

Abstract
The budding reactions of a number of enveloped viruses use the cellular machinery involved in the formation of the luminal vesicles of endosomal multivesicular bodies (MVB). Budding of these viruses is dependent on the presence of specific late-domain motifs in membrane-associated viral proteins. Such budding reactions usually involve ubiquitin and are blocked by expression of an ATPase-deficient form of VPS4, a cellular AAA+ ATPase believed to be required late in the MVB pathway for the disassembly/release of the MVB machinery. Here we examined the role of the MVB pathway in the budding of the late-domain-containing rhabdovirus vesicular stomatitis virus (VSV) and the alphavirus Semliki Forest virus (SFV). We tested early and late steps in the MVB pathway by depleting ubiquitin with the proteasome inhibitor MG-132 and by using cell lines inducibly expressing VPS4A or VPS4B protein. As previously shown, VSV budding was strongly dependent on ubiquitin. In contrast to the findings of previous studies with VPS4A, expression of ATPase-deficient mutants of either VPS4A or VPS4B inhibited VSV budding. Inhibition by VPS4 required the presence of the PPPY late domain on the VSV matrix protein and resulted in the accumulation of nonreleased VSV particles at the plasma membrane. In contrast, SFV budding was independent of both ubiquitin and the activity of VPS4, perhaps reflecting the important role of the highly organized envelope protein lattice during alphavirus budding.
A critical step in the life cycle of all enveloped viruses is the virus budding reaction, which encloses the viral core in a lipid bilayer containing virus membrane proteins that mediate receptor binding and membrane fusion. Virus budding can occur at the plasma membrane or on various intracellular membranes of the secretory system. The viral proteins involved may include transmembrane proteins, capsid proteins, and/or matrix-type proteins that associate with the inner face of the budding membrane (15). The membrane of the budding virus bends away from the cytoplasm, and a membrane fission event releases the completed virus particle. The topology of virus budding is similar to that of vesicle budding into the lumen of the late endosomal multivesicular body (MVB) compartment (reviewed in references 2, 19, and 23). Recent studies have demonstrated that a number of enveloped viruses coopt the MVB machinery of mammalian cells for their budding reactions (reviewed in references 4, 11, and 41).
The luminal vesicles of the MVB are enriched in transmembrane cargo proteins that are targeted for degradation. Although not essential for sorting in all cases (see, e.g., reference 22), most of these cargo proteins are ubiquitinated on their cytoplasmic tails (21). Sorting of cargo and formation of luminal MVB vesicles involve the sequential recruitment of a series of soluble protein complexes known as ESCRT proteins (endosomal sorting complex required for transport) (reviewed in references 2, 19, and 23). Monoubiquitinated proteins first interact with the HRS-STAM complex. Binding of this complex results in recruitment of the ESCRT-I complex, which then recruits the ESCRT-II and -III complexes. Both ESCRT-I and ESCRT-II can interact with monoubiquitinated cargoes, while ESCRT-III appears to act downstream of ubiquitin recognition. A late step in the MVB pathway involves the recruitment of VPS4, an AAA+ ATPase, to the assembled ESCRT complexes. VPS4 activity is required for vesicle budding and is thought to be involved in the disassembly and recycling of the ESCRT components (3). There are two VPS4 isoforms, VPS4A and VPS4B, which are ∼80% identical in amino acid sequence. Expression of an ATPase-deficient form of either isoform has been shown to inhibit endosomal trafficking (5, 38, 49).
Enveloped RNA viruses, including members of the retroviruses, filoviruses, and paramyxoviruses, have been shown to use components of the MVB machinery for budding (reviewed in references 4, 11, and 41). These viruses all have short sequence motifs called late domains in their membrane-associated Gag, VP40, or M proteins, respectively. The late domains recruit ESCRT complexes to the site of budding, with different late-domain motifs interacting specifically with different ESCRT components. In spite of differences in their late domains, the budding reactions of retroviruses, filoviruses, and paramyxoviruses are all inhibited by expression of ATPase-deficient VPS4. Recent studies have shown that functional VPS4 is also required for efficient budding of the DNA viruses herpes simplex virus type 1 and hepatitis B virus (9, 27). While in many cases the budding of ESCRT-dependent viruses also appears to involve ubiquitin, at least one example of ubiquitin-independent but ESCRT-dependent virus budding has been described, that of the retrovirus equine infectious anemia virus (43, 47). Conversely, the rhabdovirus vesicular stomatitis virus (VSV) has a well-defined late-domain motif, PPPY, on its M (matrix) protein and shows strongly ubiquitin dependent budding (8, 24, 26). However, studies using ATPase-deficient VPS4A did not show a role for the ESCRT machinery in VSV budding (25).
The role of the MVB machinery in the budding reaction of alphaviruses has not been characterized. Alphaviruses are highly organized enveloped RNA viruses that have T=4 icosahedral quasisymmetry in both the outer viral glycoprotein layer and the inner capsid shell (reviewed in references 32 and 45). The outer glycoprotein layer contains 240 copies of the E2 and E1 transmembrane (TM) proteins, which act as the receptor-binding and membrane fusion proteins, respectively (28). These envelope proteins are organized as 80 trimers of E2/E1 heterodimers on the virus particle. Following low-pH-dependent fusion of the virus with the endosome membrane, the nucleocapsid is released into the cytoplasm, and virus replication ensues. Capsid proteins bind newly synthesized genomic RNA to form nucleocapsid cores (32). The nascent membrane proteins are cotranslationally translocated into the endoplasmic reticulum and transported through the secretory pathway to the plasma membrane. A specific tyrosine-containing sequence on the cytoplasmic tail of the E2 protein interacts with a hydrophobic pocket on the nucleocapsid core (35, 48), and the virus buds from the plasma membrane. It is clear that alphavirus budding requires the glycoproteins and the nucleocapsid, the E2 tail-nucleocapsid interaction, E2-E1 heterodimer formation, and lateral interactions between E1 proteins in the icosahedral lattice (16, 32) and that it is promoted by the small virus membrane protein 6K (37).
While the functions of the alphavirus proteins have been studied in depth, there is essentially no information on the involvement of cellular proteins in alphavirus budding. We therefore set out to investigate whether the MVB machinery might play a role in the budding reaction of this group of enveloped RNA viruses. Because viruses can recruit ESCRT proteins at different steps, we tested the requirement for both ubiquitin, a relatively early component of the MVB pathway, and VPS4, a protein required at a late stage. In each approach we compared the results for the alphavirus Semliki Forest virus (SFV) and the late-domain-containing rhabdovirus VSV. Our findings indicated that the alphavirus budding reaction is independent of both ubiquitin and functional VPS4. In contrast, VSV budding was strongly dependent on ubiquitin, as predicted, but was also inhibited by expression of ATPase-deficient forms of either VPS4A or VPS4B.
MATERIALS AND METHODS
Viruses and plasmids.
The SFV strain used in these experiments was a well-characterized plaque-purified isolate propagated in BHK-21 cells (18). Wild-type VSV (Indiana strain) and the VSV-AAPA mutant were propagated in BHK-21 cells. The PPPY late domain in this mutant is changed to AAPA, making the virus less efficient in budding but insensitive to MG-132 (20, 26). This mutant was kindly provided by Michael A. Whitt (University of Tennessee—Memphis).
Plasmids encoding green fluorescent protein (GFP)-tagged wild-type VPS4A and the ATP hydrolysis-deficient VPS4A E228Q mutant (VPS4Awt and VPS4AEQ) were kindly provided by Wes Sundquist (University of Utah).
Cell lines.
BHK-21 cells were cultured at 37°C in Dulbecco's modified Eagle's medium containing 5% fetal calf serum, 10% tryptose phosphate broth, 100 U penicillin/ml, and 100 μg streptomycin/ml (44).
The parental T-REx HEK293 cells (Invitrogen) express the Tet repressor and were cultured at 37°C in Dulbecco's modified Eagle's medium containing 10% tetracycline-free fetal calf serum (HyClone), 100 U penicillin/ml, 100 μg streptomycin/ml, and 5 μg blasticidin/ml. Tetracycline-inducible stable cell lines expressing VPS4B were generated previously from T-REx HEK293 cells as described elsewhere (10, 38). These clonal cell lines inducibly express GFP-tagged forms of wild-type VPS4B (SKD1wt-GFP, referred to here as VPS4Bwt) or the ATP hydrolysis-deficient VPS4B E235Q mutant (SKD1EQ-GFP, referred to here as VPS4BEQ). The inducible cell lines were cultured at 37°C in T-REx HEK293 medium with the addition of 100 μg Zeocin/ml to maintain the plasmid.
Tetracycline-inducible stable cell lines expressing wild-type or mutant VPS4A were generated as follows. GFP-tagged VPS4Awt or VPS4AEQ sequences were amplified from the plasmids described above and inserted into pcDNA4/TO (Invitrogen) using the BamHI and XbaI sites. These constructs were then transfected into T-REx HEK293 cells by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Stable cell lines were selected using T-REx HEK293 medium containing 125 μg/ml Zeocin. Isolated Zeocin-resistant colonies were picked, amplified, induced with 1 μg/ml tetracycline, and screened for GFP expression by fluorescence microscopy and flow cytometry. Clonal cell lines were selected for use based on efficient and uniform GFP expression upon induction and absence of expression in the absence of induction. Selected cell lines were maintained in T-REx HEK293 medium containing 100 μg/ml Zeocin.
MG-132 treatment.
BHK-21 cells were infected with SFV or VSV for 1 h at 37°C using a multiplicity of 10 PFU/cell in medium A, pH 7.0 (minimal essential medium containing 100 U penicillin/ml and 100 μg streptomycin/ml, 0.2% bovine serum albumin, and 10 mM HEPES). Cells were washed to remove input virus and incubated in medium A for 2 h. MG-132 (Calbiochem) was then added from a stock prepared in dimethyl sulfoxide (DMSO), using final concentrations of 50 μM MG-132 and 0.1% DMSO. The culture medium was collected 5 h after addition of MG-132 (7 h postinfection), and the viral titer was determined by a plaque assay on BHK-21 cells (30).
Cell surface budding assay.
The cell surface budding assay was carried out as described previously (40), with minor modifications. Briefly, BHK-21 cells were infected with SFV or VSV at 10 PFU/cell for 1 h at 37°C in medium A, pH 7.0. Four hours postinfection, the cells were starved for 15 min by incubation in modified Eagle's medium lacking cysteine and methionine, pulse-labeled for 15 min with 100 μCi [35S]methionine-cysteine/ml (Pro-Mix Cell Labeling Mix; GE-Healthcare), and chased in medium supplemented with a 10-fold excess of cysteine and methionine for 45 min. Proteins at the cell surface were then biotin tagged by incubation at 4°C for 15 min with 0.5 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Pierce Chemicals) in phosphate-buffered saline containing 1 mM glucose. Reactive biotin was quenched by washing with cold RPMI medium containing 10 mM glycine (pH 8.0). The biotinylated cells were then transferred to after-chase medium (medium A, pH 8.0, containing NaCl instead of sodium bicarbonate) and incubated in a 37°C water bath for 1 h to allow virus budding. Cells were treated with 50 μM MG-132 during the pulse, chase, biotinylation, and budding steps. After the 1-h budding incubation, the medium was collected and the cells were solubilized in lysis buffer containing protease inhibitors and 1% Triton X-100. Biotin-tagged virus particles and lysate proteins were retrieved using BioMag streptavidin particles (Polysciences, Inc., Warrington, PA) as described previously (40). Samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% acrylamide gels.
Induction and infection of inducible VPS4B- and VPS4A-expressing cells.
VPS4Bwt, VPS4Awt, VPS4BEQ, and VPS4AEQ cells were induced with 1 μg tetracycline/ml for 1 h and then infected for 1 h at 37°C with SFV or VSV at 10 PFU/cell in medium A, pH 7.0. The cells were then washed three times to remove input virus and incubated for an additional 6 h at 37°C. The virus in the culture medium was quantitated by a plaque assay on BHK-21 cells.
Flow cytometry analyses.
To determine the timing of shutoff of host protein synthesis by SFV and VSV, VPS4BEQ or VPS4AEQ cells were induced for 6 h and infected at the indicated times with SFV or VSV at 10 PFU/cell as described above. At the end of the 6-h induction, the cells were removed from the plates by treatment with Accutase (Sigma), washed with phosphate-buffered saline, and fixed with 3% paraformaldehyde. The level of VPS4BEQ-GFP or VPS4AEQ-GFP expression in each cell population was determined by flow cytometry using a FACScan in the Fluorescence-Activated Cell Sorting Facility of the Albert Einstein College of Medicine.
To quantitate virus infection, VPS4BEQ or VPS4AEQ cells were induced at the indicated times, infected with SFV or VSV at 10 PFU/cell, and incubated overnight at 28°C in complete culture medium containing 15 mM NH4Cl to prevent secondary infection. Cells were removed from the plate using Accutase and fixed in 3% paraformaldehyde. SFV-infected cells were stained with a rabbit polyclonal serum against the SFV E1 and E2 glycoproteins (29), followed by a goat anti-rabbit antibody conjugated to phycoerythrin (PE)-Cy5.5 (Caltag Laboratories), both at dilutions of 1:300. VSV-infected cells were stained with the I1 mouse monoclonal antibody against VSV G protein (36), followed by a 1:300 dilution of goat anti-mouse antibody conjugated to PE-Cy5.5 (Caltag Laboratories). The cells were subsequently analyzed by flow cytometry for both GFP (VPS4BEQ or VPS4AEQ expression) and PE-Cy5.5 (glycoprotein expression).
Electron microscopy.
VPS4BEQ and VPS4AEQ cells were induced for 1 h as indicated and infected as described above with SFV or VSV at 10 PFU/cell. At 6 h postinfection, cells were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 40 min at room temperature, washed in 0.1 M cacodylate buffer, osmicated, dehydrated, and embedded in LX112 (Ladd Research, Burlington, VT). Thin sections were stained with uranyl acetate and lead citrate and were examined under a 1200 EX electron microscope (JEOL, Peabody, MA) in the Analytical Imaging Facility of the Albert Einstein College of Medicine.
RESULTS
Alphavirus budding is not ubiquitin dependent.
A role for ubiquitination in virus budding can be assessed by using proteasome inhibitors such as MG-132. MG-132 acts by decreasing the cellular level of free ubiquitin and thus inhibits the budding of ubiquitin-dependent viruses, including rhabdoviruses such as VSV (4, 20). To test for a role of ubiquitination in alphavirus budding, BHK-21 cells were infected with SFV for 1 h, cultured for 2 h, and then incubated for 5 h in the presence of 50 μM MG-132. Cells were similarly infected with VSV as a positive control. In keeping with previous results (20), under these conditions the titer of VSV was reduced from ∼109 PFU/ml in the absence of MG-132 to 106 PFU/ml in the presence of MG-132, a decrease of ∼300-fold (Table 1). In contrast, production of SFV was not affected by MG-132 treatment, with titers of ∼109 PFU/ml produced in both control and MG-132-treated cells. These results suggest that SFV budding does not involve ubiquitination.
TABLE 1.
Effect of MG-132 on virus production
| Treatmenta | Virus titerb (fold decreasec)
|
|
|---|---|---|
| VSV | SFV | |
| None | 1.6 × 109 | 5.2 × 109 |
| DMSO | 1.0 × 109 | 3.4 × 109 |
| MG-132 | 3.4 × 106 (290) | 2.8 × 109 (1.2) |
BHK-21 cells were infected with VSV or SFV at 10 PFU/cell for 1 h, washed, cultured for an additional 2 h, and then incubated in the presence of the carrier DMSO or 50 μM MG-132 as indicated.
In PFU per milliliter. Media from infected cells were collected 5 h after addition of MG-132. Virus titers were determined by a plaque assay. The average titer from duplicate plaque assays from one experiment is shown.
Compared to the value for the DMSO control.
We took advantage of our previously described cell surface budding assay (40) to confirm that the inhibition of VSV production by MG-132 occurred through effects on virus release rather than by nonspecific inhibition of envelope protein expression or transport. The budding assay is based on tagging radiolabeled cell surface envelope proteins with biotin and then following their incorporation into virus particles that are retrieved from the medium by using streptavidin-conjugated magnetic beads (Mag-SA) in the absence of detergent. In this way, biotin-tagged VSV envelope protein G or SFV envelope proteins E2 and E1 on the virus surface retrieve the internal viral proteins VSV N/P and M or SFV capsid (39, 40). VSV- or SFV-infected cells were treated with MG-132 during the radiolabeling, chase, and budding steps. Mag-SA retrieval and SDS-PAGE analysis of lysates prepared from control or MG-132-treated cells showed that the levels of biotin-tagged VSV G protein were similar, demonstrating comparable biosynthesis and delivery to the cell surface (Fig. 1A). As expected, VSV particles containing G, N/P, and M proteins were efficiently retrieved from the medium of control cells, and retrieval was dependent on G protein biotinylation and on incubation of the cells at 37°C (Fig. 1B). The release of biotin-tagged VSV particles was strongly decreased in MG-132-treated cells (Fig. 1B, third versus fourth lane). Phosphorimager quantitation of the results of two experiments showed that retrieval of M protein from the medium of MG-132-treated cells was ∼7% that of control cells treated with the carrier (DMSO) alone. The SFV E1 and E2 envelope proteins were also expressed at comparable levels on the surfaces of control and MG-132-treated cells (Fig. 1C). In contrast to VSV, however, SFV budding in the presence of MG-132 was comparable to that of control cells (Fig. 1D, fourth versus fifth lane). Quantitation of two experiments showed that capsid retrieval from the medium of MG-132-treated cells was 83% that of control cells. Thus, the results of the cell surface budding assay correlate well with those of assays of infectious virus production. Together, these results confirm that VSV budding is dependent on ubiquitin and demonstrate that SFV budding is ubiquitin independent.
FIG. 1.
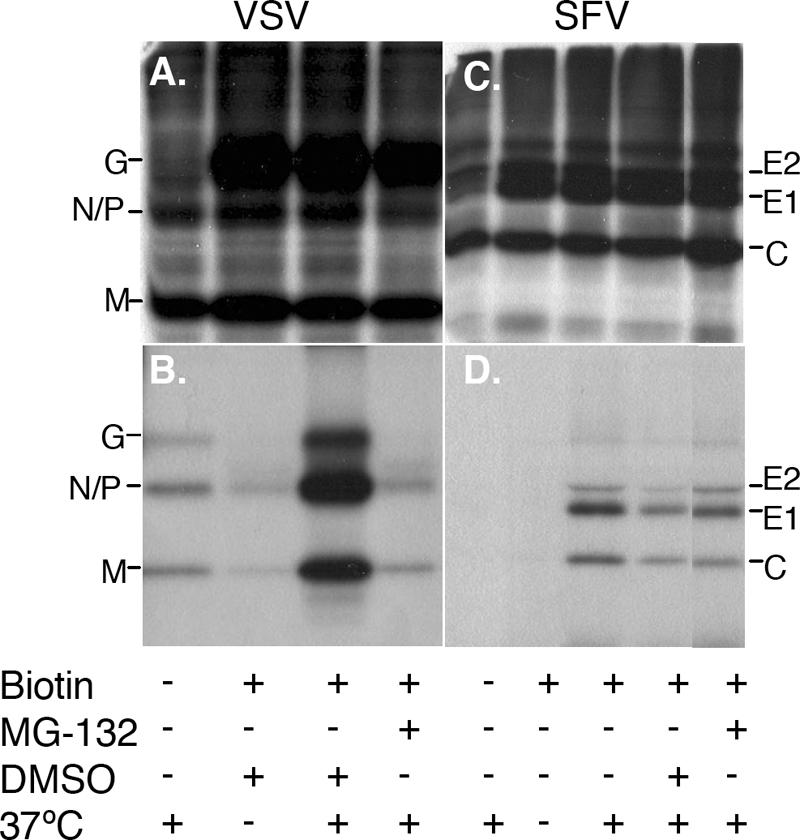
Effects of MG-132 treatment on cell surface budding of VSV and SFV. BHK-21 cells were infected for 1 h with VSV (A and B) or SFV (C and D) at a multiplicity of 10 PFU/cell, incubated for 4 h, and pulse-labeled with [35S]methionine-cysteine. The cells were chased in the absence of label, derivatized with biotin, and incubated for 1 h at 37°C to permit budding of biotin-tagged virus particles. As indicated, beginning with the pulse-labeling step, the incubations were carried out in the presence of 50 μM MG-132 or the carrier DMSO. At the end of the 1-h budding period, the biotin-tagged proteins in the cell lysates (A and C) and media (B and D) were retrieved with Mag-SA and analyzed by SDS-PAGE. Controls include 37°C incubation without biotinylation and biotinylation in the absence of 37°C incubation. Equivalent exposures of the gels are shown.
Establishing conditions for virus infection and expression of VPS4EQ mutants.
VPS4 is an AAA+ ATPase that is involved in the final disassembly and recycling of the ESCRT components (2, 3). To date, functional VSP4 is required by all viruses whose budding is dependent on the MVB machinery (4, 41), in keeping with the essential role of VPS4 late in the MVB pathway. Since not all viruses that use the MVB machinery are dependent on ubiquitination, it was important to determine the role of VPS4 in alphavirus budding. Although VSV budding is clearly dependent on ubiquitin and the viral late domain (20, 26), it has been reported to be independent of functional VPS4A (6, 25). Given this result, we also wanted to compare the roles of the two VPS4 isoforms in VSV budding.
Most tests of the role of VPS4 in virus budding have relied on transfection of cells with an expression vector encoding an ATP hydrolysis-deficient VPS4 mutant, VPS4EQ. In this system, typically cells are either cotransfected with a virus DNA clone, as in the human immunodeficiency virus type 1 studies (17), or infected with a virus, as in studies of paramyxovirus budding (46). We initially tested the transfection of VPS4AEQ-expressing plasmids as a system with which to evaluate the role of the MVB machinery. However, since both SFV and VSV will replicate to high levels in any untransfected cells, it appeared that more uniform and efficient expression of VPS4EQ would be needed for definitive results. We therefore turned to two previously developed 293 cell lines, referred to here as VPS4Bwt and VPS4BEQ, which inducibly express GFP-tagged forms of wild-type VPS4B or a VPS4BEQ mutant (see reference 38 and Materials and Methods for details). In agreement with previous reports (38), 4 h after tetracycline induction, the VPS4Bwt protein was observed in the cytoplasm while the VPS4BEQ protein was localized to typical enlarged endosome compartments (data not shown). Based on our preliminary results, and to allow comparison of the roles of the VPS4 isoforms, we also generated 293 cell lines that inducibly express GFP-tagged forms of VPS4Awt and VPS4AEQ. Tetracycline induction for 4 h gave expression patterns forVPS4Awt and VPS4AEQ similar to those for VPS4Bwt and VPS4BEQ (data not shown). We specify the VPS4 isoform used in each experiment below and use VPS4EQ as a general term to refer to both proteins in the Discussion.
Two important factors had to be considered in using these inducible cell lines for SFV and VSV studies. First, during replication each virus efficiently shuts off host protein synthesis and would thus inhibit the production of the VPS4 protein. Second, expression of VPS4BEQ or VPS4AEQ ultimately blocks late steps in endosomal traffic, which might inhibit the endocytic entry pathway used by these two viruses. Therefore, the timing of virus infection and tetracycline induction must be optimized to permit both inhibitory levels of VPS4BEQ/VPS4AEQ expression and efficient virus infection. To evaluate the effect of virus shutoff of cellular protein synthesis, we quantitated VPS4BEQ-GFP expression by flow cytometry. Initial time course studies showed that ∼60% of the cells were gated as positive at 6 h postinduction, 78% at 8 h, 83% at 10 h, 92% at 12 h, and 94% at 17 h (data not shown). Evaluation by fluorescence microscopy showed that essentially all of the cells expressed VPS4BEQ-GFP localized to enlarged endosomes by 6 h postinduction (data not shown). This is in keeping with the flow cytometry results, which showed that by 6 h postinduction, most of the cells were shifted from the negative-control (uninduced) position (Fig. 2). At later time points, VPS4BEQ-GFP saturates the endosome membrane and is also diffusely localized in the cytoplasm (reference 38 and data not shown), presumably leading to increased scoring of cells as positive at these flow cytometer settings. We picked 6 h as a sensitive induction time point to assay the effects of virus infection on VPS4BEQ expression. Cells were induced for 6 h beginning at various times prior to or during virus infection, and the level of GFP-tagged VPS4BEQ was determined (Fig. 2). In the absence of virus infection, after 6 h of induction ∼60% of the cells were scored positive for VPS4BEQ-GFP by flow cytometry (Fig. 2). When the VPS4BEQ cells were infected with virus 1 h after the beginning of induction, ∼50% of the SFV- or VSV-infected cells registered as GFP positive (Fig. 2A). In contrast, when cells were simultaneously infected with virus and induced for VPS4BEQ expression, after 6 h the level of expression was ∼25% in SFV-infected cells and ∼35% in VSV-infected cells (Fig. 2B). When the VPS4BEQ cells were infected with virus for 1 h before tetracycline addition, only ∼16% of SFV- or ∼27% of VSV-infected cells scored positive for VPS4BEQ (Fig. 2C). Thus, little effect on VPS4BEQ synthesis was observed when induction was carried out 1 h prior to virus infection, but inhibition of expression became more marked as virus infection was established. Importantly, the kinetics of host protein shutoff were comparable for SFV and VSV, enabling comparison of the results for these two viruses. Similar results were obtained for VPS4AEQ-expressing cells. In the absence of virus infection, 70 to 75% of the cells were positive for VPS4AEQ-GFP after 6 h of induction, and expression remained at ∼70% when cells were infected with SFV or VSV 1 h postinduction (data not shown). Expression was reduced to 52% (SFV) or 61% (VSV) when cells were simultaneously infected and induced, and expression was further reduced to 30% (SFV) and 54% (VSV) when VPS4AEQ was induced 1 h postinfection.
FIG. 2.

Timing of inhibition of host protein synthesis in VSV- and SFV-infected cells. VPS4BEQ cells were induced with tetracycline 1 h prior to (A), during (B), or 1 h following (C) infection with 10 PFU/cell VSV (blue) or SFV (red). An aliquot of the same induced cells that were mock infected is shown in green as a control. The expression of VPS4BEQ-GFP was monitored in each cell population by flow cytometry. For each panel, the total time of tetracycline induction was 6 h; thus, the duration of virus infection ranged from 5 h (A) to 7 h (C). The background fluorescence of uninduced VPS4BEQ cells is shown on each panel as a filled trace.
We then evaluated the effect of VPS4BEQ expression on virus infection to test for possible inhibition of endocytic uptake or other early events in virus replication. Expression was induced for 8 h, 4 h, or 1 h prior to addition of SFV or VSV at multiplicities that infect ∼20 to 30% of the cells. The cells were incubated overnight at 28°C in the presence of NH4Cl to prevent secondary infection and were analyzed by flow cytometry using antibodies to the SFV envelope proteins or the VSV G protein. The percentage of virus-infected cells was compared to that of uninduced control cells (Table 2). Some reduction in virus infection was observed when the cells were induced 1 h prior to infection: VSV infection was reduced ∼1.4-fold, and SFV infection was reduced ∼1.9-fold. Induction for 4 or 8 h produced more significant inhibition of infection, and in each case the inhibition of SFV infection was somewhat greater than that of VSV. Similar results were obtained if the cells were permeabilized prior to antibody staining. Tests of VPS4AEQ expression showed comparable reductions in virus infection: induction 1 h prior to infection decreased VSV infection 1.3-fold and SFV infection 1.4-fold, while induction 4 h prior to infection decreased VSV infection 2.1-fold and SFV infection 2.2-fold. Taken together, these two approaches indicate that the optimal time of tetracycline induction for our system is 1 h prior to virus infection, a time point that maximizes VPS4BEQ and VPS4AEQ expression and minimizes effects on virus entry and infection.
TABLE 2.
Effect of VPS4BEQ expression on virus infection
| Time of inductiona | % of cells infectedb (fold decrease)c
|
|
|---|---|---|
| VSV | SFV | |
| No induction | 19 ± 2 | 36 ± 5 |
| 1 h prior | 14 ± 0.6 (1.4) | 19 ± 3 (1.9) |
| 4 h prior | 11 ± 2 (1.7) | 15 ± 3 (2.4) |
| 8 h prior | 8 ± 0.6 (2.4) | 9 ± 2 (4.0) |
VPS4BEQ cells were induced with 1 μg/ml tetracycline at the indicated times prior to infection with VSV or SFV at 10 PFU/cell. The cells were then cultured overnight at 28°C in the presence of NH4Cl to prevent secondary infection.
Infected cells were stained with antibodies against the viral glycoproteins and detected by flow cytometry. Data are averages ± standard deviations from three independent experiments.
Compared to the value for the uninduced control.
Effects of VPS4BEQ/VPS4AEQ expression on VSV and SFV budding.
Using the optimized conditions, we then tested the effects of expression of VPS4Bwt and VPS4BEQ on VSV and SFV budding. We also took advantage of a VSV mutant with an inactivated late domain (PPPY to AAPA). This late-domain mutant shows an overall decrease in virus budding, but the budding that is observed is insensitive to MG-132 (20). Expression of VPS4Bwt or VPS4BEQ was induced for 1 h, and the cells were then infected with either VSV, VSV-AAPA, or SFV. Following a 1-h infection period, the cells were washed to remove input virus. Medium was collected for determination of viral titers either after the wash (t = 0) or after an additional 6-h incubation (t = 6) (Table 3). VSV grew efficiently in the parental T-REx cells and the uninduced VPS4BEQ cells, showing an increase in titer of ∼6 log units during the 6-h incubation. Induction of VPS4BEQ expression decreased virus production about 30-fold, while induction of the wild-type protein showed no effect. As expected, production of the VSV-AAPA mutant in uninduced cells was about 2 log units lower than that of wild-type VSV but was unaffected by VPS4BEQ expression. Thus, a functional late domain was required for the VPS4B dependence of VSV budding. Similarly, SFV was efficiently produced in the parental and uninduced cells, with an increase in titer of ∼6 log units during the 6-h incubation. However, SFV production was not significantly inhibited by either VPS4BEQ or VPS4Bwt expression, with only a 1.4-fold decrease in titer in cells expressing VPS4BEQ. This small reduction in SFV production is consistent with the slight decrease in virus infection observed in VPS4BEQ-expressing cells after a 1-h induction (Table 2).
TABLE 3.
Effects of VPS4BEQ expression on virus budding
| Cell line | Inductiona/time of infectionb | Titerc (fold decreased)
|
||
|---|---|---|---|---|
| VSV | VSV-AAPA | SFV | ||
| T-REx | Unind/t = 6 | 7.6 × 108 | 9.8 × 105 | 1.4 × 109 |
| VPS4BEQ | Unind/t = 0 | 4.0 × 102 | 5.4 × 102 | 1.2 × 103 |
| Unind/t = 6 | 9.6 × 108 | 2.2 × 106 | 1.2 × 109 | |
| Induced/t = 6 | 2.4 × 107 (28) | 1.7 × 106 (1.9) | 1.1 × 109 (1.4) | |
| VPS4Bwt | Unind/t = 6 | 4.4 × 108 | 1.5 × 106 | 1.8 × 108 |
| Induced/t = 6 | 3.8 × 108 (1.2) | 1.3 × 106 (1) | 1.7 × 108 (1.4) | |
VPS4BEQ or VPS4Bwt cells were either uninduced (Unind) or induced with 1 μg/ml tetracycline for 1 h prior to infection with VSV or SFV at 10 PFU/cell.
Following an initial 1-h infection period, the cells were washed to remove input virus, and incubation was continued for 0 h or 6 h (t = 0 or t = 6).
In PFU per milliliter. Medium was collected at the indicated times, and virus titers were determined by a plaque assay. The t = 0 time point represents input virus not removed by washing. Titers shown are data from one of three independent experiments.
Compared to the titer for the uninduced control of each set. Values are averages from three independent experiments.
We then tested the effects of VPS4Awt and VPS4AEQ expression on SFV, VSV, and VSV-AAPA budding (Table 4). The results were similar to those for the VPS4B isoform: VSV grew efficiently in uninduced VPS4AEQ cells, and virus production was inhibited (∼18-fold) by the VPS4AEQ mutant but not by VPS4Awt. Inhibition by VSP4AEQ required the functional VSV late domain. SFV production was not significantly inhibited by expression of either VPS4AEQ or VPS4Awt.
TABLE 4.
Effect of VPS4AEQ expression on virus budding
| Cell line | Inductiona/time of infectionb | Titerc (fold decreased)
|
||
|---|---|---|---|---|
| VSV | VSV-AAPA | SFV | ||
| T-REx | Unind/t = 6 | 8.4 × 108 | 1.8 × 106 | 8.2 × 108 |
| VPS4AEQ | Unind/t = 0 | 3.8 × 102 | 4.4 × 102 | 3.2 × 103 |
| Unind/t = 6 | 9.2 × 108 | 2.2 × 106 | 3.8 × 108 | |
| Induced/t = 6 | 5.4 × 107 (18) | 2.6 × 106 (1.0) | 9.2 × 108 (2.8) | |
| VPS4Awt | Unind/t = 6 | 2.5 × 109 | 5.8 × 106 | 3.6 × 109 |
| Induced/t = 6 | 8.2 × 109 (1.0) | 3.4 × 106 (1.3) | 2.0 × 109 (2.3) | |
VPS4AEQ or VPS4Awt cells were either uninduced (Unind) or induced with 1 μg/ml tetracycline for 1 h prior to infection with VSV or SFV at 10 PFU/cell.
Following an initial 1-h infection period, the cells were washed to remove input virus, and incubation was continued for 0 h or 6 h (t = 0 or t = 6).
In PFU per milliliter. Medium was collected at the indicated times, and virus titers were determined by a plaque assay. The t = 0 time point represents input virus not removed by washing. Titers shown are data from one of three independent experiments.
Compared to the titer for the uninduced control of each set. Values are averages from three independent experiments.
We used electron microscopy to assess the effects of VPS4BEQ/VPS4AEQ expression on the budding phenotypes of VSV and SFV (Fig. 3). As in the experiments for which results are shown in Tables 3 and 4, expression was induced 1 h prior to infection, and infection was continued for 6 h before fixation and processing for electron microscopy. Uninduced cells showed typical bullet-shaped VSV particles budding at the plasma membrane or already released from the cell surface (Fig. 3A and C). In contrast, cells expressing either VPS4BEQ or VPS4AEQ showed marked increases in the numbers of VSV particles lined up along the plasma membrane (Fig. 3B and D). Most particles were clearly still connected to the cell, and cellular processes containing multiple incompletely budded virus particles were apparent in many cells (Fig. 3B). SFV budding was observed at the plasma membranes of both uninduced and induced cells (shown for VPS4BEQ in Fig. 3E and F), but in contrast to VSV, no difference in the frequency of SFV particles at the cell surface or the morphology of SFV budding was detected for induced cells. SFV budding in VPS4AEQ-expressing cells was similar to that shown for VPS4BEQ-expressing cells (data not shown). Taken together, our data thus demonstrate that VSV budding is inhibited by either VPS4BEQ or VPS4AEQ in a late-domain-dependent fashion, while SFV budding is independent of functional VPS4B and VPS4A.
FIG. 3.

Electron microscopic evaluation of VSV and SFV budding in cells expressing VPS4BEQ or VPS4AEQ. (A, B, E, and F) VPS4BEQ cells; (C and D) VPS4AEQ cells. Cells were either not induced (A, C, and E) or induced 1 h prior to virus infection (B, D, and F). Cells were then infected with VSV (A to D) or SFV (E and F) at a multiplicity of 10 PFU/cell for 1 h. Infected cells were incubated for an additional 6 h and then fixed and processed for electron microscopy. In panels A to D, arrows indicate examples of tangential sections of VSV particles, and arrowheads indicate examples of cross sections. Asterisk in panel B indicates an example of a cellular process from which multiple VSV particles are budding. Arrows in panels E and F indicate examples of SFV particles. Bars, 200 nm.
DISCUSSION
Use of VPS4BEQ and VPS4AEQ cells in virus budding assays.
Our assays of alphavirus and rhabdovirus budding relied on infection of inducible VPS4BEQ and VPS4AEQ cells with SFV or VSV via the normal endocytic route. This is in contrast to experiments on the role of the MVB machinery in, for example, retrovirus budding, which generally take advantage of cotransfection of cells with a plasmid encoding ATPase-deficient VPS4 plus either provirus DNA or a plasmid encoding the Gag protein (4, 41). Since most transfected cells will receive both plasmids, the presence of VPS4EQ-negative cells is not a concern. Cotransfection experiments are not possible for SFV infection, and cotransfection of the VSV M protein may not be an optimal method to study VSV budding, as discussed more fully below. Thus, the VPS4BEQ and VPS4AEQ cells were critical reagents in characterizing the role of VPS4 in the budding reactions of these viruses. We anticipate that the VPS4BEQ and VPS4AEQ cells will be very useful tools for testing a variety of other viruses that require direct infection. However, as addressed in our experiments, the effects of VPS4EQ expression on endocytic virus entry and the inhibition of VPS4EQ expression by virus infection can be important variables. As shown in Table 2, decreased virus infection was observed with increasing times of VPS4EQ induction. This inhibition was relatively small when the cells were induced 1 h prior to infection but became more significant 8 h after induction. We believe that this effect is due primarily to a decrease in virus endocytic uptake, since it could be largely bypassed by direct low-pH-triggered fusion of SFV with the plasma membranes of the induced cells. Under these conditions, induction of VPS4BEQ expression for as long as 6 or 12 h prior to fusion resulted in only a 1.2- or 1.7-fold decrease in virus infection (data not shown).
For viruses that inhibit cellular protein expression, the timing of virus infection versus induction must also be considered. In this regard, it was very useful to compare VSV and SFV budding in parallel. The kinetics of virus growth and host protein shutoff were comparable for VSV and SFV, but only VSV budding was inhibited by VPS4EQ. By optimizing the timing, we were able to achieve inhibitory levels of VPS4EQ expression before significant host protein shutoff from virus infection.
Role of the MVB pathway in alphavirus budding.
Several key features of the alphaviruses may explain their ubiquitin- and VPS4-independent budding. Alphaviruses do not have a membrane-associated protein that interacts with the internal face of the virus membrane, and there are no currently defined late domains present in the cytoplasmic regions of alphavirus structural proteins (G. M. Taylor, unpublished data). While a number of viruses that use the MVB machinery can bud in the absence of viral TM proteins, alphavirus budding requires both the E1 and E2 TM proteins and the nucleocapsid core. The MVB-dependent viruses do not have a highly symmetrical organization. In contrast, the membrane proteins of the alphaviruses are organized with T=4 icosahedral symmetry, and the extensive lateral interactions in the glycoprotein shell may play a dominant role in driving membrane curvature during budding. Interestingly, recent studies indicate that influenza virus budding requires the viral envelope proteins rather than the matrix protein M1 and is also independent of functional VPS4 (6). Together these data suggest that virus budding can be based on either an internal protein coat similar to the M proteins or an external protein coat such as the alphavirus E1 and E2 proteins. This is in keeping with a recent comparison of extravesicular coats such as clathrin with intravesicular coats such as the M protein (31). Viruses dependent on the MVB machinery for budding may use an internal protein coat both to directly mediate membrane curvature and as a platform to recruit ESCRT components.
Membrane proteins of flaviviruses such as dengue virus and West Nile virus also form a highly organized icosahedral lattice (33, 42). In contrast to the alphaviruses, budding can be driven by the flavivirus TM proteins without the nucleocapsid core (1). While the resultant T=1 flavivirus subviral particles are smaller than the T=3 flavivirus particle (12), this example does illustrate that an external protein coat can play a significant role in inducing membrane curvature. It will be interesting to determine if viruses such as flaviviruses with highly organized external protein coats are also independent of the MVB machinery.
Role of the MVB pathway in VSV budding.
The internal membrane-associated M protein of VSV includes a PPPY late-domain motif required for efficient virus budding (8, 26). The PPPY sequence interacts with the WW domain of a Nedd4-like ubiquitin ligase and confers ubiquitin dependence on the VSV budding reaction (20). Previous results indicated that VSV budding is not inhibited by transfection of cells with a vector expressing VPS4AEQ (25). These results thus suggested that VSV's requirement for ubiquitin might be separate from involvement of the ESCRT pathway. Here we demonstrated, using both plaque assays and electron microscopy, that VSV budding was inhibited by expression of either VPS4AEQ or VPS4BEQ. As with the ubiquitin dependence of VSV, the PPPY late domain was required for VPS4EQ inhibition. EQ mutant versions of both VPS4 isoforms inhibited VSV budding, in keeping with their ∼80% sequence identity and comparable formation of swollen endosomes.
What might be responsible for the differences between our data and those of prior experiments? One approach in the previous study (25) was based on transfection of cells with VPS4AEQ and infection with VSV 24 h later. Depending on the transfection efficiency, this approach may not be sensitive enough to detect inhibition of a robustly replicating virus such as VSV (G. M. Taylor, data not shown). A second approach was based on cotransfection of plasmids encoding VSV M protein and VPS4AEQ, similar to the cotransfection studies that defined the VPS4 dependence of a number of viruses. It is possible that this approach for VSV did not show inhibition due to the vaccinia virus-driven expression of the M protein (for example, see reference 6) or that M protein alone does not completely recapitulate the VSV budding pathway (as was found for hepatitis B virus subviral particles [see reference 34]). Lastly, the prior study showed that the growth of recombinant rabies virus expressing either VPS4Awt or dominant-negative VPS4A was similar to that of the wild-type virus. With this experimental system, there is no independent evaluation of whether the level and/or kinetics of VPS4 expression is sufficient to inhibit VPS4-dependent budding. Thus, although we found the prior data compelling, consideration of current results in the field suggests a possible explanation for each experimental finding. Inducible VPS4BEQ/VPS4AEQ cells appear to provide a useful system with which to test the role of VPS4 in the budding reactions of viruses that are not amenable to cotransfection studies and/or that strongly inhibit host protein synthesis (see also reference 9). Since VPS4EQ expression is ultimately deleterious, rapid induction of VPS4EQ expression in stable cell lines may also be useful in preventing possible cellular compensatory mechanisms that could affect the experimental outcome.
Cellular proteins in the alphavirus budding pathway.
While our data indicate that alphavirus budding is not dependent on either early or late steps in the MVB pathway, the overall involvement of host proteins in budding remains an open question. The envelope proteins clearly are very important in providing the T=4 external lattice, which can even drive the organization of the T=4 nucleocapsid for capsid mutants that are defective in core formation (13, 14). However, host proteins could contribute to various stages of the alphavirus budding process. For example, the final fission/pinching off of the complete virus particle could be promoted by cellular factors, perhaps analogously to endocytic vesicle fission, in which dynamin plays an important role (7). A pool of capsid protein and/or nucleocapsids becomes localized to the plasma membrane, where budding takes place. This process might be mediated solely by passive diffusion, might involve interactions with the tail of the E2 protein, and/or might be promoted via cellular proteins. Host proteins might also play a role in the generation or maintenance of the high concentration of alphavirus membrane proteins at the plasma membrane during the assembly of the ordered virus particle. Given our limited knowledge of alphavirus budding, these and other questions involving potential roles for host proteins will be interesting areas for future investigation.
Acknowledgments
We thank Michael Whitt for providing the VSV-AAPA mutant, Douglas Lyles for providing the I1 monoclonal antibody to VSV G, and Wes Sundquist for providing the VPS4A plasmids. We thank William King of the Einstein FACS facility for advice on flow cytometry. Chantal Chanel-Vos initiated and contributed to experiments on the effect of MG-132 on SFV budding. We thank Alice Guo for excellent technical assistance and the members of the Kielian lab for helpful discussions and comments on the manuscript.
This work was supported by a grant to M.K. from the Public Health Service (R01 GM057454) and by Cancer Center Core Support grant NIH/NCI P30-CA13330. G.M.T. was supported in part by a training grant from the Public Health Service (T32 AI07506).
Footnotes
Published ahead of print on 3 October 2007.
REFERENCES
- 1.Allison, S. L., K. Stadler, C. W. Mandl, C. Kunz, and F. X. Heinz. 1995. Synthesis and secretion of recombinant tick-borne encephalitis virus protein E in soluble and particulate form. J. Virol. 69:5816-5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babst, M. 2005. A protein's final ESCRT. Traffic 6:2-9. [DOI] [PubMed] [Google Scholar]
- 3.Babst, M., T. K. Sato, L. M. Banta, and S. D. Emr. 1997. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO J. 16:1820-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieniasz, P. D. 2006. Late budding domains and host proteins in enveloped virus release. Virology 344:55-63. [DOI] [PubMed] [Google Scholar]
- 5.Bishop, N., and P. Woodman. 2000. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell 11:227-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, B. J., G. P. Leser, E. Morita, and R. A. Lamb. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 81:7111-7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conner, S. D., and S. L. Schmid. 2003. Regulated portals of entry into the cell. Nature 422:37-44. [DOI] [PubMed] [Google Scholar]
- 8.Craven, R. C., R. N. Harty, J. Paragas, P. Palese, and J. W. Wills. 1999. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol. 73:3359-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crump, C. M., C. Yates, and T. Minson. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 81:7380-7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalal, S., M. F. Rosser, D. M. Cyr, and P. I. Hanson. 2004. Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell 15:637-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demirov, D. G., and E. O. Freed. 2004. Retrovirus budding. Virus Res. 106:87-102. [DOI] [PubMed] [Google Scholar]
- 12.Ferlenghi, I., M. Clarke, T. Ruttan, S. L. Allison, J. Schalich, F. X. Heinz, S. C. Harrison, F. A. Rey, and S. D. Fuller. 2001. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol. Cell 7:593-602. [DOI] [PubMed] [Google Scholar]
- 13.Forsell, K., G. Griffiths, and H. Garoff. 1996. Preformed cytoplasmic nucleocapsids are not necessary for alphavirus budding. EMBO J. 15:6495-6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsell, K., L. Xing, T. Kozlovska, R. H. Cheng, and H. Garoff. 2000. Membrane proteins organize a symmetrical virus. EMBO J. 19:5081-5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garoff, H., R. Hewson, and D.-J. E. Opstelten. 1998. Virus maturation by budding. Microbiol. Mol. Biol. Rev. 62:1171-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garoff, H., M. Sjoberg, and R. H. Cheng. 2004. Budding of alphaviruses. Virus Res. 106:103-116. [DOI] [PubMed] [Google Scholar]
- 17.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55-65. [DOI] [PubMed] [Google Scholar]
- 18.Glomb-Reinmund, S., and M. Kielian. 1998. fus-1, a pH-shift mutant of Semliki Forest virus, acts by altering spike subunit interactions via a mutation in the E2 subunit. J. Virol. 72:4281-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gruenberg, J., and H. Stenmark. 2004. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 5:317-323. [DOI] [PubMed] [Google Scholar]
- 20.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hicke, L., and R. Dunn. 2003. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 19:141-172. [DOI] [PubMed] [Google Scholar]
- 22.Hislop, J. N., A. Marley, and M. Von Zastrow. 2004. Role of mammalian vacuolar protein-sorting proteins in endocytic trafficking of a non-ubiquitinated G protein-coupled receptor to lysosomes. J. Biol. Chem. 279:22522-22531. [DOI] [PubMed] [Google Scholar]
- 23.Hurley, J. H., and S. D. Emr. 2006. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 35:277-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irie, T., J. M. Licata, H. R. Jayakar, M. A. Whitt, P. Bell, and R. N. Harty. 2004. Functional analysis of late-budding domain activity associated with the PSAP motif within the vesicular stomatitis virus M protein. J. Virol. 78:7823-7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irie, T., J. M. Licata, J. P. McGettigan, M. J. Schnell, and R. N. Harty. 2004. Budding of PPxY-containing rhabdoviruses is not dependent on host proteins TSG101 and VPS4A. J. Virol. 78:2657-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jayakar, H. R., K. G. Murti, and M. A. Whitt. 2000. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J. Virol. 74:9818-9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kian Chua, P., M. H. Lin, and C. Shih. 2006. Potent inhibition of human hepatitis B virus replication by a host factor Vps4. Virology 354:1-6. [DOI] [PubMed] [Google Scholar]
- 28.Kielian, M. 2006. Class II virus membrane fusion proteins. Virology 344:38-47. [DOI] [PubMed] [Google Scholar]
- 29.Kielian, M., S. Jungerwirth, K. U. Sayad, and S. DeCandido. 1990. Biosynthesis, maturation, and acid activation of the Semliki Forest virus fusion protein. J. Virol. 64:4614-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kielian, M. C., S. Keränen, L. Kääriäinen, and A. Helenius. 1984. Membrane fusion mutants of Semliki Forest virus. J. Cell Biol. 98:139-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozlov, M. M., and L. V. Chernomordik. 2002. The protein coat in membrane fusion: lessons from fission. Traffic 3:256-267. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn, R. J., and M. G. Rossmann. 2005. Structure and assembly of icosahedral enveloped RNA viruses. Adv. Virus Res. 64:263-284. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn, R. J., W. Zhang, M. G. Rossman, S. V. Pletnev, J. Corver, E. Lenches, C. T. Jones, S. Mukhopadhyay, P. R. Chipman, E. G. Strauss, T. S. Baker, and J. H. Strauss. 2002. Structure of Dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert, C., T. Doring, and R. Prange. 2007. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and γ2-adaptin. J. Virol. 81:9050-9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee, S., K. E. Owen, H.-K. Choi, H. Lee, G. Lu, G. Wengler, D. T. Brown, M. G. Rossmann, and R. J. Kuhn. 1996. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 4:531-541. [DOI] [PubMed] [Google Scholar]
- 36.Lefrancois, L., and D. S. Lyles. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121:157-167. [PubMed] [Google Scholar]
- 37.Liljeström, P., S. Lusa, D. Huylebroeck, and H. Garoff. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 65:4107-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin, Y., L. A. Kimpler, T. V. Naismith, J. M. Lauer, and P. I. Hanson. 2005. Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7-1 with itself, membranes, and the AAA+ ATPase SKD1. J. Biol. Chem. 280:12799-12809. [DOI] [PubMed] [Google Scholar]
- 39.Lu, Y. E., C. H. Eng, S. G. Shome, and M. Kielian. 2001. In vivo generation and characterization of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 75:8329-8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu, Y. E., and M. Kielian. 2000. Semliki Forest virus budding: assay, mechanisms and cholesterol requirement. J. Virol. 74:7708-7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morita, E., and W. I. Sundquist. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20:395-425. [DOI] [PubMed] [Google Scholar]
- 42.Mukhopadhyay, S., R. J. Kuhn, and M. G. Rossmann. 2005. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3:13-22. [DOI] [PubMed] [Google Scholar]
- 43.Patnaik, A., V. Chau, F. Li, R. C. Montelaro, and J. W. Wills. 2002. Budding of equine infectious anemia virus is insensitive to proteasome inhibitors. J. Virol. 76:2641-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phalen, T., and M. Kielian. 1991. Cholesterol is required for infection by Semliki Forest virus. J. Cell Biol. 112:615-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlesinger, S., and M. J. Schlesinger. 2001. Togaviridae: the viruses and their replication, p. 895-916. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 46.Schmitt, A. P., G. P. Leser, E. Morita, W. I. Sundquist, and R. A. Lamb. 2005. Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J. Virol. 79:2988-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shehu-Xhilaga, M., S. Ablan, D. G. Demirov, C. Chen, R. C. Montelaro, and E. O. Freed. 2004. Late domain-dependent inhibition of equine infectious anemia virus budding. J. Virol. 78:724-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skoging, U., M. Vihinen, L. Nilsson, and P. Liljeström. 1996. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure 4:519-529. [DOI] [PubMed] [Google Scholar]
- 49.Yoshimori, T., F. Yamagata, A. Yamamoto, N. Mizushima, Y. Kabeya, A. Nara, I. Miwako, M. Ohashi, M. Ohsumi, and Y. Ohsumi. 2000. The mouse SKD1, a homologue of yeast Vps4p, is required for normal endosomal trafficking and morphology in mammalian cells. Mol. Biol. Cell 11:747-763. [DOI] [PMC free article] [PubMed] [Google Scholar]