

Abstract
E2F transcription factors play pivotal roles in controlling the expression of genes involved in cell viability as well as genes involved in cell death. E2F1 is an important constituent of this protein family, which thus far contains eight members. The interaction of E2F1 with its major regulator, retinoblastoma protein (Rb), has been studied extensively in the past two decades, concentrating on the role of E2F1 in transcriptional regulation and the role of Rb in cell replication and cancer formation. Additionally, the effect of viral infections on E2F1/Rb interactions has been analyzed for different viruses, concentrating on cell division, which is essential for viral replication. In the present study, we monitored E2F1-Rb interactions during human herpesvirus 6A (HHV-6A) and HHV-6B infections of SupT1 T cells. The results have shown the following dramatic alterations in E2F1-Rb pathways compared to the pathways of parallel mock-infected control cultures. (i) The E2F1 levels were elevated during viral infections. (ii) The cellular localization of E2F1 was dramatically altered, and it was found to accumulate both in the cytoplasmic and nuclear fractions, as opposed to the strict nuclear localization seen in the mock-infected cells. (iii) Although E2F1 expression was elevated, two exemplary target genes, cyclin E and MCM5, were not upregulated. (iv) The Rb protein was dephosphorylated early postinfection, a trait that also occurred with UV-inactivated virus. (v) Infection was associated with significant reduction of E2F1/Rb complexing. (vi) HHV-6 infections were accompanied by cell cycle arrest. The altered E2F1-Rb interactions and functions might contribute to the observed cell cycle arrest.
Human herpesvirus 6A (HHV-6A) and HHV-6B were initially isolated from peripheral blood mononuclear cells of immunologically deprived AIDS patients and patients with lymphoproliferative disorders (42, 58). They were recognized as distinct viruses by employing restriction enzyme analyses, antigenicity, and epidemiology (1, 60). Together with HHV-7, they constitute the group of roseoloviruses, a subgroup of the betaherpesvirus subfamily, possessing a colinear gene arrangement (54, 64). HHV-6A, HHV-6B, and HHV-7 associations with diseases have been reviewed recently (24, 37). Both HHV-6A and HHV-6B variants use CD46 as a receptor (59) to gain entry into various types of cells. They replicate productively in cultured CD4+ T cells but also are known to have central nervous system involvement, as reviewed previously (24, 37). HHV-6B infects the majority of children by the age of 2, causing either asymptomatic infections or roseola infantum, characterized by high fever and skin rash. The virus can be isolated from peripheral blood mononuclear cells of the sick children (78, 79). In more rare cases, HHV-6 and HHV-7 infections were reported to cause seizures, convulsions, and encephalopathy (6, 49, 66, 70, 77, 78, 80, 82, 84). Furthermore, HHV-6B strains were found to be activated from latency in bone marrow, kidney, liver, and other transplantations (9, 25, 41, 55, 65, 74, 81, 83-88). Of interest is our previous study (55), in which HHV-6B reactivation was prophylactically inhibited by ganciclovir treatment at the onset of transplantation. There is no acute disease currently known to be caused by HHV-6A. Viral isolates were implicated in the aggravation of symptoms of chronic fatigue syndrome and also in a subset of relapsing-remitting multiple sclerosis exacerbations. Recent studies have tested these associations (2-4, 14, 23, 34, 44, 56, 76).
In the present paper, we describe the interaction of HHV-6A and HHV-6B with the host cell, concentrating on changes in E2F1/Rb pathways in infected SupT1 T cells. There are eight known members of the E2F family, which function to activate or repress the transcription of different combinations of genes, promoting or inhibiting cell cycle progression (18). The E2F1 transcription factor, originally identified as the cellular protein capable of binding to the adenovirus E2 gene promoter (36), is an extensively studied member of the family (18, 48). The activation of the factor induces a rapid response to intracellular signaling pathways, resulting in gene expression instead of repression of selected target genes. The retinoblastoma protein (Rb) was documented to have growth suppression activity through its interaction with E2F1 (28, 39, 69). The ability of Rb to bind and inactivate E2F1 is an important downstream function of the protein, sequestering E2F1 from affecting cell proliferation through transcription of genes involved in the cell cycle as well as inducing apoptosis, either by p53-dependent transcription or by alternative pathways (11, 12, 18, 46, 69, 75). This interaction was first recognized by the binding of the adenovirus E1A protein to Rb, releasing E2F1 from the inhibitory complex, so as to start active transcription (7, 16, 27, 32, 62). Proteins encoded by different viruses were found to interact with Rb, including E7 of the human papillomavirus type 16, the large T antigen of simian virus 40, and the E1A protein of adenovirus 12. These oncoproteins, as well as other Rb-binding proteins encoded by plant and additional animal viruses, use a conserved LXCXE motif to interact with Rb (15, 19, 38). The activation of E2F1 by viral proteins may promote cell cycle arrest at the S phase (26), facilitating the replication of small viruses that depend heavily on cellular DNA replication machinery. A known mechanism that releases E2F1 from the Rb inhibitory complex functions by Rb phosphorylation, eliminating its ability to inhibit E2F1, as shown for human cytomegalovirus (HCMV) (31, 67). For herpes simplex virus type 1, the formation of E2F1-Rb complexes inhibited cell cycle progression at the G1 phase (21, 29). In this paper, we describe HHV-6A- and HHV-6B-induced alterations in E2F1-Rb interactions, including protein levels, localization, phosphorylation, and the expression of exemplary target genes.
MATERIALS AND METHODS
Cells and viruses.
The human T-cell line SupT1 was cultured in RPMI 1640 medium, 10% fetal calf serum (FCS). For the preparation of virus stocks, HHV-6A (U1102)- and HHV-6B (Z29)-infected cells were incubated with uninfected cells until they reached maximal cytopathic effect (CPE) (cell-to-cell passaging). Cell-free virus was concentrated from infected cell medium by centrifugation at 21,000 rpm for 2.25 h. The pellet was suspended in a small amount of medium, frozen, and used for infection. In the experiments described, the infections were routinely done using cell-free virus stocks of 1 × 106 to 3 × 106 50% tissue culture infectious doses to infect 4 × 106 cells. Following 2 h of virus adsorption in minimal volumes, the cells were incubated in growth medium at a concentration of 2 × 105 cells per ml to allow optimal culturing devoid of cell stress due to excessive cell accumulation per milliliter.
Transfection.
The SupT1 cells were electroporated using an MP-100 microporator (Digital Bio Technology) with the parameters 1,300 V, two pulses, and 20 ms according to the manufacturer's instructions.
Plasmids.
The E2F1-expressing plasmid was a gift of Yoel Kloog, Tel Aviv University, Israel.
Antibodies.
Rabbit polyclonal antibodies to E2F1 (C-20), Rb (C-15), pRb (ser-759), eIF2α (FL-315), MCM5 (H-300), and γ-tubulin (H-300) and mouse monoclonal antibody (MAb) to cyclin E (clone HE-12) were purchased from Santa Cruz Biotechnology. Mouse MAb 9A5, reactive to the HHV-6 P41 processivity factor encoded by the U27 gene, was a kind gift of N. Balachandran. Horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit 111-035-144 and goat anti-mouse 115-035-146) were purchased from Jackson ImmunoResearch Laboratories.
Immunofluorescence.
After a phosphate-buffered saline (PBS) rinse, concentrated cells were placed on glass slides coated with poly-l-lysine (1 mg/ml), fixed with 4% paraformaldehyde for 3 h, rinsed with PBS, and incubated with 0.7% Triton X-100 for 15 min. After being rinsed in PBS and blocked by incubation for 30 min in PBS, 20% FCS, the cells were incubated for 1 h at room temperature (RT) with the primary antibodies and rinsed three times with PBS at RT for 10 min. Secondary antibody (1:200 goat anti-mouse immunoglobulin G Cye2/5) was added for 30 min, and the cells were rinsed three times for 10 min in PBS. Cytox orange was used (1:100,000) to dye the nucleus, and the cells were rinsed three times in PBS. Galvanol mounting reagent (Calbiochem, LaJolla, CA) was added, and the solutions were covered with a coverslip. The cells were viewed by using an Axiovert 135 M confocal microscope (Carl Zeiss) equipped with an argon-krypton laser using a ×100 objective lens.
Immunoprecipitation.
The cells were lysed for 1 h at 4°C in 10 mM Tris, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40, and 1/20 protease inhibitor (Roche), followed by spinning at 14,000 rpm. Lysate supernatants were incubated with anti-E2F1 at 4°C overnight, and protein A-agarose beads (sc-2001; Santa Cruz Biotechnology Inc.) were added and shaken for 1 h at 4°C. The beads were rinsed twice with cold lysis buffer, resuspended in Laemmli reducing sample buffer (50 mM Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 50 mM dithiothreitol, 0.5% bromophenol blue), and boiled for 5 min. The immunoprecipitants were analyzed by SDS-polyacrylamide gel electrophoresis (10% polyacrylamide).
Western blotting.
Cells were lysed in whole-cell lysis buffer (50 mM Tris [pH 7.5], 280 mM NaCl, 0.5% NP-40, 0.2 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol [DTT], 10 mM NaF, 10 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride [PMSF] in isopropanol, 1/20 protease inhibitor cocktail) for 30 min at 4°C. Extracts were spun at 14,000 rpm, and samples were loaded onto SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Bio-Rad) for Western blotting. The blots were blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline (TBS) containing 0.02% Tween 20 (TTBS) for 1 h at RT and were incubated for 2 h at 4°C with primary antibody in TTBS containing 3% BSA and 0.05% sodium azide. After being rinsed with TTBS, they were incubated with secondary antibody, conjugated with horseradish peroxidase in TTBS containing 3% skim milk, rinsed with TTBS, and developed using an ECL detection kit (Pierce) according to the manufacturer's instructions. For the analyses of cytoplasmic and nuclear proteins, samples were first incubated on ice for 10 min in Cyto lysis buffer (10 mM HEPES, pH 7.5, 10 mM KCl, 3 mM MgCl2, 0.05% NP-40, 1 mM EDTA, 1 mM DTT, 10 mM NaF, 10 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM PMSF, 1/20 protease inhibitor cocktail). They were then centrifuged at 500 × g for 5 min at 4°C. The supernatant and cytoplasmic extracts were frozen at −80°C. The nuclear pellets were rinsed once in Cyto lysis buffer and lysed with nuclear lysis buffer (50 mM HEPES, pH 7.9, 250 mM KCl, 1% NP-40, 5% glycerol, 0.1 mM EDTA, 1 mM DTT, 10 mM NaF, 10 mM β-glycerophosphate, 0.1 mM sodium orthovandate, 1 mM PMSF, 1/20 protease inhibitor cocktail). The samples were frozen and thawed three times and were incubated on ice for 30 min. The insoluble material was pelleted at 14,000 rpm for 10 min at 4°C. The supernatant fluids with the nuclear extract were frozen at −80°C prior to Western blot analyses.
FACS analyses.
For fluorescent-activated cell sorter (FACS) analyses, each sample contained 1 × 106 to 2 × 106 cells, which were fixed in cold methanol and stored at −20°C. For nuclear labeling with antibody, the methanol-fixed cells were rinsed in FACS medium (cell medium plus 5% serum), incubated in 50 μl with primary antibody for 1 h at 4°C, and then rinsed three times with FACS medium and incubated with secondary antibody in a total volume of 50 μl at 4°C for 30 min. Cells were washed three times with FACS medium and resuspended in 500 μl PBS and 0.01% azide before FACS analyses.
RESULTS
E2F1 levels increase in HHV-6-infected cells.
Due to its pivotal role in cell cycle progression and in gene regulation, it was of interest to examine E2F1 regulation during viral infections. Earlier studies by microarray analyses revealed that the level of E2F1 mRNA increased during HHV-6 infections of T cells (45, 71). To verify and extend this information, cultures of SupT1 T cells were mock infected or were infected with HHV-6A (U1102) and HHV-6B (Z29). Protein extracts prepared 12, 24, 48, 72, and 96 h postinfection (hpi) were tested by Western blotting employing the E2F1, P41, and γ-tubulin antibodies (Fig. 1). The 9A5 MAb, produced by Balachandran and coworkers (8, 13), was chosen to monitor infection progression; it recognizes the HHV-6 41-kDa phosphoprotein (P41) encoded by the U27 gene, which functions as a DNA polymerase accessory protein. The protein is highly conserved in HHV-6A and HHV-6B strains (8, 13). As shown in Fig. 1A and quantified in Fig. 1B, the E2F1 levels increased in both HHV-6A (U102) and HHV-6B (Z29) infections, paralleling the accumulation of P41. To test whether the E2F1 increase took place in the infected cells or in neighboring uninfected cells, SupT1 cells were infected with HHV-6B (Z29) and tested at 24, 48, and 72 hpi for reactivity with the E2F1 and the 9A5 antibodies. The nuclei were stained with Cytox orange. The triple staining showed that the elevated levels of E2F1 coincided with the HHV-6 P41 staining, starting at 48 hpi (Fig. 2). Thus, the E2F1 levels increased specifically in the infected cells.
FIG. 1.

E2F1 expression during HHV-6A (U11O2) and HHV-6B (Z29) infections. (A) Western blot analyses of proteins from mock-infected and virus-infected cells, employing antibodies to E2F1, HHV-6, P41, and γ-tubulin. The antibody used for P41 is the MAb 9A5 of N. Balachandran (8, 13). (B) Quantitative representation of the results. Shown are E2F1 and P41 protein accumulation relative to the protein level at the highest point of accumulation of the HHV-6A samples, at 72 hpi. The P41 and E2F1 levels were corrected for loading variations by the intensity of the γ-tubulin staining.
FIG. 2.

E2F1 is increased in the infected cells. Cultures of 4 × 106 cells were infected with HHV-6B (Z29) and reacted with primary antibodies for E2F1 (rabbit), P41 (mouse), and Cytox orange for nuclear localization. Cye2 (anti-rabbit) and Cye5 (anti-mouse) secondary antibodies were used to mark E2F1 in green and P41 in purple. Each time frame contains a Cye5 field (P41), Cye2 field (E2F1), Cytox orange field, bright field, and a merging of fields 1 to 3.
E2F1 cytoplasmic and nuclear localization is altered in the infected cells.
The functions of the E2F transcription factors depend on their cellular localization, and alterations in protein localization were found to play a role in their transcriptional activity (5, 40). While some members of the E2F family can be found in the cytoplasmic as well as in the nuclear fractions, E2F1 is regularly confined to the nucleus. To examine whether E2F1 undergoes alterations in its localization, proteins were prepared from uninfected and HHV-6-infected cells using cytoplasmic and nuclear lysis. Since P41, which was used to monitor the infection, is a nuclear viral protein, it also could serve as a control for nuclear contaminations of the cytoplasmic fractions. The eIF2α and γ-tubulin proteins were used to equalize protein loading. The results (Fig. 3) revealed the following. (i) The P41 protein was recovered from the infected cells by 24 hpi, revealing the expected progress of the infections. Furthermore, the P41 protein was recovered solely from the nuclei, in accordance with its function in viral DNA replication, and it could be concluded that there was no nuclear contamination of the cytoplasmic fraction and no nuclear protein spillage due to nonspecific virally induced nuclear breakage. (ii) In the mock-infected cells, E2F1, which has a nuclear localization signal (43, 47, 75), was recovered mostly in the nuclear fraction, as expected. Strikingly, in the infected cells it was found in both cytoplasmic and nuclear fractions, starting at 72 hpi (Fig. 3). (iii) All differences in E2F1 nuclear and cytoplasmic localizations in the mock-infected and virus-infected cells, starting from 48 hpi, were statistically significant, as determined by t tests. The finding that the P41 protein was recovered solely in the nuclear fraction, whereas E2F1 was recovered in both nuclear and cytoplasmic fractions postinfection, attested to the specificity of the E2F1 cytoplasmic and nuclear recovery in the infected cultures.
FIG. 3.

Alterations in E2F1 cytoplasmic (cyto) and nuclear (nuc) localization. (A) Recovery of E2F1 and P41 proteins in the cytoplasmic or nuclear fractions. (B) Quantitative representation of the results shown relative to the highest level of E2F1 expression in the cytoplasmic and nuclear fractions. The E2F1-nuc analyses represent averages from four experiments, and the E2F1 cytoplasmic data represented the averages from two experiments. All differences were statistically significant by t tests.
The total amount of the Rb protein and its localization did not change during the infection, but the protein was dephosphorylated.
Following the finding that E2F1 was upregulated and could be recovered from the cytoplasmic fraction of infected cells, we tested for the presence of Rb, known to be a major regulator of E2F1. Both E2F1 and its regulator Rb are known to act in the nuclear fraction. Rb levels, Rb phosphorylation, and cellular localization were of interest, inasmuch as hypophosphorylated Rb was previously shown to bind E2F1, affecting its transcriptional activity (48). The cytoplasmic and nuclear lysates of 12 × 106 mock-infected, HHV-6A-infected, and HHV-6B-infected cells were tested by Western blotting, employing antibodies to Rb and phosphorylated Rb (pRb) (Fig. 4A). The data were corrected for loading variations by employing the eIF2α protein. The quantitative results can be summarized as follows. (i) The levels of Rb recovered from the cytoplasmic fractions were similar between the mock-infected, HHV-6A-infected, and HHV-6B-infected cells. Furthermore, pRb was not detected in the cytoplasmic fractions (data not shown). (ii) Similar levels of the Rb protein were detected in the nuclear fractions of the mock-infected and virus-infected cells (Fig. 4B). (iii) Most significantly, compared to the levels of pRb in mock-infected cells, the levels of pRb were greatly reduced in the nuclear fractions of the HHV-6A and HHV-6B infections starting at 24 hpi (Fig. 4C). (iv) Starting from 48 hpi, the differences in pRb levels between mock-infected and virus-infected cells were statistically significant, as determined by t tests. Figure 5 summarizes the alterations observed in E2F1 and pRb nuclear recovery.
FIG. 4.

Rb hypophosphorylation in HHV-6A- and HHV-6B-infected SupT1 cells. (A) Western blots of mock-infected and virus-infected nuclear lysates were reacted with antibodies to Rb and pRb proteins. (B and C) Graphic representation of the results relative to the highest level of Rb or pRb protein found in the samples. The intensities of Rb and pRb were corrected for loading variations by the intensity of eIF2α. The measurements for the Rb were based on results from two experiments. The comparison of the pRb in the mock infection versus that of HHV-6B (Z29) infection was based on results from a single experiment. pRb in the cytoplasmic fractions of mock-infected and virus-infected cells was not detected (data not shown). The quantitations of pRb in the mock and HHV-6A (U1102) infections were based on results from three separate experiments. nuc, nuclear.
FIG. 5.
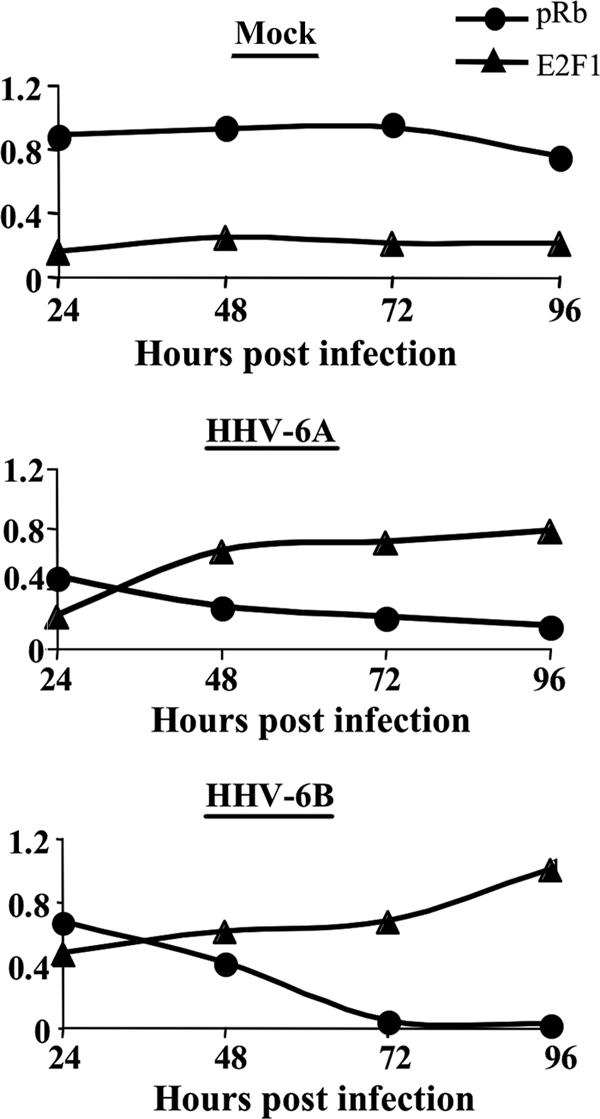
Alterations of pRb and E2F1 in mock-infected and virus-infected cells. Graphic representations of E2F1 and pRb in the nuclei of mock-infected and virus-infected cells are shown. pRb and E2F1 values are shown relative to the highest protein level detected: E2F1 in the 96-hpi sample of HHV-6B infections and pRb at 72 hpi in the mock-infected cells.
Rb dephosphorylation also occurs in cells infected with UV-inactivated virus.
Following the finding that Rb was dephosphorylated during HHV-6A and HHV-6B infections, it was of interest to test whether the effect required viral gene expression postinfection. To evaluate this question, 4 × 106 cells were either (i) mock infected, (ii) treated with medium that was UV irradiated, (iii) infected with cell-free HHV-6A and HHV-6B, or (iv) treated with an equal quantity of cell-free virus that was UV irradiated to inactivate the virus. Protein extracts prepared at 72 hpi were tested in Western blots employing the pRb, P41, and eIF2α antibodies. The results (Fig. 6) are shown with low and high exposures of the Western blots and can be summarized as follows. (i) Significant Rb dephosphorylation was observed in the cells infected with live, untreated viruses, as described above. (ii) Significant Rb dephosphorylation was also apparent with the UV-irradiated viruses compared to that of the mock-infected cells, although this occurred to a lesser extent than in the cells receiving the equivalent live -virus. The quantitative analyses (Fig. 6B) revealed that the infections with live HHV-6A and HHV-6B reduced Rb phosphorylation to 5.3 and 3.7%, respectively, of their levels in the mock-infected cells. The UV-irradiated HHV-6A and HHV-6B reduced the Rb phosphorylation to 10.3 and 22.3%, respectively, compared to that of the mock-infected cells treated with the UV-irradiated medium. (iii) Comparison of P41 expression in cells infected with live HHV-6A and HHV-6B to that of the UV-irradiated virus showed that viral inactivation in the HHV-6A UV-treated samples was incomplete, and there were still low levels of P41 protein expression (Fig. 6). Taken together, these results indicated that Rb dephosphorylation in HHV-6A and HHV-6B infections might be caused by protein(s) brought into the cells within the structural particles or might result from viral absorption or entry into the cells. There was no requirement for elaborate viral gene expression and replication.
FIG. 6.

Dephosphorylation of Rb in cells infected with inactivated virus. (A) The viruses were irradiated by UV or were untreated, and Western blots from mock-infected and virus-infected cell lysates were reacted with antibodies to the pRb and P41 proteins (9A5 MAb). Two exposures of the blots (low-exp and high-exp) are shown for the pRb and the P41 proteins. (B) Graphic representation of the results relative to the highest pRb levels (mock-infected cells). The intensities of pRb were corrected for loading variations by the intensity of eIF2α. nuc, nuclear.
E2F1 accumulation does not result in higher levels of cyclin E and MCM5, two known E2F1-transcribed target genes.
E2F1 acts as a transcription factor for a large number of genes initiated in late G1 phase, when E2F1 accumulates to promote cell cycle progression by various mechanisms (33). To test whether the levels of E2F1-targeted genes also increased during viral infections, two target genes were chosen, cyclin E and MCM5, both transcribed by E2F1 (30, 35, 50, 51). Cyclin E is an evolutionarily conserved protein that plays an essential role in promoting cell cycle transition from the G1 to the S phase. It binds to and activates the cyclin-dependent kinase Cdk2, with consequent effects on cell cycle progression (63). In this state it is able to phosphorylate Rb, which is first phosphorylated by cyclin D. Altogether, the phosphorylation of Rb results in its release from E2F1. MCM5 is one of seven minichromosome maintenance family members, which are highly conserved in eukaryotes. They function as DNA helicases, forming heterohexamer complexes that bind to the DNA replication origins, moving along with the DNA polymerase during DNA replication (57). To determine the level of cyclin E and MCM5, 12 × 106 SupT1 cells were mock infected or were infected with HHV-6A (U1102). eIF2α was employed to correct for equal protein loading. As shown in Fig. 7, although E2F1 levels increased during infection, neither cyclin E nor MCM5 levels changed compared to those of the uninfected cells. Furthermore, there were no changes in their cytoplasmic and nuclear localizations.
FIG. 7.

Levels of cyclin E and MCM5 proteins in HHV-6A (U11O2)-infected SupT1 cells. (A) Western blots of proteins prepared from the cytoplasmic (cyto) and nuclear (nuc) fractions of mock-infected and HHV-6A-infected cells tested with cyclin E, MCM5, and eIF2α antibodies. (B) Quantitative representation of the results for the nuclear fractions relative to the highest level of cyclin E and MCMS protein expression in the mock-infected cultures. The data are based on results from a single experiment for cyclin E and the averages from two experiments for MCM5.
E2F1 activation pathway is intact in the SupT1 cells.
To test whether E2F1 accumulation can cause upregulation of its known target genes in the cell line used in this work, SupT1 cells were transfected by electroporation with an E2F1 expression plasmid driven by the HCMV promoter. Extracts prepared 1 day posttransfection were tested in Western blots using E2F1, cyclin E, and eIF2α antibodies. As seen in Fig. 8, after transfection with 5 μg of the E2F1 plasmid, the E2F1 levels were found to increase fivefold in a manner similar to that of the increase observed following the HHV-6A and HHV-6B infections (Fig. 1 and 3). In contrast to the infected cells, in which no upregulation of E2F1 target genes was found, 5 μg of the E2F1 plasmid caused upregulation of cyclin E. These results confirm that E2F1 can induce the expression of cyclin E in SupT1 cells, suggesting a different route for E2F1 in the infection.
FIG. 8.
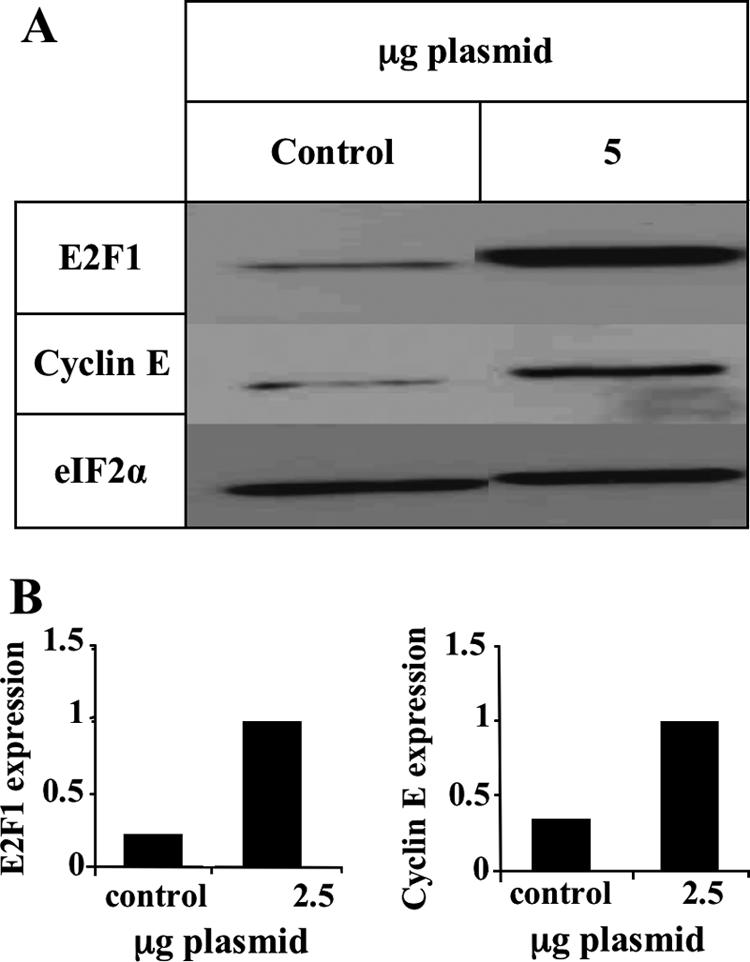
E2F1 can initiate cyclin E expression in SupT1 cells. (A) Western blots tested with E2F1, cyclin E, and eIF2α antibodies after transfection of SupT1 cells with an E2F1-expressing plasmid. (B) Graphic representation of the results relative to the highest level of protein expression in panel A.
Rb and MCM5 have reduced complexing with E2F1 in the infected cells.
Following the finding that E2F1 was upregulated in the infected cells, it was of interest to examine whether E2F1 was in its free form or was complexed to Rb. Additionally, MCM5 is known to interact with E2F1 through Rb and to play critical roles in the formation of complexes functioning in host DNA replication, cell cycle regulation, and chromosomal stabilization (10, 22, 61, 68). Disruption of the complexing upon viral replication might be related to the inhibition of cell DNA replication, as we have previously shown (20). To address these questions, protein lysates prepared from mock-infected or HHV-6A-infected SupT1 cells were immunoprecipitated, employing E2F1 antibody coupled to protein A beads. Resultant precipitates were subjected to Western blotting employing Rb, MCM5, and γ-tubulin antibodies. The results revealed the following (Fig. 9). (i) As expected, γ-tubulin, which is not known to interact with E2F1, was not recovered in the immunoprecipitate. (ii) In the mock-infected samples, both Rb and MCM5 proteins were recovered in the immunoprecipitated E2F1 samples. (iii) Based on two independent experiments, there were lower levels of Rb (35%) and MCM5 protein (60%) recovered in the E2F1 precipitates than those recovered in the mock-infected samples. (iv) The differences in the Rb and MCM5 coprecipitations were statistically significant, as determined by t tests. We conclude that the upregulated E2F1 in the infected cells was mostly uncoupled from its main regulator, Rb, and there were somewhat lower quantities of MCM5-containing E2F1 complexes.
FIG. 9.
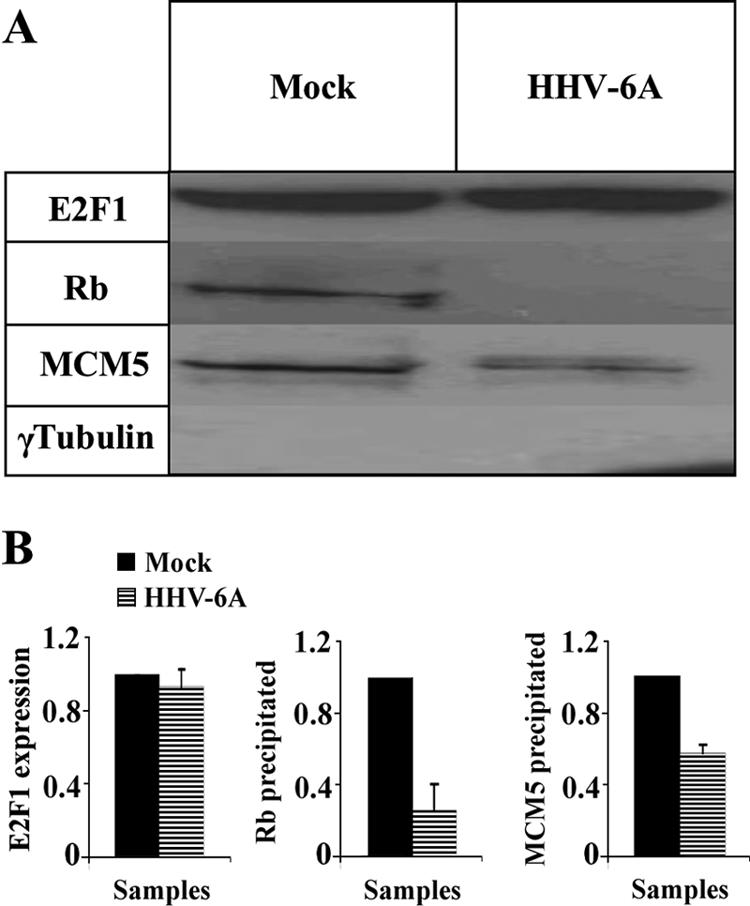
Reduced Rb and MCM5 complexes with E2F1 in the infected cells. (A) E2F1 was immunoprecipitated with anti-E2F1 antibody, starting with 500 μg lysate. Proteins, which were coprecipitated, were tested using antibodies to Rb, MCM5, and γ-tubulin. (B) Graphic representation of the results relative to the highest level of protein expression in the mock-infected cells. The quantitative analyses were based on results from two independent experiments. The differences of Rb and MCM5 between mock-infected and virus-infected cells were statistically significant, as determined by t tests.
HHV-6 infection in SupT1 cells causes cell cycle arrest.
E2F1 and Rb play significant roles in the regulation of the cell cycle. Free E2F1 promotes cell cycle progression past G1 into the S phase, whereas E2F1 bound to Rb promotes cell cycle arrest at G1. The increased E2F1 levels postinfection predicted progression into the S phase, but the E2F1 target genes cyclin E and MCM5 were unaffected during the infection. Moreover, there was reduced coupling of Rb and E2F1. To test whether these alterations induced changes in cell cycle progression, 4 × 106 cells were mock infected or were infected with HHV-6A (U11O2). At 24 and 48 hpi, prior to the appearance of pronounced CPE, the cultures were fixed and subjected to FACS analysis: 2 × 106 cells were incubated with the anti-P41 9A5 MAb and a secondary antibody, fluorescein isothiocyanate (FITC) goat anti-mouse antibody, in order to mark and determine the percentage of the infected cells. The remaining 2 × 106 cells were subjected to cell cycle analysis using propidium iodide. Prior to cell cycle analysis by the FACS, the cells were marked with the 9A5 MAb and a secondary FITC goat anti-mouse antibody, analyzing only the infected cells by gating for FITC in the FACS cell cycle assay (Fig. 10). The choice of the time points 24 and 48 hpi, prior to the appearance of pronounced CPE, was dictated by the inability to adequately sort cell cultures containing the large syncytial cells at late time points postinfection. The results revealed the following. (i) More than 95% of the cells were infected by 48 hpi and contained the P41 protein, marked by the 9A5 antibody (Fig. 10A). (ii) Cell cycle arrest was evident at the G2 phase starting at 24 hpi (Fig. 10B). There were reduced numbers of cells in the G1 and S phases and an increased fraction of cells in the G2/M phase. (iii) The differences in the ratios of the G2/M phases between the mock-infected and virus-infected cells at 24 and 48 hpi were statistically significant, as determined by t tests, based on the FACS analyses of three experiments.
FIG. 10.

Effect of the infection on the cell cycle. (A) FACS analyses of mock-infected and virus-infected cells prepared at 24 and 48 hpi and labeled with the P41 9A5 MAb (gray line). (B) Cell cycle analyses at 48 hpi. (C) Graphic representations of the FACS results based on results from three separate experiments. The differences in the ratios of G2/M phases between mock-infected and virus-infected cells at 24 and 48 h were statistically significant, as determined by t tests. Ap, apoptotic cells.
DISCUSSION
E2F1 and its main repressor, Rb, serve as major regulators for the transcription of genes that function in cell cycle progression, viability, and apoptosis. The studies described in this paper revealed significant alterations in the E2F1/Rb pathways at different points of viral infection. (i) E2F1 levels increased during HHV-6A and HHV-6B infections and could be recovered in both the cytoplasmic and nuclear fractions, unlike its strict nuclear localization in the uninfected cells. The cytoplasmic recovery of E2F1 starting at 72 hpi was intriguing in its specificity, unlike the nuclear recovery of the P41 viral protein, indicating the absence of random nuclear leakage. It is noteworthy that the p53 protein, also possessing a nuclear localization signal, was reported to have cytoplasmic recovery in HHV-6-infected cells that was attributed to its interaction with the U14 viral protein and its encapsidation within structural virions (52, 53, 72, 73). Since high levels of E2F1 promote apoptosis, the diversion of excess E2F1 into the cytoplasm might avoid early apoptosis, securing cell survival for viral replication. Further studies are needed to determine the reason(s) for the increased cytoplasmic E2F1. (ii) In the infected cells there was a significant reduction in Rb complexing with the E2F1 protein and also some reduction in the MCM5 protein coprecipitating with E2F1 antibodies. Both reductions were statistically significant by t tests. The release of E2F1 from Rb is known to result from Rb phosphorylation or following binding of viral proteins to Rb, disrupting the E2F1/Rb complexes. In the HHV-6-infected cells, the total levels of Rb did not change, but its phosphorylation was significantly reduced. Moreover, Rb dephosphorylation also occurred with UV-irradiated virus, indicating that it was mediated by virion protein(s) coming into the cells with the infecting virus or by viral attachment and/or interaction with the viral receptor at the cell surface. Considering the kinetics of E2F1, Rb, and pRb during the infection, the lower level of recovery of E2F1/Rb complexes could reflect the upregulation of E2F1 along with the steady unincreased levels of Rb, resulting in free E2F1. (iii) The increased free E2F1 did not induce higher levels of accumulation of cyclin E and MCM5, known to be induced by E2F1, although such an increase was possible in the infected SupT1 cells. (iv) In the HHV-6A (U1102)-infected cells there was some arrest of cell cycle progression. A similar cell cycle arrest also was observed for HHV-6B (Z29)-infected cells (data not shown). Different viruses were found to affect cell replication, and cell cycle arrest previously was reported to occur in certain herpesvirus infections, including herpes simplex virus type 1 and HCMV, which were reported to undergo arrest at the G1/S phase. Furthermore, earlier reports have documented arrest in HHV-6-infected cells at different levels of the cell cycle. De Bolle and coworkers reported cell cycle arrest at the G2 phase during HHV-6A infection of cord blood mononuclear cells (17). Hollsberg and coworkers have shown that HHV-6B infection of MOLT 3 cells caused cell cycle arrest at the G1 phase concomitantly with p53 phosphorylation and accumulation (52). More recently, they have shown that the arrest at the G1/S phase involved a pathway independent of p53 (53). In the present study, we have found arrest at the G2 phase as early as 24 and 48 hpi, prior to the appearance of pronounced CPE. The differences in cell cycle profiles of the mock-infected and virus-infected cells were reproduced in three separate experiments and were statistically significant by t tests (Fig. 10). The arrest might have resulted from both p53 and the presence of complexes of E2F1/Rb early postinfection upon Rb dephosphorylation. Cell cycle arrest at early points in the infection may be advantageous for virus replication in dividing lymphocytes, inasmuch as with this slowly replicating virus, arresting cell replication might enable the formation of syncytia and more efficient viral spread. At later times the infection was accompanied by total shutoff of host DNA replication, as shown in previous studies quantitating [32P]phosphate incorporation into viral and host DNA in infected phytohemagglutinin-activated peripheral blood lymphocytes at 65 to 86 hpi (20). Eventually, the infection might lead to apoptosis and/or necrosis. Overall, the data presented in this study shed new light on HHV-6 interaction with the infected cells, suggesting different manipulations of key factors relevant to cell proliferation and death.
Acknowledgments
We thank Ester Michael and Shulamit Grinshtein for valuable technical assistance. We thank N. Balachandran from the Chicago Medical School for his kind contribution of the 9A5 MAb. We thank Yoel Kloog, Tel Aviv University, for the E2F1-expressing plasmid.
Our research was supported by the S. Daniel Abraham Institute for Molecular Virology and the S. Daniel Abraham Chair for Molecular Virology and Gene Therapy, Tel Aviv University, Israel.
Footnotes
Published ahead of print on 3 October 2007.
REFERENCES
- 1.Ablashi, A. H., Z. Berneman, et al. 1993. Human herpesvirus-6 strain groups: a nomenclature. Arch. Virol. 129:363-366. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Lafuente, R., V. De Las Heras, M. Bartolome, M. Garcia-Montojo, and R. Arroyo. 2006. Human herpesvirus 6 and multiple sclerosis: a one-year follow-up study. Brain Pathol. 16:20-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez-Lafuente, R., V. de Las Heras, M. Garcia-Montojo, M. Bartolome, and R. Arroyo. 2007. Human herpesvirus-6 and multiple sclerosis: relapsing-remitting versus secondary progressive. Mult. Scler. 13:578-583. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez-Lafuente, R., M. Garcia-Montojo, V. De las Heras, M. Bartolome, and R. Arroyo. 2006. Clinical parameters and HHV-6 active replication in relapsing-remitting multiple sclerosis patients. J. Clin. Virol. 37(Suppl. 1):S24-S26. [DOI] [PubMed] [Google Scholar]
- 5.Apostolova, M. D., I. A. Ivanova, C. Dagnino, S. J. D'Souza, and L. Dagnino. 2002. Active nuclear import and export pathways regulate E2F-5 subcellular localization. J. Biol. Chem. 277:34471-34479. [DOI] [PubMed] [Google Scholar]
- 6.Asano, Y., T. Yoshikawa, Y. Kajita, R. Ogura, S. Suga, T. Yazaki, T. Nakashima, A. Yamada, and T. Kurata. 1992. Fatal encephalitis/encephalopathy in primary human herpesvirus-6 infection. Arch. Dis. Child. 67:1484-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bagchi, S., R. Weinmann, and P. Raychaudhuri. 1991. The retinoblastoma protein copurifies with E2F-I, an E1A-regulated inhibitor of the transcription factor E2F. Cell 65:1063-1072. [DOI] [PubMed] [Google Scholar]
- 8.Balachandran, N., S. Tirawatnapong, B. Pfeiffer, D. V. Ablashi, and S. Z. Salahuddin. 1991. Electrophoretic analysis of human herpesvirus 6 polypeptides immunoprecipitated from infected cells with human sera. J. Infect. Dis. 163:29-34. [DOI] [PubMed] [Google Scholar]
- 9.Boeckh, M., V. Erard, D. Zerr, and J. Englund. 2005. Emerging viral infections after hematopoietic cell transplantation. Pediatr. Transplant 9(Suppl. 7):48-54. [DOI] [PubMed] [Google Scholar]
- 10.Bosco, G., W. Du, and T. L. Orr-Weaver. 2001. DNA replication control through interaction of E2F-RB and the origin recognition complex. Nat. Cell Biol. 3:289-295. [DOI] [PubMed] [Google Scholar]
- 11.Bracken, A. P., M. Ciro, A. Cocito, and K. Helin. 2004. E2F target genes: unraveling the biology. Trends Biochem. Sci. 29:409-417. [DOI] [PubMed] [Google Scholar]
- 12.Cam, H., and B. D. Dynlacht. 2003. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell 3:311-316. [DOI] [PubMed] [Google Scholar]
- 13.Chang, C. K., and N. Balachandran. 1991. Identification, characterization, and sequence analysis of a cDNA encoding a phosphoprotein of human herpesvirus 6. J. Virol. 65:2884-2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapenko, S., A. Millers, Z. Nora, I. Logina, R. Kukaine, and M. Murovska. 2003. Correlation between HHV-6 reactivation and multiple sclerosis disease activity. J. Med. Virol. 69:111-117. [DOI] [PubMed] [Google Scholar]
- 15.Chau, B. N., C. W. Pan, and J. Y. Wang. 2006. Separation of anti-proliferation and anti-apoptotic functions of retinoblastoma protein through targeted mutations of its A/B domain. PLoS 1:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chellappan, S. P., S. Hiebert, M. Mudryj, J. M. Horowitz, and J. R. Nevins. 1991. The E2F transcription factor is a cellular target for the RB protein. Cell 65:1053-1061. [DOI] [PubMed] [Google Scholar]
- 17.De Bolle, L., S. Hatse, E. Verbeken, E. De Clercq, and L. Naesens. 2004. Human herpesvirus 6 infection arrests cord blood mononuclear cells in G2 phase of the cell cycle. FEBS Lett. 560:25-29. [DOI] [PubMed] [Google Scholar]
- 18.DeGregori, J., and D. G. Johnson. 2006. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 6:739-748. [DOI] [PubMed] [Google Scholar]
- 19.Dick, F. A., E. Sailhamer, and N. J. Dyson. 2000. Mutagenesis of the pRb pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20:3715-3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Luca, D., G. Katsafanas, E. C. Schirmer, N. Balachandran, and N. Frenkel. 1990. The replication of viral and cellular DNA in human herpesvirus 6-infected cells. Virology 175:199-210. [DOI] [PubMed] [Google Scholar]
- 21.Ehmann, G. L., T. I. McLean, and S. L. Bachenheimer. 2000. Herpes simplex virus type 1 infection imposes a G1/S block in asynchronously growing cells and prevents G1 entry in quiescent cells. Virology 267:335-349. [DOI] [PubMed] [Google Scholar]
- 22.Forsburg, S. L. 2004. Eukaryotic MCM proteins: beyond replication initiation. Microbiol. Mol. Biol. Rev. 68:109-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fotheringham, J., and S. Jacobson. 2005. Human herpesvirus 6 and multiple sclerosis: potential mechanisms for virus-induced disease. Herpes 12:4-9. [PubMed] [Google Scholar]
- 24.Frenkel, N., and R. Borenstein. 2006. Characterization of the lymphotropic amplicons-6 and tamplicon-7 vectors derived from HHV-6 and HHV-7. Curr. Gene Ther. 6:399-420. [DOI] [PubMed] [Google Scholar]
- 25.Frenkel, N., G. C. Katsafanas, L. S. Wyatt, T. Yoshikawa, and Y. Asano. 1994. Bone marrow transplant recipients harbor the B variant of human herpesvirus 6. Bone Marrow Transplant. 14:839-843. [PubMed] [Google Scholar]
- 26.Grand, R. J., A. P. Ibrahim, A. M. Taylor, A. E. Milner, C. D. Gregory, P. H. Gallimore, and A. S. Turnell. 1998. Human cells arrest in S phase in response to adenovirus 12 E1A. Virology 244:330-342. [DOI] [PubMed] [Google Scholar]
- 27.Helin, K., J. A. Lees, M. Vidal, N. Dyson, E. Harlow, and A. Fattaey. 1992. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell 70:337-350. [DOI] [PubMed] [Google Scholar]
- 28.Helin, K., C. L. Wu, A. R. Fattaey, J. A. Lees, B. D. Dynlacht, C. Ngwu, and E. Harlow. 1993. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 7:1850-1861. [DOI] [PubMed] [Google Scholar]
- 29.Hilton, M. J., D. Mounghane, T. McLean, N. V. Contractor, J. O'Neil, K. Carpenter, and S. L. Bachenheimer. 1995. Induction by herpes simplex virus of free and heteromeric forms of E2F transcription factor. Virology 213:624-638. [DOI] [PubMed] [Google Scholar]
- 30.Hwang, H. C., and B. E. Clurman. 2005. Cyclin E in normal and neoplastic cell cycles. Oncogene 24:2776-2786. [DOI] [PubMed] [Google Scholar]
- 31.Jault, F. M., J. M. Jault, F. Ruchti, E. A. Fortunato, C. Clark, J. Corbeil, D. D. Richman, and D. H. Spector. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J. Virol. 69:6697-6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaelin, W. G., Jr., W. Krek, W. R. Sellers, J. A. DeCaprio, F. Ajchenbaum, C. S. Fuchs, T. Chittenden, Y. Li, P. J. Farnham, M. A. Blanar, et al. 1992. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell 70:351-364. [DOI] [PubMed] [Google Scholar]
- 33.Kel, A. E., O. V. Kel-Margoulis, P. J. Farnham, S. M. Bartley, E. Wingender, and M. Q. Zhang. 2001. Computer-assisted identification of cell cycle-related genes: new targets for E2F transcription factors. J. Mol. Biol. 309:99-120. [DOI] [PubMed] [Google Scholar]
- 34.Komaroff, A. L. 2006. Is human herpesvirus-6 a trigger for chronic fatigue syndrome? J. Clin. Virol. 37(Suppl. 1):S39-S46. [DOI] [PubMed] [Google Scholar]
- 35.Koutsami, M. K., P. K. Tsantoulis, M. Kouloukoussa, K. Apostolopoulou, I. S. Pateras, Z. Spartinou, A. Drougou, K. Evangelou, C. Kittas, J. Bartkova, J. Bartek, and V. G. Gorgoulis. 2006. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J. Pathol. 209:512-521. [DOI] [PubMed] [Google Scholar]
- 36.Kovesdi, I., R. Reichel, and J. R. Nevins. 1986. Identification of a cellular transcription factor involved in E1A trans-activation. Cell 45:219-228. [DOI] [PubMed] [Google Scholar]
- 37.Krueger, G., and D. V. Ablashi (ed.). 2006. Human herpesvirus-6, general virology, epidemiology and clinical pathology. Perspectives in medical virology, vol. 12. Elsevier Science B.V., Amsterdam, The Netherlands.
- 38.Lee, C., and Y. Cho. 2002. Interactions of SV40 large T antigen and other viral proteins with retinoblastoma tumour suppressor. Rev. Med. Virol. 12:81-92. [DOI] [PubMed] [Google Scholar]
- 39.Lees, J. A., M. Saito, M. Vidal, M. Valentine, T. Look, E. Harlow, N. Dyson, and K. Helin. 1993. The retinoblastoma protein binds to a family of E2F transcription factors. Mol. Cell. Biol. 13:7813-7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindeman, G. J., S. Gaubatz, D. M. Livingston, and D. Ginsberg. 1997. The subcellular localization of E2F-4 is cell-cycle dependent. Proc. Natl. Acad. Sci. USA 94:5095-5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ljungman, P., and N. Singh. 2006. Human herpesvirus-6 infection in solid organ and stem cell transplant recipients. J. Clin. Virol. 37(Suppl. 1):S87-S91. [DOI] [PubMed] [Google Scholar]
- 42.Lopez, C., P. Pellett, J. Stewart, C. Goldsmith, K. Sanderlin, J. Black, D. Warfield, and P. Feorino. 1988. Characteristics of human herpesvirus-6. J. Infect. Dis. 157:1271-1273. [DOI] [PubMed] [Google Scholar]
- 43.Magae, J., C. L. Wu, S. Illenye, E. Harlow, and N. H. Heintz. 1996. Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. J. Cell Sci. 109:1717-1726. [DOI] [PubMed] [Google Scholar]
- 44.Mameli, G., V. Astone, G. Arru, S. Marconi, L. Lovato, C. Serra, S. Sotgiu, B. Bonetti, and A. Dolei. 2007. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not human herpesvirus 6. J. Gen. Virol. 88:264-274. [DOI] [PubMed] [Google Scholar]
- 45.Mayne, M., C. Cheadle, S. S. Soldan, C. Cermelli, Y. Yamano, N. Akhyani, J. E. Nagel, D. D. Taub, K. G. Becker, and S. Jacobson. 2001. Gene expression profile of herpesvirus-infected T cells obtained using immunomicroarrays: induction of proinflammatory mechanisms. J. Virol. 75:11641-11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Müller, H., A. P. Bracken, R. Vernell, M. C. Moroni, F. Christians, E. Grassilli, E. Prosperini, E. Vigo, J. D. Oliner, and K. Helin. 2001. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 15:267-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Müller, H., M. C. Moroni, E. Vigo, B. O. Petersen, J. Bartek, and K. Helin. 1997. Induction of S-phase entry by E2F transcription factors depends on their nuclear localization. Mol. Cell. Biol. 17:5508-5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mundle, S. D., and G. Saberwal. 2003. Evolving intricacies and implications of E2F1 regulation. FASEB J. 17:569-574. [DOI] [PubMed] [Google Scholar]
- 49.Nagasawa, T., I. Kimura, Y. Abe, and A. Oka. 2007. HHV-6 encephalopathy with cluster of convulsions during eruptive stage. Pediatr. Neurol. 36:61-63. [DOI] [PubMed] [Google Scholar]
- 50.Ohtani, K., J. DeGregori, and J. R. Nevins. 1995. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 92:12146-12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohtani, K., R. Iwanaga, M. Nakamura, M. Ikeda, N. Yabuta, H. Tsuruga, and H. Nojima. 1999. Cell growth-regulated expression of mammalian MCM5 and MCM6 genes mediated by the transcription factor E2F. Oncogene 18:2299-2309. [DOI] [PubMed] [Google Scholar]
- 52.Øster, B., B. Bundgaard, and P. Hollsberg. 2005. Human herpesvirus 6B induces cell cycle arrest concomitant with p53 phosphorylation and accumulation in T cells. J. Virol. 79:1961-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oster, B., M. D. Kaspersen, E. Kofod-Olsen, B. Bundgaard, and P. Hollsberg. 2006. Human herpesvirus 6B inhibits cell proliferation by a p53-independent pathway. J. Clin. Virol. 37(Suppl. 1):S63-S68. [DOI] [PubMed] [Google Scholar]
- 54.Pellett, D. A. 2002. Human herpesviruses 6A, 6B, and 7 and their replication, p. 2769-2784. In B. N. Fields and D. M. Knipe (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 55.Rapaport, D., D. Engelhard, G. Tagger, R. Or, and N. Frenkel. 2002. Antiviral prophylaxis may prevent human herpesvirus-6 reactivation in bone marrow transplant recipients. Transplant. Infect. Dis. 4:10-16. [DOI] [PubMed] [Google Scholar]
- 56.Rotola, A., I. Merlotti, L. Caniatti, E. Caselli, E. Granieri, M. R. Tola, D. Di Luca, and E. Cassai. 2004. Human herpesvirus 6 infects the central nervous system of multiple sclerosis patients in the early stages of the disease. Mult. Scler. 10:348-354. [DOI] [PubMed] [Google Scholar]
- 57.Ryu, S., J. Holzschuh, S. Erhardt, A. K. Ettl, and W. Driever. 2005. Depletion of minichromosome maintenance protein 5 in the zebrafish retina causes cell-cycle defect and apoptosis. Proc. Natl. Acad. Sci. USA 102:18467-18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salahuddin, S. Z., D. V. Ablashi, P. D. Markham, S. F. Josephs, S. Sturzenegger, M. Kaplan, G. Halligan, P. Biberfeld, F. Wong-Staal, B. Kramarsky, et al. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596-601. [DOI] [PubMed] [Google Scholar]
- 59.Santoro, F., P. E. Kennedy, G. Locatelli, M. S. Malnati, E. A. Berger, and P. Lusso. 1999. CD46 is a cellular receptor for human herpesvirus 6. Cell 99:817-827. [DOI] [PubMed] [Google Scholar]
- 60.Schirmer, E. C., L. S. Wyatt, K. Yamanishi, W. J. Rodriguez, and N. Frenkel. 1991. Differentiation between two distinct classes of viruses now classified as human herpesvirus 6. Proc. Natl. Acad. Sci. USA 88:5922-5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwed, G., N. May, Y. Pechersky, and B. R. Calvi. 2002. Drosophila minichromosome maintenance 6 is required for chorion gene amplification and genomic replication. Mol. Biol. Cell 13:607-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shan, B., X. Zhu, P. L. Chen, T. Durfee, Y. Yang, D. Sharp, and W. H. Lee. 1992. Molecular cloning of cellular genes encoding retinoblastoma-associated proteins: identification of a gene with properties of the transcription factor E2F. Mol. Cell. Biol. 12:5620-5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singer, J. D., M. Gurian-West, B. Clurman, and J. M. Roberts. 1999. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 13:2375-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singer, O., and N. Frenkel. 1997. Human herpesvirus 7 (HHV-7) DNA: analyses of clones spanning the entire genome. Arch. Virol. 142:287-303. [DOI] [PubMed] [Google Scholar]
- 65.Smith, J. M., and R. A. McDonald. 2006. Emerging viral infections in transplantation. Pediatr. Transplant. 10:838-843. [DOI] [PubMed] [Google Scholar]
- 66.Soldan, S. S., A. D. Goodman, and S. Jacobson. 2006. HHV-6 and the central nervous system, p. 213-224. In G. Krueger and D. V. Ablashi (ed.), Human herpesvirus-6, general virology, epidemiology and clinical pathology. Perspectives in medical virology, vol. 12. Elsevier Science B.V., Amsterdam, The Netherlands. [Google Scholar]
- 67.Song, Y. J., and M. F. Stinski. 2002. Effect of the human cytomegalovirus IE86 protein on expression of E2F-responsive genes: a DNA microarray analysis. Proc. Natl. Acad. Sci. USA 99:2836-2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stagljar, I., U. Hubscher, and A. Barberis. 1999. Activation of DNA replication in yeast by recruitment of the RNA polymerase II transcription complex. Biol. Chem. 380:525-530. [DOI] [PubMed] [Google Scholar]
- 69.Stanelle, J., and B. M. Putzer. 2006. E2F1-induced apoptosis: turning killers into therapeutics. Trends Mol. Med. 12:177-185. [DOI] [PubMed] [Google Scholar]
- 70.Suga, S., K. Suzuki, M. Ihira, T. Yoshikawa, Y. Kajita, T. Ozaki, K. Iida, Y. Saito, and Y. Asano. 2000. Clinical characteristics of febrile convulsions during primary HHV-6 infection. Arch. Dis. Child. 82:62-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takaku, T., J. H. Ohyashiki, Y. Zhang, and K. Ohyashiki. 2005. Estimating immunoregulatory gene networks in human herpesvirus type 6-infected T cells. Biochem. Biophys. Res. Commun. 336:469-477. [DOI] [PubMed] [Google Scholar]
- 72.Takemoto, M., M. Koike, Y. Mori, S. Yonemoto, Y. Sasamoto, K. Kondo, Y. Uchiyama, and K. Yamanishi. 2005. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J. Virol. 79:13037-13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takemoto, M., Y. Mori, K. Ueda, K. Kondo, and K. Yamanishi. 2004. Productive human herpesvirus 6 infection causes aberrant accumulation of p53 and prevents apoptosis. J. Gen. Virol. 85:869-879. [DOI] [PubMed] [Google Scholar]
- 74.Tomonari, A., S. Takahashi, J. Ooi, T. Iseki, K. Takasugi, M. Uchiyama, T. Konuma, M. Futami, N. Ohno, K. Uchimaru, A. Tojo, and S. Asano. 2005. Human herpesvirus 6 variant B infection in adult patients after unrelated cord blood transplantation. Int. J. Hematol. 81:352-355. [DOI] [PubMed] [Google Scholar]
- 75.Trimarchi, J. M., and J. A. Lees. 2002. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3:11-20. [DOI] [PubMed] [Google Scholar]
- 76.Tuke, P. W., S. Hawke, P. D. Griffiths, and D. A. Clark. 2004. Distribution and quantification of human herpesvirus 6 in multiple sclerosis and control brains. Mult. Scler. 10:355-359. [DOI] [PubMed] [Google Scholar]
- 77.Ward, K. N., N. J. Andrews, C. M. Verity, E. Miller, and E. M. Ross. 2005. Human herpesviruses-6 and -7 each cause significant neurological morbidity in Britain and Ireland. Arch. Dis. Child 90:619-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamanishi, K. 2002. Human herpesvirus 6 and human herpesvirus 7, p. 2785-2801. In B. N. Fields and D. M. Knipe (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 79.Yamanishi, K., T. Okuno, K. Shiraki, M. Takahashi, T. Kondo, Y. Asano, and T. Kurata. 1988. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet i:1065-1067. [DOI] [PubMed] [Google Scholar]
- 80.Yamashita, N., and T. Morishima. 2005. HHV-6 and seizures. Herpes 12:46-49. [PubMed] [Google Scholar]
- 81.Yao, K., N. Akyani, D. Donati, N. Sengamalay, J. Fotheringham, E. Ghedin, M. Bishop, J. Barrett, F. Kashanchi, and S. Jacobson. 2006. Detection of HHV-6B in post-mortem central nervous system tissue of a post-bone marrow transplant recipient: a multi-virus array analysis. J. Clin. Virol. 37(Suppl. 1):S57-S62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshikawa, T., and Y. Asano. 2000. Central nervous system complications in human herpesvirus-6 infection. Brain Dev. 22:307-314. [DOI] [PubMed] [Google Scholar]
- 83.Yoshikawa, T., Y. Asano, M. Ihira, K. Suzuki, M. Ohashi, S. Suga, K. Kudo, K. Horibe, S. Kojima, K. Kato, T. Matsuyama, and Y. Nishiyama. 2002. Human herpesvirus 6 viremia in bone marrow transplant recipients: clinical features and risk factors. J. Infect. Dis. 185:847-853. [DOI] [PubMed] [Google Scholar]
- 84.Yoshikawa, T., T. Nakashima, S. Suga, Y. Asano, T. Yazaki, H. Kimura, T. Morishima, K. Kondo, and K. Yamanishi. 1992. Human herpesvirus-6 DNA in cerebrospinal fluid of a child with exanthem subitum and meningoencephalitis. Pediatrics 89:888-890. [PubMed] [Google Scholar]
- 85.Yoshikawa, T., S. Suga, Y. Asano, T. Nakashima, T. Yazaki, Y. Ono, T. Fujita, K. Tsuzuki, S. Sugiyama, and S. Oshima. 1992. A prospective study of human herpesvirus-6 infection in renal transplantation. Transplantation 54:879-883. [DOI] [PubMed] [Google Scholar]
- 86.Yoshikawa, T., S. Suga, Y. Asano, T. Nakashima, T. Yazaki, R. Sobue, M. Hirano, M. Fukuda, S. Kojima, and T. Matsuyama. 1991. Human herpesvirus-6 infection in bone marrow transplantation. Blood 78:1381-1384. [PubMed] [Google Scholar]
- 87.Zerr, D. M. 2006. Human herpesvirus 6 and central nervous system disease in hematopoietic cell transplantation. J. Clin. Virol. 37(Suppl. 1):S52-S56. [DOI] [PubMed] [Google Scholar]
- 88.Zerr, D. M. 2006. Human herpesvirus 6: a clinical update. Herpes 13:20-24. [PubMed] [Google Scholar]