

Abstract
The heat shock sigma factor (σ32 in Escherichia coli) directs the bacterial RNA polymerase to promoters of a specific sequence to form a stable complex, competent to initiate transcription of genes whose products mitigate the effects of exposure of the cell to high temperatures. The histidine at position 107 of σ32 is at the homologous position of a tryptophan residue at position 433 of the main sigma factor of E. coli, σ70. This tryptophan is essential for the strand separation step leading to the formation of the initiation-competent RNA polymerase-promoter complex. The heat shock sigma factors of all gammaproteobacteria sequenced have a histidine at this position, while in the alpha- and deltaproteobacteria, it is a tryptophan. In vitro the alanine-for-histidine substitution at position 107 (H107A) destabilizes complexes between the GroE promoter and RNA polymerase containing σ32, implying that H107 plays a role in formation or maintenance of the strand-separated complex. In vivo, the H107A substitution in σ32 impedes recovery from heat shock (exposure to 42°C), and it also leads to overexpression at lower temperatures (30°C) of the Flu protein, which is associated with biofilm formation.
Organisms from bacteria (4, 8, 20, 28, 29) to humans (e.g., reference 24) exhibit the heat shock response, aimed at mitigating the effects of exposure to temperatures higher than those normally encountered by the organism. In bacteria, the control of this response is exerted by the heat shock sigma factor, σH (4, 14, 18, 26, 30). The complex of a sigma factor and the bacterial RNA polymerase (RNAP) core enzyme (E), the so-called holoenzyme (Eσ), can bind promoter DNA to form the transcription-competent open complex where strand separation has occurred over 14 base pairs, including the start site of transcription (3, 8). Sigma factors have four functionally important regions of high sequence conservation. One subregion, designated 2.3, is involved in carrying out the RNAP-mediated strand separation of promoter DNA (9, 10, 15), leading to formation of the open, initiation-competent, RNAP-promoter complex. In Escherichia coli σ70, this region contains four basic and seven aromatic amino acids, which by mutational analysis have been shown to participate in DNA binding and promoter strand separation, respectively (6, 19, 25). Region 2.3 of the E. coli heat shock sigma factor, usually referred to as σ32, has only three aromatic amino acids (12, 15).
It is of interest to determine whether the nonaromatic amino acids of σ32 that are at homologous positions to the aromatic amino acids in σ70 have similar functions in facilitating the strand separation process. Here, we have focused on the H107 residue of σ32, homologous to W433 in σ70. We previously showed that the H107A substitution in the region 2.3 of σ32 had but a small effect on the ability of the Eσ32 holoenzyme to initiate transcription from the strong GroE promoter (PgroE) in vivo (12), calling into question whether H107 of σ32 was important for open complex formation. Here, we demonstrate that with PgroE, the H107A substitution in σ32 leads to formation of an RNAP-promoter complex in vitro with increased sensitivity to a challenge by the polyanion, heparin. This reflects defects in formation and/or maintenance of open RNAP-promoter complexes. The H107A substitution leads to diminished ability of the cell to survive exposure to heat shock. Surprisingly, it also causes increased levels of expression of the Flu protein, thus revealing a possible link between σ32 and biofilm formation (21).
MATERIALS AND METHODS
Materials.
Commercial sources were used for oligodeoxyribonucleotide sequencing and random primers (Invitrogen), DNA purification (Qiagen), site-directed mutagenesis (Stratagene), His-Bind resin (Novagen), E. coli RNAP core enzyme (Epicenter Biotechnologies), RNA purification (Ambion), ReadyMix Taq PCR mix (Sigma), and radioactive nucleotides (PerkinElmer and MP Biomedicals). Medium reagents were purchased from Gibco-Brl; other chemicals were from Sigma, Roche, Amersham, Promega, and Fisher.
Bacterial strains and plasmids.
An E. coli mutant strain lacking a functional heat shock sigma factor, CAG9301 (MG1655 rpoH120::kan zhg-21::Tn10), has been described previously (31). This strain was obtained from Carol Gross and is here also referred to as the “minus-σ32” strain. The CAG9301 strain has been reported to be able to grow and divide at about 20°C but not 30°C. It does display an increase in the optical density at 600 nm (OD600) at 30°C but without a concomitant increase in the number of CFU (31). The properties of the strain we used here are compatible with those findings. We observed an increase in the OD600 in liquid medium at 32°C (see Fig. 4); however, on LB plates the strain will grew colonies at room temperature but not at 32°C (data not shown). The plasmid pSAKT32 (27) was used as an extrachromosomal, intracellular source of the rpoH gene, encoding wild-type (WT) or H107A mutant σ32. For purification of σ32, the plasmid pCL412 (pET28) containing the σ32 gene with an N-terminal His6 tag was employed. Plasmids pQF50KgroE (containing positions −47 to −9 of PgroE, wild type or with substitutions [27]) and pCR-BluntII-TOPOgroE (with a cloned DNA fragment including the entire PgroE, with the UP element as well as part of the groES gene) were used to compare in vitro transcription activities of WT and mutant RNAP holoenzyme.
FIG. 4.
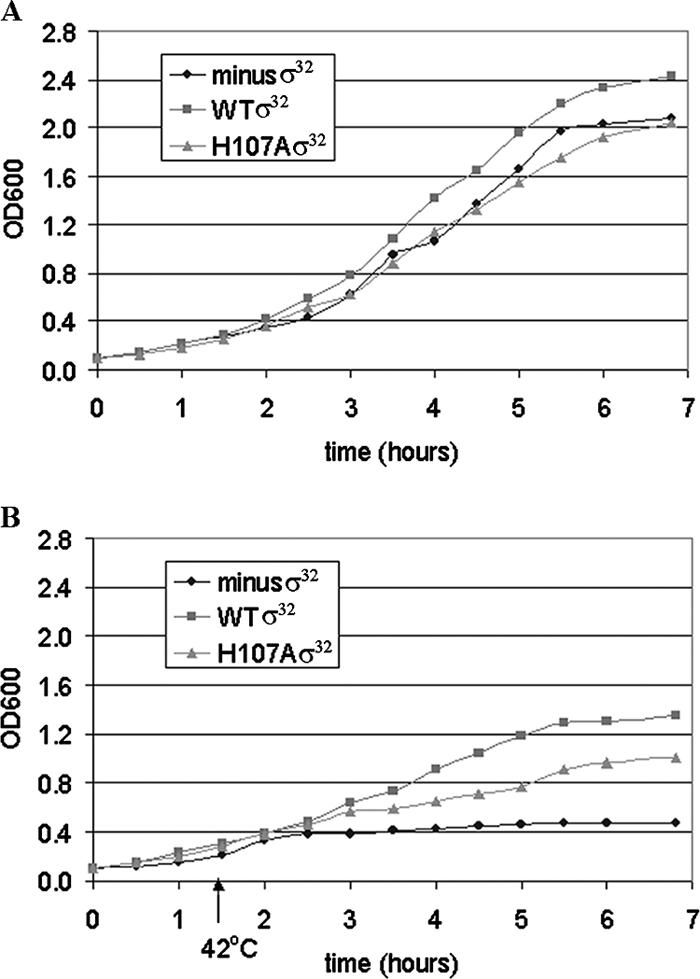
The effect of the H107A substitution in σ32 on cell growth. All growth curves are for E. coli strain CAG9301 in which the endogenous σ32 was inactivated (minus-σ32 strain). The strains differ in whether they contain plasmid expressing WT σ32, plasmid expressing H107A σ32, or no plasmid. Expression of the σ32 genes was driven by the basal transcription activity from the lac promoter; no IPTG inducer was added. (A) Growth at 32°C. (B) Initial growth at 32°C, followed by a shift in temperature from 32 to 42°C 1.5 h after inoculation of the culture.
Growth under heat shock conditions.
Overnight cultures in LB medium (at 23°C with shaking at 230 rpm) were used to inoculate fresh enriched M9 minimal medium (i.e., supplemented with 1% glucose and 0.2% Casamino Acids) at a starting OD600 of 0.1. No isopropyl-β-d-thiogalactopyranoside (IPTG) was added at any time during these experiments. Cultures were initially grown at 32°C. In order to avoid nonlinear OD600 readings, samples from cultures grown to an OD600 greater than 0.4 were diluted in fresh medium before measurement. To induce heat shock, the temperature was shifted to 42°C when the OD600 reached 0.3.
Purification of σ32.
E. coli BL21(DE3) cells transformed with plasmid pCL412 were grown in LB medium to an OD600 of 0.5 and induced with 1 mM IPTG. After 3 h the cells were harvested by centrifugation. Isolation of σ32 followed Novagen protocol TB054 (similar to a procedure previously used in our laboratory) (19). Briefly, cells were lysed by treatment with lysozyme, and this step was followed by sonication. Inclusion bodies were solubilized in buffer containing 6 M guanidine-HCl, and denatured proteins were bound to the His-Bind resin. Proteins were refolded while bound to the column by step-wise washes with buffers containing decreasing concentrations of denaturant (6 to 0 M guanidine-HCl). After a wash with 25 mM imidazole, bound σ32 was eluted with 250 mM imidazole, and fractions containing σ32 were dialysed against storage buffer (30 mM Tris-HCl, pH 7.5, 50% glycerol, 100 mM NaCl, 0.1 mM EDTA).
Runoff transcription.
DNA templates for in vitro transcription were obtained by amplification of plasmid DNA containing PgroE as well as sequences up- and downstream from it. To reconstitute holoenzyme containing σ32, core RNAP was incubated with a fivefold excess of purified WT and H107A σ32 in binding buffer (40 mM Tris, pH 7.9, 35% glycerol, 250 mM NaCl, 0.1 mM EDTA) on ice for 1 h. After incubation of DNA and RNAP for 3 min at 37°C, transcription buffer was added to give a 20-μl reaction mixture containing 7.5 nM promoter DNA, 150 nM reconstituted RNAP, 40 mM Tris-HCl, pH 7.9, 9% glycerol, 100 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol, 0.5 mg/ml bovine serum albumin, 250 μM ATP, 250 μM GTP, 250 μM CTP, 25 mM UTP, and 0.15 μM [α-32P]UTP. This reaction mixture was incubated for an additional 5 min at 37°C. Then, a 200 μM concentration of cold UTP was added, and incubation was continued for 2 min. Reactions were stopped by the addition of Tris-saturated phenol. Phenol extraction was followed by phenol-chloroform-isoamyl alcohol extraction; nucleic acids were ethanol precipitated and redissolved in formamide gel loading buffer (80% formamide, 10 mM NaOH, 1 mM EDTA, and 0.1% each of xylene cyanol FF and bromophenol blue). After incubation at 90°C for 3 min, the samples were subjected to electrophoresis in an 8% acrylamide-7 M urea-TBE (Tris-borate-EDTA) gel, followed by phosphorimaging analysis to determine relative intensities of the RNA product bands. Heparin challenge experiments were performed under the same conditions as above, except that 5 μg/ml heparin was added, and incubation was continued for different lengths of time before transcription was initiated. To determine the total number of DNA-RNAP complexes, reactions were also carried out without heparin challenge. We also carried out experiments where the RNAP was incubated with different concentrations of heparin for 2 min, added prior to the addition of the DNA. Then the reaction mixture was incubated an additional 5 min at 37°C, followed by runoff transcription as described above.
RNA isolation and cDNA synthesis.
Aliquots from overnight cultures of WT and H107A σ32 grown at 32°C with shaking at 230 rpm in MOPS (morpholinepropanesulfonic acid) minimal medium supplemented with thiamine at 0.1 μg/ml (16) were inoculated in 5 ml of the same growth medium to an OD600 of 0.1. Growth was continued at 32°C until the OD600 reached 0.6. The cells were collected by brief centrifugation and flash-frozen in a dry ice-ethanol bath. RNA was isolated using a RiboPure-Bacteria kit from Ambion, as recommended by the manufacturer. The quality of RNA was assessed from the appearance and relative intensities of the 16S and 23S rRNA bands. The yield was determined by spectrophotometry. cDNA synthesis was performed using random hexamer primers and Superscript II reverse transcriptase (both from Invitrogen) as recommended for prokaryotic target preparation by Affymetrix. cDNA yield was determined by spectrophotometry.
Semiquantitative reverse transcription-PCR.
cDNA generated as described above was used as a template for PCR. Primers for the flu and rrsH (16S RNA) genes were as described previously (1). Amplification was performed with ReadyMix Taq PCR mix (Sigma) under the following conditions: following the initial denaturation at 94°C for 3 min, each PCR cycle was 60 s at 92°C, 60 s at 57°C, and 130 s at 72°C. The PCR products were loaded onto a 2% agarose ethidium bromide-stained gel. DNA molecular weight markers were also included to determine whether the amplification product had the expected size. The optimal number of PCR cycles was determined empirically to ensure that the amplification product is clearly visible on an agarose gel and can be quantified and also that amplification is still in the exponential range prior to reaching a plateau. For these reasons, only the data from the 19th and 22nd cycles were used for calculations. Images of the agarose gels were acquired with a Syngene image analysis system, and quantification of the bands was performed. Band intensity was expressed as relative absorbance units. Mean and standard deviations of relative flu cDNA amounts were calculated after normalization to rrsH (16S RNA) (1).
Autoaggregation and sand column adhesion assays.
For direct observation of the cells, strains were grown overnight in enriched M9 medium at 30°C. From these cultures 2 μl was taken and spotted on glass microslides. Microscopic observation and image acquisition were performed on a Leica DM 2500 oil immersion digital microscope with a 100× objective and were processed for display using Adobe Photoshop software.
For the autoaggregation assay, the cells were grown overnight in enriched M9 minimal medium. After the OD600 of the cultures was adjusted to 0.1 by dilution with fresh medium, the cells were allowed to grow until the OD600 reached 0.7 to 0.8. At this point 4-ml portions of each culture were transferred to 5-ml tubes so that the extent of autoaggregation could be determined from the sedimentation during static incubation at room temperature. At intervals of 30 min, 75 μl of the upper layer was removed from each tube, and the OD600 was determined. A final reading was taken after 18 h of static incubation.
To determine the ability of the bacteria to adhere to a stationary matrix, the cells were grown at 30°C in enriched M9 medium to an OD600 of 0.7. The cells were collected by centrifugation, resuspended in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 100 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) to an OD600 of 1.0, and passed over a sand column. The percentage of attached bacteria was determined from the reduction in the OD600 as described previously (1).
RESULTS
Conservation across proteobacteria of residues homologous to H107 of E. coli σ32.
We determined the extent to which the identity of the H107 is conserved in heat shock sigma factors by using the collection of heat shock sigma factors previously analyzed by Green and Donahue (7). The results are displayed in Fig. 1 according to the groups of proteobacteria assigned by these investigators. With few exceptions, the penultimate amino acid residue at the C-terminal end of region 2.3 is seen to be a histidine (H), as in E. coli σ32, or a tryptophan (W), as in E. coli σ70. In the σ32 of all alpha- and deltaproteobacteria, a W is found at this position, while all gammaproteobacteria have an H. The small group of four betaproteobacteria have either a histidine (2) or another amino acid residue (T and Y are seen once each). Further examination of the sequence database indicates similar behavior for other amino acids: Y104 tracks with W107, as these residues also do in the primary sigma factor of E. coli, σ70 (Fig. 1, bottom line), while V102 and F104 track with H107. In view of its apparent importance to the group of gammaproteobacteria, which includes E. coli as well as several other pathogens, and our previous extensive characterization of W433 in E. coli, we chose to subject the H107 of E. coli σ32 to further study.
FIG. 1.

Comparison of sequences of the conserved region 2.3 among sigma factors of the proteobacteria. The 43 heat shock sigma factors were grouped according to Green and Donohue (7). The sequence of the E. coli heat shock sigma factor σ32 is underlined. Bold lettering highlights either the Y--W pattern seen in the alpha- and deltaproteobacteria or the V-F--H pattern characteristic of the gammaproteobacteria and some betaproteobacteria.
In vitro effects of the H107A substitution in E. coli σ32.
As the activities of WT and H107A σ32 factors in directing transcription from the GroE promoter in vivo were similar, it seemed likely that the H107A substitution did not affect the interaction of the core with σ32(12). With purified H107A σ32 and core RNAP, we have confirmed that this is indeed the case in vitro (data not shown). To probe whether effects of the H107A substitution could be observed on transcription from PgroE in vitro, we carried out runoff assays. The results are shown in Fig. 2A. As expected, the substitution does not affect the end-to-end transcription from the fragment used (band near top of gel). No effect of the substitution is seen on transcription from PgroE: the intensity of the band for the H107A RNAP was about 114% that for WT RNAP (Fig. 2B). By either DNase I footprinting or KMnO4 probing (data not shown), we were unable to detect any differences between complexes of WT and H107A σ32 containing holoenzymes with the PgroE variant promoter (Fig. 2C), which is identical to PgroE from −47 to −9 (27). However, significant effects were evident for another RNA band (Fig. 2C, Minor promoter): in transcription by H107A RNAP this band had 45% of the intensity compared to transcription by WT RNAP (Fig. 2B). The likely origin of this band is a σ32 promoter of unknown function within the GroES gene. Such intragenic promoters, albeit not this particular one, had previously been described (26). In comparison with PgroE, significant sequence differences are seen both in the upstream and in the downstream areas of the −10 region. The upstream and downstream differences were introduced separately as well as jointly into PgroE to obtain the promoter variants shown in the bottom three lines of Fig. 2C. With these promoters, greater differences were seen between WT and H107A RNA polymerases: 70% of WT RNAP transcription was detected for the H107A RNAP with the PgroE variant with a C or T mutation and 50% was detected for PgroE with joint C and T mutations (Fig. 2D and E). The latter level is similar to that observed for the putative intragenic promoter that formed the basis for the substitutions we made (Fig. 2A and B). Thus, the effects of the H107A substitution are promoter dependent.
FIG. 2.

The effect of substitutions in σ32 and promoter DNA on runoff transcription from a PgroE-like promoter and its variants. Runoff transcription was carried out on a DNA fragment containing PgroE and RNAP holoenzyme containing WT σ32 or H107A σ32. (A) Runoff transcription from PgroE with WT and H107A. Bands were assigned as indicated. (B) Quantification of the effect of the H107A substitution on transcription from PgroE and the minor promoter. Each bar represents the ratio of transcription by RNAP containing H107A σ32 and WT σ32 for that particular promoter. (C) Promoters used in this work. Base identities in the −10 and −35 regions are shown in bold. The top line shows the sequence of the WT E. coli PgroE; the DNA template used, extending upstream to position −109 and downstream to position +390 in relation to the transcription start site also contained a minor σ32 promoter, shown in the second line (important differences with the PgroE [above] are indicated by underlining). The bottom four templates have a PgroE-like promoter, identical to PgroE through position −9, used in prior work (12, 27), as well as variants of this promoter (with base sequence differences underlined). Sequences downstream of −9, derived from the pQF50K vector (27), are shown in lowercase letters. (D) Runoff transcription assays. Pairwise comparisons of transcription by RNAP containing WT (lanes 2, 4, 6, and 8) and H107A (lanes 3, 5, 7, and 9) σ32 from the bottom four templates indicated in panel C. Shown is the gel analysis of the RNA products, labeled by the inclusion of [32P]UTP in the reaction mixtures. The bands were assigned as indicated. (E) Normalized levels of radioactivity in RNA bands resulting from transcription by RNAP containing WT and H107A σ32 on the promoters shown in panel D above. For each bar, representing one promoter, transcription with WT RNAP is 100%. mut, mutant.
To determine whether complexes of PgroE with WT and H107A RNAP were differentially stable, they were challenged with 5 μg/ml heparin for various time intervals before the transcription reactions were carried out (Fig. 3A and B). The data presented in Fig. 3A and B show that complexes formed with RNAP containing both WT and H107A σ32 dissociated within 2 min to levels of 70% and 30%, respectively, of the complexes present prior to the heparin challenge. While this result is indicative of a clear defect of the H107A substitution, the nature of the complexes that stayed transcription competent in the presence of the heparin remained unclear. Were they heparin resistant, or was the small amount of heparin added sufficient to inactivate only a fraction of the RNAP in the solution? To distinguish between these alternatives, we added heparin (at various concentrations) to the RNAP prior to incubation with the DNA, followed by runoff transcription. The results are shown in Fig. 3C. When heparin was added to a concentration of 5 μg/ml, the fraction of active RNAP was similar to that when heparin was added after the DNA (Fig. 3B), and it decreased upon addition of higher heparin concentrations. However, in each case more complexes containing WT Eσ32 survived the heparin addition than with H107A Eσ32, thus revealing an effect of the H107A substitution in σ32 on the affinity of Eσ32 for PgroE. In addition, our observations are consistent with both an unstable Eσ32-promoter complex and a nonsaturating concentration of heparin in the experiments for which the results are shown in Fig. 3A and B. It is evident that complexes of Eσ32 and promoter DNA are considerably less stable than those of Eσ70 and its cognate promoters, which can have half-lives of hours (see, e.g., reference 19).
FIG. 3.
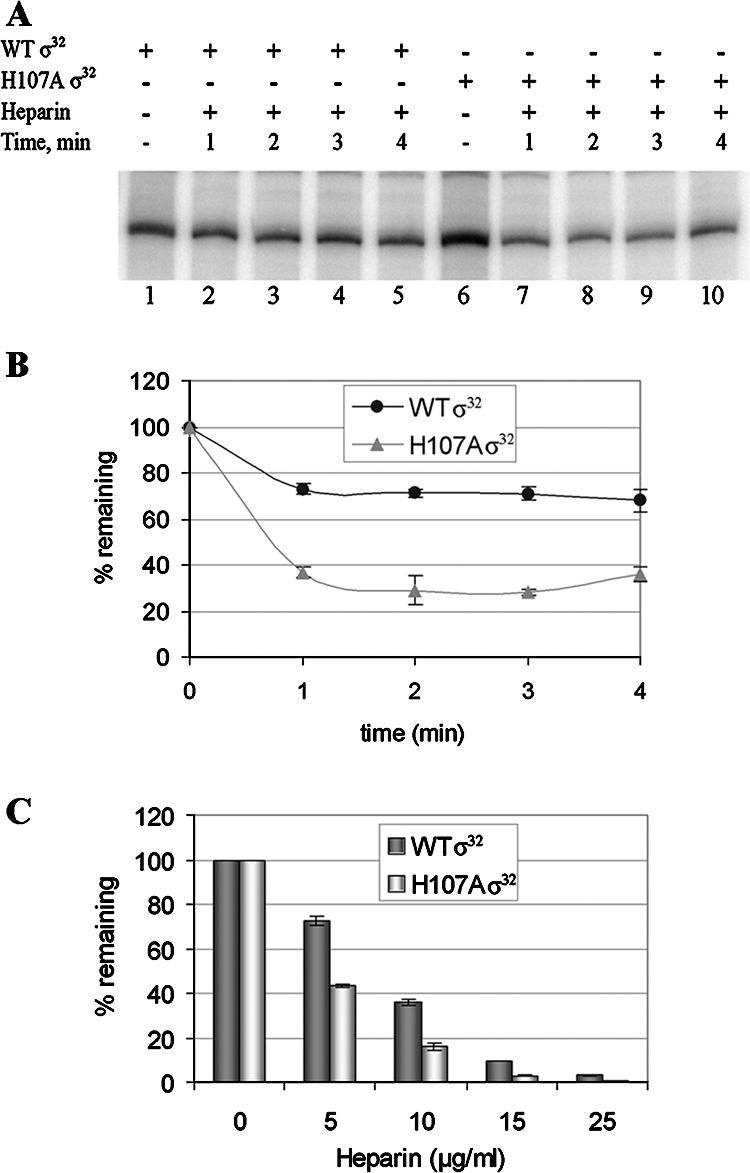
The H107A substitution affects formation of stable open complexes at PgroE: runoff transcription following heparin challenge. Comparison of transcription by RNAP containing WT or H107A σ32. (A) Image of the gel. Lanes 1 to 5, WT σ32; lanes 6 to 10, H107A σ32. The heparin challenge was for the times indicated in the boxes above the lanes. (B) Band intensities as a function of time. The zero time point (just prior to the heparin challenge) was defined as having 100% of transcription activity; the values on the y axis refer to the percentage of intensity of the band at each time point compared to the band at time zero for each of the two RNAPs. (C) The effects of heparin added at different concentrations to reaction mixtures containing WT and H107A RNAP prior to the addition of promoter DNA.
In vivo effects of the H107A substitution in E. coli σ32.
Despite the fact that H107 is highly conserved, with the GroE promoter we found only relatively small consequences of the H107A substitution in vitro (see above) and no effects in prior experiments in vivo (12). However, an alanine substitution for the homologous amino acid W433 in σ70 (19) resulted in about a twofold reduction in activity in vivo. To determine whether the H107A substitution might affect cellular function despite the lack of a notable effect on PgroE activity, we compared the growth curves of the strain CAG9301, a derivative of MG1655 bearing an endogenous σ32 gene inactivated by insertion of a kanamycin cassette (31), and the same strain containing the WT and H107A σ32 on the low-copy-number plasmid pSAKT32. As induced cells had been found to be compromised for growth, we carried out all subsequent experiments without inducing σ32 synthesis, relying instead on the basal levels of its expression, which are still clearly detectable by Western analysis (data not shown). The results are shown in Fig. 4. In Fig. 4A, nearly superimposing curves are obtained for the three strains tested. However, subsequent to heat shock (42°C), a gradation is evident in the final OD600 values attained: in descending order of the height of their growth curves, the strains are CAG9301 with WT σ32, CAG9301 with H107A σ32, and CAG9301 (minus-σ32 strain) (Fig. 4B). We had previously shown that the levels of WT and H107A σ32 were similar both prior to heat shock (12) and 2 h after the temperature upshift (data not shown), when the difference in growth between the cells with WT and H107A Eσ32 is already evident (Fig. 4). We conclude from these results that the H107A substitution in σ32 must have significant consequences on transcription from some promoter(s) in vivo.
To determine a more complete spectrum of effects of the H107A substitution in vivo, microarray experiments were carried out (data not shown). The greatest effect of the H107A substitution in σ32 was enhanced expression of the flu gene, encoding the outer membrane fluffing protein involved in biofilm formation (21), and gene b2001, immediately downstream. We carried out these experiments under noninducing conditions, as the increases in flu mRNA levels in cells containing H107A σ32 were about fivefold whether σ32 expression was induced or not. Under these conditions the elevated expression of the flu and b2001 genes was the only difference between cells with the H107A and WT σ32. In induced cells expression of 12 other genes was increased twofold or more, and expression of 16 genes was decreased twofold or more in H107A cells (data not shown). We have independently confirmed the microarray result for flu expression by carrying out PCR amplifications by a method (1) akin to that of reverse transcription-PCR (see Materials and Methods). The levels for the flu mRNA were 2.5-fold higher in the E. coli cells carrying H107A σ32 than in those containing WT σ32 (Fig. 5), which is in reasonably good agreement with the microarray experiments.
FIG. 5.

Differential expression of flu mRNA in cells containing WT or H107A σ32. The mRNA was first reverse transcribed as indicated in Materials and Methods and then amplified with flu- or 16S rRNA-specific primers (the latter for normalization) for the numbers of cycles indicated. Only the observed bands after 19 and 22 cycles were used to calculate the relative amounts of flu mRNA in cells with WT σ32 and H107A σ32.
The biological consequences of the overexpression of Flu protein were found to manifest themselves in a variety of ways. First, the cells containing only the σ32 gene inactivated by insertion of the kanamycin cassette are seen to be “filamenting” (Fig. 6, left), as previously shown with this strain (31); in contrast, the bacteria additionally containing the plasmid with the WT σ32 gene (Fig. 6, center) are observed to be present mostly as individual cells, while those with the H107A σ32 gene display a tendency to aggregate (Fig. 6, right). Second, the cells containing the H107A σ32 sediment over a period of 2 h, in contrast to cells containing WT σ32 or those that express no active σ32 (Fig. 7A), for which no significant sedimentation was observed in 18 h. This is consistent with the autoaggregation of the cells containing the H107A σ32 gene, seen above (Fig. 6, right). Remarkably, cells bearing the H107W substitution which causes a 40% reduction in PgroE expression in vivo (12) behave similarly to those expressing the WT σ32 gene in this regard (data not shown). Third, the cells expressing the H107A σ32 have an increased penchant for attaching themselves to a sand matrix (Fig. 7B). Thus, the cells expressing H107A σ32 are more prone than those expressing WT σ32 to interact with not only each other but also a stationary matrix (e.g., sand).
FIG. 6.

σ32 Dependence of the properties of cells growing in enriched M9 medium at logarithmic phase. All cells were grown at 30°C in the absence of IPTG: CAG9301 cells for which the chromosomal σ32 gene was inactivated by insertion of a kanamycin cassette (left), CAG9301 cells containing a plasmid-borne gene expressing WT σ32 (center), and CAG9301 cells containing a plasmid-borne gene expressing H107A σ32 (right).
FIG. 7.
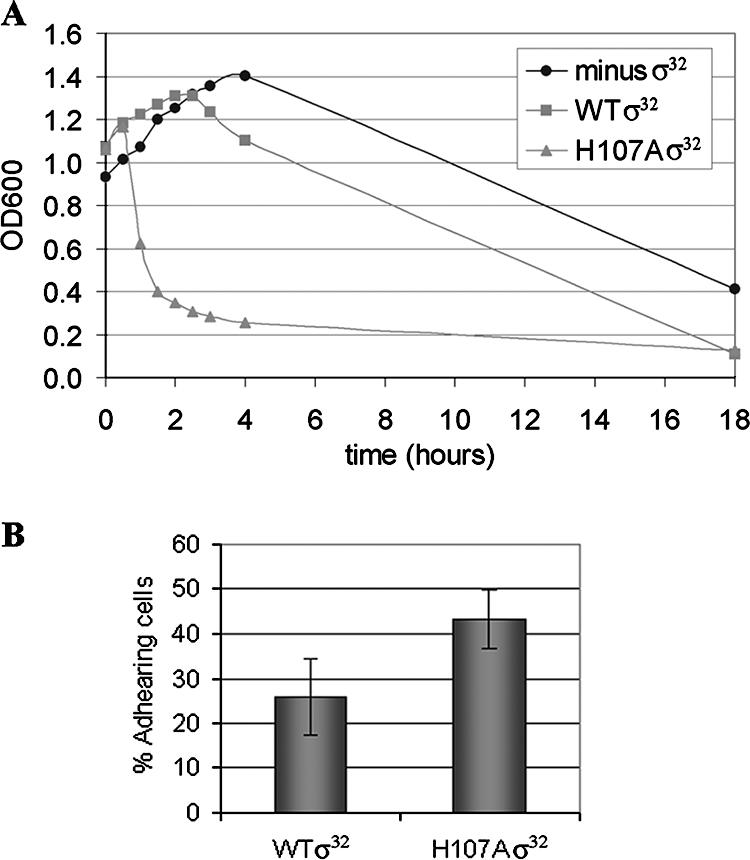
Properties of CAG9301 cells expressing H107A σ32 are consistent with overexpression of the Flu protein. All cells were grown at 30°C in the absence of any added IPTG. (A) Increased sedimentation due to aggregation of cells containing the H107A σ32 gene. After the cells had been grown to an OD600 of about 0.8, agitation was ceased. Sedimentation was determined from the decrease in the OD600 of the upper layer of the cultures. (B) Increased surface adhesion of cells containing the H107A σ32 gene. Cells grown to an OD600 of 0.7 to 0.8 were passed over a bed of sand, and the adhesion was determined from the reduction in the OD600 of the effluent. The results are averages of three independent experiments. The observed difference between WT and H107A σ32 was found to be significant with a P value of 0.007 in a paired t test.
DISCUSSION
A role for the H107 residue of E. coli σ32 in open complex formation.
The H107 residue in region 2.3 of E. coli σ32 is strictly conserved in the heat shock sigma factors of the gammaproteobacteria. Interestingly, it occurs as part of the pattern V-F--H. On the other hand, the alpha- and deltaproteobacteria have Y and W at the positions homologous to the 104 and 107 of σ32 (i.e., the pattern Y-W). The latter are identical to the sequence that is also found in E. coli σ70 as Y430 and W433 (Fig. 1), residues that play important roles in open complex formation (6, 25). The finding that the H107A substitution renders holoenzyme containing σ32 more sensitive to heparin challenge demonstrates that the H107 of σ32 is important in the formation and/or maintenance of the open complex. The W433A substitution has been reported to affect the kinetics of both association (19) and dissociation (23) of RNAP and promoter DNA. Thus, in σ32 the nonaromatic residue H107 may have a function similar to that of the homologous residue, W433, in σ70, although the role of W433 in open complex formation is not yet fully understood (5, 23). This is an important result that was not evident from our previous assays of the effects of the H107A substitution with PgroE (12). In studies of the effects of substitutions for W433 of σ70, its replacement by histidine was tolerated to some extent: it was not as deleterious as leucine or alanine but more so than the aromatic amino acid phenylalanine or tyrosine. While histidine lacks a planar aromatic group, it would be able to interact electrostatically with a base that was rotated (flipped) out of the double-helical DNA while remaining coplanar with it (13), an orientation similar to that of aromatic amino acids which are stacking to a DNA base. Thus, there may not be a unique solution to the problem of how to initiate promoter DNA strand separation in formation of the open complex.
Separate substitutions in PgroE, not only in the melted region but also upstream of it in the conserved C residues, were found to increase sensitivity to the σ32 H107A substitution. The fact that the H107A substitution has an effect on mutant promoters with substitutions outside the melted region is not inconsistent with specific contacts between the H107 of σ32 and DNA bases in the melted region. It may be that the different kinetic characteristics of the promoters, changed by the substitutions, make them sensitive or insensitive to the disruption by the H107A. PgroE (essentially a consensus promoter for Eσ32 [27]) may be so strong that it can effectively overcome the defect of the H107A substitution. In this respect, the results would be similar to those obtained with σ70 bearing substitutions in region 2.3. We showed that for strong promoters, the effects of single (23) or even triple (2) substitutions in region 2.3 of σ70 were mitigated.
Potential links between σ32 and expression of the flu gene.
The connection between the H107A substitution in σ32 and its effects on in vivo transcription of the flu gene is as yet not clear. The expression of Flu (previously called Ag43) is known to be under OxyR control (11): the reduced form of OxyR represses Flu expression (22). No involvement of σ32 in the regulation of OxyR levels has yet been documented, and our microarray experiments did not provide any indication of such control (data not shown). However, our microarray data were consistent with the observation that cells containing the H107A σ32 experience oxidative stress (data not shown), and oxidized OxyR has been reported to be unable to repress expression of the flu gene (22), establishing a possible link between H107A σ32 and Flu expression. Another intriguing possibility is that the holoenzyme containing the H107A σ32 has acquired the novel feature of recognizing a non-σ32 promoter. It is known that holoenzymes containing σ70 can recognize σ32 promoters (17, 26), reflecting the evident homology between the two classes of promoters in the −35 region and some more subtle homology in the −10 region. To the best of our knowledge, the inverse has not been described, but possibly the H107A substitution would facilitate such cross-recognition. This interesting possibility is compatible with the lack of flu expression in the strain that does not express σ32 and merits further investigation. The putative promoter may be biologically relevant even if the RNAP containing the H107A substitution enhances its expression to nonphysiological levels, resulting in a readily observable increase in Flu expression.
Acknowledgments
This work was supported by NIH grant GM 31808 to P.L.dH.
We thank John Campbell (UIUC) for advice with the interpretation of the microarray results, Jeff Coller and T. J. Sweet (Case Western Reserve University) for help with acquisition of the images shown in Fig. 6, our laboratory colleagues for advice, Lisa Schroeder and Vicki Cook for critically reading the manuscript, and two anonymous reviewers for their valuable comments.
Footnotes
Published ahead of print on 5 October 2007.
REFERENCES
- 1.Beloin, C., K. Michaelis, K. Lindner, P. Landini, J. Hacker, J.-M. Ghigo, and U. Dobrindt. 2006. The transcriptional antiterminator RfaH represses biofilm formation in Escherichia coli. J. Bacteriol. 188:1316-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook, V. M., and P. L. deHaseth. 2007. Strand opening-deficient E. coli RNA polymerase facilitates investigation of closed complexes with promoter DNA: effects of DNA sequence and temperature. J. Biol. Chem. 282:21319-21326. [DOI] [PubMed] [Google Scholar]
- 3.deHaseth, P. L., M. Zupancic, and M. T. Record, Jr. 1998. RNA polymerase-promoter interaction: the comings and goings of RNA polymerase. J. Bacteriol. 180:3019-3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson, J. W., V. Vaughn, W. A. Walter, F. C. Neidhardt, and C. A. Gross. 1987. Regulation of the promoters and transcripts of rpoH, the Escherichia coli heat shock regulatory gene. Genes Dev. 1:419-432. [DOI] [PubMed] [Google Scholar]
- 5.Fenton, M. S., and J. D. Gralla. 2003. Roles for inhibitory interactions in the use of the −10 promoter element by σ70 holoenzyme. J. Biol. Chem. 278:39669-39674. [DOI] [PubMed] [Google Scholar]
- 6.Fenton, M. S., H. J. Lee, and J. D. Gralla. 2000. Escherichia coli promoter opening and −10 recognition: mutational analysis of σ70. EMBO J. 19:1130-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green, H. A., and T. J. Donohue. 2006. Activity of Rhodobacter sphaeroides RpoHII, a second member of the heat shock sigma factor family. J. Bacteriol. 188:5712-5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross, C., C. Chan, A. Dombroski, T. Gruber, M. Sharp, J. Tupy, and B. A. Young. 1998. The functional and regulatory roles of sigma factors in transcription. Cold Spring Harb. Symp. Quant. Biol. 63:141-155. [DOI] [PubMed] [Google Scholar]
- 9.Helmann, J. D. 1994. Bacterial sigma factors, p. 1-17. In R. C. Conaway and J. W. Conaway (ed.), Transcription: mechanisms and regulation. Raven Press, New York, NY.
- 10.Helmann, J. D., and J. Chamberlin. 1988. Structure and function of bacterial sigma factors. Annu. Rev. Biochem. 57:839-872. [DOI] [PubMed] [Google Scholar]
- 11.Henderson, I. R., and P. Owen. 1999. The major phase-variable outer membrane protein of Escherichia coli structurally resembles the immunoglobulin A1 protease class of exported protein and is regulated by a novel mechanism involving Dam and OxyR. J. Bacteriol. 181:2132-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kourennaia, O. V., L. Tsujikawa, and P. L. deHaseth. 2005. Mutational analysis of Escherichia coli heat shock transcription factor sigma 32 reveals similarities with sigma 70 in recognition of the −35 promoter element and differences in promoter DNA melting and −10 recognition. J. Bacteriol. 187:6762-6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar, N. V., and G. Govil. 1984. Theoretical studies on protein-nucleic acid interactions. III. Stacking of aromatic amino acids with bases and base pairs of nucleic acids. Biopolymers 23:2009-2024. [DOI] [PubMed] [Google Scholar]
- 14.Landick, R., V. Vaughn, E. T. Lau, R. A. VanBogelen, J. W. Erickson, and F. C. Neidhardt. 1984. Nucleotide sequence of the heat shock regulatory gene of E. coli suggests its protein product may be a transcription factor. Cell 38:175-182. [DOI] [PubMed] [Google Scholar]
- 15.Lonetto, M., M. Gribskov, and C. A. Gross. 1992. The sigma 70 family: sequence conservation and evolutionary relationships. J. Bacteriol. 174:3843-3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neidhardt, F. C., P. L. Bloch, and D. F. Smith. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newlands, J. T., T. Gaal, J. Mecsas, and R. L. Gourse. 1993. Transcription of the Escherichia coli rrnB P1 promoter by the heat shock RNA polymerase (E sigma 32) in vitro. J. Bacteriol. 175:661-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nonaka, G., M. Blankschien, C. Herman, C. A. Gross, and V. A. Rhodius. 2006. Regulon and promoter analysis of the E. coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes Dev. 20:1776-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panaghie, G., S. E. Aiyar, K. L. Bobb, R. S. Hayward, and P. L. deHaseth. 2000. Aromatic amino acids in region 2.3 of Escherichia coli sigma 70 participate collectively in the formation of an RNA polymerase-promoter open complex. J. Mol. Biol. 299:1217-1230. [DOI] [PubMed] [Google Scholar]
- 20.Rosen, R., and E. Z. Ron. 2002. Proteome analysis in the study of the bacterial heat-shock response. Mass Spectrom. Rev. 21:244-265. [DOI] [PubMed] [Google Scholar]
- 21.Schembri, M. A., K. Kjoergaard, and P. Klemm. 2003. Global gene expression in Escherichia coli biofilms. Mol. Microbiol. 48:253-267. [DOI] [PubMed] [Google Scholar]
- 22.Schembri, M. A., and P. Klemm. 2001. Coordinate gene regulation by fimbriae-induce signal transduction. EMBO journal. 20:3074-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroeder, L. A., A.-J. Choi, and P. L. deHaseth. 2007. The −11A of promoter DNA and two conserved amino acids in the melting region of σ70 both directly affect the rate limiting step in formation of the stable RNA polymerase-promoter complex, but they do not necessarily interact. Nucleic Acids Res. 35:4141-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shamovsky, I., M. Ivannikov, E. S. Kamdel, D. Gershon, and E. Nudler. 2006. RNA-mediated response to heat shock in mammalian cells. Nature 440:556-560. [DOI] [PubMed] [Google Scholar]
- 25.Tomsic, M., L. Tsujikawa, G. Panaghie, Y. Wang, J. Azok, and P. L. deHaseth. 2001. Different roles for basic and aromatic amino acids in conserved region 2 of Escherichia coli σ70 in the nucleation and maintenance of the single-stranded DNA bubble in open RNA polymerase-promoter complexes. J. Biol. Chem. 276:31891-31896. [DOI] [PubMed] [Google Scholar]
- 26.Wade, J. T., D. C. Roa, D. C. Grainger, D. Hurd, S. J. W. Busby, K. Struhl, and E. Nudler. 2006. Extensive functional overlap between σ factors in Escherichia coli. Nat. Struct. Mol. Biol. 13:806-814. [DOI] [PubMed] [Google Scholar]
- 27.Wang, Y., and P. L. deHaseth. 2003. Sigma 32-dependent promoter activity in vivo: sequence determinants of the groE promoter. J. Bacteriol. 185:5800-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yura, T., H. Nagai, and H. Mori. 1993. Regulation of the heat-shock response in bacteria. Annu. Rev. Biochem. 47:321-350. [DOI] [PubMed] [Google Scholar]
- 29.Yura, T., and K. Nakahigashi. 1999. Regulation of the heat-shock response. Curr. Opin. Microbiol. 2:153-158. [DOI] [PubMed] [Google Scholar]
- 30.Zhao, K., M. Liu, and R. R. Burgess. 2005. The global transcriptional response of Escherichia coli to induced σ32 protein involves σ32 regulon activation followed by inactivation and degradation of σ32 in vivo. J. Biol. Chem. 280:17758-17768. [DOI] [PubMed] [Google Scholar]
- 31.Zhou, Y. N., N. Kusukawa, J. W. Erickson, C. A. Gross, and T. Yura. 1988. Isolation and characterization of Escherichia coli mutants that lack the heat shock sigma factor σ32. J. Bacteriol. 170:3640-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]