

Abstract
Viral myocarditis is an important human disease, and reovirus-induced murine myocarditis provides an excellent model system for study. Cardiac myocytes, like neurons in the central nervous system, are not replenished, yet there is no cardiac protective equivalent to the blood-brain barrier. Thus, cardiac myocytes may have evolved a unique antiviral response relative to readily replenished cell types, such as cardiac fibroblasts. Our previous comparisons of these two cell types revealed a conundrum: reovirus T3D induces more beta-interferon (IFN-β) mRNA in cardiac myocytes, yet there is a greater induction of IFN-stimulated genes (ISGs) in cardiac fibroblasts. Here, we investigated possible underlying molecular determinants. We found that greater basal expression of IFN-β in cardiac myocytes results in greater basal activated nuclear STAT1 and STAT2 and greater basal ISG mRNA expression and provides greater basal antiviral protection relative to cardiac fibroblasts. Conversely, cardiac fibroblasts express greater basal IFN-α/β receptor 1 (IFNAR1) and greater basal cytoplasmic Jak1, Tyk2, STAT2, and IRF9, leading to a greater increase in reovirus T3D- or IFN-induced nuclear activated STAT1 and STAT2 and greater induction of ISGs for a greater IFN-induced antiviral protection relative to cardiac myocytes. Our results suggest that high basal IFN-β expression in cardiac myocytes prearms this vulnerable, nonreplenishable cell type, while high basal expression of IFNAR1 and latent Jak-STAT components in adjacent cardiac fibroblasts renders these cells more responsive to IFN and prevents them from inadvertently serving as a reservoir for viral replication and spread to cardiac myocytes. These studies provide the first indication of an integrated network of cell-type-specific innate immune components for organ protection.
Viral myocarditis affects an estimated 5 to 20% of the human population. It can be fatal in infants and, although usually resolved in adults, can progress to chronic myocarditis and/or dilated cardiomyopathy with concomitant cardiac failure (11, 28). Recent clinical studies have indicated that alpha interferon (IFN-α) (11, 21, 35, 36) and IFN-β (29) can reduce the severity of viral myocarditis, inhibiting viral replication and improving cardiac function. Nonetheless, viral myocarditis remains a disease without reliable treatment, and the prognosis remains poor for more than half of patients presenting with clinical symptoms (45).
A wide variety of viruses have been implicated in human myocarditis, with the majority of cases being associated with enteroviruses and adenoviruses (4, 30, 34). While enteroviruses induce both immune-mediated damage to (10, 41) and a direct cytopathogenic effect on (8, 23) the heart, adenovirus-induced myocarditis is most likely not immune mediated (34). Reovirus-induced myocarditis is not immune mediated (47, 48) but instead reflects virally induced apoptosis of cardiac cells (13). Thus, reovirus infection in a mouse model provides an excellent model for investigating the cardiac response to viral infection.
Previously, we demonstrated that nonmyocarditic reoviruses induce more IFN-β and/or are more sensitive to the antiviral effects of IFN-β than myocarditic reoviruses in primary cardiac myocyte cultures (49). In addition, nonmyocarditic reoviruses induce myocarditis in mice pretreated with anti-IFN-α/β antibody, demonstrating directly that IFN-α/β is a determinant of protection against viral myocarditis (49). Interestingly, addition of anti-IFN-α/β antibody enhances reovirus spread in primary cardiac myocyte cultures but not in differentiated C2C12 (skeletal muscle) cell cultures (49), suggesting a unique role for IFN-α/β in the heart.
IFN-α/β exerts its antiviral effects through the induction of IFN-stimulated genes (ISGs), whose promoters are activated through a signal transduction pathway following binding of IFN-α/β to the IFN-α/β receptor, composed of IFNAR1 and IFNAR2 subunits. Receptor binding activates tyrosine kinases, Jak1 and Tyk2, which in turn phosphorylate signal transducers and activators of transcription (STATs), STAT1 (STAT1α and STAT1β) and STAT2 (22, 39, 44, 52). Upon phosphorylation, STAT1 and STAT2 heterodimerize and associate with p48/IRF9, forming the multimeric protein complex ISG factor 3 (ISGF3) (17, 57). ISGF3 complexes translocate to the nucleus and initiate the transcription of many ISGs, including ISG561 and the transcription factor IRF7, leading to the induction of an antiviral state (12).
Cardiac myocytes, like neurons in the central nervous system, are not replenishable. However, there is no cardiac equivalent to the blood-brain barrier, leaving the heart uniquely vulnerable to damage from viral infection. Accordingly, we have hypothesized that cardiac myocytes may have a unique antiviral protective response, and indeed, we have found differences in responses between cardiac myocytes and cardiac fibroblasts, a readily replenished cardiac cell type. Specifically, in comparing the two cardiac cell types, reovirus T3D induces more IFN-β mRNA in cardiac myocytes, yet there is a greater induction of ISG561 and IRF7 mRNA in cardiac fibroblasts (53). This conundrum led us to our current study: to identify the molecular differences between cardiac myocytes and cardiac fibroblasts that determine their protective responses to viral infection. Here, we report that cardiac myocytes rely on greater basal expression of IFN-β, resulting in greater basal activated nuclear ISGF3 components and greater basal ISG mRNA expression, as a prearming mechanism for protection. In contrast, cardiac fibroblasts express higher basal IFNAR1 and cytoplasmic Jak-STAT components, rendering them better prepared to respond to IFN induced by T3D infection, leading to the observed greater increase in nuclear activated STAT1 and STAT2 and providing a mechanism for greater induction of ISGs in this cell type. The results of this study suggest that nonreplenishable cardiac myocytes express an innate basal protective response, while adjacent cardiac fibroblasts are highly responsive to IFN, limiting their use as a reservoir for viral replication in this essential organ.
MATERIALS AND METHODS
Viruses and cell lines.
Mouse L929 cells were maintained in minimal essential medium (SAFC Biosciences, Denver, PA), supplemented with 5% fetal calf serum (Atlanta Biologicals, Atlanta, GA), and 2 mM l-glutamine (Mediatech, Inc., Herndon, VA). Reovirus type 3 Dearing (T3D) and type 1 Lang (T1L) were plaque purified and amplified in L929 cells, purified on a CsCl gradient (50), and stored in diluted aliquots at −80°C. Reovirus T3D was chosen for study because of its strong induction of IFN-β in cardiac myocytes (49). Not surprisingly, it does not induce myocarditis in mice (48). In contrast to T3D, reovirus T1L was chosen for study due to its low or negligible induction of IFN-β in cardiac myocytes (49).
Mice and primary cell cultures.
Timed pregnant Cr:NIH(S) mice were obtained from the National Cancer Institute. IFN-α/β-receptor-null mice (IFN-α/β-RKO mice [38]) were maintained as a colony and used for timed matings as necessary. Mice were housed according to the recommendations of the Association for Assessment and Accreditation of Laboratory Animal Care, and all procedures were approved by the North Carolina State University institutional animal care and use committee.
To generate primary cardiac myocyte and fibroblast cultures from Cr:NIH(S) or IFN-α/β-RKO mice, full-term fetuses or 1-day-old neonates were euthanized and the apical two-thirds of the hearts were excised and trypsinized (3). Cells were plated at a density of 1.25 × 106 per well in six-well clusters (Costar, Cambridge, MA) and incubated for 2 h at 37°C in 5% CO2 in order to separate cardiac myocytes from cardiac fibroblasts by rapid adherence of the latter. Cardiac myocytes were resuspended in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Gaithersburg, MD) supplemented with 7% fetal calf serum, 0.06% thymidine (Sigma Co., St. Louis, MO), and 10 μg of gentamicin (Sigma Co.) per ml. Cardiac myocytes were plated at 106 cells per well in 24-well clusters (for RNA and protein harvests, enzyme-linked immunosorbent assay [ELISA], and fluorescence-activated cell sorting [FACS]) or 1.5 × 105 cells per well in 96-well clusters (for viral replication studies). Cardiac fibroblasts were trypsinized from six-well clusters and resuspended in DMEM supplemented with 7% fetal calf serum and 10 μg of gentamicin per ml. Fibroblasts were then plated at 5 × 105 cells per well in 24-well clusters (for RNA and protein harvests, ELISA, and FACS) or 7.5 × 104 cells per well in 96-well clusters (for replication studies). Fibroblasts plated at these densities were confluent by the day of infection and were assumed to have doubled from the initial plating density. Immunofluorescence microscopy of these primary cultures using antimyomesin and antivimentin antibodies (as described below) revealed <5% fibroblast contamination in myocyte cultures and <1% myocyte contamination in fibroblast cultures (data not shown). Cells were never passaged before use.
Infections for viral replication studies.
In studies to determine relative replication in wild-type and IFN-α/β-RKO cells (see Fig. 3), cardiac myocyte and cardiac fibroblast cultures were generated from Cr:NIH(S) and IFN-α/β-RKO mice. Two days post-plating in 96-well clusters, cells were infected with T3D or T1L at 10 PFU per cell for the indicated times postinfection. In studies to determine relative sensitivity to IFN-β (see Fig. 9), cells were plated in 96-well clusters, and 24 h later IFN-β (10, 100, or 1,000 U/ml; PBL Biomedical Laboratories, Piscataway, NJ) was added to half of the wells. Two days postplating, cells were infected with T3D at 5 PFU per cell, and rabbit anti-mouse IFN-β antibody (1,000 neutralizing units/ml; PBL Biomedical Laboratories) was added to “control” wells (to neutralize virus-induced IFN-β and thereby provide cultures devoid of IFN-β for comparison). In studies to determine protective effects of uninfected myocytes on infected fibroblasts (see Fig. 11), cardiac fibroblasts (7.5 × 104 cells per well) were plated in 96-well clusters, and 48 h later 10 PFU per cell of T3D was added. After 1 h of incubation at 4°C or 37°C, inoculum was removed and cultures were washed three times. Medium, IFN-β (dose indicated), or uninfected cardiac myocytes (cell number indicated) were added, and cultures were incubated for an additional 18 h before harvest. Light microscopy confirmed that cardiac myocytes had adhered to the fibroblast layer and were beating. In all studies, cell cultures were frozen at −80°C at the indicated times postinfection and then subjected to two additional freeze-thaw cycles. Cultures were then lysed in 0.5% Nonidet P-40, and titers were determined by plaque assay on mouse L929 cell monolayers as previously described (46).
FIG. 3.

Basal expression of IFN provides greater protection for cardiac myocytes than for cardiac fibroblasts. Cardiac myocyte (Myo) and cardiac fibroblast (Fib) cultures were generated from either wild-type or IFN-α/β-RKO mice. Two days postplating, cultures were infected with T3D or T1L at 10 PFU per cell. At the indicated times postinfection, cultures were frozen and titers were determined by plaque assay. (A) Viral titers from a subset of experiments, expressed as PFU per well ± standard deviations. (B and C) Data from panel A and additional experiments are expressed as a ratio of titers: viral titers from IFN-α/β-RKO cultures relative to viral titers from wild-type cultures.
FIG. 9.

IFN-β-dependent inhibition of T3D replication is greater in cardiac fibroblasts than in cardiac myocytes. One day postplating, primary cardiac cell cultures were pretreated with IFN-β (10, 100, or 1,000 U/ml). Twenty-four hours post-IFN-β treatment, cultures were infected with T3D at 5 PFU per cell. Control-treated cultures received 1,000 neutralizing units/ml of rabbit polyclonal anti-mouse IFN-β antibody. At 12 h and 24 h postinfection, cell cultures were frozen and titers were determined by plaque assay. (A) Viral titers, expressed as PFU per well ± standard deviations. (B) Data from panel A are expressed as percentages of PFU in IFN-β-treated cultures relative to control (anti-IFN-β)-treated cultures ± standard deviations. At every time point, differences between myocytes and fibroblasts were statistically significant (P < 0.05).
FIG. 11.

Cardiac myocytes do not inhibit viral replication in adjacent cardiac fibroblasts. Two days post-plating of cardiac fibroblasts, 10 PFU per cell of T3D was added. After incubation for 1 h on ice (left panel) or at 37°C (right panel), inoculum was removed, cultures were washed extensively, and the indicated reagent or cells were added. Each well contained 1.5 × 105 fibroblasts; 1.5 × 105 or 1.5 × 106 myocytes were added as indicated for a 1:1 or 10:1 ratio, respectively. Cultures were incubated for an additional 18 h and then harvested for viral titration by plaque assay. Results are expressed as PFU per well ± standard deviation. Supernatants from cultures contained no IFN-β that was detectable by ELISA (data not shown), indicating that myocytes were not inadvertently infected and secreting IFN-β for modulation of viral replication in fibroblasts during the 18-hour coculture.
Infections for RNA and protein harvests and for ELISA.
Two days post-plating in 24-well clusters, primary cardiac myocyte and cardiac fibroblast cultures were washed twice with supplemented DMEM and then immediately infected with reovirus T3D or T1L at 10 PFU per cell in 700 μl of supplemented DMEM. After 1 h at 37°C, 1 ml of supplemented DMEM was added, and cultures were harvested at the indicated times. “Mock-infected” cultures received medium only, were washed with supplemented DMEM, and were harvested immediately after addition of 1 ml of supplemented DMEM.
IFN-α/β treatment for RNA and protein harvests.
Two days post-plating in 24-well clusters, primary cardiac myocyte and cardiac fibroblast cultures were washed twice with supplemented DMEM and treated with IFN-α/β (Access Biomedical, San Diego, CA) at the indicated doses. “Mock-treated” cultures received medium only and were washed with supplemented DMEM.
Real-time reverse transcription-PCR (RT-PCR).
At the indicated times postinfection, supernatants were removed and saved for ELISA. Total RNA was harvested by direct lysis of cells from culture plates using an RNeasy kit (Qiagen, Inc., Valencia, CA). Contaminating genomic DNA was removed using RNase-free DNase I (Qiagen, Inc.). One-third of the RNA from each well was then converted to cDNA by reverse transcription in a total reaction volume of 100 μl containing 5 μM oligo(dT); 1× Taq buffer; 7.5 mM MgCl2; 1 mM dithiothreitol; 1 mM (each) deoxynucleoside triphosphate, 0.67 U/μl RNasin (Promega Corp., Madison, WI); and 0.40 U/μl of avian myeloblastosis virus reverse transcriptase (Promega Corp.). Approximately 5% of the reverse transcription reaction product was then amplified by real-time PCR to determine the relative abundances of specific mRNA sequences by comparison to a standard curve generated from serial dilutions of a DNA standard and normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Real-time PCR experiments were performed in duplicate 25-μl reaction mixtures in 96-well plates using Quantitech master mix (Qiagen, Inc.) spiked with 10 nM fluorescein (Invitrogen Corp., Carlsbad, CA) to optimize fluorescence data quality and analysis. Amplification, quantification, and melting curve analysis (validation of a single amplification product) were performed on an iCycler iQ fluorescence thermocycler (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's recommended protocol. Primer sequences for reactions were as published previously (53).
ELISA.
IFN-β in cardiac myocyte and cardiac fibroblast culture supernatants was measured using a commercially available IFN-β ELISA (PBL Biomedical Laboratories). Samples (100 μl each) were analyzed in accordance with the manufacturer's instructions. All samples taken at different times postinfection were analyzed on the same ELISA plate in duplicate. Quantified mouse IFN-β (PBL Biomedical Laboratories) was used as a standard, and IFN-β concentrations were calculated based upon a standard curve. Mock-infected cultures and medium alone generated equivalent values and were therefore interpreted as background and subtracted from the values indicated for infected cultures.
Antibodies for Western blot analysis.
Primary antibodies were purchased from the following suppliers and used at the indicated final dilutions: mouse monoclonal anti-STAT1α/β (1:1,000; no. 610186; BD Biosciences, San Jose, CA), rabbit polyclonal anti-phospho-STAT1 Tyr701 (1:1,000; no. 07-307; Millipore Corp., Billerica, MA), rabbit polyclonal anti-STAT2 (1:1,000, no. 07-140; Millipore Corp.), rabbit polyclonal anti-phospho-STAT2 (1:1,000; no. 07-224; Millipore Corp.), rabbit polyclonal anti-IRF9 (1:100; sc-10793; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rabbit polyclonal anti-Tyk2 (1:100; sc-169; Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti-Jak1 (1:1,000; no. 44-400G; Invitrogen), goat polyclonal antiactin conjugated to horseradish peroxidase (HRP) (1:500; sc-1615-HRP; Santa Cruz Biotechnology, Inc.), and goat polyclonal HRP-conjugated anti-GAPDH (1:200; sc-20357-HRP; Santa Cruz Biotechnology, Inc.).
SDS-PAGE and Western blot analysis.
Total cellular protein extracts were prepared using RIPA lysis buffer (50 mM Tris HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) containing a cocktail of protease and phosphatase inhibitors (Sigma Co.; P8340 and P2850), by incubation on ice for 30 min and centrifugation at 10,000 × g for 10 min to remove cellular debris. Cytoplasmic and nuclear extracts were obtained using the NE-PER kit (Pierce, Rockford, IL) following the instructions provided by the manufacturer. Protein concentrations were quantified using a bicinchoninic acid protein assay kit (Pierce), and 20 μg of protein from each lysate was boiled for 5 min in 1× Laemmli sample buffer and subjected to 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following protein transfer, the nitrocellulose membranes were blocked for 60 min with 5% milk in Tris-buffered saline (20 mM Tris [pH 7.6], 137 mM NaCl) containing 0.05% Tween 20 (TBS-T) and probed with the indicated primary antibodies overnight at 4°C. Blots were washed three times with TBS-T buffer, followed by 90 min of incubation with the appropriate HRP-conjugated, species-specific secondary antibodies at room temperature. The membranes were washed in TBS-T buffer, and antibody-labeled proteins were detected using protocols and reagents contained in the enhanced chemiluminescence (ECL Plus) kit (GE Healthcare, Buckinghamshire, United Kingdom). Immunoblots were exposed to film and converted to digital format using an HP Scanjet 5470c.
Immunohistochemistry.
Formalin-fixed, paraffin-embedded hearts from Cr:NIH(S) and IFN-α/β-RKO mice were sectioned (5 μm) and then deparaffinized in xylene and rehydrated in descending grades of alcohol to distilled water (dH2O). Antigen retrieval was accomplished by immersing slides in target retrieval solution, citrate buffer, pH 6.1 (Dako, Carpinteria, CA), in a pressure cooker. After slides were rinsed in dH2O, endogenous peroxidase activity was blocked in 3% hydrogen peroxide in dH2O for 20 min at room temperature. After being rinsed in dH2O for 2 min, the sections were incubated overnight at 4°C with anti-phospho-STAT1 Tyr701 (no. 9167; Cell Signaling Technology, Inc., Danvers, MA) diluted 1:500 in common antibody diluent (BioGenex, San Ramon, CA). For a negative control, supersensitive rabbit negative control (normal rabbit immunoglobulin [Ig]; BioGenex) was used in place of the primary antibody. Following three washes in wash buffer (Dako), sections were incubated with EnVision-Plus polymer conjugated to HRP (Dako) for 30 min at room temperature. To identify positive staining, 3,3′-diaminobenzidine (DAB; Vector Laboratories Ltd., Burlingame, CA) was used as a substrate to visualize the bound Ig-peroxidase complexes in brown. Tissue sections were washed in dH2O and counterstained with methyl green-alcian blue, dehydrated in ascending grades of alcohol, and mounted from xylene using Permount (Fisher Scientific Co., Suwanee, GA) mounting medium. Sections were imaged digitally by light microscopy. Positive nuclei were characterized by brown DAB labeling, whereas negative nuclei displayed only the methyl green-alcian blue staining.
Indirect immunofluorescence.
Hearts from Cr:NIH(S) mice were excised, snap frozen in liquid nitrogen-cooled isopentane (Fisher Scientific Co.), and stored at −80°C. Tissue samples were mounted frozen on a metal chuck using a small volume of Tissue-Tek OCT (Ted Pella, Inc., Redding, CA) compound in a cryostat (Leica CM1850) running at −20°C. Transverse cryosections (1 μm) were collected on SuperFrost/Plus slides (Fisher Scientific) and stored at −80°C. Sections were thawed and blocked for 1 h at room temperature in 10% rabbit serum (Sigma Co.)-10% donkey serum (Sigma Co.) in phosphate-buffered saline (PBS) containing 0.1% Triton X-100. Primary and secondary antibodies were diluted in PBS containing 0.3% IgG-free, protease-free bovine serum albumin (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). After blocking, samples were incubated with a mix of goat antimyomesin (1:50; Santa Cruz Biotechnology, Inc.) and chicken antivimentin (1:5,000; Affinity Bioreagents Inc., Golden, CO) antibodies overnight at 4°C, washed three times in PBS, and then incubated with a combination of Alexa 594-conjugated rabbit anti-goat IgG (1:1,000; Invitrogen) and fluorescein isothiocyanate-conjugated donkey anti-chicken IgG (1:1,000; Jackson ImmunoResearch Laboratories, Inc.) secondary antibodies for 2 h at room temperature. Following a final wash in PBS, coverslips were mounted with Prolong Gold reagent (Invitrogen) and analyzed using a Nikon TE-200 inverted epifluorescence microscope equipped with the proper optics and filter blocks. Images taken using a 100× objective under oil immersion were collected digitally (SPOT, Jr.; Diagnostics Instruments, Inc., Sterling Heights, MI) and processed using the manufacturer's instructions and Adobe Photoshop.
FACS assessment of IFNAR1 and IFNAR2 expression.
Two days post-plating in 24-well clusters, primary cardiac myocyte and cardiac fibroblast cultures were washed with PBS and treated with trypsin-EDTA (50 μl/well; BioWhittaker) for 5 min at 37°C in 5% CO2. Cells were removed from the plate by being washed with PBS containing 0.5 mM EDTA to prevent aggregation (PBS-EDTA). All washes unless otherwise indicated were completed using PBS-EDTA. Myocytes or fibroblasts from approximately 12 wells were combined, washed, and centrifuged at 250 × g for 10 min at 16°C. Myocytes and fibroblasts were aliquoted into individual tubes and washed again at 250 × g for 10 min. Samples were fixed for 20 min on ice, and primary, secondary, and isotype control antibodies were stained according to the manufacturer's instructions using the BD Cytofix/Cytoperm kit. The antibodies used were those against an intracellular epitope of IFNAR1 (sc-7391; Santa Cruz Biotechnology) and an extracellular epitope of IFNAR2 (BAF1083; R&D Systems). Secondary and control antibodies were goat anti-mouse IgG1-allophycocyanin (APC) (no. 1070-11), streptavidin-APC (no. 7100-11), and APC-conjugated isotype control (clone 15H6; Southern Biotechnology). After the final wash, all samples were resuspended in PBS-2% fetal calf serum and run immediately on a BD LSRII flow cytometer. All samples were stained and run in duplicate with 50,000 to 100,000 gated events collected for each sample. Analysis and histograms were completed using FACS Diva and FCS Express software.
Statistical analysis.
A Student one-tailed t test and pooled variance were used for statistical analysis using Systat 9.0. In all cases, differences were considered significant when P was <0.05.
RESULTS
Basal IFN-β mRNA, nuclear activated ISGF3 components, and ISG expression are higher in cardiac myocytes than in cardiac fibroblasts.
Basal expression of genes involved in the innate response to viral infection may be critical for initial antiviral protection. To determine whether cardiac cell type specificity exists in this basal expression, we compared mock-infected primary cultures of cardiac myocytes and cardiac fibroblasts. Basal IFN-β mRNA was sixfold higher (Fig. 1A, P = 0.007) and basal ISG561 mRNA was 4.7-fold higher (Fig. 1B, P < 0.001) in cardiac myocytes than in cardiac fibroblasts, consistent with our previous results (53). To determine whether basal nuclear activated ISGF3 components are also higher in cardiac myocytes, protein extracts were harvested for Western blot analysis. Basal nuclear phosphorylated STAT1 (Tyr701) and phosphorylated STAT2 were higher in cardiac myocytes than in cardiac fibroblasts (Fig. 1C), consistent with higher IFN-β and ISG expression in cardiac myocytes. Greater basal nuclear IRF9 in cardiac fibroblasts suggests that IRF9 is not a limiting ISGF3 component in those cells. Not unexpectedly, basal nuclear phosphorylated STAT1 (Tyr701) and phosphorylated STAT2 were undetectable in cardiac myocytes prepared from IFN-α/β-receptor-null (IFN-α/β-RKO) mice, confirming that basal activation of these components is dependent on type I IFN signaling. To verify basal IFN expression in vivo, nuclear STAT1 phosphorylation in both wild-type and IFN-α/β-RKO mice was evaluated by immunohistochemical analysis (Fig. 2). STAT1 phosphorylation was observed in nuclei of cardiac sections from wild-type mice (Fig. 2A and B) but, as expected, not in cardiac sections from IFN-α/β-RKO mice (Fig. 2C). These results confirm that basal nuclear STAT1 phosphorylation in cardiac sections, as in cardiac myocyte cultures, is dependent on type I IFN signaling and therefore confirm basal expression of IFN in the heart.
FIG. 1.

Cardiac myocytes express higher basal IFN-β mRNA and nuclear activated ISGF3 components and have higher ISG561 mRNA expression than cardiac fibroblasts. RNA and nuclear protein extracts were harvested from primary cardiac myocyte (Myo) and cardiac fibroblast (Fib) cultures at 2 days postplating. Cultures were generated from wild-type mice, unless indicated otherwise (IFN-α/β-RKO mice). RNA was analyzed by real-time RT-PCR, and copy number was normalized to GAPDH. (A) Basal IFN-β mRNA. (B) Basal ISG561 mRNA. Graphs are representatives of three independent experiments from three independently derived myocyte and fibroblast cultures ± standard deviations. The asterisk indicates a difference at P < 0.05. (C) Western blot analysis of basal nuclear activated ISGF3 components.
FIG. 2.

Basal expression of phosphorylated STAT1 in cardiac sections. Immunohistochemical analysis of paraffin-embedded cardiac tissue from uninfected wild-type mice (A, B, and D) or uninfected IFN-α/β-RKO mice (C), using phospho-STAT1 Tyr701 (A, B, and C) or normal rabbit Ig (D). Positive nuclei displayed brown DAB labeling, whereas negative nuclei displayed only methyl green-alcian blue staining.
Basal expression of IFN provides greater protection for cardiac myocytes than for cardiac fibroblasts.
To determine the biological significance of basal IFN in cardiac myocytes relative to cardiac fibroblasts, viral replication in primary cardiac cell cultures generated from IFN-α/β-RKO mice was compared to that in cultures generated from wild-type mice. Two days postplating, cultures were infected with T3D or T1L at 10 PFU per cell. At the indicated times postinfection, viral titers were quantitated by plaque assay (Fig. 3A). Both T1L and T3D replicated to higher titers in cardiac myocytes than in cardiac fibroblasts. Because these differences in replication could reflect any parameter of the virus-host interaction, titers in IFN-α/β-RKO cells were normalized to those in wild-type cells, to directly determine the role of IFN in restricting viral replication (Fig. 3B and C). Between 6 and 18 h postinfection, viral replication in cardiac myocytes derived from IFN-α/β-RKO mice was 3.7-fold to 10-fold higher for T3D and 4.9-fold to 10.5-fold higher for T1L than in wild-type cardiac myocytes. These data suggest a protective role for basal expression of IFN in cardiac myocytes for two reasons. First, while T3D induces significant IFN-β in cardiac myocytes during this time period, T1L does not (Fig. 4A), leaving only basal expression as the possible mediator of expression. Second, given the relatively high multiplicity of infection used for this infection and the early times of harvest, viral replication reflected primary infections rather than secondary infections, providing minimal opportunity for induced IFN to be protective. In contrast to the case in cardiac myocytes, viral replication in cardiac fibroblasts was approximately equivalent for IFN-α/β-RKO and wild-type cultures, suggesting that basal IFN does not play a significant antiviral role in cardiac fibroblasts.
FIG. 4.

Cardiac fibroblasts are more responsive than cardiac myocytes to IFN for induction of ISG561 mRNA. Supernatants and RNA were harvested from primary cardiac myocyte and cardiac fibroblast cultures at 2 days postplating at the indicated times postinfection or at 1 h post-IFN-α/β treatment. (A) T3D and T1L induction of IFN-β. Supernatants were evaluated by ELISA for secreted IFN-β (mean of two samples ± standard deviation). At every time point, differences between myocytes and fibroblasts were statistically significant (P < 0.05). Data are representative of multiple independent experiments. (B) T3D induction of ISG561 mRNA. RNA was analyzed by real-time RT-PCR and normalized to GAPDH expression. Results are expressed relative to mock-infected cultures (average of two samples). Statistical significance is as for panel A. (C) IFN-α/β induction of ISG561 and IRF7 mRNA. Increasing doses of IFN-α/β were administered to primary cardiac cultures, and RNA was harvested at either 1 h (ISG561) or 6 h (IRF7) post-IFN-α/β treatment. Analysis is as for panel B, and statistical significance is as for panel A.
Cardiac fibroblasts are more responsive than cardiac myocytes to IFN: induction of ISG mRNA.
Higher basal expression of IFN-β mRNA, activated ISGF3 components, and ISGs suggest an overall greater protective response in cardiac myocytes than in cardiac fibroblasts. Indeed, when primary cardiac myocyte and cardiac fibroblast cultures were infected with reovirus T3D and supernatants were harvested for ELISA, cardiac myocytes produced more IFN-β than did cardiac fibroblasts (Fig. 4A), consistent with our previous IFN-β mRNA results (53). In contrast to T3D induction of IFN-β, the induction of ISG561 mRNA was greater in cardiac fibroblasts than in cardiac myocytes (Fig. 4B). Similar results were seen for IRF7 (data not shown). Induction (n-fold) of ISG561 is more relevant here than absolute copy number, because it reflects the effects of IFN-β on the cell (the difference between initial and final states) rather than only the outcome. Together these data suggest that cardiac fibroblasts are more responsive than cardiac myocytes to IFN. To directly test this, cardiac myocyte and cardiac fibroblast cultures were treated with increasing doses of IFN-α/β, and ISG561 and IRF7 mRNAs were quantitated by real-time RT-PCR (Fig. 4C). Cardiac fibroblasts required only 33 U/ml IFN-α/β to achieve approximately the same induction (n-fold) of ISG561 mRNA as that for cardiac myocytes treated with 1,000 U/ml IFN-α/β, and at that highest IFN dose, induction was ∼9-fold greater in cardiac fibroblasts than in cardiac myocytes. Similar results were obtained for IRF7, with ∼5-fold-greater expression in cardiac fibroblasts than in cardiac myocytes at the highest dose of IFN. Therefore, cardiac fibroblasts are more responsive than cardiac myocytes to IFN induction of ISGs.
Basal expression of IFNAR and basal cytoplasmic expression of multiple Jak-STAT components are higher in cardiac fibroblasts than in cardiac myocytes.
The Jak-STAT pathway is an essential signaling cascade for the transcription of many ISGs. To determine whether the heightened cardiac fibroblast responsiveness to IFN reflects greater basal cytoplasmic expression of components in the Jak-STAT signaling pathway, cytoplasmic protein extracts were harvested from primary cardiac myocyte and cardiac fibroblast cultures for Western blot analysis. Basal cytoplasmic expression of Jak1, Tyk2, STAT2, and IRF9 was higher in cardiac fibroblasts than in cardiac myocytes (Fig. 5). The equivalent expression of basal cytoplasmic STAT1 in the two cell types suggests that STAT1 is not a limiting factor in cardiac myocytes. It is worth noting that while equivalent protein concentrations were loaded in each lane, cytoplasmic GAPDH and actin expression is cell type specific and therefore cannot be used for normalizations. In sum, the greater cytoplasmic expression of multiple Jak-STAT components in cardiac fibroblasts provides a mechanism for the greater IFN responsiveness observed in the cardiac fibroblasts.
FIG. 5.

Multiple basal cytoplasmic Jak-STAT components are expressed at higher levels in cardiac fibroblasts than in cardiac myocytes. Cytoplasmic protein lysates were harvested from mock-treated cardiac myocytes and cardiac fibroblasts, separated by SDS-PAGE, transferred to a nylon membrane, and probed using the indicated antibodies. Results are for duplicate samples and are representative of multiple experiments.
To determine basal IFNAR1 and IFNAR2 expression in cardiac myocyte and cardiac fibroblasts, primary cell cultures were harvested and examined by flow cytometry (Fig. 6). Over 99% of cardiac fibroblasts expressed an intracellular IFNAR1 epitope, whereas it was detected in only 53% of the cardiac myocytes. Mean fluorescence intensities for cardiac fibroblasts and cardiac myocytes were 14,800 and 3,628, respectively, indicating that IFNAR1 was expressed at higher levels in cardiac fibroblasts than in cardiac myocytes. Our strategy for identifying IFNAR2 was to stain for the extracellular epitope as it is the only portion of the receptor expressed by all three splice variants. By utilizing an intracellular staining technique, both the soluble intracellular form (serving as a reservoir for later interactions with IFNAR1 on the cell surface [40]) and the membrane-bound form of the IFNAR2 receptor would be identified. Approximately 11% of cardiac myocytes expressed IFNAR2, whereas 38% of cardiac fibroblasts expressed IFNAR2, with similar overall mean fluorescence intensities. The smaller fraction of cardiac myocytes than cardiac fibroblasts expressing IFNAR2 could limit the IFN responsiveness of cardiac myocytes as a population. Together, greater IFNAR1 basal expression in cardiac fibroblasts than in cardiac myocytes and IFNAR2 basal expression in a greater fraction of cardiac fibroblasts than cardiac myocytes provide an additional mechanism for the heightened IFN responsiveness of cardiac fibroblasts.
FIG. 6.

Basal IFNAR expression in cardiac fibroblasts and in cardiac myocytes. Basal levels of IFNAR1 (A and B) and IFNAR2 (C and D) were assessed 2 days postplating in cardiac myocytes (A and C) and cardiac fibroblasts (B and D). Cell suspensions were fixed, permeabilized, and stained with control (solid black line) or with the indicated monoclonal antibody (filled gray histogram). Representative histograms are shown; all samples were stained and run in duplicate.
Cardiac fibroblasts are more responsive than cardiac myocytes to IFN: induction of STAT phosphorylation.
To determine whether higher basal cytoplasmic Jak-STAT components in cardiac fibroblasts result in their greater activation upon IFN stimulation, STAT1 and STAT2 phosphorylation was evaluated by Western blot analysis. Primary cardiac myocyte and fibroblast cultures were treated with increasing doses of IFN-α/β, and total protein extracts were harvested at the indicated times (Fig. 7). Not unexpectedly, basal STAT phosphorylation was undetectable in these total cell lysates (in contrast to high levels of basal phosphorylated STAT in nuclear extracts [Fig. 1]). STAT phosphorylation was greater in cardiac fibroblasts than in cardiac myocytes at most doses and time points. For example, at 30 min posttreatment, both 100 U/ml and 1,000 U/ml induced significantly more STAT1 phosphorylation in cardiac fibroblasts than in cardiac myocytes, and the same was true for STAT2 phosphorylation. The difference between these cell types at 2 h posttreatment was most pronounced at 100 and 1,000 U/ml for STAT1 phosphorylation and at 10 and 100 U/ml for STAT2 phosphorylation. As expected, GAPDH expression was higher in myocytes than in fibroblasts, while the converse was true for actin expression (although not as pronounced in these total cell extracts as in cytoplasmic cell extracts [Fig. 5]).
FIG. 7.

Cardiac fibroblasts are more responsive than cardiac myocytes to IFN for induction of STAT phosphorylation. Two days postplating, primary cardiac cell cultures were treated with increasing doses of IFN-α/β and at 30 min and 2 h post-IFN treatment total protein extracts were harvested. Proteins were separated by SDS-PAGE, transferred to a nylon membrane, and probed using the indicated antibodies. Results are representative of multiple experiments.
Reovirus activation of multiple nuclear ISGF3 components is higher in cardiac fibroblasts than in cardiac myocytes.
Given that reovirus induces IFN-β poorly in cardiac fibroblasts (Fig. 4A) but that cardiac fibroblasts are highly responsive to IFN (Fig. 4C and 7), subsequent activation of ISGF3 components in the nucleus of cardiac fibroblasts was investigated relative to that in cardiac myocytes. Primary cardiac cultures were infected with reovirus (T3D) at 10 PFU per cell, and nuclear protein extracts were harvested at the indicated times postinfection. T3D-induced STAT1 and STAT2 phosphorylation was greater in cardiac fibroblasts than in cardiac myocytes and most clearly evident at 4 h postinfection in comparison to mock infections (Fig. 8). T3D failed to induce STAT1 and STAT2 phosphorylation in IFN-α/β-RKO cardiac myocytes and cardiac fibroblasts, indicating that the phosphorylation in wild-type cells was mediated thru IFN-α/β signaling (data not shown). Although T3D induces less IFN-β in cardiac fibroblasts than in cardiac myocytes, reovirus-induced IFN results in a more robust activation of multiple nuclear ISGF3 components in cardiac fibroblasts, thus providing further evidence that cardiac fibroblasts are more responsive to IFN-α/β.
FIG. 8.

Reovirus activation of multiple nuclear ISGF3 components is greater in cardiac fibroblasts than in cardiac myocytes. Nuclear protein extracts were harvested from primary cardiac cell cultures at the indicated times post-T3D infection (10 PFU/cell), resolved by SDS-PAGE, transferred to a nylon membrane, and probed with the indicated antibodies. Results are representative of multiple experiments.
IFN-β inhibition of viral replication is greater in cardiac fibroblasts than in cardiac myocytes.
To investigate the biological significance of cell-type-specific responsiveness to IFN-β, we evaluated viral replication in primary cardiac cell cultures after treatment with increasing doses of IFN-β. One day postplating, cultures were pretreated with the indicated doses of IFN-β for 24 h, followed by T3D infection at 5 PFU per cell for 12 or 24 h. At the indicated times postinfection, viral titers were evaluated by plaque assay. At every dose and time tested, IFN-β inhibited T3D replication in cardiac fibroblasts more than in cardiac myocytes (Fig. 9). For example, at 10 U/ml IFN-β, 50% of PFU remained in cardiac fibroblasts compared to 80% of PFU remaining in cardiac myocytes at 12 h post-T3D infection.
Myocytes and fibroblasts are intimately associated in the heart.
Data presented here suggest that cardiac myocytes and cardiac fibroblasts respond uniquely to reovirus infection and that while cardiac myocytes secrete more IFN-β (Fig. 4A), cardiac fibroblasts are more responsive to IFN (Fig. 4C and 7). To determine whether these cells are likely to interact in vivo, we performed indirect immunofluorescence on normal mouse myocardium using cell-specific markers to detect cardiac myocytes (antimyomesin) and cardiac fibroblasts (antivimentin) simultaneously. Cardiac fibroblasts are abundant and lie directly adjacent to cardiac myocytes (Fig. 10). This dense network of cells is consistent with efficient communication between the two cell types, both as a threat (spread of virus) and as a protective mechanism (secretion of IFN-β).
FIG. 10.
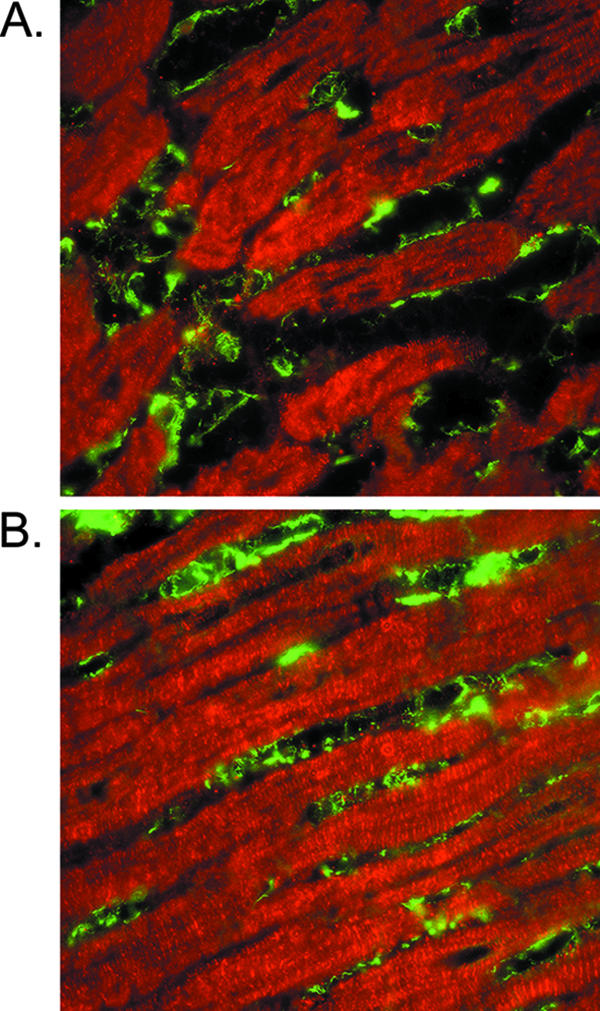
Association of myocytes and fibroblasts in normal mouse myocardium. Immunofluorescence microscopy of cross sections (A and B) of mouse myocardium, immunostained with antimyomesin to identify cardiac myocytes (red) and antivimentin to mark cardiac fibroblasts (green).
Basal IFN expression in myocytes is insufficient to protect adjacent fibroblasts.
Given the high basal expression of IFN-β in cardiac myocytes (Fig. 1) and the autocrine protection that it provides (Fig. 3) and given the sensitivity of cardiac fibroblasts to low levels of IFN (Fig. 9) and their intimate association with cardiac myocytes in the heart (Fig. 10), it seemed possible that basal expression of IFN in cardiac myocytes might also provide paracrine protection for adjacent fibroblasts. To determine whether uninfected cardiac myocytes could inhibit viral replication in adjacent cardiac fibroblasts, cells were cocultured as follows. Cardiac fibroblasts were incubated with T3D for 1 h at 37°C or on ice to prevent viral penetration, and then inoculum was removed and cultures were washed extensively. Medium, IFN, or uninfected cardiac myocytes were added; cultures were incubated for an additional 18 h; and then viral replication was determined by plaque assay (Fig. 11). While IFN reduced viral replication in cardiac fibroblasts 3.2- to 15.1-fold even though it was added post-viral attachment, cardiac myocytes failed to reduce viral replication in cardiac fibroblasts even when present at a ratio of 10 myocytes to one fibroblast. These data suggest that even though cardiac fibroblasts are highly sensitive to the antiviral effects of IFN, basal IFN secreted by adjacent cardiac myocytes provides only autocrine and not paracrine protection.
DISCUSSION
Cardiac myocytes are continually exposed to viruses, are not replenished, and yet are critical to survival, suggesting that this differentiated cardiac cell type may have evolved a unique intrinsic protective response against viral infection. In particular, cardiac myocytes may be more dependent on the innate immune response than cell types that are rapidly replenished, such as cardiac fibroblasts. We have previously shown that the innate IFN response is a critical determinant of viral spread in cardiac myocyte cultures but not in differentiated skeletal muscle cells, suggesting a cell-type-specific role for IFN-β in antiviral protection against reovirus-induced myocarditis (49).
The traditional view of IFN-β in antiviral protection begins with viral induction of this cytokine for subsequent induction of ISGs expressing direct antiviral activity. Our results strongly suggest that basal expression of IFN-β plays an important protective role as well. Specifically, basal IFN-β mRNA, basal nuclear expression of phosphorylated STAT1 (Tyr701) and phosphorylated STAT2, and basal ISG561 mRNA were higher in cardiac myocytes than in cardiac fibroblasts (Fig. 1). The absence of basal nuclear STAT1 and STAT2 phosphorylation in IFN-α/β-RKO cultures indicates that their basal activation in wild-type cardiac myocytes is dependent on signaling through the IFN-α/β receptor. Together, the data suggest that basal IFN-β in cardiac myocytes, but not in cardiac fibroblasts, is sufficient for activation of the Jak-STAT pathway and induction of ISGs. Surprisingly however, basal secreted IFN-β in cardiac myocyte cultures was no greater than medium alone in ELISA (data not shown), suggesting that IFN-β was bound to receptors on the cells from which it was secreted and was therefore unavailable for detection by ELISA. Indeed, uninfected cardiac myocytes failed to inhibit viral replication in cocultured infected cardiac fibroblasts, even when present at a ratio of 10 myocytes to one fibroblast (Fig. 11). This suggests a model (Fig. 12, part 1) where basal IFN-β expressed in cardiac myocytes is sufficient for autocrine, but not paracrine, activation of the Jak-STAT pathway. To our knowledge, this is the first report of high basal STAT activation in a normal cell type.
FIG. 12.

Model for cell-type-specific IFN responses in the heart.
Basal STAT activation is not uncommon in diseased cells and tissues. Several studies have demonstrated basal activation of STATs, in particular STAT1, STAT3, and STAT5, in a number of diverse human tumor cell lines (5, 7, 19, 56). In addition, a wide variety of human tumors have been shown to display basal phosphorylation of STATs, including both hematologic malignancies (leukemias, lymphomas, and multiple myelomas) and solid tumors (head and neck, breast, and prostate) (5, 9, 18, 24, 33, 37, 51). Basal activation of STATs has also been shown to be cell type specific. For example, basal phosphorylation of STAT1 was observed selectively in airway epithelial cells brushed from asthmatic subjects, while in contrast, bronchoalveolar macrophages from the same subjects showed no evidence of basal STAT1 activation. The biological relevance of constitutive activation of epithelial STAT1 in asthmatic subjects was linked to the pathogenesis of this inflammatory disease (42). Here, we provide evidence that basal nuclear phosphorylation of STATs in cardiac myocytes likely serves a functional role, too, as a prearming mechanism for protection. Specifically, viral replication was increased to a greater extent in IFN-α/β-RKO cardiac myocytes than in IFN-α/β-RKO cardiac fibroblasts relative to their wild-type counterparts (Fig. 3). Therefore, basal IFN expression in cardiac myocytes provides partial autocrine protection against viral infection. As discussed above, however, this basal IFN expression is insufficient to protect adjacent cardiac fibroblasts from infection (Fig. 11). Therefore, cardiac myocyte basal IFN expression provides autocrine, but not paracrine, protection (Fig. 12, part 1).
Induction of IFN-β involves a positive feedback loop (32). Cells primed with small amounts of IFN-α/β and then virally infected express more IFN-α/β than nonprimed cells (43), most likely reflecting IFN-β induction of the transcription factor IRF7 during priming and subsequent viral activation of IRF7 for further induction of IFN-β (54). Cardiac myocytes have higher basal IRF7 (53), consistent with basal IFN-β activating the Jak-STAT pathway for induction of IRF7 as a second component of the prearming protective mechanism (Fig. 12, part 1). The role of basal IFN-β expression in determining basal IRF7 expression and subsequent viral amplification of IFN-β has been investigated previously (20). In those studies of primary cultures of mouse embryo fibroblasts and splenocytes, differences in basal IFN-β and IRF7 expression between cell types dominated the innate response to viral infection, determining cell-type-specific differences in viral induction of IFN in the absence of the critical transcription factor IRF3. However, they found that basal IRF7 expression was not completely IFN dependent, as cells expressing similar IFN-β levels expressed significantly different IRF7 levels. This is in contrast to cardiac myocytes and cardiac fibroblasts, where basal IFN-β expression determines basal IRF7 expression (53). The greater expression of basal IRF7 in cardiac myocytes renders these cells poised for greater induction of IFN-β upon viral infection, resulting in paracrine protection of adjacent fibroblasts (Fig. 12, part 2). The direct proximity of myocytes and fibroblasts in the heart (Fig. 10) supports this model of efficient paracrine communication.
High basal expression of IFN-β would appear to be beneficial to cells, providing high basal antiviral ISGs and high IRF7 for rapid amplification of IFN-β upon viral infection. IFN-β, however, induces a large number of genes (14, 15), many of which have antiproliferative functions (52). Therefore, high basal IFN-β expression sufficient for autocrine, but not paracrine, effects may be an effective strategy only for nondividing cells such as cardiac myocytes. The lack of high basal IFN-β in splenocytes (20) and other lymphoid cells that express high IRF7 (2) in contrast to cardiac myocytes suggests a possible divergence of mechanisms regulating the innate response depending on cell potential for proliferation. Components of the innate response have been shown to differ in basal expression and in response to type I IFN in cells arrested in different stages of the cell cycle (31), suggesting an additional complexity to their regulation.
While cardiac myocytes may benefit from basal autocrine stimulation, this relatively low level of protection would likely be inadequate for protection against a flood of progeny virus from adjacent infected cardiac fibroblasts. Therefore, cardiac fibroblasts must have a strong innate response as well, to preclude their serving as a reservoir for viral replication. Yet cardiac fibroblast proliferation would likely be compromised by high basal IFN-β expression, interfering with their reparative role following cardiac tissue damage (6). We demonstrate here that cardiac fibroblasts are more responsive than cardiac myocytes to IFN signaling, supporting a model where IFN secreted from cardiac myocytes efficiently protects neighboring cardiac fibroblasts from infection (Fig. 12, part 2). Specifically, cardiac fibroblasts have higher basal IFNAR1 (Fig. 6) and higher basal cytoplasmic Jak1, Tyk2, STAT2, and IRF9 (Fig. 5), resulting in greater activation of STAT1 and STAT2 following IFN stimulation (Fig. 7) or reovirus T3D infection (Fig. 8) and greater IFN-induced antiviral protection (Fig. 9). Basal STAT1 expression determines responsiveness to type I IFN in melanoma cells (59) and a breast tumor cell line (27), and basal STAT1, STAT2, and IRF9 modulate responsiveness to type I IFN in a melanoma cell line (59), implicating this as a general mechanism for regulating the innate immune response. The molecular mechanism for high basal expression of IFNAR1 and latent Jak-STAT components is likely specific to each component.
Why might cardiac myocytes, the highly differentiated cells essential for cardiac function, have evolved comparatively lower basal cytoplasmic Jak-STAT expression and, consequently, lower sensitivity to IFN stimulation? First, high basal IFN-β expression in cardiac myocytes already induces relatively high basal STAT activation and ISG expression. High basal Jak-STAT components could increase basal ISG expression to detrimental levels. The relatively high basal expression of activated STATs in cardiac myocytes, compared with the relatively low basal expression of latent Jak-STAT components in those cells, may reflect an optimal balance. Second, STAT1 is a critical component of angiotensin II-mediated signaling (16), regulating responses ranging from cardiac myocyte hypertrophy to apoptosis. High basal expression of Jak-STAT components could render cardiac myocytes hyperresponsive to angiotensin II, with likely damaging consequences. Finally, STAT1 physically interacts with and modulates the function of the cardiac transcription factor GATA-4 (58), which regulates numerous pathways critical for cardiac development and stress responses (55), as well as cardiac myocyte survival (1, 26). High basal Jak-STAT components could interfere with signaling essential for cardiac remodeling in myocardial infarction and other pathological states.
By 24 h post-T3D infection, STAT1 and STAT2 phosphorylation returned to basal levels or lower in cardiac myocytes but not in cardiac fibroblasts (Fig. 8). Continued STAT phosphorylation in the face of sustained IFN stimulation can be halted by suppressors of cytokine signaling (SOCS) (25), at least two of which are known to function in cardiac myocytes (60, 61). Data here suggest that SOCS or other inhibitors are more rapidly activated in cardiac myocytes than in cardiac fibroblasts, consistent with arguments above for the possible detrimental effects of excessive STAT activation in cardiac myocytes. Signaling in cardiac fibroblasts cannot be sustained indefinitely, however, or their proliferation and contribution to cardiac tissue repair would likely be impaired.
In sum, the data presented here support a model (Fig. 12) whereby high basal IFN-β expression in cardiac myocytes prearms this vulnerable and essential cell type, while high basal expression of IFNAR1 and latent Jak-STAT components in adjacent cardiac fibroblasts prevents them from inadvertently serving as a reservoir for viral replication and spread to adjacent myocytes. The relatively high basal expression of two of the three activated components of ISGF3 in cardiac myocytes and four of the five latent components of the Jak-STAT pathway and IFNAR1 in cardiac fibroblasts suggests a strong selection for this strategic variation in cell-type-specific innate responses to viral infection. These studies provide the first indication of an integrated network of cell-type-specific innate immune components for organ protection.
Acknowledgments
We thank Lianna Li, Susan Irvin, Sandra Horton, and Tim Petty for insightful discussions; Wrennie Edwards for technical assistance; and Mac Law for photography assistance.
This work was supported by NIH grant AI062657.
Footnotes
Published ahead of print on 17 October 2007.
REFERENCES
- 1.Aries, A., P. Paradis, C. Lefebvre, R. J. Schwartz, and M. Nemer. 2004. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 101:6975-6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Au, W. C., P. A. Moore, D. W. LaFleur, B. Tombal, and P. M. Pitha. 1998. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J. Biol. Chem. 273:29210-29217. [DOI] [PubMed] [Google Scholar]
- 3.Baty, C. J., and B. Sherry. 1993. Cytopathogenic effect in cardiac myocytes but not in cardiac fibroblasts is correlated with reovirus-induced acute myocarditis. J. Virol. 67:6295-6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowles, N. E., J. Ni, D. L. Kearney, M. Pauschinger, H. P. Schultheiss, R. McCarthy, J. Hare, J. T. Bricker, K. R. Bowles, and J. A. Towbin. 2003. Detection of viruses in myocardial tissues by polymerase chain reaction. Evidence of adenovirus as a common cause of myocarditis in children and adults. J. Am. Coll. Cardiol. 42:466-472. [DOI] [PubMed] [Google Scholar]
- 5.Bowman, T., R. Garcia, J. Turkson, and R. Jove. 2000. STATs in oncogenesis. Oncogene 19:2474-2488. [DOI] [PubMed] [Google Scholar]
- 6.Camelliti, P., T. K. Borg, and P. Kohl. 2005. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 65:40-51. [DOI] [PubMed] [Google Scholar]
- 7.Catlett-Falcone, R., W. S. Dalton, and R. Jove. 1999. STAT proteins as novel targets for cancer therapy. Signal transducer an activator of transcription. Curr. Opin. Oncol. 11:490-496. [DOI] [PubMed] [Google Scholar]
- 8.Chow, L. H., K. W. Beisel, and B. M. McManus. 1992. Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab. Investig. 66:24-31. [PubMed] [Google Scholar]
- 9.Coffer, P. J., L. Koenderman, and R. P. de Groot. 2000. The role of STATs in myeloid differentiation and leukemia. Oncogene 19:2511-2522. [DOI] [PubMed] [Google Scholar]
- 10.Cook, D. N., M. A. Beck, T. M. Coffman, S. L. Kirby, J. F. Sheridan, I. B. Pragnell, and O. Smithies. 1995. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science 269:1583-1585. [DOI] [PubMed] [Google Scholar]
- 11.Daliento, L., F. Calabrese, F. Tona, A. L. Caforio, G. Tarsia, A. Angelini, and G. Thiene. 2003. Successful treatment of enterovirus-induced myocarditis with interferon-alpha. J. Heart Lung Transplant. 22:214-217. [DOI] [PubMed] [Google Scholar]
- 12.Darnell, J. E., Jr., I. M. Kerr, and G. R. Stark. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415-1421. [DOI] [PubMed] [Google Scholar]
- 13.DeBiasi, R. L., C. L. Edelstein, B. Sherry, and K. L. Tyler. 2001. Calpain inhibition protects against virus-induced apoptotic myocardial injury. J. Virol. 75:351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Der, S. D., A. Zhou, B. R. Williams, and R. H. Silverman. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 95:15623-15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Veer, M. J., M. Holko, M. Frevel, E. Walker, S. Der, J. M. Paranjape, R. H. Silverman, and B. R. Williams. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912-920. [PubMed] [Google Scholar]
- 16.El-Adawi, H., L. Deng, A. Tramontano, S. Smith, E. Mascareno, K. Ganguly, R. Castillo, and N. El-Sherif. 2003. The functional role of the JAK-STAT pathway in post-infarction remodeling. Cardiovasc. Res. 57:129-138. [DOI] [PubMed] [Google Scholar]
- 17.Fu, X. Y., D. S. Kessler, S. A. Veals, D. E. Levy, and J. E. Darnell, Jr. 1990. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. USA 87:8555-8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia, R., T. L. Bowman, G. Niu, H. Yu, S. Minton, C. A. Muro-Cacho, C. E. Cox, R. Falcone, R. Fairclough, S. Parsons, A. Laudano, A. Gazit, A. Levitzki, A. Kraker, and R. Jove. 2001. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 20:2499-2513. [DOI] [PubMed] [Google Scholar]
- 19.Garcia, R., and R. Jove. 1998. Activation of STAT transcription factors in oncogenic tyrosine kinase signaling. J. Biomed. Sci. 5:79-85. [DOI] [PubMed] [Google Scholar]
- 20.Hata, N., M. Sato, A. Takaoka, M. Asagiri, N. Tanaka, and T. Taniguchi. 2001. Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem. Biophys. Res. Commun. 285:518-525. [DOI] [PubMed] [Google Scholar]
- 21.Heim, A., M. Stille-Siegener, R. Kandolf, H. Kreuzer, and H. R. Figulla. 1994. Enterovirus-induced myocarditis: hemodynamic deterioration with immunosuppressive therapy and successful application of interferon-alpha. Clin. Cardiol. 17:563-565. [DOI] [PubMed] [Google Scholar]
- 22.Heim, M. H. 1999. The Jak-STAT pathway: cytokine signalling from the receptor to the nucleus. J. Recept. Signal Transduct. Res. 19:75-120. [DOI] [PubMed] [Google Scholar]
- 23.Herzum, M., V. Ruppert, B. Kuytz, H. Jomaa, I. Nakamura, and B. Maisch. 1994. Coxsackievirus B3 infection leads to cell death of cardiac myocytes. J. Mol. Cell. Cardiol. 26:907-913. [DOI] [PubMed] [Google Scholar]
- 24.Huang, M., J. F. Dorsey, P. K. Epling-Burnette, R. Nimmanapalli, T. H. Landowski, L. B. Mora, G. Niu, D. Sinibaldi, F. Bai, A. Kraker, H. Yu, L. Moscinski, S. Wei, J. Djeu, W. S. Dalton, K. Bhalla, T. P. Loughran, J. Wu, and R. Jove. 2002. Inhibition of Bcr-Abl kinase activity by PD180970 blocks constitutive activation of Stat5 and growth of CML cells. Oncogene 21:8804-8816. [DOI] [PubMed] [Google Scholar]
- 25.Knisz, J., and P. B. Rothman. 2007. Suppressor of cytokine signaling in allergic inflammation. J. Allergy Clin. Immunol. 119:739-745. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi, S., T. Lackey, Y. Huang, E. Bisping, W. T. Pu, L. M. Boxer, and Q. Liang. 2006. Transcription factor gata4 regulates cardiac BCL2 gene expression in vitro and in vivo. FASEB J. 20:800-802. [DOI] [PubMed] [Google Scholar]
- 27.Kolla, V., D. J. Lindner, W. Xiao, E. C. Borden, and D. V. Kalvakolanu. 1996. Modulation of interferon (IFN)-inducible gene expression by retinoic acid. Up-regulation of STAT1 protein in IFN-unresponsive cells. J. Biol. Chem. 271:10508-10514. [DOI] [PubMed] [Google Scholar]
- 28.Kuhl, U., M. Pauschinger, M. Noutsias, B. Seeberg, T. Bock, D. Lassner, W. Poller, R. Kandolf, and H. P. Schultheiss. 2005. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 111:887-893. [DOI] [PubMed] [Google Scholar]
- 29.Kuhl, U., M. Pauschinger, P. L. Schwimmbeck, B. Seeberg, C. Lober, M. Noutsias, W. Poller, and H. P. Schultheiss. 2003. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107:2793-2798. [DOI] [PubMed] [Google Scholar]
- 30.Kuhl, U., M. Pauschinger, B. Seeberg, D. Lassner, M. Noutsias, W. Poller, and H. P. Schultheiss. 2005. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112:1965-1970. [DOI] [PubMed] [Google Scholar]
- 31.Kumar, R., L. Korutla, and K. Zhang. 1994. Cell cycle-dependent modulation of alpha-interferon-inducible gene expression and activation of signaling components in Daudi cells. J. Biol. Chem. 269:25437-25441. [PubMed] [Google Scholar]
- 32.Levy, D. E., I. Marie, E. Smith, and A. Prakash. 2002. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 22:87-93. [DOI] [PubMed] [Google Scholar]
- 33.Lin, T. S., S. Mahajan, and D. A. Frank. 2000. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene 19:2496-2504. [DOI] [PubMed] [Google Scholar]
- 34.Martin, A. B., S. Webber, F. J. Fricker, R. Jaffe, G. Demmler, D. Kearney, Y. H. Zhang, J. Bodurtha, B. Gelb, J. Ni, et al. 1994. Acute myocarditis. Rapid diagnosis by PCR in children. Circulation 90:330-339. [DOI] [PubMed] [Google Scholar]
- 35.Miric, M., A. Miskovic, J. D. Vasiljevic, N. Keserovic, and M. Pesic. 1995. Interferon and thymic hormones in the therapy of human myocarditis and idiopathic dilated cardiomyopathy. Eur. Heart J. 16(Suppl. O):150-152. [DOI] [PubMed] [Google Scholar]
- 36.Miric, M., J. Vasiljevic, M. Bojic, Z. Popovic, N. Keserovic, and M. Pesic. 1996. Long-term follow up of patients with dilated heart muscle disease treated with human leucocytic interferon alpha or thymic hormones: initial results. Heart 75:596-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mora, L. B., R. Buettner, J. Seigne, J. Diaz, N. Ahmad, R. Garcia, T. Bowman, R. Falcone, R. Fairclough, A. Cantor, C. Muro-Cacho, S. Livingston, J. Karras, J. Pow-Sang, and R. Jove. 2002. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 62:6659-6666. [PubMed] [Google Scholar]
- 38.Muller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918-1921. [DOI] [PubMed] [Google Scholar]
- 39.Pellegrini, S., and I. Dusanter-Fourt. 1997. The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). Eur. J. Biochem. 248:615-633. [DOI] [PubMed] [Google Scholar]
- 40.Prejean, C., and O. R. Colamonici. 2000. Role of the cytoplasmic domains of the type I interferon receptor subunits in signaling. Semin. Cancer Biol. 10:83-92. [DOI] [PubMed] [Google Scholar]
- 41.Rose, N. R., and S. L. Hill. 1996. The pathogenesis of postinfectious myocarditis. Clin. Immunol. Immunopathol. 80:S92-S99. [DOI] [PubMed] [Google Scholar]
- 42.Sampath, D., M. Castro, D. C. Look, and M. J. Holtzman. 1999. Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J. Clin. Investig. 103:1353-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539-548. [DOI] [PubMed] [Google Scholar]
- 44.Schindler, C., and J. E. Darnell, Jr. 1995. Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu. Rev. Biochem. 64:621-651. [DOI] [PubMed] [Google Scholar]
- 45.Sheppard, R., M. Bedi, T. Kubota, M. J. Semigran, W. Dec, R. Holubkov, A. M. Feldman, W. D. Rosenblum, C. F. McTiernan, and D. M. McNamara. 2005. Myocardial expression of fas and recovery of left ventricular function in patients with recent-onset cardiomyopathy. J. Am. Coll. Cardiol. 46:1036-1042. [DOI] [PubMed] [Google Scholar]
- 46.Sherry, B., C. J. Baty, and M. A. Blum. 1996. Reovirus-induced acute myocarditis in mice correlates with viral RNA synthesis rather than generation of infectious virus in cardiac myocytes. J. Virol. 70:6709-6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherry, B., X. Y. Li, K. L. Tyler, J. M. Cullen, and H. W. Virgin IV. 1993. Lymphocytes protect against and are not required for reovirus-induced myocarditis. J. Virol. 67:6119-6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherry, B., F. J. Schoen, E. Wenske, and B. N. Fields. 1989. Derivation and characterization of an efficiently myocarditic reovirus variant. J. Virol. 63:4840-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherry, B., J. Torres, and M. A. Blum. 1998. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J. Virol. 72:1314-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith, R. E., H. J. Zweerink, and W. K. Joklik. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791-810. [DOI] [PubMed] [Google Scholar]
- 51.Song, J. I., and J. R. Grandis. 2000. STAT signaling in head and neck cancer. Oncogene 19:2489-2495. [DOI] [PubMed] [Google Scholar]
- 52.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 53.Stewart, M. J., K. Smoak, M. A. Blum, and B. Sherry. 2005. Basal and reovirus-induced beta interferon (IFN-β) and IFN-β-stimulated gene expression are cell type specific in the cardiac protective response. J. Virol. 79:2979-2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taniguchi, T., and A. Takaoka. 2001. A weak signal for strong responses: interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2:378-386. [DOI] [PubMed] [Google Scholar]
- 55.Temsah, R., and M. Nemer. 2005. GATA factors and transcriptional regulation of cardiac natriuretic peptide genes. Regul. Pept. 128:177-185. [DOI] [PubMed] [Google Scholar]
- 56.Turkson, J., and R. Jove. 2000. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19:6613-6626. [DOI] [PubMed] [Google Scholar]
- 57.Veals, S. A., C. Schindler, D. Leonard, X. Y. Fu, R. Aebersold, J. E. Darnell, Jr., and D. E. Levy. 1992. Subunit of an alpha-interferon-responsive transcription factor is related to interferon regulatory factor and Myb families of DNA-binding proteins. Mol. Cell. Biol. 12:3315-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang, J., P. Paradis, A. Aries, H. Komati, C. Lefebvre, H. Wang, and M. Nemer. 2005. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Mol. Cell. Biol. 25:9829-9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wong, L. H., K. G. Krauer, I. Hatzinisiriou, M. J. Estcourt, P. Hersey, N. D. Tam, S. Edmondson, R. J. Devenish, and S. J. Ralph. 1997. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J. Biol. Chem. 272:28779-28785. [DOI] [PubMed] [Google Scholar]
- 60.Yajima, T., H. Yasukawa, E. S. Jeon, D. Xiong, A. Dorner, M. Iwatate, M. Nara, H. Zhou, D. Summers-Torres, M. Hoshijima, K. R. Chien, A. Yoshimura, and K. U. Knowlton. 2006. Innate defense mechanism against virus infection within the cardiac myocyte requiring gp130-STAT3 signaling. Circulation 114:2364-2373. [DOI] [PubMed] [Google Scholar]
- 61.Yasukawa, H., T. Yajima, H. Duplain, M. Iwatate, M. Kido, M. Hoshijima, M. D. Weitzman, T. Nakamura, S. Woodard, D. Xiong, A. Yoshimura, K. R. Chien, and K. U. Knowlton. 2003. The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury. J. Clin. Investig. 111:469-478. [DOI] [PMC free article] [PubMed] [Google Scholar]