, without (lane 1) or after (lane 2) GSH treatment. Lane 4 shows the gB Western blot of lane 3 after stripping.
Imaging analysis of gB endocytosis during infection.
Since the biotinylation method does not provide an accurate quantitative estimation of internalized glycoproteins (44), we visualized endocytosis of gB during HSV-1 infection (as previously in transfection experiments) by performing an internalization assay of exogenously added antibodies. 143B cells were chosen because in these cells the Golgi bodies and the TGN are not disrupted during early steps of HSV-1 infection (5, 73). An anti-gB antibody was added to the culture medium 7 h after infection with KOS. After 45 min at 0°C, the cells were either immediately fixed or incubated at 37°C for various time intervals to allow and monitor the internalization of the gB-antibody complexes from the plasma membrane. In cells that were not shifted to 37°C, referred to as time zero cells, the gB-antibody complex was detected exclusively at the surface of cells (Fig. 2M and Q). Fifteen minutes after the shift, vesicles containing internalized gB were clearly detected under the cell membrane, and some of these vesicles were seen proximal to the nucleus and, in particular, in close apposition to the TGN, as identified with a TGN 46 antibody (Fig. 2J, N, and R). Thirty minutes after the shift, numerous vesicles were visible under the cell membrane, displaying a punctuate pattern. In some infected cells, gB was detected in larger vesicles or patches under the cell membrane. Most importantly, a significant amount of internalized gB-antibody complex had reached the TGN. In fact, most of the TGN contained internalized gB, as revealed by almost complete colocalization of TGN-specific and gB stainings (Fig. 2K, O, and S). At 60 min, a major amount of the antibody-linked glycoprotein was concentrated in the TGN, albeit small vesicles were still seen dispersed in the cytoplasm of cells. At this time of the assay, remarkably little antibody-linked gB was detectable at the cell surface (Fig. 2L, P, and T). These results demonstrate that HSV-1 gB was internalized from the cell surface to concentrate in the TGN, as previously shown in transfected cells. At the early stage of infection investigated (7 h postinfection), the capsid-specific Cy2 staining was mainly concentrated in the nucleus, although some faint fluorescence was visible in the cytoplasm (Fig. 2E, F, G, and H). Colocalization of capsids with gB was, however, not obviously detected in particular in the TGN (Fig. 2O, P, S, and T).
FIG. 2.

Confocal microscopy analysis of gB internalization in KOS-infected cells. 143B cells were infected with wild-type KOS virus at an MOI of 0.1. At 7 h postinfection, the cells were incubated with an anti-gB antibody at 4°C for 45 min and then placed at 37°C for 0 min (A, E, I, M, and Q), 15 min (B, F, J, N, and R), 30 min (C, G, K, O, and S), or 60 min (D, H, L, P, and T). Cells were fixed in methanol and labeled with anti-TGN 46 and anti-VP5 antibodies. Panels A to D correspond to phase-contrast images of infected cells, whereas panels E to P show indirect immunofluorescence from anti-VP5 and Cy2-labeled secondary antibodies (green) (E to H), from anti-TGN 46 and Cy5-labeled secondary antibodies (pseudo-colored in blue) (I to L), or from anti-gB and Cy3-labeled secondary antibodies (red) (M to P). (Q to T) Merged images. Fluorescence was visualized with a Leica TCS SP2 AOBS confocal microscope.
Internalization of mutated gBs in infected cells.
In transfected cells, disruption of either of the YTQV or LL motifs impaired the endocytosis of gB. To investigate whether this remains true during infection, we repeated the internalization assay described above in cells infected with the gB-mutated viruses. At 0 min, in cells infected with KgBY889A that were not shifted to 37°C, gB was detected abundantly at the surface of cells, with a thick and continuous pattern of staining lining the cell periphery (Fig. 3M and Q). In sharp contrast to observations in KOS-infected cells, virtually the whole amount of antibody-linked glycoprotein remained at the cell surface 15 to 30 min after the shift (Fig. 3N, O, R, and S). In particular, no vesicle containing gB was detected inside the cells. At the last time point of the assay, very few sparse gB-stained vesicles were actually visible under the cell membrane (Fig. 3P and T). However, the TGN 46 staining and antibody-linked gB did not colocalize, revealing that the TGN contained no detectable gB internalized from the cell surface (Fig. 3S and T). These results indicate that disruption of the YTQV motif prevented the internalization of gB from the cell surface and its concentration in the TGN after internalization in infected cells as previously described in transfection assays. Most of the capsid-specific staining was seen inside the nuclei of infected cells (Fig. 3E, F, G, and H). However, some of the capsid staining was visualized in close apposition to the TGN and/or in this latter compartment, as suggested by merged images (see Fig. 3R). The pattern observed in cells infected with KgBLL871AA was quite different. At 0 min, the antibody stained the glycoprotein at the cell surface (Fig. 4M and Q). At 15 min after the shift to 37°C, some vesicles containing antibody-linked gB were actually observed under the cell surface (Fig. 4N and R). Careful observation revealed, however, that frank colocalization of gB with TGN 46 did not occur, although gB-containing vesicles were seen in a compartment close to the TGN (Fig. 4J, N, and R). At 30 and 60 min, these vesicles remained accumulated under the cell surface, most of them homogenously surrounding the nuclei of infected cells. As observed above after infection with KOS, dense accumulations or patches of antibody-linked gB were present at or under the cell surface at 15 and 30 min after the shift (Fig. 4N, O, R, and S). In contrast to KOS, however, no internalized gB had yet gained the TGN compartment at these time points (Fig. 4J, K, R, and S), and very few reached the TGN at the end of the assay (Fig. 4L, P, and T). These results suggest that gB was effectively endocytosed from the surface of KgBLL871AA-infected cells, but most of the internalized protein did not reach the TGN, accumulating instead in an intermediate vesicular compartment. Overall, these observations in infected cells were strikingly similar to our previous findings in the context of transfection (7).
FIG. 3.

Internalization of gB in KgBY889A-infected cells. The same experiment as that described in Fig. 1 was reproduced in 143B cells infected with a virus expressing a mutated YTQV→ATQV gB protein. Subcellular localization of surface-stained gB was analyzed by confocal microscopy as described in the legend for Fig. 1. (A, E, I, M, and Q) Incubation for 0 min at 37°C; (B, F, J, N, and R) 15-min incubation at 37°C; (C, G, K, O, and S) 30-min incubation at 37°C; (D, H, L, P, and T) 60-min incubation at 37°C. Cells were fixed with methanol and then stained with anti-TGN 46 and anti-VP5 antibodies. Panels A to D show phase-contrast images of the different time intervals analyzed. Panels E to H show immunostaining with VP5 (green). Panels I to L show immunostaining with TGN 46 (blue). Panels M to P show immunostaining with anti-gB antibodies (red). (Q to T) Merged images.
FIG. 4.

Internalization of gB in KgBLL871AA-infected cells. Seven hours after infection of 143B cultures by a mutant virus in which the gB LL motif was replaced by AA, cells were processed as described in the legend of Fig. 1. (A, E, I, M, and Q) Incubation for 0 min at 37°C; (B, F, J, N, and R) 15-min incubation at 37°C; (C, G, K, O, and S) 30-min incubation at 37°C; (D, H, L, P, and T) 60-min incubation at 37°C. Panels A to D show phase-contrast images. Anti-VP5, anti-TGN 46, and anti-gB staining is shown in panels E to H, I to L, and M to P, respectively. (Q to T) Merged images.
To characterize more precisely the vesicles where gB was observed and verify that they corresponded to early recycling endosomes, the same experiment was repeated except that the cells were incubated with an anti-transferrin receptor (CD71) antibody in addition to the anti-gB antibody. The transferrin receptor is known to be internalized during the first hours of infection with herpesviruses (68) and labels endosomes of the early and recycling pathway. After 15 min of internalization and up to the end of the assay, most of the intracellular vesicles containing gB in cells infected with wild-type or LL871AA-mutated virus were identified as early recycling endosomes, as visualized by colocalization of gB with CD71 (Fig. 5C, D, K, and L). As expected, gB did not colocalize with internalized CD71 in KgBY889A-infected cells (Fig. 5G and H).
FIG. 5.

Cointernalization of gB and transferrin receptor in infected cells. To characterize the vesicular compartment where gB was internalized, the same experiments as in Fig. 2, 3, and 4 were repeated except that the cells were incubated with a CD71-specific antibody, in addition to the anti-gB antibody, for 60 min. After fixation, cells were stained with an anti-TGN antibody. Merged images of the anti-TGN (blue), anti-CD71 (green), and anti-gB (red) stains are shown for KOS-, KgBY889A-, and KgBLL871AA-infected cells in panels D, H, and L, respectively.
Subcellular distribution of gB in infected cells.
To verify whether endocytosis of gB had an effect on the overall subcellular distribution of the glycoprotein at the steady-state of infection, 143B cells were infected with KOS, KgBY889A, or KgB871LLAA and then fixed 7 h postinfection. Permeabilized cells were stained with anti-CD71, anti-TGN, and anti-gB (Fig. 6). To analyze the fraction of total gB in each cellular compartment, confocal imaging quantification was performed on eight cells for each series of infection. As seen in Table 1, a significantly lower fraction of gB was present in the TGN of cells infected with the KgBLL871AA compared to cells infected with KOS, whereas the difference between KgBY889 and KOS did not reach statistical significance. Conversely, a significantly lower fraction of the tyrosine-mutated gB colocalized with early recycling endosomes compared to KOS. Since the subcellular localization of mutant gBs differed from that of wild-type gB at 7 h postinfection, a time when the internalization of mutant gBs was significantly impaired compared to wild-type gB, and since gB transits through the TGN during infection before reaching the cell surface, these results overall suggest that part of gB observed in the TGN of infected cells was internalized from the cell surface.
FIG. 6.

Subcellular distribution of gB in infected cells. 143B cells were infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 0.1, fixed 7 h postinfection; permeabilized; and stained with anti-TGN, anti-CD71, and anti-gB antibodies. Immunostaining of TGN (blue), CD71 (green), and gB (red) is shown in panels A to C, D to E, and F to H, respectively. (I to K) Merged images.
TABLE 1.
Quantification of gB subcellular distribution in infected cellsa
| Site | gB colocalization (%) in cells infected with:
|
||
|---|---|---|---|
| KOS | KgBY889A | KgBLL871AA | |
| ER endosomes | 69 | 46b | 64 |
| TGN | 28 | 16 | 10c |
143B cells infected with KOS, KgBY889A, or KgBLL871AA were stained with anti-gB, anti-CD71, or anti-TGN antibodies and visualized with fluorescent secondary antibodies. Confocal imaging quantification was performed on eight cells for each series of infection. The results are expressed as the mean value of the fraction of total gB colocalizing with TGN46 and CD71. Statistical analysis was performed using the Student t test.
P = 0.004.
P = 0.006.
Growth characteristics of gB mutant viruses.
To analyze the consequences of gB mutations on viral replication, the growth of KgBY889A and KgBLL871AA was compared to that of KOS, in both Cos-7 and 143B cells. Both mutant viruses exhibited final decreased titers of released progeny virions relative to the wild-type virus, in single-step or multistep growth curves. A more marked reduction was reproducibly observed in cells infected with KgBY889A (Fig. 7), which exhibited the lowest intra- and extracellular infectious titers. High titers obtained with KgBLL871AA at some intermediate points of the multistep growth curve were probably due to the rapid syncytial and lytic ECP observed with that mutant. In addition, results from four independent assays showed that KOS and KgBY889A intracellular titers were markedly reduced by low pH treatment, since 90 and 120% reductions were observed after treatment at 16 and 20 h postinfection, respectively, proving that a significant fraction of the cell-associated infectivity corresponded to virions at the cell surface. In contrast, low-pH washing reduced KgBLL871AA cell-associated infectivity by only 16 and 20% at the same times of the assay (P < 0.05), suggesting that most of KgBLL871AA infectivity was intracellular and not associated with virus budding at the cell surface. Altogether, these results suggest that the production of mutant infectious particles is impaired at different intracellular steps of virus assembly.
FIG. 7.

Virus growth. 143B cells were infected with KOS, KgBY889A, or KgBLL871AA, and the infectivity of the progeny viruses was analyzed at different times postinfection by titration on Vero cells. (A) Cells were infected at an MOI of 5, and extra- and intracellular infectivities were assayed at 6, 9, 12, 16, 20, and 24 h postinfection. (B) Multiple-step growth analysis. Cells were infected at an MOI of 0.001, and progeny viruses were harvested 24, 48, and 72 h postinfection. The data shown correspond to averages of two or three independent experiments. Vertical lines indicate the standard deviations.
Incorporation of gB into virion particles.
To ensure that the recombinant gB molecules were incorporated into virion particles in the same amounts as wild-type gB, a quantification analysis of gB immunostaining was performed by Western blotting on extracellular virions preparations. The membrane was labeled with gB and capsid-specific antibodies and visualized with secondary HRP-coupled antibodies. Quantification of the bands corresponding to gB and ICP5, respectively, demonstrated that the ratio of gB relative to that of ICP5 was similar in KgBY889A, KgBLL871AA, and in KOS particles (Fig. 8), indicating that incorporation of gB into particles was not modified by alteration of the endocytic signals.
FIG. 8.
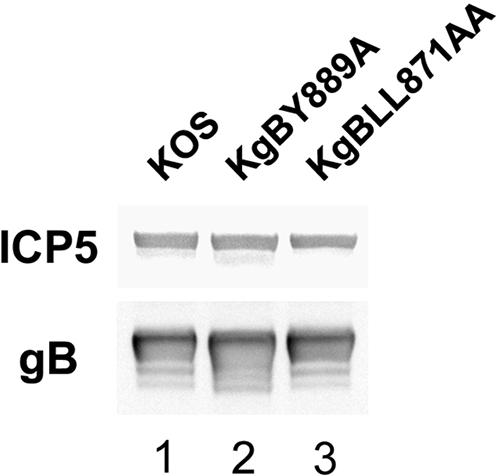
Incorporation of gB in virions. Extracellular virus recovered from infected cell supernatants at 8 h postinfection were subjected to polyacrylamide gel electrophoresis under denaturing conditions and transferred onto nitrocellulose membrane. The membrane was treated with anti-gB and anti-ICP5 antibodies and then with HRP-coupled secondary antibodies visualized by enhanced chemiluminescence. Quantification analysis was performed with Fujifilm Multi-Gauge software.
Cell-cell fusion in infected cultures.
Syncytium formation is an unusual event upon HSV-1 infection, which has been reported to depend on both cellular and viral factors. For instance, syncytia are more readily observed after infection of Cos than Vero cells (3). Among viral factors involved in syncytium formation, mutations in UL24, gK, UL20, and gB have been described (64). In particular, most gB mutations causing a syncytial phenotype are located in the cytoplasmic tail of the protein (24). To investigate whether mutations in YTQV and LL motifs of gB affect cell-cell fusion, Cos-7 cells were infected with KOS, KgBY889A, or KgBLL871AA at an MOI of 1 and then fixed 20 h after infection and visualized by phase-contrast microscopy. In KOS-infected cells, small polycaryocytes containing an average of three nuclei (Fig. 9A) were counted in each field, i.e., 30% of cells belonged to such small syncytia. At the same time postinfection, cells infected with KgBY889A exhibited virtually no visible polycaryocyte (Fig. 9B), suggesting that an increased presence of gB at the surface of cells did not enhance cell-cell fusion. In contrast, giant syncytia were readily observed in cells infected with KgBLL871AA, some of which contained more than 100 nuclei (Fig. 9C), i.e., 85% of the cells belonged to syncytia. Thus, a maximal syncytial effect was associated with a gB mutation that allows internalization of gB from the cell surface but prevents its subsequent transport to the TGN.
FIG. 9.

Infection phenotypes of wild-type and gB-mutated viruses in Cos-7 cells. Confluent cell monolayers were infected with KOS (A), KgBY889A (B), and KgBLL871AA (C) at an MOI of 1. Live cells were visualized by phase-contrast microscopy at 20 h postinfection.
Effect of drugs on virus-induced cell-cell fusion.
Our observations on gB transport during infection and on virus-induced cell-cell fusion suggested that both processes might be related. We hypothesized that if gB trafficking is indeed involved in syncytium formation, drugs known to affect specific steps of the retrograde transport from the cell surface back to the TGN should affect cell-cell fusion in infected cell cultures. Since inhibition of gB internalization was associated with a nonsyncytial phenotype, we investigated the effects on cell-cell fusion of a drug that blocks protein internalization from the cell surface. Chlorpromazine is a cationic amphiphilic compound, which induces the redistribution of clathrin-coated components by removing the adaptor protein AP-2 from the plasma membrane (36, 66). Thus, in chlorpromazine-treated cells, clathrin-dependent internalization of proteins is blocked. Two hours after infection with KOS, KgBY889A, or KgBLL871AA at an MOI of 1, Cos-7 cells were either treated with chlorpromazine or left untreated and then fixed 20 h postinfection and visualized under a phase-contrast microscope. We first checked that internalization of gB was blocked in chlorpromazine-treated cells infected with KOS or KgBLL871AA at the latest time point of our assay (data not shown). As seen in Fig. 10A and B, treatment with chlorpromazine prevented the formation of small syncytia otherwise observed in KOS-infected cells. In contrast, the phenotype of KgBY889A-infected cells was not modified by the drug (Fig. 10C and D). Remarkably, chlorpromazine completely abolished the formation of giant syncytia in cells infected with KgBLL871AA since polycaryocytes were not observed (compare Fig. 10E and F). Therefore, inhibition of internalization abolished the cell fusion phenotype associated with the wild-type gB and the gB dileucine mutant (KgBLL871AA), thus reproducing the phenotype of infection with KgBY889A that results from the gB tyrosine motif mutation.
FIG. 10.

Effects of chlorpromazine on infection phenotypes. Confluent Cos-7 cell monolayers were infected with KOS (A and B), KgBY889A (C and D), or KgBLL871AA (E and F) or were mock infected (G and H). At 2 h after infection, cells were either treated with 5 μg of chlorpromazine/ml (A, C, E, and G) or left untreated (B, D, F, and H). Cells were observed at 20 h postinfection.
The gB dileucine mutation alters the retrograde transport of gB to the TGN and increases cell-cell fusion. In transfected cells, this mutation was associated with an increased recycling of gB to the cell surface. We wondered whether inhibition of recycling would prevent cell-cell fusion. Bafilomycin A1 is a potent inhibitor of vacuolar proton ATPases and blocks trafficking from early to late endosomes (74, 82). In live cells, the drug inhibits acidification of endosomes and lysosomes (74, 82). This results in the inhibition of recycling of proteins to the cell surface without preventing their internalization (6). We first verified that internalization of gB was not blocked in bafilomycin-treated cells infected with KOS or KgBLL871AA at the latest time point of our assay (Fig. 11). Two hours after infection with KOS, KgBY889A, or KgBLL871AA at an MOI of 1, Cos-7 cells were either treated with bafilomycin A1 or DMSO as a control or left untreated. Live cells were visualized by phase-contrast microscopy at 20 h postinfection and then fixed. Immunostaining with anti-ICP5 confirmed that all cells were infected in the presence of bafilomycin A1 (compare Fig. 12D, H, and L and Fig. 7O). Treatment with bafilomycin A decreased the number of small polycaryocytes from 35 to 7% in KOS-infected cells (compare Fig. 12A, B, and C), which therefore had the same phenotype as KgBY889A-infected cells (see Fig. 12E, F, and G). Moreover, the drug reduced the size (from 80 to 4 nuclei) and the number of syncytia (from 84 to 25%) in cells infected with KgBLL871AA (compare Fig. 12I, J, and K). These results suggested that inhibition of acidification of endosomes (34, 42) and/or the inhibition of recycling to the cell surface abolished the formation of syncytia, particularly those associated with the KgBLL871AA mutation.
FIG. 11.
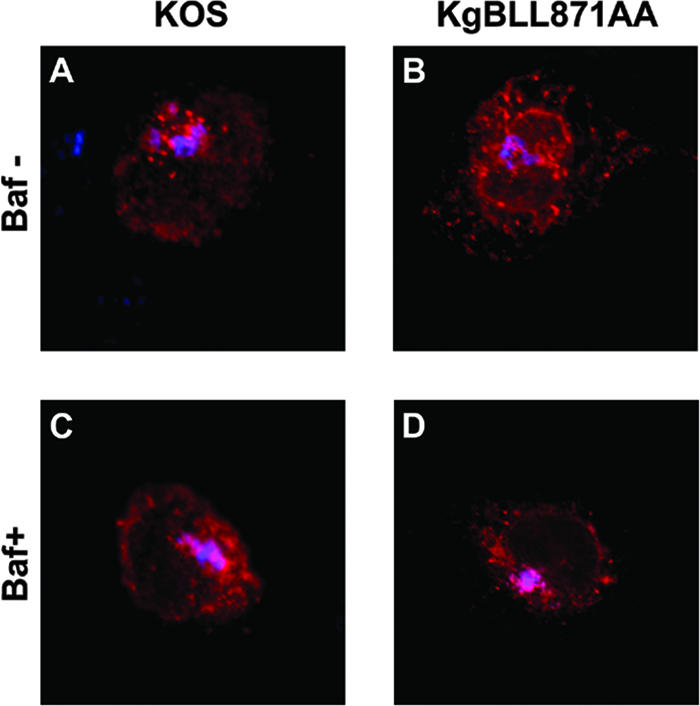
Internalization assay of bafilomycin A1-treated infected cells. The internalization assay was repeated in KOS- or KgBLL871AA-infected cells as described in Fig. 2 and 4, except that the cells were treated with bafilomycin A1 at 2 h postinfection. Images taken at the latest time point of the assay (60 min) correspond to the merge of the gB (red) and TGN (blue) stainings. (A and B) Control untreated cells; (C and D) treated cells.
FIG. 12.

Effects of bafilomycin A1 on infection phenotypes. Confluent Cos-7 cell monolayers were infected with KOS (A to D), KgBY889A (E to H), or KgBLL871AA (I to L) or were mock infected (M to O). At 2 h after infection, cells were either left untreated (A, E, I, and M) or treated with 1 μg of DMSO/ml (B, F, and J) or 250 nM bafilomycin A (C, D, G, H, K, L, N, and O). Live cells were observed 20 h postinfection by phase-contrast microscopy (A to C, E to G, I to K, and M and N). To check for infection, cells were fixed with methanol and then incubated successively with an anti-VP5 antibody, a biotin-coupled secondary antibody, and β-galactosidase-coupled streptavidin; the staining was revealed with X-Gal (D, H, L, and O).
Effect of expression of a dominant-negative form of Rab11 on virus-induced cell fusion.
The drastic effect induced by bafilomycin A treatment could be due to inactivation of a mechanism of fusion requiring low pH and/or to inhibition of recycling to the cell surface. Rab11, a member of the Rab family of small GTPases, localizes to the endocytic compartment and has been shown to regulate membrane distribution inside the early endosomal pathway (80). In that study, overexpression of a mutant form of Rab11, Rab11S25N, prevented both recycling to the cell membrane and access to the TGN. To further investigate whether recycling to the cell surface could be involved in cell fusion, Cos-7 cells were transfected with an ECFP-tagged dominant-negative Rab11S25N construct or a control ECFP-expressing plasmid and then infected 24 h posttransfection with KOS, KgBY889A, or KgBLL871AA at an MOI of 2. Cells were fixed at 20 h postinfection and visualized by phase-contrast and classical fluorescence microscopy. In these experiments, the expression of ECFP permits easy recognition of syncytium formation. To check the effect of the dominant-negative protein on gB, transfected infected cells were fixed 7 h postinfection and stained with the gB antibody. As seen in Fig. 13, the paranuclear TGN localization of wild-type and mutated gB was totally disrupted in Rab11S25N-transfected cells compared to controls. Infected cells previously transfected with the control ECFP construct displayed the same infection phenotype as that previously observed in untransfected cells. In particular, small polycaryocytes were observed in KOS-infected cells (compare Fig. 14E and H), whereas 80% of KgBLL871AA-infected cells were in large or giant syncytia (Fig. 14G). In cells transfected with the Rab11S25N construct, expression of the dominant-negative protein was accompanied by a drastic diminution of cell fusion compared to control cells. In fact, giant syncytia were no longer detected in KgBLL871AA-infected-cells (compare Fig. 14G and O), although 29% of the cells still belonged to small polycaryocytes containing fewer than five nuclei (see Fig. 14M, N, O, and P), probably because the dominant-negative protein was expressed in 50% of the cells at most, and the remaining cells still have the potential to initiate fusion. Overall, the results suggested that impaired recycling of internalized proteins significantly reduced cell-cell fusion in KgBLL871AA-infected cells.
FIG. 13.

Subcellular distribution of gB in Rab11S25N-transfected infected cells. Cells were transfected with the control or Rab11 mutant-expressing plasmids as for Fig. 13, infected at 0.1 PFU, fixed at 7 h postinfection, permeabilized, and stained with anti-gB antibody (red).
FIG. 14.

Phenotypes of infections in infected cells expressing a transdominant-negative form of Rab11. Cos-7 cells were transfected with plasmids expressing either ECFP (A to H) or an ECFP-tagged transdominant-negative Rab11 mutant, ECFP-Rab11S25N (I to P). Cells were infected 24 h after transfection with KOS (A, E, I, and M), KgBY889A (B, F, J, and N), or KgBLL871AA (C, G, K, and O) at an MOI of 2 or were mock infected (D, H, L, and P). Cells were fixed 20 h postinfection and visualized under phase-contrast and conventional fluorescence microscopy (E to H and M to P). (A to D and E to L) Merged images.
DISCUSSION
Many viruses take advantage of the cell endocytic mechanisms in multiple aspects of their replication cycles (45). Endocytosis of membrane proteins is generally mediated by consensus tyrosine-based YXXΦ or dileucine motifs (8, 18). Endocytosis of viral glycoproteins expressed in transfected cells has been described in several models (9, 23). In particular, the gBs of HSV-1, HSV-2, PRV, VZV, and CMV are transported to the cell surface to be subsequently internalized (7, 17, 19, 31, 63, 71). In addition to gB, the gD, gE, gH, gI, and gM proteins of several herpesviruses contain functional endocytosis motifs (1, 2, 14, 56, 57, 68). Endocytosis during infection has been demonstrated for PRV, CMV, and VZV gB (44, 55, 63) and PRV, HSV-1, and VZV gE (8, 44, 46, 67) and gH (30, 44). The fact that most herpesvirus gBs contain more than one trafficking signal questions the role of this apparent redundancy. We showed here that in HSV-1-infected cells the YTQV motif located at the carboxy-terminal end of the gB tail played a central role in endocytosis since its disruption totally prevented internalization of the protein. In PRV-infected cells, the carboxy-terminal YQRL motif (residues 902 to 905) of gB was shown to play a similar role (20) via its interaction with AP-2 (75). In transfection experiments at least, suppression of the YSRV motif (aa 857 to 860) of VZV gB also suppressed internalization of the protein (31). The role of the LL motif present in HSV-2, VZV, PRV, and CMV gB homologues has been less deeply investigated. In VZV, previous studies reported that neither the internalization nor the concentration of gB at the Golgi were impaired when the LL motif was mutated (17, 31). In HSV-2 gB, mutation of the LL motif did not affect the overall distribution of the protein (17). Our results differ from these previous reports. Careful confocal fluorescence imaging of gB mutated on the LL motif in HSV-1-infected cells revealed a pattern of retrograde transport to the TGN surprisingly similar to what we had reported in transiently transfected cells expressing gB (7). Indeed, internalization of gB from the surface of cells infected with the gB dileucine mutant virus did occur, but a further step of its transport to the TGN was blocked. Colocalization of internalized gB with Rab5a, a marker of early endosomes, but not with Rab7, a marker of late endosomes, was demonstrated in transfection assays (3). We show here that after internalization wild-type gB was mainly concentrated in early/sorting or recycling endosomes and in the TGN compartment. This trafficking pattern is strikingly reminiscent of that of a transmembrane protein, the cation-independent mannose phosphate receptor (CI-MPR). After endocytosis, CI-MPR passes through the early recycling compartment before reaching the TGN. With each round of recycling, 15% of the receptors are delivered to the TGN and to late endosomes, while 85% are recycled to the cell surface (70). We showed previously in transfected cells that HSV-1 gB is internalized and recycles to the cell surface and that disruption of the dileucine motif enhances recycling. In infected cells, the dileucine mutation significantly lowered the fraction of total gB in the TGN, and gB was mainly associated with early recycling endosomes. A dileucine motif in the cytoplasmic tail of the CI-MPR was similarly shown to play a critical role in sorting of the receptor from the early recycling compartment to the TGN (70).
One of the proposed roles of endocytosis during virus replication is the targeting of viral components to the appropriate cell compartment for viral assembly (9, 18, 45). Several herpesvirus glycoproteins, including gB, are targeted to the TGN (2, 14, 18, 22, 29, 46, 84). Moreover, incorporation of internalized glycoproteins has been demonstrated for PRV gE (68); for CMV gB (63); and, more recently, for VZV gB, gE, and gH (44). These data favor a long-debated model which states that assembly and final envelopment of virions take place in the TGN (reviewed in reference 49). Using triple-labeling experiments of infected cells, we observed that gB accumulated in the TGN, but merged images showed few superposition of capsids and gB in this compartment. Interestingly, by using immunofluorescence analysis of synchronized HSV-1 infection, Turcotte et al. also reported that most capsids were localized immediately adjacent to the TGN46 marker, whereas some perfectly colocalized with the TGN (73). It was suggested that this might be related to the kinetics of envelopment. Whether internalization of glycoproteins affects the production of infectious particles has not been completely determined and seems to vary according to the virus and the glycoprotein investigated. For instance, endocytosis of VZV gE is essential for the virus life cycle, since a single mutation of the gE YAGL endocytosis motif is lethal for replication (51). In contrast, endocytosis of PRV gE is dispensable for virus growth (68). The data concerning gB are complex and contradictory. Inhibition of endocytosis of HCMV gB using a dominant-negative dynamin I molecule did not affect the production of infectious CMV (15, 38). However, small interfering RNA silencing of PACS-1, a protein required for transport of HCMV gB to the TGN after internalization in infected cells, slightly decreased HCMV titers (15). Moreover, the increased transport to the TGN of a mutated HCMV gB resulted in increased levels of virus production in infected fibroblasts (39). In the case of PRV gB, mutation of the YQRL motif (aa 902 to 905) did not modify virus growth (20), whereas truncation of the last 28 last carboxy-terminal amino acids, which removes both the YQRL and the LL motifs, slightly reduced virus production (54). Similarly, a VZV recombinant virus that lacks the C-terminal 36 aa of gB, including the YSRV motif and an LL motif (aa 840 to 841), displayed a slight reduction in virus production. Here, we showed that substitution of gB tyrosine or dileucine motifs diminished the infectivity of HSV-1 in a range comparable to that previously reported in ambB1, an HSV-1 mutant that lacks the 41 carboxy-terminal amino acids of gB (35). Altogether, these observations suggest that although endocytosis of gB is not essential to virus growth it contributes to the production of infectious virus and that each of the tyrosine and dileucine motifs differentially contributes to this effect. Further studies will be necessary to determine whether this is different in other cell types. Endocytosis is involved in the sorting of specific glycoproteins to subcellular compartments of polarized cells. For instance, endocytosis of HSV-1 gE is required for gE targeting to lateral surfaces and cell junctions during intermediate to late stages of HSV infection (40). The cytoplasmic tail of CMV gB contains determinants of vectorial sorting in polarized epithelial cells (72). In PRV, the tyrosine motif of gB was involved in the sorting to basolateral surfaces of infected cells, and this signal was supposedly involved in the level of viral cell-cell spread (20). HSV-1 gB has been shown to relocalize to the basolateral cell membrane of infected cells (81). Whether the YTQV or the LL motifs are involved in this polarized targeting and influence the production of viral particles in these cells requires further investigation.
Another proposed role of envelope glycoprotein endocytosis in the replication cycle of viruses consists of the regulation of cell-cell fusion (37, 64, 76). In the case of HSV, fusion for entry and cell-cell fusion require the functional participation of gB, gD, and gH/gL (reviewed in reference 65). The four glycoproteins are sufficient to induce cell-cell fusion in transfected cells provided these express HSV receptors (10, 52, 60, 64). Although the precise mechanisms of fusion have not been completely deciphered, gB and gH/gL are supposed to be the most probable effectors (26, 64). However, HSV-induced cell fusion is usually limited in vitro and in vivo (17), which may result from regulation mechanisms aimed to prevent enhanced pathogenesis (27). It is generally assumed that the increased cell surface expression of viral fusogenic glycoproteins may increase virus-mediated cell-cell fusion (37). Endocytosis could contribute to counteract this process. For instance, transfection studies showed that gH endocytosis leads to decreased cell-cell fusion in association with decreased surface density of gH (58). Interestingly, some of the mutations in the cytoplasmic tail of herpesvirus gBs that enhance cell-cell fusion also affect trafficking signals (17, 21, 24, 27, 64, 77). We have shown that endocytosis of gB does occur in cells infected with wild-type HSV-1, which could account for the limited cell-cell fusion phenotype observed in these cells. Surprisingly, however, the increased presence of gB at the cell surface does not necessarily correlate with increased cell fusion. In HSV-2, the increased cell surface expression of a mutated Y867A gB did not induce a syncytial phenotype (17), and in PRV gB the Y902A mutation induced a small-plaque phenotype (20). Similarly, in our experiments disruption of the YTQV motif of gB abolished fusion in HSV-1-infected cells, and this effect was reproduced by a drug which blocks AP-2-mediated internalization from the cell surface and which prevented gB endocytosis. This suggests that, after access to the cell surface, internalization of gB is required for fusion. Interestingly, a similar correlation between endocytosis and fusion was reported for Nipah virus, a member of the Paramyxoviridae family. The fusion envelope protein F of this virus is internalized in infected and transfected cells. Disruption of the YSRL motif of protein F abolished both internalization and cell fusion in a transfection assay, suggesting that blocking of the protein at the cell surface negatively regulates fusion (76).
In contrast, in HSV-1-infected cells, we observed an upregulation of cell-cell fusion upon mutation of the gB dileucine motif. Similarly, in HSV-2 gB, the LL849/850AA mutation, which did not increase cell surface expression, did produce a syncytial phenotype (17). Replacement of the LL motif in PRV gB also resulted in the formation of large syncytia, and this effect was supposed to result from an enhancement of the fusogenic activity of gB (20). Since internalization of gB seems necessary for cell-cell fusion, and fusion most probably requires the presence of the glycoprotein at the cell membrane (64), we hypothesize that the enhanced fusion phenotype associated with the LL mutation could result from increased recycling of gB to the cell surface after endocytosis. Indeed, we previously demonstrated that in transfected cells the LL mutation is associated with an increased recycling of gB to the cell membrane. Furthermore, we showed in the present study that syncytium formation was inhibited by a drug or by a dominant-negative protein which impair recycling to the surface.
In addition to mutations in HSV-1 gB, mutations in gK, UL20, UL24, and UL45 enhance cell-cell fusion in infected and transfected cells (3, 28, 47). This is thought to reflect a role for these proteins in the regulation of the process of fusion, probably through direct or indirect interactions with the fusogenic glycoproteins. Syncytial mutations in HSV-1, HSV-2, and PRV gBs have been suggested to modify the structure of the alpha-helical domain II of gB cytoplasmic domain (4, 17, 20, 21), thereby impairing functional interactions with proteins regulating fusion (20, 47). Our results favor the hypothesis that regulation of gB fusogenic properties through these interactions also depends on gB intracellular trafficking. Modifications of gB intracellular transport might prevent interactions with other viral and cellular proteins involved in fusion and/or neutralize their functional effects, thus leading to enhanced cell-cell fusion. Further studies will be necessary to characterize proteins involved in this process and investigate their putative physical and functional interactions with gB.
Acknowledgments
We gratefully thank Roselyn J. Eisenberg and Gary H. Cohen for providing the anti-gB antibody and J. Salamero for the Rab11S25N construct. We are indebted to Prashant Desai and Stanley Person for the gift of the K082 virus and the D6 cells. We thank Jacques Coppey, Maïté Coppey-Moisan, and Christiane Durieux for their constant interest and help in our work. We also thank Roxane Beauvoir, Gregory Camus, and Sandra López-Vergès for helpful discussions. We acknowledge the technical support of Pierre Bourdoncle from the Cochin Institute confocal microscopy facility.
This study was supported in part by grants from the French GIS Maladies Rares and the Action Concertée Incitative de Microbiologie. I.B.O.D.Z. was a recipient of a Scientist Training Fellowship from the Department of Education, Universities, and Research of the Basque Government.
Footnotes
Published ahead of print on 3 October 2007.
REFERENCES
- 1.Alconada, A., U. Bauer, L. Baudoux, J. Piette, and B. Hoflack. 1998. Intracellular transport of the glycoproteins gE and gI of the varicella-zoster virus: gE accelerates the maturation of gI and determines its accumulation in the trans-Golgi network. J. Biol. Chem. 273:13430-13436. [DOI] [PubMed] [Google Scholar]
- 2.Alconada, A., U. Bauer, B. Sodeik, and B. Hoflack. 1999. Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 73:377-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avitabile, E., G. Lombardi, T. Gianni, M. Capri, and G. Campadelli-Fiume. 2004. Coexpression of UL20p and gK inhibits cell-cell fusion mediated by herpes simplex virus glycoproteins gD, gH-gL, and wild-type gB or an endocytosis-defective gB mutant and downmodulates their cell surface expression. J. Virol. 78:8015-8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baghian, A., L. Huang, S. Newman, S. Jayachandra, and K. G. Kousoulas. 1993. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J. Virol. 67:2396-2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baines, J. D., P. L. Ward, G. Campadelli-Fiume, and B. Roizman. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414-6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balbis, A., G. Baquiran, V. Dumas, and B. I. Posner. 2004. Effect of inhibiting vacuolar acidification on insulin signaling in hepatocytes. J. Biol. Chem. 279:12777-12785. [DOI] [PubMed] [Google Scholar]
- 7.Beitia Ortiz de Zarate, I., K. Kaelin, and F. Rozenberg. 2004. Effects of mutations in the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B on intracellular transport and infectivity. J. Virol. 78:1540-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonifacino, J. S., and L. M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395-447. [DOI] [PubMed] [Google Scholar]
- 9.Brideau, A. D., L. W. Enquist, and R. S. Tirabassi. 2000. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. 17:69-82. [DOI] [PubMed] [Google Scholar]
- 10.Browne, H., B. Bruun, and T. Minson. 2001. Plasma membrane requirements for cell fusion induced by herpes simplex virus type 1 glycoproteins gB, gD, gH, and gL. J. Gen. Virol. 82:1419-1422. [DOI] [PubMed] [Google Scholar]
- 11.Bzik, D. J., B. A. Fox, N. A. DeLuca, and S. Person. 1984. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology 137:185-190. [DOI] [PubMed] [Google Scholar]
- 12.Cai, W. H., B. Gu, and S. Person. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai, W. Z., S. Person, C. DebRoy, and B. H. Gu. 1988. Functional regions and structural features of the gB glycoprotein of herpes simplex virus type 1. An analysis of linker insertion mutants. J. Mol. Biol. 201:575-588. [DOI] [PubMed] [Google Scholar]
- 14.Crump, C. M., B. Bruun, S. Bell, L. E. Pomeranz, T. Minson, and H. M. Browne. 2004. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 85:3517-3527. [DOI] [PubMed] [Google Scholar]
- 15.Crump, C. M., C. H. Hung, L. Thomas, L. Wan, and G. Thomas. 2003. Role of PACS-1 in trafficking of human cytomegalovirus glycoprotein B and virus production. J. Virol. 77:11105-11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engel, J. P., E. P. Boyer, and J. L. Goodman. 1993. Two novel single amino acid syncytial mutations in the carboxy terminus of glycoprotein B of herpes simplex virus type 1 confer a unique pathogenic phenotype. Virology 192:112-120. [DOI] [PubMed] [Google Scholar]
- 17.Fan, Z., M. L. Grantham, M. S. Smith, E. S. Anderson, J. A. Cardelli, and M. I. Muggeridge. 2002. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J. Virol. 76:9271-9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Favoreel, H. W. 2006. The why's of Y-based motifs in alphaherpesvirus envelope proteins. Virus Res. 117:202-208. [DOI] [PubMed] [Google Scholar]
- 19.Favoreel, H. W., H. J. Nauwynck, H. M. Halewyck, P. Van Oostveldt, T. C. Mettenleiter, and M. B. Pensaert. 1999. Antibody-induced endocytosis of viral glycoproteins and major histocompatibility complex class I on pseudorabies virus-infected monocytes. J. Gen. Virol. 80(Pt. 5):1283-1291. [DOI] [PubMed] [Google Scholar]
- 20.Favoreel, H. W., G. Van Minnebruggen, H. J. Nauwynck, L. W. Enquist, and M. B. Pensaert. 2002. A tyrosine-based motif in the cytoplasmic tail of pseudorabies virus glycoprotein B is important for both antibody-induced internalization of viral glycoproteins and efficient cell-to-cell spread. J. Virol. 76:6845-6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster, T. P., J. M. Melancon, and K. G. Kousoulas. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18-29. [DOI] [PubMed] [Google Scholar]
- 22.Foster, T. P., J. M. Melancon, T. L. Olivier, and K. G. Kousoulas. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 78:13262-13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fraile-Ramos, A., A. Pelchen-Matthews, T. N. Kledal, H. Browne, T. W. Schwartz, and M. Marsh. 2002. Localization of HCMV UL33 and US27 in endocytic compartments and viral membranes. Traffic 3:218-232. [DOI] [PubMed] [Google Scholar]
- 24.Gage, P. J., M. Levine, and J. C. Glorioso. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J. Virol. 67:2191-2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerdts, V., J. Beyer, B. Lomniczi, and T. C. Mettenleiter. 2000. Pseudorabies virus expressing bovine herpesvirus 1 glycoprotein B exhibits altered neurotropism and increased neurovirulence. J. Virol. 74:817-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gianni, T., A. Piccoli, C. Bertucci, and G. Campadelli-Fiume. 2006. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol. 80:2216-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodman, J. L., and J. P. Engel. 1991. Altered pathogenesis in herpes simplex virus type 1 infection due to a syncytial mutation mapping to the carboxy terminus of glycoprotein B. J. Virol. 65:1770-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haanes, E. J., C. M. Nelson, C. L. Soule, and J. L. Goodman. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J. Virol. 68:5825-5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hambleton, S., M. D. Gershon, and A. A. Gershon. 2004. The role of the trans-Golgi network in varicella-zoster virus biology. Cell Mol. Life Sci. 61:3047-3056. [DOI] [PubMed] [Google Scholar]
- 30.Heineman, T. C., and S. L. Hall. 2002. Role of the varicella-zoster virus gB cytoplasmic domain in gB transport and viral egress. J. Virol. 76:591-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heineman, T. C., and S. L. Hall. 2001. VZV gB endocytosis and Golgi localization are mediated by YXXΦ motifs in its cytoplasmic domain. Virology 285:42-49. [DOI] [PubMed] [Google Scholar]
- 32.Highlander, S. L., W. H. Cai, S. Person, M. Levine, and J. C. Glorioso. 1988. Monoclonal antibodies define a domain on herpes simplex virus glycoprotein B involved in virus penetration. J. Virol. 62:1881-1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Highlander, S. L., D. J. Dorney, P. J. Gage, T. C. Holland, W. Cai, S. Person, M. Levine, and J. C. Glorioso. 1989. Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration. J. Virol. 63:730-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holland, T. C., and S. Person. 1977. Ammonium chloride inhibits cell fusion induced by syn mutants of herpes simplex virus type 1. J. Virol. 23:213-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huff, V., W. Cai, J. C. Glorioso, and M. Levine. 1988. The carboxy-terminal 41 amino acids of herpes simplex virus type 1 glycoprotein B are not essential for production of infectious virus particles. J. Virol. 62:4403-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Husain, M., and B. Moss. 2003. Intracellular trafficking of a palmitoylated membrane-associated protein component of enveloped vaccinia virus. J. Virol. 77:9008-9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Husain, M., and B. Moss. 2005. Role of receptor-mediated endocytosis in the formation of vaccinia virus extracellular enveloped particles. J. Virol. 79:4080-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarvis, M. A., K. N. Fish, C. Soderberg-Naucler, D. N. Streblow, H. L. Meyers, G. Thomas, and J. A. Nelson. 2002. Retrieval of human cytomegalovirus glycoprotein B from cell surface is not required for virus envelopment in astrocytoma cells. J. Virol. 76:5147-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarvis, M. A., T. R. Jones, D. D. Drummond, P. P. Smith, W. J. Britt, J. A. Nelson, and C. J. Baldick. 2004. Phosphorylation of human cytomegalovirus glycoprotein B (gB) at the acidic cluster casein kinase 2 site (Ser900) is required for localization of gB to the trans-Golgi network and efficient virus replication. J. Virol. 78:285-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson, D. C., M. Webb, T. W. Wisner, and C. Brunetti. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kostal, M., I. Bacik, J. Rajcani, and H. C. Kaerner. 1994. Replacement of glycoprotein B gene in the herpes simplex virus type 1 strain ANGpath DNA by that originating from nonpathogenic strain KOS reduces the pathogenicity of recombinant virus. Acta Virol. 38:77-88. [PubMed] [Google Scholar]
- 42.Kousoulas, K. G., S. Person, and T. C. Holland. 1978. Timing of some of the molecular events required for cell fusion induced by herpes simplex virus type 1. J. Virol. 27:505-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manservigi, R., P. G. Spear, and A. Buchan. 1977. Cell fusion induced by herpes simplex virus is promoted and suppressed by different viral glycoproteins. Proc. Natl. Acad. Sci. USA 74:3913-3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maresova, L., T. J. Pasieka, E. Homan, E. Gerday, and C. Grose. 2005. Incorporation of three endocytosed varicella-zoster virus glycoproteins, gE, gH, and gB, into the virion envelope. J. Virol. 79:997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marsh, M., and A. Pelchen-Matthews. 2000. Endocytosis in viral replication. Traffic 1:525-532. [DOI] [PubMed] [Google Scholar]
- 46.McMillan, T. N., and D. C. Johnson. 2001. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 75:1928-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melancon, J. M., R. E. Luna, T. P. Foster, and K. G. Kousoulas. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167-180. [DOI] [PubMed] [Google Scholar]
- 49.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mettenleiter, T. C. 2003. Pathogenesis of neurotropic herpesviruses: role of viral glycoproteins in neuroinvasion and transneuronal spread. Virus Res. 92:197-206. [DOI] [PubMed] [Google Scholar]
- 51.Moffat, J., C. Mo, J. J. Cheng, M. Sommer, L. Zerboni, S. Stamatis, and A. M. Arvin. 2004. Functions of the C-terminal domain of varicella-zoster virus glycoprotein E in viral replication in vitro and skin and T-cell tropism in vivo. J. Virol. 78:12406-12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muggeridge, M. I. 2000. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH, and gL in transfected cells. J. Gen. Virol. 81:2017-2027. [DOI] [PubMed] [Google Scholar]
- 53.Navarro, D., P. Paz, and L. Pereira. 1992. Domains of herpes simplex virus I glycoprotein B that function in virus penetration, cell-to-cell spread, and cell fusion. Virology 186:99-112. [DOI] [PubMed] [Google Scholar]
- 54.Nixdorf, R., B. G. Klupp, A. Karger, and T. C. Mettenleiter. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J. Virol. 74:7137-7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nixdorf, R., B. G. Klupp, and T. C. Mettenleiter. 2001. Role of the cytoplasmic tails of pseudorabies virus glycoproteins B, E, and M in intracellular localization and virion incorporation. J. Gen. Virol. 82:215-226. [DOI] [PubMed] [Google Scholar]
- 56.Olson, J. K., and C. Grose. 1997. Endocytosis and recycling of varicella-zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J. Virol. 71:4042-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasieka, T. J., L. Maresova, and C. Grose. 2003. A functional YNKI motif in the short cytoplasmic tail of varicella-zoster virus glycoprotein gH mediates clathrin-dependent and antibody-independent endocytosis. J. Virol. 77:4191-4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pasieka, T. J., L. Maresova, K. Shiraki, and C. Grose. 2004. Regulation of varicella-zoster virus-induced cell-to-cell fusion by the endocytosis-competent glycoproteins gH and gE. J. Virol. 78:2884-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pereira, L. 1994. Function of glycoprotein B homologues of the family herpesviridae. Infect. Agents Dis. 3:9-28. [PubMed] [Google Scholar]
- 60.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313-324. [DOI] [PubMed] [Google Scholar]
- 61.Potel, C., K. Kaelin, L. Danglot, A. Triller, C. Vannier, and F. Rozenberg. 2003. Herpes simplex virus type 1 glycoprotein B sorting in hippocampal neurons. J. Gen. Virol. 84:2613-2624. [DOI] [PubMed] [Google Scholar]
- 62.Potel, C., K. Kaelin, I. Gautier, P. Lebon, J. Coppey, and F. Rozenberg. 2002. Incorporation of green fluorescent protein into the essential envelope glycoprotein B of herpes simplex virus type 1. J. Virol. Methods 105:13-23. [DOI] [PubMed] [Google Scholar]
- 63.Radsak, K., M. Eickmann, T. Mockenhaupt, E. Bogner, H. Kern, A. Eis-Hubinger, and M. Reschke. 1996. Retrieval of human cytomegalovirus glycoprotein B from the infected cell surface for virus envelopment. Arch. Virol. 141:557-572. [DOI] [PubMed] [Google Scholar]
- 64.Ruell, N., A. Zago, and P. G. Spear. 2006. Alanine substitution of conserved residues in the cytoplasmic tail of herpes simplex virus gB can enhance or abolish cell fusion activity and viral entry. Virology 346:229-237. [DOI] [PubMed] [Google Scholar]
- 65.Spear, P. G. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 6:401-410. [DOI] [PubMed] [Google Scholar]
- 66.Sun, X., V. K. Yau, B. J. Briggs, and G. R. Whittaker. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 338:53-60. [DOI] [PubMed] [Google Scholar]
- 67.Tirabassi, R. S., and L. W. Enquist. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J. Virol. 73:2717-2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tirabassi, R. S., and L. W. Enquist. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J. Virol. 72:4571-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tirabassi, R. S., R. A. Townley, M. G. Eldridge, and L. W. Enquist. 1998. Molecular mechanisms of neurotropic herpesvirus invasion and spread in the CNS. Neurosci. Biobehav. Rev. 22:709-720. [DOI] [PubMed] [Google Scholar]
- 70.Tortorella, L. L., F. B. Schapiro, and F. R. Maxfield. 2007. Role of an acidic cluster/dileucine motif in cation-independent mannose 6-phosphate receptor traffic. Traffic 8:402-413. [DOI] [PubMed] [Google Scholar]
- 71.Tugizov, S., E. Maidji, J. Xiao, and L. Pereira. 1999. An acidic cluster in the cytosolic domain of human cytomegalovirus glycoprotein B is a signal for endocytosis from the plasma membrane. J. Virol. 73:8677-8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tugizov, S., E. Maidji, J. Xiao, Z. Zheng, and L. Pereira. 1998. Human cytomegalovirus glycoprotein B contains autonomous determinants for vectorial targeting to apical membranes of polarized epithelial cells. J. Virol. 72:7374-7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turcotte, S., J. Letellier, and R. Lippe. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Umata, T., Y. Moriyama, M. Futai, and E. Mekada. 1990. The cytotoxic action of diphtheria toxin and its degradation in intact Vero cells are inhibited by bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase. J. Biol. Chem. 265:21940-21945. [PubMed] [Google Scholar]
- 75.Van Minnebruggen, G., H. W. Favoreel, and H. J. Nauwynck. 2004. Internalization of pseudorabies virus glycoprotein B is mediated by an interaction between the YQRL motif in its cytoplasmic domain and the clathrin-associated AP-2 adaptor complex. J. Virol. 78:8852-8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vogt, C., M. Eickmann, S. Diederich, M. Moll, and A. Maisner. 2005. Endocytosis of the Nipah virus glycoproteins. J. Virol. 79:3865-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walev, I., K. Weise, and D. Falke. 1991. Differentiation of herpes simplex virus-induced fusion from without and fusion from within by cyclosporin A and compound 48/80. J. Gen. Virol. 72(Pt. 6):1377-1382. [DOI] [PubMed] [Google Scholar]
- 78.Weise, K., H. C. Kaerner, J. Glorioso, and C. H. Schroder. 1987. Replacement of glycoprotein B gene sequences in herpes simplex virus type 1 strain ANG by corresponding sequences of the strain KOS causes changes of plaque morphology and neuropathogenicity. J. Gen. Virol. 68(Pt. 7):1909-1919. [DOI] [PubMed] [Google Scholar]
- 79.White, J. M. 1990. Viral and cellular membrane fusion proteins. Annu. Rev. Physiol. 52:675-697. [DOI] [PubMed] [Google Scholar]
- 80.Wilcke, M., L. Johannes, T. Galli, V. Mayau, B. Goud, and J. Salamero. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J. Cell Biol. 151:1207-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wisner, T. W., and D. C. Johnson. 2004. Redistribution of cellular and herpes simplex virus proteins from the trans-golgi network to cell junctions without enveloped capsids. J. Virol. 78:11519-11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshimori, T., A. Yamamoto, Y. Moriyama, M. Futai, and Y. Tashiro. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266:17707-17712. [PubMed] [Google Scholar]
- 83.Yuhasz, S. A., and J. G. Stevens. 1993. Glycoprotein B is a specific determinant of herpes simplex virus type 1 neuroinvasiveness. J. Virol. 67:5948-5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu, Z., M. D. Gershon, Y. Hao, R. T. Ambron, C. A. Gabel, and A. A. Gershon. 1995. Envelopment of varicella-zoster virus: targeting of viral glycoproteins to the trans-Golgi network. J. Virol. 69:7951-7959. [DOI] [PMC free article] [PubMed] [Google Scholar]