

Abstract
Homology requirements for Moloney murine leukemia virus recombination were addressed in this study by monitoring titer defects observed when acceptor/donor template identity lengths were systematically reduced. Recombination acceptors with at least 16 contiguous bases of donor template identity were recognized as efficiently as longer acceptors. In contrast, a sharp 1-log titer drop was observed for an acceptor of only 15 bases long, with an additional 1-log titer decline for an 8-base acceptor and further decreases for shorter acceptors. Eighty-three independent nonhomologous recombination products were sequenced to examine recombination template selection in the absence of significant sequence identity. These replication products contained a total of 152 nonhomologous crossover junctions. Forced copy choice models predict that forced nonhomologous recombination should result in DNA synthesis to the donor template's 5′ end, followed by microidentity-guided acceptor template selection. However, only a single product displayed this structure. The majority of examined nonhomologous recombination products contained junction-associated sequence insertions. Most insertions resulted from the use of one or more tertiary templates, recognizable as discontiguous portions of viral or host RNA or minus-strand DNA. The donor/acceptor template microidentity evident at most crossovers reconfirmed the remarkable capability of the reverse transcription machinery to recognize short regions of sequence identity. These results demonstrate that recruitment of discontiguous host or viral sequences is a common way for retroviruses to resolve nonhomologous recombination junctions and provide experimental support for the role of splinting templates in the generation of retroviral insertions.
Retroviral genetic recombination does not involve nucleic acid breakage and rejoining but instead results from template switching by reverse transcriptase (RT) during viral DNA synthesis (2). Early work demonstrated that the reassortment of retroviral genes is so frequent that even markers approximately 1 kilobase apart behave essentially as if they are unlinked (29). More-recent studies assessing crossover frequencies provide the basis for these observations by demonstrating that recombinogenic template switching likely occurs several times during the synthesis of nearly all retroviral DNAs (21, 37, 49, 72).
How elongating reverse transcription complexes recognize and recruit secondary templates and how a growing DNA strand is transferred to an acceptor template are areas of active investigation. Models for recombination during both plus- and minus-strand DNA synthesis have been proposed, with experimental evidence supporting a dominant role for minus-strand recombination (9, 22, 67). Forced copy choice models for minus-strand recombination suggest that RT/primer-template complex dissociation at a broken template end precedes recombinogenic template switching (9). However, evidence that factors other than template integrity can modulate recombination frequency demonstrates that template breakage is not a prerequisite for recombinogenic template switching (42, 44). An alternate model for minus-strand homologous recombination—the acceptor invasion model—suggests that minus-strand DNA sequences behind the growing point for DNA synthesis, which are unmasked by the removal of RNA template segments by RNase H, recruit acceptor templates by base pairing with them and that template switching itself involves a subsequent repositioning of the primer strand's 3′ end from the donor to the acceptor template (5, 8, 50).
Most retroviral recombination occurs in regions of donor/acceptor template identity, with nonhomologous recombination occurring roughly 100- to 1,000-fold less frequently than homologous recombination (69, 70). The frequencies of retroviral recombination are roughly proportional to the lengths of sequence identity (70). Recombination between similar, or “homologous,” sequences occurs less frequently than recombination between identical sequences, with recombination becoming less frequent the more dissimilar templates are to one another (1). When nonhomologous recombination is monitored, short regions of sequence microidentity are observed at the crossover junctions in some instances, due to the retroviral replication machinery's remarkable capability of recognizing short patches of sequence identity in the context of otherwise nonhomologous sequence (19, 60, 65). However, many nonhomologous crossovers display no microidentity or even have sequence insertions at the crossover junctions (41). Based on the similarity between certain sequence insertions in human immunodeficiency virus type 1 clinical isolates and segments of the human genome, we have previously proposed that many of the insertions observed in retroviral genomes may result from the use of short sections of discontiguous nucleic acids as tertiary templates to bridge nonhomologous crossover junctions (3, 57).
The current work employs a novel Moloney murine leukemia virus (MMLV) forced template switching assay to examine how systematic variation in lengths of donor/acceptor identity affects recombinogenic template switching. Acceptor template selection in the absence of appreciable donor/acceptor identity and the mechanisms employed by the reverse transcription machinery to resolve nonhomologous recombination junctions were addressed by sequencing and analyzing a large number of individual forced nonhomologous recombination products.
MATERIALS AND METHODS
Plasmids.
The retroviral inside-out (RIO) vectors used in this report were based on a previously described forced homologous recombination vector called 30TM (for 30-terminal match) (33). RIO vector structures are introduced in Fig. 1. For the experiments here, overlap extension PCR was used to modify the 30-base acceptor (5′-GCGCCAGTCCTCCGATTGACTGAGTCGCCC-3′) of the 30TM vector to create the 1-terminal mismatch (1TMM) and shortened acceptor 27, 24, 21, 20, 19, 18, 17, 16, 15, 12, 10, 8, 5, and 0TM vectors (33). pRSVpuro (pSF116-1), which contains an intronless puromycin resistance gene, was used as a standard non-RIO (that is, not inside-out; possessing two long terminal repeats [LTRs]) MMLV-based vector control (33).
FIG. 1.

Forced recombination assay. (A) Two-vector recombination assay as described in reference 43. Newly synthesized DNA undergoes a recombinogenic strand transfer from the 5′ end of an RNA donor (D) template to the 3′ end of a distinct RNA acceptor (A) template. PRSV, Rous sarcoma virus promoter; pbs, primer binding site; ppt, polypurine track. (B) RIO template. DNA is shown as a complete circle. The RIO template contains a single, modified, central LTR (ΔU U5). An SV40 promoter-driven puromycin resistance gene containing an artificial intron is separated into 5′ (puro-, in-) and 3′ (-tron, -mycin) portions. Homologous D and A regions are designated within the intron. (C) Recombination during reverse transcription using RIO vector. Upon reaching the 5′ end of the donor template, the newly synthesizing DNA is forced to undergo a recombinogenic strand transfer to a region of sequence identity within the acceptor template. After completing plus-strand synthesis and transfer, the final provirus resembles a retroviral provirus. The puromycin resistance gene and intron are reassembled, and two LTRs flank the provirus. Ψ is the packaging signal, and PSV40 indicates the SV40 late promoter. Primers shown in the bottom row are those used to analyze products as for Fig. 2D.
The forced nonhomologous recombination assay vector used here, designated “humanTM,” was derived from the 5TM vector by modifying the splice donor region and inserting a fragment of human sequence to serve as a nonhomologous recombination acceptor template. In previous studies with the 5TM vector, some products appeared to result from the use of limited identity in the vicinity of the splice donor (33). To reduce this unwanted alternate target product, the RIO vector acceptor region was modified by PCR to ablate this microidentity in humanTM. To introduce a human sequence acceptor, DNA isolated from 293T cells by use of a DNeasy tissue kit (QIAGEN) was digested with EcoRI, shotgun cloned into pBluescript SK+, and sequenced. A 590-base fragment from human chromosome 14 (map position 21710256 to position 21710845 of GenBank accession no. NT_0264737) was selected for further use and engineered into humanTM, thus replacing the limited acceptor template fragment of the parental 5TM vector.
Cells and virus.
293T cells (human embryonic kidney cells expressing simian virus 40 [SV40] T antigen) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 1% penicillin-streptomycin. RIO vector plasmids were cotransfected with MMLV gag-pol and MMLV ecotropic env helper plasmids (pNGVL3 gag-pol and pAM 178-2, respectively [44, 64]) into 293T cells by calcium phosphate precipitation as previously described (44). Medium containing virions was harvested beginning 24 h posttransfection and filtered through 0.2-μm filters. D17/pJET cells (canine osteosarcoma cells expressing murine ecotropic receptor) were infected with serial dilutions of vector-containing virions in the presence of 0.8 μg/ml Polybrene (hexadymethrine bromide; Sigma) for 2 h and selected 48 h postinfection in Dulbecco's modified Eagle's medium with 10% calf serum, 1% penicillin-streptomycin (GIBCO/Invitrogen), and 1 μg/ml puromycin (25, 39). Titers were determined by the number of CFU per ml of virion-containing medium after 12 days of puromycin selection. Titers were averaged from four independent infections using virus from at least two independent transfections for each vector. CFU/ml values were then normalized to RT protein levels, as determined by RT activity (58), relative to a reference sample. Error bars were calculated as standard deviation of the mean.
Analysis of strand transfer products.
For homologous recombination products, after 12 days of selection in puromycin, colonies were pooled, and provirus-containing genomic DNA was isolated using a quick lysis plus proteinase K digestion protocol modified from the work of Woffendin et al. (63). The artificial intron within the puromycin-coding sequence was PCR amplified using primers SF304 (5′-GGCCTAGGCTTTTGCAAAAAGCTTAC-3′) and MK204 (5′-TCGGCGGTGACGGTGAA-3′) for 35 cycles. For product mobility analyses, SF304 was 5′ end labeled with [32P]ATP and T4 polynucleotide kinase, and radioactive PCR products were digested with AscI, separated on a 5% acrylamide gel, and quantified by phosphorimager. Alternately, product bands were resolved on 1% agarose gels and purified using a QIAquick gel extraction kit (QIAGEN) for sequencing by the University of Michigan DNA Sequencing Core Facility.
For nonhomologous recombination product analysis, individual puromycin-resistant colonies containing integrated humanTM vector products were expanded for DNA extraction, and genomic DNA was isolated using a DNeasy tissue kit (QIAGEN). Intron-containing puromycin sequences were PCR amplified and sequenced as described above.
Sequences were analyzed by aligning parental vector and product sequences with DNAStar SeqBuilder (Lasergene). When a likely template could not be identified in the vector, sequences were compared to GenBank using NCBI BLASTn. Sequences including and flanking the insertions were queried, with the longest identified putative template segment designated the best candidate template. Sequences were considered to be likely templates if sequence identities shared by inserts and putative templates were 8 bases or greater in the case of vector-derived sequences and 19 bases or greater in the case of host sequences. Matches of these lengths would be predicted to arise roughly 10-fold less frequently than by random chance.
RESULTS
A single-RNA forced recombination assay.
We previously examined the effects of limiting donor/acceptor identity on forced copy choice recombination by use of a two-vector assay (43). In that study, one vector was designed to mimic the 3′ portion of an MMLV genomic RNA, and the second was designed to mimic a genomic RNA's 5′ end (Fig. 1A). When one copy of each of these two vectors was copackaged, template switching between them led to the production of an intact selectable provirus. Such two-vector assays yield low proviral titers (43, 47), at least partly because coexpressed RNAs in MMLV preferentially self-associate for packaging (15, 23, 48).
Here, we revisited forced copy choice recombination by use of a single-vector RIO approach, which circumvents the need for the copackaging of two coexpressed vectors. RIO vectors contain primary (donor) and secondary (acceptor) template sequences like those used in two-vector forced recombination assays, but these two recombination templates are contained on single RNAs. RIO vectors have been called “prejumped” because their first DNA intermediate resembles a reverse transcription product that results after minus-strand strong-stop strand transfer (33). The recombination donor was engineered to reside at the RNA's 5′ end, as in two-vector forced homologous recombination assays. However, the acceptor was engineered into the 3′ portion of a single RIO vector RNA. Here, the two halves of the RIO vector each contained a portion of the puromycin resistance gene separated by an artificial intron that served as the recombination target sequence (Fig. 1B). Generation of an intact puromycin resistance gene, which requires recombination between 5′ donor and 3′ acceptor sequences, proceeds readily, and thus RIO vectors provide a highly tractable one-vector approach for studying retroviral recombination (Fig. 1C).
Previous two-vector forced homologous recombination experiments suggested that 30-nucleotide secondary templates are recognized as well as longer acceptors, and it has been determined that the reverse transcription machinery can extend a single mismatched nucleotide fairly efficiently (43). Thus, to address how frequently RT proceeded to the primary (donor) template's 5′ end before switching to a secondary template, the extent of single-base mismatch extension was determined using a forced homologous recombination assay. Donor and acceptor sequences were designed so that recombination occurring precisely from the donor template's 5′ end—which would require mismatch extension—would yield a diagnostic restriction site in the product DNA, while premature template switching would not generate the diagnostic site. An overview of the experimental approach is presented in Fig. 2A.
FIG. 2.

One-base mismatch extension by RIO vector. (A) Experimental scheme. (B) 30TM acceptor. Newly synthesized DNA transfers from a donor template to an acceptor template with 30 bases of identity. If the DNA transfers at any position within the 30 bases of identity on the donor template, an AscI site will be generated in the provirus. (C) 1TMM acceptor. The 1TMM acceptor contains 29 bases of identity and 1 mismatched base. If DNA transfers from the 5′ end of the donor and the mismatch extends, an AscI site will be generated. If the DNA transfer occurs prior to the template end and no mismatch extension occurs, the template base will be incorporated and no AscI site will be formed. Acceptor region abbreviations are given in the legend to Fig. 1. (D) PCR amplification of puromycin resistance gene from pooled proviral DNA by use of primers indicated in Fig. 1C and analysis with AscI digestion. pRSVpuro, a vector containing an intronless puromycin resistance gene amplified with primers SF304 and MK204. RIO vectors, with their artificial introns, yield a longer amplification product. PCR products from 30TM and 1TMM, undigested and digested with AscI, are shown. Alternate targets that appear in the 1TMM product pool are indicated. puro, puromycin.
Virions containing RIO vectors with either 30TM (Fig. 2B) between acceptor and donor templates or 29 bases of match and a single terminal mismatch (1TMM; Fig. 2C) displayed similar titers (0.8 × 104 ± 0.2 × 104 CFU/ml when normalized as described in Materials and Methods). Infected cells were selected in puromycin, >100 puromycin-resistant colonies per vector were pooled, and integrated proviral DNA was amplified by PCR. Radiolabeled amplification products, both cleaved with the diagnostic restriction enzyme and uncleaved, were separated on polyacrylamide gels, and products were quantified by phosphorimager.
Figure 2D compares products of the terminally mismatched and terminally matched RIO vectors. Digestion with the diagnostic restriction site for products of the matched template vector was >95% complete. For the single-base mismatch vector, 80% of the products were digestible with the diagnostic enzyme. This suggests that roughly 80% of the products of this vector resulted from RT proceeding to the donor template's 5′ end, transferring to the mismatched acceptor template, and extending the 1-base mismatch that would result upon template switching. About one-half of the products that did not cut with the diagnostic enzyme migrated slightly faster than the correctly targeted product (Fig. 2D, rightmost lane). Low levels of this alternate target product were detectable among terminally matched vector products as well. Sequencing revealed that this product resulted from the use of 5 bases of fortuitous microidentity between the donor template 5′ terminus and an internal portion of the vector, which thereby served as an alternate acceptor template. These findings suggest that this vector system successfully generated forced recombination products, as a large majority of puromycin-resistant proviruses resulted from RT proceeding to the donor template's 5′ end.
Donor/acceptor identity required for homologous recombination.
A series of RIO vectors with incremental differences in donor/acceptor identity length was next used to assess acceptor template recognition. As shown in Fig. 3A, vectors with stepwise reductions in donor terminal identity were generated in series, and their titers were compared to those of the parental vector with 30 bases of acceptor/donor identity, as described in Materials and Methods.
FIG. 3.

Minimal sequence identity required for efficient and accurate recombinogenic strand transfer. (A) Acceptor sequences. The series of stepwise shortened acceptors from 27 bases to 0 bases of identity at the engineered acceptor position. (B) Titer/RT for 30 to 0 bases of identity vectors, determined as described in Materials and Methods. Note that the titer in the 5TM column, indicated with an open circle and connected to dashed lines, is the titer for a 5TM vector variant in which the major alternate target sites were eliminated by mutation. This alternate 5TM vector was generated to assess titers in the absence of the major alternate targets but is not used elsewhere in this report. (C) PCR products of pooled proviral products of the limited identity vectors, with or without AscI digestion. Product mobilities are indicated at left; some alternate target products are indicated at right. puro, puromycin.
Titer data for these limiting identity vectors are presented in Fig. 3B. The results show that all vectors with acceptors of between 30 and 16 nucleotides in length displayed similarly high titers. In contrast, the loss of a single additional nucleotide of donor identity—a 15-nucleotide acceptor—resulted in a 10-fold decrease in the puromycin resistance titer. Titers for acceptors with 15, 12, and 10 bases of donor identity were similar to one another. A further sharp 1-log drop in titer was observed for an acceptor with only 8 bases of donor identity, and vectors with shorter regions of donor template identity displayed further titer decreases.
Provirus-containing genomic DNA from pooled puromycin-resistant colonies produced by each vector was PCR amplified and analyzed on acrylamide gels as described above (Fig. 3C). The results revealed that nearly all of the puromycin-resistant products of acceptors of ≥10 bases in length were correctly targeted to the engineered acceptor. In contrast, limited use of the fortuitous 5-base alternate acceptor (Fig. 3C, bottom right arrow) was observed for the 8-base acceptor. This and other alternate acceptors predominated for the shorter acceptors in this vector series.
Forced nonhomologous recombination.
For the following experiments, RIO vectors were modified to assess forced nonhomologous recombination. A RIO vector (humanTM) was constructed in which the major and ectopic acceptor sites used during reverse transcription of the above-described vectors were mutationally ablated. In their place, a human genome fragment that provided a relatively large number of candidate alternate acceptors with very limited identity to the donor was introduced. The nonhomologous acceptor template region selected for use here was a 590-base shotgun-cloned nonrepetitive noncoding fragment of human DNA (Fig. 4A). This human fragment acceptor included one region with 3 bases of identity to the donor template's 5′ end and many candidate 2-base acceptors but lacked identity of 4 bases or longer. The puromycin resistance-conferring titer of humanTM, when produced by transient transfection with helper function plasmids, was 500-fold lower than that of 30TM (1 × 101 CFU/ml versus 6 × 103 CFU/ml; Fig. 4B).
FIG. 4.

Forced nonhomologous recombination. (A) RIO template containing a human sequence nonhomologous acceptor. The acceptor region included a 590-base fragment of human chromosome 14 that contains no region of identity greater than 3 bases with the 5′ end of the donor. Note that polyadenylation signal readthrough resulted in the appendage of RNA copies of plasmid backbone sequences to a subset of the vector RNAs' 3′ ends. Acceptor region abbreviations are given in the legend to Fig. 1. (B) Vector titers determined as described in Materials and Methods. 30 represents titers for the 30TM vector; human indicates humanTM values; 0 indicates the value for 0TM vector. (C) PCR amplification of cell DNA from individual puromycin resistance cell clones. The amplification product of a correctly targeted clonal 5-base acceptor product is shown, as are PCR products from representative individual humanTM product cell clones. The leftmost two lanes contain radiolabeled size standards. puro, puromycin.
Cells containing individual integrated humanTM reverse transcription products were cloned and expanded. Nonhomologous recombination junctions were PCR amplified and sequenced (Fig. 4C). Cell clones that were not expandable or that did not yield PCR products were not analyzed further. A total of 83 independent clones were analyzed, as summarized in Table 1. To verify that the detected nonhomologous recombination junctions were generated during vector reverse transcription and not by PCR, the PCR products from 22 of these cell clones were reisolated from clonal cell samples and resequenced, and in all cases products were found to be identical to those isolated initially.
TABLE 1.
Nonhomologous recombination products
| Recombinant type | Clone no. | Donor departure | Acceptor entry | 3° Template
|
4° Template
|
5° Template | Micro-identity (bases)a | 3° Template lengthb | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Departure | Entry | Departure | |||||||
| Simple nonhomologous recombinants | 1 | 60 | 3073 | 1 | ||||||
| 6 | 39 | 2407 | 1 | |||||||
| 8 | 63 | 2543 | 2 | |||||||
| 9 | 51 | 2368 | 0 | |||||||
| 12 | 24 | 2552 | 3 | |||||||
| 13 | 28 | 2712 | 1 | |||||||
| 16 | 55 | 2480 | 0 | |||||||
| 17, 30, 112 | 39 | 2761 | 6 | |||||||
| 20 | 62 | 2604 | 2 | |||||||
| 21 | 60 | 3032 | 0 | |||||||
| 23 | 15 | 2322 | 3 | |||||||
| 24 | 59 | 2368 | 0 | |||||||
| 32 | 62 | 2811 | 1 | |||||||
| 37 | 61 | 2781 | 1 | |||||||
| 54 | 63 | 2492 | 2 | |||||||
| 68, 93 | 42 | 2654 | 1 | |||||||
| 76 | 60 | 2494 | 1 | |||||||
| 77 | 55 | 2366 | 1 | |||||||
| 80 | 51 | 2819 | 0 | |||||||
| 88 | 69 | 2608 | 1 | |||||||
| 97 | 47 | 2573 | 1 | |||||||
| 98 | 1 | 2408 | 1 | |||||||
| 134 | 53 | 2566 | 0 | |||||||
| 137 | 65 | 2564 | 1 | |||||||
| Complex nonhomologous recombinants | ||||||||||
| Bridged by vector-internal sequences | 10 | 50 | 2754 | 948 | 884 | 3, 7 | 65 | |||
| 19 | 42 | 2754 | 944 | 884 | 4, 7 | 61 | ||||
| 39 | 48 | 3074 | 2887 | 2880 | 0, 2 | 8 | ||||
| 50 | 21 | 2754 | 951 | 884 | 0, 7 | 68 | ||||
| 118 | 22 | 2754 | 950 | 884 | 1, 7 | 67 | ||||
| Bridged by polyadenylation readthrough sequences | 2, 120, 133 | 48 | 2567 | 6263 | 6108 | 3, 10 | 156 | |||
| 14, 38, 119 | 48 | 2765 | 6263 | 6180 | 3, 7 | 83 | ||||
| 29, 41 | 50 | 2535 | 4073 | 3972 | 2, 8 | 101 | ||||
| 36 | 50 | 2533 | 5081 | 5047 | 2, 8 | 35 | ||||
| 43 | 50 | 2787 | 4073 | 3928 | 2, 5 | 145 | ||||
| 45 | 48 | 2499 | 6263 | 6165 | 3, 5 | 99 | ||||
| 53, 67, 121 | 52 | 2374 | 4007 | 4000 | 2, 3 | 8 | ||||
| 66 | 50 | 2351 | 4894 | 4830 | 2, 6 | 65 | ||||
| 71, 84 | 48 | 2506 | 6263 | 6201 | 3, 6 | 63 | ||||
| 79 | 50 | 2565 | 4894 | 4851 | 2, 2 | 44 | ||||
| 83 | 49 | 2535 | 4189 | 3972 | 3, 8 | 218 | ||||
| 90 | 50 | 2560 | 4894 | 4867 | 2, 0 | 28 | ||||
| 94 | 50 | 2883 | 4894 | 4867 | 2, 0 | 28 | ||||
| 99, 127 | 49 | 2535 | 4163 | 3972 | 3, 8 | 192 | ||||
| 122 | 48 | 3074 | 6263 | 6249 | 3, 6 | 15 | ||||
| Bridged by minus-strand DNA sequences | 5, 81 | 41 | 2368 | 797 | 816 | 2, 7 | 20 | |||
| 7 | 45 | 2368 | 804 | 816 | 1, 7 | 13 | ||||
| 11 | 50 | 2368 | 797 | 816 | 10, 7 | 20 | ||||
| 47, 72 | 52 | 2368 | 796 | 816 | 7, 7 | 21 | ||||
| 49 | 46 | 2368 | 793 | 816 | 3, 7 | 24 | ||||
| Bridged by host sequences | 18, 65 | 1 | 2771 | tRNALeuc | tRNALeuc | 4, 1 | 24 | |||
| 130 | 25 | 2374 | U1d,e | U1d,e | 2, 6 | 21 | ||||
| Bridged by undetermined sequences | 31 | 1 | 2509 | GGGCGTTGGf | ≥9 | |||||
| 33 | 67 | 2368 | TTCATf | ≥5 | ||||||
| 46 | 49 | 2374 | Tf | ≥1 | ||||||
| 82 | 24 | 2378 | TTGGGGACCCAGATTGf | ≥16 | ||||||
| 123 | 1 | 2482 | GGCf | ≥3 | ||||||
| Bridged by 2 tertiary template segments | 34 | 50 | 2516 | 4868g | 4686g | 4071g | 3974g | 6, 2, 0 | 183, 98 | |
| 52 | 50 | 2535 | 4894g | 4686g | 4071g | 3972g | 8, 2, 0 | 209, 100 | ||
| 57 | 50 | 2369 | 4894g | 4877g | 1381e | 1399 | 7, 2, 2 | 18, 19 | ||
| 58, 74 | 50 | 2506 | 4894g | 4686g | 4161g | 3974g | 6, 2, 1 | 209, 187 | ||
| 78 | 51 | 2360 | Dogh | Dogh | GAAGf | 2 | 221, ≥4 | |||
| 89 | 16 | 2509 | 951i | 918i | 28S rRNAj | 28S rRNAj | 8, 0, 9 | 34, 61 | ||
| 100 | 50 | 2535 | 4894g | 4686g | 4246g | 3972g | 8, 2, 2 | 209, 275 | ||
| 124 | 50 | 2506 | 4894g | 4686g | 4187g | 3974g | 6, 2, 1 | 209, 214 | ||
| 129 | 1 | 2368 | tRNALysk | tRNALysk | 794e | 816 | 7, 4, 0 | 56, 23 | ||
| 135 | 50 | 2535 | 4890g | 4686g | 4161g | 3972g | 8, 2, 1 | 205, 190 | ||
| Bridged by 3 tertiary template segments | 51 | 48 | 2374 | 6263g | 6193g | 1126i | 847i | CGCCAAGTf | 3, 3, 8, 3 | 71, 280, ≥8 |
For products with multiple crossover junctions, presented in sequential order.
Where template lengths are presented as “≥x,” x is the length of inserted sequences. Multiple values are in sequential order.
From map position 72 to position 94 of GenBank accession no. X04117 (human tRNA leu).
From map position 95 to position 115 of GenBank accession no. K007851 (human U1A RNA).
Minus-strand DNA.
Sequence insertion of undetermined origins. Template entry and departure sites are unknown; the entire insert sequence is provided in the entry column.
Polyadenylation site readthrough sequences.
From map position 38820306 to position 38820526 of GenBank accession no. NC_006589 (Canis familiaris chromosome 7).
Vector-internal sequences.
From map position 3809 to position 3869 of GenBank accession no. M11167 (human 28S rRNA).
From map position 3998 to position 3943 of GenBank accession no. AL3555505.16 (human tRNA lys).
Analysis of forced nonhomologous recombination products.
The strategy for isolating crossover junctions used here placed several constraints upon which nonhomologous recombination products could be recovered. For example, because only products that conferred puromycin resistance were detected, all proviruses needed to incorporate both 3′ and 5′ portions of the puromycin acetyltransferase gene, and only recombinants that contained functional-length (≥80-base) introns that lacked strong cryptic splice acceptors or polyadenylation signals in the intronic region would survive the imposed selection (62). Nonetheless, the sequences of the 83 products revealed that the reverse transcription machinery employed a remarkable variety of strategies to resolve forced nonhomologous recombination. These products can be divided into classes based on the nature of the crossover junctions and on the number of template segments employed.
Forced copy choice nonhomologous recombinants.
Based on the very low frequency of premature jumps observed above during RIO vector forced homologous recombination, as well as on previous observations of donor/acceptor microidentity at many nonhomologous crossover junctions, we had anticipated that most of this vector's products would result from reverse transcription to the vector's 5′ end and then use of donor terminus microidentity in the acceptor as the site of transfer. Remarkably, only 1 of the 83 reverse transcription products analyzed here had that structure. Even though the acceptor region included 1 trinucleotide match to the vector's 5′ end and 34 matching dinucleotides, this single product of transfer from the vector's 5′ end to the acceptor template region (clone 98 in Table 1; Fig. 5A) utilized only a single base of donor terminal/acceptor microidentity. An additional five products (clones 18, 31, 65, 123, and 129 in Table 1), described below in the discussion of splinting tertiary template segments, resulted from forced template switching at the donor 5′ end. However, all of these latter were complex products for which the initial template switch was to a discontiguous tertiary template sequence, rather than a direct transfer to the engineered human chromosome 14 acceptor.
FIG. 5.
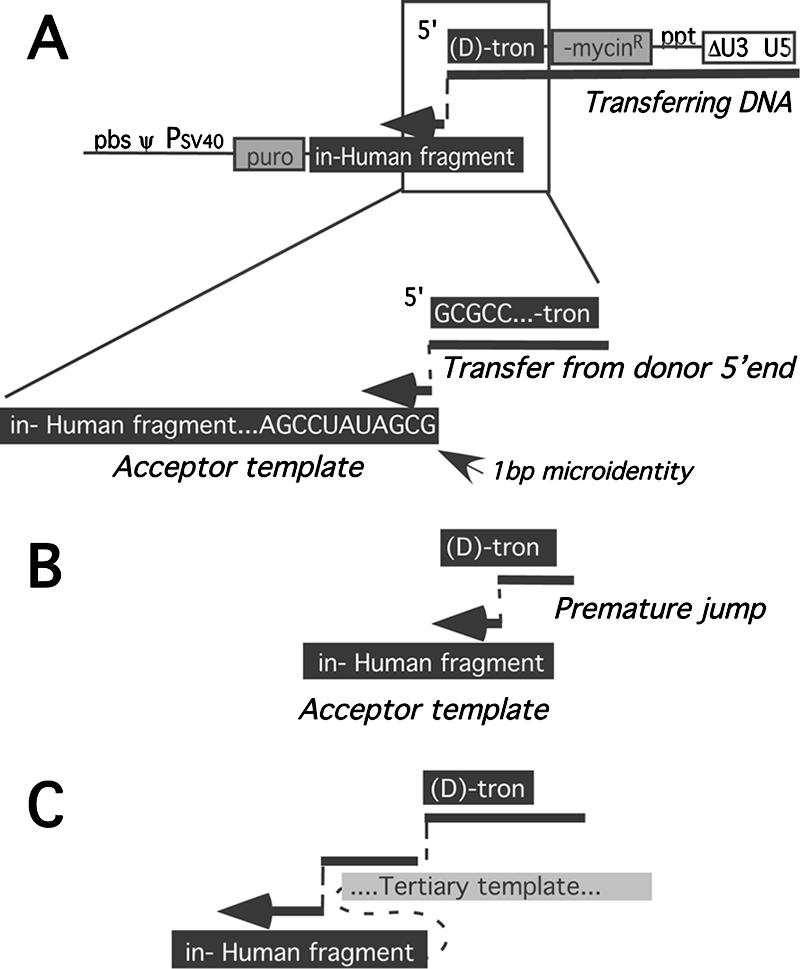
Categories of nonhomologous recombination products. (A) Forced copy choice nonhomologous recombination product. DNA synthesized to the 5′ end of the donor template transfers to an acceptor template position with primer terminal microidentity. (B) Premature jump. DNA synthesis transfers from the donor to the acceptor template before reaching the 5′ end of the donor template. (C) Incorporation of tertiary template sequences. Nascent DNA transfers from the donor template to a tertiary template before subsequent transfer to the acceptor template. Illustrated here, the tertiary template used is a segment of polyadenylation signal readthrough RNA. Acceptor region abbreviations are given in the legend to Fig. 1.
Additional simple nonhomologous recombinants.
RT switched to the acceptor template prior to reaching the donor template's 5′ end the during the synthesis of 26 of the remaining 77 products (Fig. 5B; Table 1). These “premature” transfer products did not contain the entire 71 nucleotides of artificial intron from the donor template (Fig. 6A) but instead contained 5′ truncations of this, fused to sequences in the acceptor template region. Among these “simple” nonhomologous recombinants, so called because a single nonhomologous template switch was involved in generating the provirus, 80% of the junctions included donor/acceptor microidentity of 1 to 6 bases. The remaining 20% resulted from donor-to-acceptor transfer with no microidentity. Simple nonhomologous recombinants' sites of donor departure and acceptor entry are indicated in Fig. 6B. The broad spectrum of observed donor departure and acceptor entry sites confirmed that the use of sequences throughout the engineered acceptor and donor sequences generated puromycin-selectable products. Although strong transfer hot spots for simple recombinants were observed in neither the donor nor the acceptor template, some sites were used more than once. For example, one junction with 6 bases of donor/acceptor identity arose in three independent clones (Table 1, clones 17, 30, and 112), and a second crossover was shared by two independent isolates (clones 68 and 93).
FIG. 6.

Spectra of donor departure and acceptor entry sites observed in analyzed clonal products. (A) Schematic representation of humanTM vector, including polyadenylation readthrough region. Numbers at top indicate map positions on the RNA and indicate that the schematic is not to scale. Note that polyadenylation signal readthrough resulted in the appendage of RNA copies of plasmid backbone sequences to a subset of the vector RNAs' 3′ ends. (B) Compendium of donor departure and acceptor entry sites for all 83 nonhomologous recombination products. Diagrammed as in Fig. 5C. Each open circle indicates a simple recombination donor departure or acceptor entry site; each black bar indicates a corresponding site for a complex recombinant. Acceptor sites for 20-base intervals are binned; stacked circles and/or bars indicate multiple entries into a single acceptor site within the interval. The RNA strand drawn in the center of Fig. 6B represents splinting templates. Crossover site locations for those recombinants that used sequences on the same or copackaged vector RNA are shown. Horizontal lines above the splinting template indicate the locations of splinting RNA template segments; lines below indicate splinting DNA templates. Each horizontal line represents a separate tertiary template segment. Each encompasses the template segments used, but these are not precisely to scale. polyA site, polyadenylation site. Acceptor region abbreviations are given in the legend to Fig. 1.
Complex nonhomologous recombinants.
Fully two-thirds of all analyzed products included additional sequences between the donor template departure and acceptor template entry sites and were thus designated “complex” recombinants (Fig. 5C and 6; Table 1). Insertions in retroviral crossover junctions have been described previously (13, 36, 40, 41, 46, 59, 68). RT rarely adds more than a single nontemplated nucleotide during DNA synthesis, and thus the majority of crossover-associated sequence insertions are likely templated by tertiary nucleic acids used after the primary (donor) template but prior to association with the secondary (acceptor) template segment (41, 45).
Identification of splinting tertiary template segments.
Sequence analysis programs and database searches were used to identify candidate noncontiguous segments of RNA or DNA that served as tertiary templates for the insertions (see Materials and Methods). The sequence analyses suggested that a total of 69 segments of nucleic acid contributed as tertiary templates in the synthesis of the 56 insert-containing clones, with likely templates identified for 90% of the insert segments. Most inserts with unidentified templates were too short (median length of 5 bases) to implicate a specific template with confidence (see Materials and Methods).
Nonhomologous junctions bridged by single segments of vector-internal or polyadenylation site readthrough RNA.
In the first and largest class of splinted recombinants, the gaps between donor and acceptor regions were bridged by single discontiguous portions of RIO vector RNA sequence or of sequences that were appended to the vectors' 3′ ends due to vector polyadenylation signal readthrough. For five of these (Table 1, clones 10, 19, 39, 50, and 118), the bridging template was derived from discontiguous portions of vector-internal sequences (Fig. 6B). This resulted in a duplication of sequences from one location between the products' LTRs to a second remote location also between the viral LTRs: rare instances of such duplications of viral genome segments have been reported previously (31, 36, 46, 56).
For most of the splinted recombinants analyzed here, inserts were templated by sequences downstream of the vector polyadenylation site (Fig. 6B). The mechanism likely to have been used to generate these insertions (a nonhomologous template switch to a discontiguous template, limited reverse transcription on that tertiary template, and then a subsequent nonhomologous template switch) was identical to that used for the patching in of the vector-internal sequences described above. However, the outcome here was to relocate vector-external sequences into internal portions of the vector rather than to duplicate vector-internal sequences at an ectopic site. The polyadenylation cassette used in humanTM is subject to up to 20% readthrough (3), thus suggesting that as many as one-third of all humanTM RIO vector-containing virions may have contained readthrough sequences. Sequences housed on readthrough RNAs are known to be packageable and to serve readily as “patch repair” recombination templates (3, 20, 37, 53, 56, 69).
Insertions templated by nascent vector DNA, host sequences, or undetermined templates.
The inserts observed in a second class of splinted recombinants were derived from the RIO vector but appeared in the product DNAs in reverse orientation, suggesting that they were templated by nascent minus-strand DNA rather than by vector RNA (clones 5, 81, 7, 11, 47, 72, and 49). “Backwards” insertions have previously been described for nonhomologous recombination products and can be explained by models for plus-strand recombination, a key feature of which is template switching between nascent DNA templates (22, 56, 66). Fairly extensive (7-base) fortuitous microidentity between the most commonly utilized minus-strand template and sequences in the acceptor template region likely contributed to the high frequency of minus-strand templated insertions observed here.
The third class of inserts was host-derived sequences. Among these were human (producer cell) leucine and lysine tRNAs (clones 18, 65, and 129), a human U1 spliceosomal RNA cDNA (clone 130), a portion of human 28S rRNA (clone 89), and canine sequences derived from the recipient cell (clone 78).
Likely templates were not identified by sequence analyses for the final class of putative template segments (7 of the 69 total insert segments). These ranged in length from 1 to 16 bases. The possibility that these segments were encoded by tertiary templates cannot be ruled out, and in each case, tertiary templates too short to fulfill the significance criteria described in Materials and Methods could be identified and may have been the source of the inserts (not shown). On the other hand, especially for the shortest of these, the insertion may have resulted from nontemplated base addition prior to nonhomologous recombination. Single-base frameshifting is a common class of reverse transcription error, and under some circumstances RT can add and extend single nontemplated bases (27, 41, 45). However, it is interesting that retrospective analysis of previous insertion-in-deletion mutations suggests that even single-base insertions may in some instances be tertiary template directed (40) (Fig. 7).
FIG. 7.

Retrospective alternate model for generation of single-base insertion plus flanking hypermutations. Based on nonhomologous recombination product 72.P8 in reference 40. (A) Model proposed in reference 40. A single-base insertion occurs during nonhomologous template switching, followed by junction-associated hypermutation by RT as it copies the acceptor template. (B) Splinted recombination model for single-base insertion. RT uses 2 bases of microidentity to switch to the tertiary template and then 6 bases of microidentity to switch back to the acceptor template. This model calls for one base substitution between the candidate splinting template, identified by BLASTn as GenBank accession no. V01204.1, which mapped to the U3 region of a spleen necrosis virus. Note that TK gene expression in the vector used (40) was driven by a spleen necrosis virus promoter, and thus it seems likely that the apparent 1-base insertion in a nonhomologous crossover junction here arose via splinting recombination with a distal portion of the vector RNA.
Complex inserts derived from more than one template segment.
A similar broad spectrum of templates was also observed among the 12 proviral products that displayed evidence of use of more than one tertiary template segment. Two separate segments of viral RNA spanned the nonhomologous recombination junctions in some clones. Other clones' two or three bridging templates included various combinations of host and vector RNA sequences, vector RNA and vector DNA sequences, host RNA and vector DNA, or vector RNA and undetermined sequences. One clone, clone 78, contained recipient cell sequences—as reported previously for nonhomologous recombination products (13)—that joined to a short segment of undetermined origin before rejoining the acceptor template.
Crossover site distribution and hot spots for nonhomologous recombination.
Figure 6B displays the donor departure and acceptor entry sites for each of the 83 analyzed nonhomologous recombination products, whether or not tertiary templates were used prior to acceptor template association. In this figure, the donor RNA's 5′ end was assigned position 1.
Analysis of crossover junctions indicated that whereas a broad range of donor departure sites was used (28 different departure sites were used among the 83 clones), two departure sites (positions 48 and 50) were used significantly more frequently than others (14% and 22% of the total clones, respectively), and fully one-half of all products resulted from template departure in the interval from donor template position 48 through position 52 (Fig. 6). Unlike the simple recombinant junctions, which were fairly widely distributed, the initial template switch for all but two of these donor departure hot spot products was to a tertiary template segment (see Table 1).
Many of the observed hot spot donor sites directed template switching to acceptor positions that were themselves hot spots. For example, 10 out of 18 template switches from position 50 used 2 bases of microidentity to arrive at splinting template hot spot acceptor position 4894. Similarly, 10 of 12 jumps from hot spot position 48 were directed to the hot spot acceptor at position 6263, using 3 bases of identity. Note however that although donor departure sites and splinting template entry sites displayed hot spots, there was significant variation in the length of splinting tertiary template that was reverse transcribed after hot spot transfer and before subsequent template switching to the engineered human acceptor. For example, clones 2 and 71 share a donor/tertiary template crossover junction, but 156 bases of tertiary template were copied in clone 2 and only 63 bases of the tertiary template were reverse transcribed in clone 71.
In other cases, a single donor hot spot was joined to many different acceptors (for example, donor position 60 was joined alternately to acceptors 2494, 3032, and 3073), and in some cases, a single acceptor hot spot was joined to many different donors (e.g., a minus DNA splint, donor sites 51 and 59, and an undetermined splinting template were all observed joined to acceptor position 2358 in different clones).
Among hot spot products, some pairs and triples possessed identical sequences. Confirming that these were not sibling integrants but instead independent reverse transcription products, the colonies containing the independent identical sequences were isolated in each instance from separate plates of infected cells; in most cases, they were products of virions produced by separate transfections.
General properties of crossover junctions and template segments.
Of the 83 nonhomologous recombination products analyzed here, about one-third were simple recombinants and two-thirds were complex splinted recombinants. In addition to the initial tertiary template, at least one additional template contributed to the synthesis of 20% of the splinted complex nonhomologous recombinants. One product (clone 51 in Table 1) appears to have been synthesized using a total of five template segments. The distribution of crossover-associated donor/acceptor microidentity lengths is presented in Fig. 8A. As shown in Fig. 8B, splinting template segments varied in length from 1 to 280 bases (median length, 63 bases).
FIG. 8.
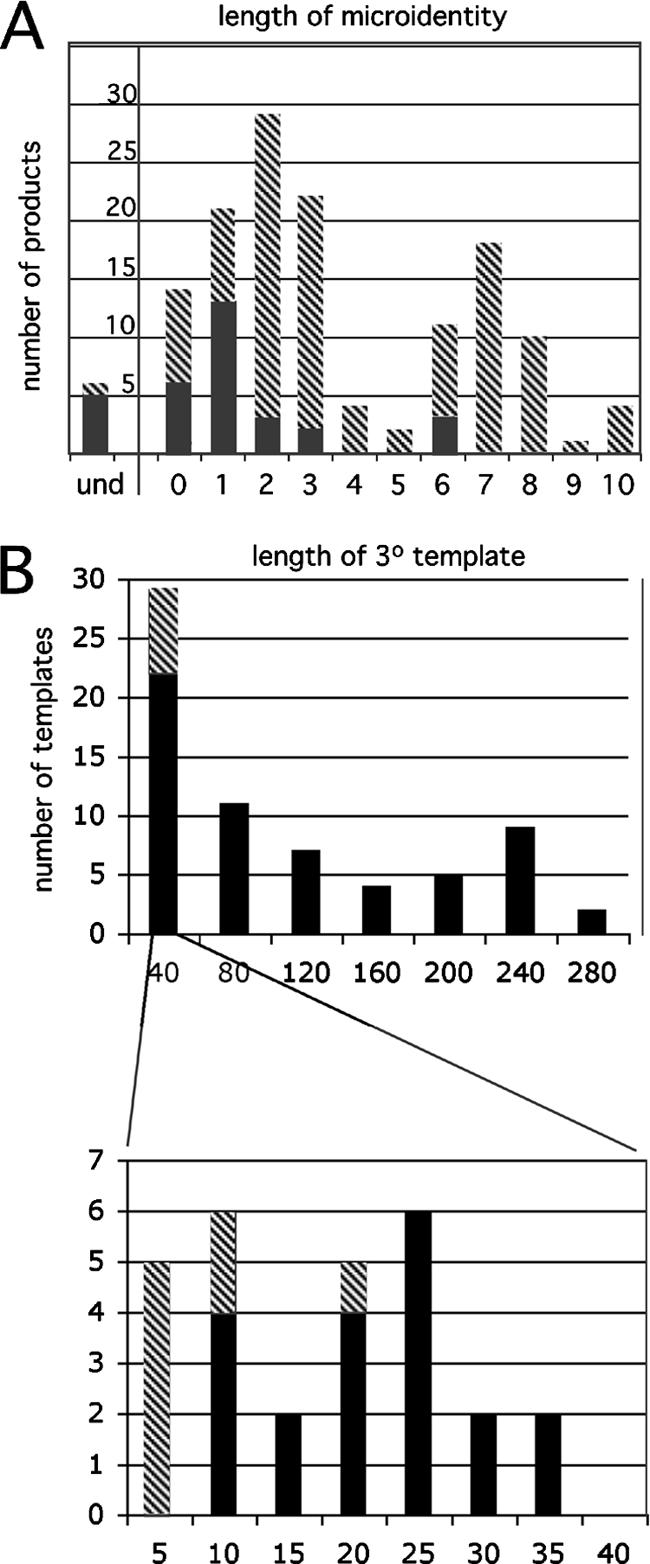
Properties of crossover junctions and splinting templates. (A) Distribution of junctional microidentity lengths. Black bars represent identities observed at simple recombinants' crossover junctions; hatched bars are complex recombinants' junctional microidentities; und represents junctions for which identity lengths were not determined because a template segment was not identified. (B) Splinting (3°) templates' lengths. Hatched bars indicate minimal template lengths for inserts with unidentified templates (e.g., the length of the insert, whereas for identified templates most included flanking microidentity residues); black bars represent assigned template segments. (Bottom) An expanded view of template length distribution for shorter insert segments shown at top.
DISCUSSION
In this report, we examined the effects of varying acceptor template identity lengths on MMLV forced genetic recombination. Here, with recombination forced to occur principally from a single-template position, 16 bases of identity were sufficient to target accurate and efficient recombinogenic switching. Similar trends in acceptor recognition have previously been reported using different sequences and more-limited sets of length variation, suggesting that whereas some interactions between RT and nucleic acids differ by sequence, the observations here are not specific to the target sequences used (10, 43, 52).
An interesting finding here was the sharp cutoff in acceptor recognition observed with incremental changes in identity lengths. Specifically, while 16 bases served as efficiently as 30 bases, a 15-base acceptor template displayed a sharp 1-log decrease in titer. Titers for acceptors of 10 to 15 bases in length were similar to one another, but a further sharp 1-log drop was observed for an 8-base acceptor. These template lengths are suggestive of the way RT is known to interact with its primer/templates. When RT reaches the end of a template, template degradation by its RNase H activity produces a residual 15- to 19-base oligoribonucleotide, and subsequent slower secondary cleavages can produce 8- to 10-base RNA products (4, 11, 17).
These findings regarding acceptor length can be interpreted in the context of the acceptor invasion model for minus-strand recombination, in which RNase H cleavage of the template strand is a required step (5, 8). Template degradation unmasks nascent minus-strand DNA to serve as a docking site for the invading acceptor template, with the region between 10 and 19 bases behind the RT growing point being especially critical to acceptor template engagement in reconstituted reactions in vitro (7, 50). Thus, speculatively, the results here may suggest that acceptor template engagement and physical interactions within RT's footprint are critical intermediates in template switching. As postulated by the model presented in Fig. 9, these interactions are most effective at engaging a productive acceptor template when RT's polymerase active site is still engaged at the primer terminus (Fig. 9A), but interactions retained after the enzyme's repositioning to generate RNase H secondary cleavages also can serve, to a lesser degree, in acceptor template recruitment (Fig. 9B).
FIG. 9.

Model for RT interactions during acceptor invasion-mediated recombination. (A) RT (represented as a peanut-shaped object) after reaching the 5′ end of the donor template. The figure represents the distance between RNase H and DNA polymerase active sites as 15 bases; donor template cleavage is represented with a dashed line; the invading acceptor is shown as a gray line. (B) RT translocated after secondary RNase H cleavages. The acceptor template is shown base pairing with nucleotides 9 to 13 from the 3′ end of the nascent pretransfer DNA. Models for acceptor invasion call for subsequent branch migration to subsequently realign the primer strand 3′ end onto the acceptor template (5).
Both titer defects and the use of alternate transfer sites confirmed the very low accuracy and efficiency of homologous recombination when acceptors were 8 bases long or shorter. With a 29-base-matched, single-terminal-mismatched template, most products resulted from template switching after RT reached the donor template's 5′ end (Fig. 2). However, below a threshold of roughly 8 bases, this major pathway of homologous acceptor template recognition was apparently no longer operational and most transfer was nonhomologous. With very limited identity acceptors (<8 bases), titers decreased dramatically and the product pool was heavily populated by premature jump and other alternate target products.
Several previous studies have addressed nonhomologous recombination (12, 19, 40, 56, 60, 69). This one differs both in attempting to force recombination at a discrete position and in the extensive collection of products analyzed. It was surprising that only a single product herein resulted from synthesis to the end of the donor template followed by microidentity-guided switch to the human sequence acceptor. The copy choice model for genetic recombination suggests that when RT reaches a broken template end, the enzyme:primer-template complex dissociates and acceptor templates are located by primer terminal complementarity (8). However, the decreased titers and the low levels of transfer observed using limiting identity templates suggest that predominantly dead-end products form if RT reaches a donor template's 5′ end in the absence of an acceptor template with subterminal identity.
The use of tRNAs as bridging templates that span nonhomologous recombination junctions has been reported previously (6, 19, 26). tRNAs and sequences from other host RNAs known to be packaged by retroviruses were observed for some insertions here (18, 38, 61). Low levels of host messenger RNAs are also packaged by retroviruses (30, 51), but the absence of producer cell mRNA sequences from any of the 83 clones is suggestive of the possibility that within individual virions, copackaging of genomic and host mRNAs is a rare occurrence. In contrast, patching in of sequences encoded downstream of the vector's polyadenylation signal—a process analogous to prevailing models for oncogene transduction—was observed for over one-third of all products analyzed (54).
One product's insertion was derived from intronic sequences from the recipient cell. Although using a recipient cell intronic sequence as a recombination substrate does not readily reconcile with our understanding of how and where reverse transcription takes place, recombination with infected recipient cell sequences has previously been described (36, 60), and recombination with integration-site unlinked noncoding sequences also has been reported (55). Thus, there is precedence for the low-frequency incorporation of recipient cell and noncoding host genomic sequences into defective retroviruses.
Previous work has identified hot spots for retroviral recombination in homologous and homologous recombination templates (14, 71). This study's nonhomologous crossovers occurred at many positions, but some template departure and entry sites were used more than others, with hot spots in both donor and acceptor sequences. Microidentity was involved in ∼90% of this study's nonhomologous crossovers, which is a greater fraction than previously implicated (68), and some frequently used crossover sites displayed junctional microidentity. However, other crossover sites that were used multiple times were identity independent. Some “warm spots” appeared half site autonomous, in that a single donor departure site was used with any of several entry sites, or one acceptor was joined to any of several donor departure sites. RNA structures have been implicated for some but not all hot spots for template departure, presumably due to pausing during DNA synthesis, and other work has addressed homopolymeric regions and other features as promoting homologous template switching (28).
Parameters governing nonhomologous transfer hot spots here were not rigorously explored. However, observations such as the strong use of the alternative 5-base acceptor in the presence of acceptors of less than 10 bases in length provided clear evidence of positional advantages. The nature of these is unclear. Previous studies have reported RNA secondary structures as biased positions for nonhomologous recombination (12), and RNA structures have also been implicated as contributing to acceptor template hot spots (35). Here, there were some instances where such factors appeared operational but others where they were not. This suggests that many different parameters can lead to recombination hot spots and/or that some other unifying property of these positions—for example, their induction of sequence context-dependent biases in the RT translocation state (32)—led to the observed recombination hot spots.
Interestingly, whereas the average distance that RT copies a viral genomic RNA segment before switching to the copackaged RNA is roughly 1 to 3 kilobases (21, 37, 49), the median splinting template length in the current report was only 63 bases. Pathak and Temin postulated that oligoribonucleotide remnants of genomic RNA generated the short insertions they observed in nonhomologous crossover junctions (41). Here, however, most implicated template segments were in readthrough RNA regions unlikely to be subject to RNase H digestion, suggesting that some factor(s) other than template length contributed to the apparent low processivity of DNA synthesis on these tertiary template segments.
We previously proposed that short sequence insertion may be the typical signature of transductive recombination and that this process may generate many of the insertions observed in retroviral genomes (3, 57). In the present study, this hypothesis was addressed by examining products generated during forced nonhomologous recombination. Some instances of incorporation of distal genome segments at nonhomologous crossover junctions have been reported previously, and “patch repair” is often observed under selective conditions (31, 34, 36, 56). The assay used here revealed that using short portions of tertiary templates is a common way for the reverse transcription machinery to resolve forced nonhomologous recombination. The frequent use of tertiary template segments that was observed here provides experimental support for the possibility that some of the length variation observed for human immunodeficiency virus type 1 clinical isolates may reflect the patching in of short segments from discontiguous templates (16, 24, 57).
Acknowledgments
We thank Eric Garcia, Adewunmi Nuga, and Mary Jane Wieland for critical reading of the manuscript and valuable discussions and Sharon Fodor-Marr for the construction of prototype RIO vectors.
This work was supported by NIH grant GM063479 to A.T.
Footnotes
Published ahead of print on 5 September 2007.
REFERENCES
- 1.An, W., and A. Telesnitsky. 2002. Effects of varying sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 76:7897-7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An, W., and A. Telesnitsky. 2002. HIV-1 genetic recombination: experimental approaches and observations. AIDS Rev. 4:195-212. [PubMed] [Google Scholar]
- 3.An, W., and A. Telesnitsky. 2004. Human immunodeficiency virus type 1 transductive recombination can occur frequently and in proportion to polyadenylation signal readthrough. J. Virol. 78:3419-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Artzi, H., E. Zeelon, B. Amit, A. Wortzel, M. Gorecki, and A. Panet. 1993. RNase H activity of reverse transcriptases on substrates derived from the 5′ end of retroviral genome. J. Biol. Chem. 268:16465-16471. [PubMed] [Google Scholar]
- 5.Brincat, J. L., J. K. Pfeiffer, and A. Telesnitsky. 2002. RNase H activity is required for high-frequency repeat deletion during Moloney murine leukemia virus replication. J. Virol. 76:88-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrasco, M. L., M. Duch, and F. S. Pedersen. 2004. Strand transfer to the 5′ part of a tRNA as a mechanism for retrovirus patch-repair recombination in vivo. J. Gen. Virol. 85:1965-1969. [DOI] [PubMed] [Google Scholar]
- 7.Chen, Y., M. Balakrishnan, B. P. Roques, and R. A. Bambara. 2003. Steps of the acceptor invasion mechanism for HIV-1 minus strand strong stop transfer. J. Biol. Chem. 278:38368-38375. [DOI] [PubMed] [Google Scholar]
- 8.Coffin, J. M. 1990. Retroviridae and their replication, p. 1437-1500. In B. N. Fields, D. M. Knipe, et al. (ed.), Virology, 2nd ed. Raven Press, New York, NY.
- 9.Coffin, J. M. 1979. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 42:1-26. [DOI] [PubMed] [Google Scholar]
- 10.Dang, Q., and W. S. Hu. 2001. Effects of homology length in the repeat region on minus-strand DNA transfer and retroviral replication. J. Virol. 75:809-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeStefano, J. J., L. M. Mallaber, P. J. Fay, and R. A. Bambara. 1993. Determinants of the RNase H cleavage specificity of human immunodeficiency virus reverse transcriptase. Nucleic Acids Res. 21:4330-4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duch, M., M. L. Carrasco, T. Jespersen, L. Aagaard, and F. S. Pedersen. 2004. An RNA secondary structure bias for non-homologous reverse transcriptase-mediated deletions in vivo. Nucleic Acids Res. 32:2039-2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunn, M. M., J. C. Olsen, and R. Swanstrom. 1992. Characterization of unintegrated retroviral DNA with long terminal repeat-associated cell- derived inserts. J. Virol. 66:5735-5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dykes, C., M. Balakrishnan, V. Planelles, Y. Zhu, R. A. Bambara, and L. M. Demeter. 2004. Identification of a preferred region for recombination and mutation in HIV-1 gag. Virology 326:262-279. [DOI] [PubMed] [Google Scholar]
- 15.Flynn, J. A., W. An, S. R. King, and A. Telesnitsky. 2004. Nonrandom dimerization of murine leukemia virus genomic RNAs. J. Virol. 78:12129-12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frost, S. D., T. Wrin, D. M. Smith, S. L. Kosakovsky Pond, Y. Liu, E. Paxinos, C. Chappey, J. Galovich, J. Beauchaine, C. J. Petropoulos, S. J. Little, and D. D. Richman. 2005. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc. Natl. Acad. Sci. USA 102:18514-18519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu, T. B., and J. Taylor. 1992. When retroviral reverse transcriptases reach the end of their RNA templates. J. Virol. 66:4271-4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hajjar, A. M., and M. L. Linial. 1993. Characterization of a unique retroviral recombinant containing 7S L sequences. J. Virol. 67:7677-7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajjar, A. M., and M. L. Linial. 1993. A model system for nonhomologous recombination between retroviral and cellular RNA. J. Virol. 67:3845-3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herman, S. A., and J. M. Coffin. 1987. Efficient packaging of readthrough RNA in ALV: implications for oncogene transduction. Science 236:845-848. [DOI] [PubMed] [Google Scholar]
- 21.Jetzt, A. E., H. Yu, G. J. Klarmann, Y. Ron, B. D. Preston, and J. P. Dougherty. 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 74:1234-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junghans, R. P., L. R. Boone, and A. M. Skalka. 1982. Retroviral DNA H structures: displacement-assimilation model of recombination. Cell 30: 53-62. [DOI] [PubMed] [Google Scholar]
- 23.Kharytonchyk, S. A., A. I. Kireyeva, A. B. Osipovich, and I. K. Fomin. 2005. Evidence for preferential copackaging of Moloney murine leukemia virus genomic RNAs transcribed in the same chromosomal site. Retrovirology 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitrinos, K. M., N. G. Hoffman, J. A. Nelson, and R. Swanstrom. 2003. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J. Virol. 77:6811-6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kizhatil, K., and L. M. Albritton. 1997. Requirements for different components of the host cell cytoskeleton distinguish ecotropic murine leukemia virus entry via endocytosis from entry via surface fusion. J. Virol. 71:7145-7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konstantinova, P., P. de Haan, A. T. Das, and B. Berkhout. 2006. Hairpin-induced tRNA-mediated (HITME) recombination in HIV-1. Nucleic Acids Res. 34:2206-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulpa, D., R. Topping, and A. Telesnitsky. 1997. Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J. 16:856-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanciault, C., and J. J. Champoux. 2006. Pausing during reverse transcription increases the rate of retroviral recombination. J. Virol. 80:2483-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linial, M., and S. Brown. 1979. High-frequency recombination within the gag gene of Rous sarcoma virus. J. Virol. 31:257-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linial, M. L., and A. D. Miller. 1990. Retroviral RNA packaging: sequence requirements and implications. Curr. Top. Microbiol. Immunol. 157:125-152. [DOI] [PubMed] [Google Scholar]
- 31.Lobato, R. L., E. Y. Kim, R. M. Kagan, and T. C. Merigan. 2002. Genotypic and phenotypic analysis of a novel 15-base insertion occurring between codons 69 and 70 of HIV type 1 reverse transcriptase. AIDS Res. Hum. Retrovir. 18:733-736. [DOI] [PubMed] [Google Scholar]
- 32.Marchand, B., and M. Gotte. 2003. Site-specific footprinting reveals differences in the translocation status of HIV-1 reverse transcriptase. Implications for polymerase translocation and drug resistance. J. Biol. Chem. 278:35362-35372. [DOI] [PubMed] [Google Scholar]
- 33.Marr, S. F., and A. Telesnitsky. 2003. Mismatch extension during strong stop strand transfer and minimal homology requirements for replicative template switching during Moloney murine leukemia virus replication. J. Mol. Biol. 330:657-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikkelsen, J. G., and F. S. Pedersen. 2000. Genetic reassortment and patch repair by recombination in retroviruses. J. Biomed. Sci. 7:77-99. [DOI] [PubMed] [Google Scholar]
- 35.Moumen, A., L. Polomack, T. Unge, M. Veron, H. Buc, and M. Negroni. 2003. Evidence for a mechanism of recombination during reverse transcription dependent on the structure of the acceptor RNA. J. Biol. Chem. 278:15973-15982. [DOI] [PubMed] [Google Scholar]
- 36.Olsen, J. C., C. Bova-Hill, D. P. Grandgenett, T. P. Quinn, J. P. Manfredi, and R. Swanstrom. 1990. Rearrangements in unintegrated retroviral DNA are complex and are the result of multiple genetic determinants. J. Virol. 64:5475-5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Onafuwa, A., W. An, N. D. Robson, and A. Telesnitsky. 2003. Human immunodeficiency virus type 1 genetic recombination is more frequent than that of Moloney murine leukemia virus despite similar template switching rates. J. Virol. 77:4577-4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onafuwa-Nuga, A. A., S. R. King, and A. Telesnitsky. 2005. Nonrandom packaging of host RNAs in Moloney murine leukemia virus. J. Virol. 79:13528-13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Reilly, L., and M. J. Roth. 2000. Second-site changes affect viability of amphotropic/ecotropic chimeric enveloped murine leukemia viruses. J. Virol. 74:899-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parthasarathi, S., A. Varela-Echavarria, Y. Ron, B. D. Preston, and J. P. Dougherty. 1995. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: characterization and implications. J. Virol. 69:7991-8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pathak, V. K., and H. M. Temin. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc. Natl. Acad. Sci. USA 87:6024-6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfeiffer, J. K., M. M. Georgiadis, and A. Telesnitsky. 2000. Structure-based Moloney murine leukemia virus reverse transcriptase mutants with altered intracellular direct-repeat deletion frequencies. J. Virol. 74:9629-9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfeiffer, J. K., and A. Telesnitsky. 2001. Effects of limiting homology at the site of intermolecular recombinogenic template switching during Moloney murine leukemia virus replication. J. Virol. 75:11263-11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfeiffer, J. K., R. S. Topping, N. H. Shin, and A. Telesnitsky. 1999. Altering the intracellular environment increases the frequency of tandem repeat deletion during Moloney murine leukemia virus reverse transcription. J. Virol. 73:8441-8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Preston, B. D., and J. P. Dougherty. 1996. Mechanisms of retroviral mutation. Trends Microbiol. 4:16-21. [DOI] [PubMed] [Google Scholar]
- 46.Pulsinelli, G. A., and H. M. Temin. 1991. Characterization of large deletions occurring during a single round of retrovirus vector replication: novel deletion mechanism involving errors in strand transfer. J. Virol. 65:4786-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rasmussen, S., and F. S. Pedersen. 2004. Complementarity between RNA dimerization elements favors formation of functional heterozygous murine leukemia viruses. Virology 329:440-453. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen, S. V., and F. S. Pedersen. 2006. Co-localization of gammaretroviral RNAs at their transcription site favours co-packaging. J. Gen. Virol. 87:2279-2289. [DOI] [PubMed] [Google Scholar]
- 49.Rhodes, T., H. Wargo, and W. S. Hu. 2003. High rates of human immunodeficiency virus type 1 recombination: near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 77:11193-11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roda, R. H., M. Balakrishnan, J. K. Kim, B. P. Roques, P. J. Fay, and R. A. Bambara. 2002. Strand transfer occurs in retroviruses by a pause-initiated two-step mechanism. J. Biol. Chem. 277:46900-46911. [DOI] [PubMed] [Google Scholar]
- 51.Rulli, S. J., Jr., C. S. Hibbert, J. Mirro, T. Pederson, S. Biswal, and A. Rein. 2007. Selective and nonselective packaging of cellular RNAs in retrovirus particles. J. Virol. 81:6623-6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schultz, S. J., M. Zhang, and J. J. Champoux. 2006. Sequence, distance, and accessibility are determinants of 5′-end-directed cleavages by retroviral RNases H. J. Biol. Chem. 281:1943-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin, N. H., D. Hartigan-O'Connor, J. K. Pfeiffer, and A. Telesnitsky. 2000. Replication of lengthened Moloney murine leukemia virus genomes is impaired at multiple stages. J. Virol. 74:2694-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugden, B. 1993. How some retroviruses got their oncogenes. Trends Biochem. Sci. 18:233-235. [DOI] [PubMed] [Google Scholar]
- 55.Sun, G., P. K. O'Neil, H. Yu, Y. Ron, B. D. Preston, and J. P. Dougherty. 2001. Transduction of cellular sequence by a human immunodeficiency virus type 1-derived vector. J. Virol. 75:11902-11906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swain, A., and J. M. Coffin. 1992. Mechanism of transduction by retroviruses. Science 255:841-845. [DOI] [PubMed] [Google Scholar]
- 57.Takebe, Y., and A. Telesnitsky. 2006. Evidence for the acquisition of multi-drug resistance in an HIV-1 clinical isolate via human sequence transduction. Virology 351:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Telesnitsky, A., S. Blain, and S. P. Goff. 1995. Assays for retroviral reverse transcriptase. Methods Enzymol. 262:347-362. [DOI] [PubMed] [Google Scholar]
- 59.Temin, H. M. 1993. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci. USA 90:6900-6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varela-Echavarria, A., N. Garvey, B. D. Preston, and J. P. Dougherty. 1992. Comparison of Moloney murine leukemia virus mutation rate with the fidelity of its reverse transcriptase in vitro. J. Biol. Chem. 267:24681-24688. [PubMed] [Google Scholar]
- 61.Waters, L. C., and B. C. Mullin. 1977. Transfer RNA into RNA tumor viruses. Prog. Nucleic Acid Res. Mol. Biol. 20:131-160. [DOI] [PubMed] [Google Scholar]
- 62.Wieringa, B., E. Hofer, and C. Weissmann. 1984. A minimal intron length but no specific internal sequence is required for splicing the large rabbit beta-globin intron. Cell 37:915-925. [DOI] [PubMed] [Google Scholar]
- 63.Woffendin, C., U. Ranga, Z. Yang, L. Xu, and G. J. Nabel. 1996. Expression of a protective gene-prolongs survival of T cells in human immunodeficiency virus-infected patients. Proc. Natl. Acad. Sci. USA 93:2889-2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang, S. L., R. Delgado, S. R. King, C. Woffendin, C. S. Barker, Z. Y. Yang, L. Xu, G. P. Nolan, and G. J. Nabel. 1999. Generation of retroviral vector for clinical studies using transient transfection. Hum. Gene Ther. 10:123-132. [DOI] [PubMed] [Google Scholar]
- 65.Yin, P. D., V. K. Pathak, A. E. Rowan, R. J. Teufel II, and W. S. Hu. 1997. Utilization of nonhomologous minus-strand DNA transfer to generate recombinant retroviruses. J. Virol. 71:2487-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu, H., A. E. Jetzt, Y. Ron, B. D. Preston, and J. P. Dougherty. 1998. The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem. 273:28384-28391. [DOI] [PubMed] [Google Scholar]
- 67.Zhang, J., L. Y. Tang, T. Li, Y. Ma, and C. M. Sapp. 2000. Most retroviral recombinations occur during minus-strand DNA synthesis. J. Virol. 74:2313-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang, J., and H. M. Temin. 1993. 3′ junctions of oncogene-virus sequences and the mechanisms for formation of highly oncogenic retroviruses. J. Virol. 67:1747-1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang, J., and H. M. Temin. 1993. Rate and mechanism of nonhomologous recombination during a single cycle of retroviral replication. Science 259:234-238. [DOI] [PubMed] [Google Scholar]
- 70.Zhang, J., and H. M. Temin. 1994. Retrovirus recombination depends on the length of sequence identity and is not error prone. J. Virol. 68:2409-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhuang, J., A. E. Jetzt, G. Sun, H. Yu, G. Klarmann, Y. Ron, B. D. Preston, and J. P. Dougherty. 2002. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 76:11273-11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhuang, J., S. Mukherjee, Y. Ron, and J. P. Dougherty. 2006. High rate of genetic recombination in murine leukemia virus: implications for influencing proviral ploidy. J. Virol. 80:6706-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]