

Abstract
Human noroviruses are positive-sense RNA viruses and are the leading cause of epidemic acute viral gastroenteritis in developed countries. The absence of an in vitro cell culture model for human norovirus infection has limited the development of effective antivirals and vaccines. Human histo-blood group antigens have been regarded as receptors for norovirus infection, and expression of the α(1,2) fucosyltransferase gene (FUT2) responsible for the secretor phenotype is required for susceptibility to Norwalk virus (NV) infection. We report for the first time that transfection of NV RNA, isolated from stool samples from human volunteers, into human hepatoma Huh-7 cells leads to viral replication, with expression of viral antigens, RNA replication, and release of viral particles into the medium. Prior treatment of the RNA with proteinase K completely abolishes RNA infectivity, suggesting a key role of an RNA-protein complex. Although overexpression of the human FUT2 gene enhances virus binding to cells, it is not sufficient to allow a complete viral infection, and viral spread from NV-transfected cells to naïve cells does not occur. Finally, no differences in NV RNA replication are observed between Huh-7 and Huh-7.5.1 cells, which contain an inactivating mutation in retinoic acid-inducible gene I (RIG-I), suggesting that the RIG-I pathway does not play a role in limiting NV replication. Our results strongly suggest that the block(s) to NV replication in vitro is at the stage of receptor and/or coreceptor binding and/or uncoating, either because cells lack some specific factor or activation of cellular antiviral responses independent of RIG-I inhibits virus replication.
The human pathogen Norwalk virus (NV) is the prototype strain of the Norovirus genus in the family Caliciviridae. Noroviruses are responsible for the majority of outbreaks of nonbacterial gastroenteritis in developed countries, and it is estimated that they have a significant impact in developing countries as well. Although human noroviruses were originally identified more than 30 years ago, our understanding of their replication cycle and mechanisms of pathogenicity has been limited because these viruses are noncultivatable in established cell lines and a small animal model to study viral infection is not available. Only recently, it has been reported that both genogroup I (GI) and GII strains of human noroviruses can be passaged several times with limited replication in a differentiated three-dimensional cell culture system derived from a human small intestinal cell line (40). In addition, gnotobiotic pigs can support replication of a human norovirus GII strain, with occurrence of mild diarrhea and virus shedding and immunofluorescent detection of both structural and nonstructural proteins in enterocytes (10). Although these results are promising, it remains unclear whether these systems are robust enough to be widely used to efficiently propagate human noroviruses in vitro, and the factors responsible for the block(s) of viral replication using standard cell culture systems remain unknown.
The NV genome is a positive-sense, polyadenylated, single-stranded RNA molecule of 7.7 kb and contains three open reading frames (ORFs): ORF1 encodes a nonstructural polyprotein, and ORF2 and ORF3 encode the major and minor capsid proteins, VP1 and VP2, respectively (14, 24). Due to the lack of an in vitro system to propagate human noroviruses, features of their life cycle have been inferred from studies using other animal caliciviruses that can grow in mammalian cell cultures. A 3′ coterminal polyadenylated subgenomic RNA is produced within infected cells, and it is believed that both genomic and subgenomic RNAs are covalently linked to the nonstructural protein VPg at their 5′ ends. Upon infection of cells, nonstructural proteins are expressed from genomic RNA and form an RNA replication complex, which generates new genomic RNA molecules as well as subgenomic RNAs encoding VP1 and VP2. After expression of the structural proteins from subgenomic RNA molecules, the capsid is assembled and viral RNA encapsidated prior to progeny release. Some of these features have been confirmed using recombinant systems to express the native NV genome in mammalian cells by using vaccinia virus expression systems (2, 25).
Studies with human volunteers have shown that some individuals are either repeatedly susceptible or resistant to NV infection (36) and led to the identification of a genetically determined factor that predicts a person's susceptibility to infection and disease (19, 30). Binding experiments using recombinant NV virus-like particles (VLPs) demonstrated attachment of VLPs to surface epithelial cells of the gastroduodenal junction on biopsies from secretors but not to cells from nonsecretors, showing that the expression pattern of ABH histo-blood group antigens may influence susceptibility to NV (32). The gene responsible for the secretor phenotype encodes an α(1,2)fucosyltransferase (FUT2) that produces H antigens on the surface of epithelial cells and in mucosal secretions (27). Since it was observed that transfection of the FUT2 gene into nonpermissive cells enhances NV binding (31), it has been hypothesized that H antigens or related blood group antigens may function as a receptor for NV.
The main goal of our study was to understand the molecular basis of the restricted growth of NV in cultured cells by transfecting wild-type NV RNA into human cells. Our studies show for the first time that transfection of wild-type NV RNA isolated from human stool samples can lead to the production of viral particles, indicating that wild-type NV RNA is infectious and replicates. However, a block to NV spread to other cells in the culture remains, indicating that the block(s) exists at the cell entry and/or uncoating steps.
MATERIALS AND METHODS
Cell lines and NV stool specimens.
The human intestinal CaCo-2 cell line was obtained from the American Type Culture Collection and was maintained in Earle's minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS). The hepatic Huh-7 cell line was kindly provided by S. Makino. Huh-7.5.1 cells were kindly provided by F. Chisari (46). Huh-7 and Huh-7.5.1 cells were maintained in Dulbecco's MEM (DMEM) supplemented with 10% FBS.
Stool specimens from human volunteers 715 (100521) and 722 (100619) experimentally infected with NV (A. R. Opekun, R. A. Atmar, M. A. Gilger, M. K. Estes, F. H. Neill, A. Chandrasekaran, C. L. Ugarte, and D. Y. Graham, presented at the Annual Meeting of the American Gastroenterological Association Institute and Digestive Disease Week, 2007; R. Atmar, unpublished data) were used throughout the study. NV titers of stool specimens were higher than 1 × 1011 RNA genomic copies per gram.
Antibodies and plasmids.
Polyclonal hyperimmune antiserum to recombinant NV VLPs raised in rabbits and mouse monoclonal antibodies (MAbs) NV3901 and NV8812 have been described elsewhere (16, 23, 35, 43). Antiserum to VPg was made by hyperimmunizing rabbits with a His-tagged VPg expressed and purified in Escherichia coli Bl-21(DE3) cells. The BG-4 anti-H type 1-specific MAb was purchased from Signet Pathology Systems (Dedham, MA), the BRIC 231 anti-H type 2 MAb was purchased from Biogenesis (United Kingdom), and the anti-Leb MAb was purchased from Gamma Biologicals (Houston, TX).
An expression vector containing the human secretor blood group FUT2 gene (accession no. U17894) was kindly provided by John B. Lowe from Case Western Reserve University School of Medicine (27), and pcDNAI (Invitrogen) was used as the corresponding empty vector negative control.
Purification of NV from stool samples.
Ten percent stool suspensions in 0.1 M phosphate-buffered saline (PBS)-0.5% Zwittergent detergent (Calbiochem) were extracted with Vertrel XF (Miller-Stephenson) and centrifuged at 12,400 × g for 10 min. The supernatant was collected, and virus was precipitated by adding a 3× polyethylene glycol-NaCl (24% polyethylene glycol 8000, 0.12 M NaCl) solution to a final concentration of 1× and incubating the mixture for 2 h at 4°C. The precipitated virus was pelleted at 10,000 × g for 15 min, and the pellet was suspended in 0.1 M PBS. The virus suspension was pelleted through a 40% sucrose cushion for 3 h at 124,000 × g and further purified by isopycnic CsCl gradient centrifugation in milli-Q water (1.36 g/ml) for 24 h at 150,000 × g in a Beckman SW55 Ti rotor. After gradient fractionation, each fraction was diluted 10 times in milli-Q water and viruses were recovered by ultracentrifugation (3 h at 150,000 × g). The presence of virus in each fraction was analyzed by enzyme-linked immunosorbent assay (ELISA) specific for the NV VP1 capsid protein as previously described (3), by quantitative real-time reverse transcription-PCR (qRT-PCR) (see below), and by electron microscopy after staining with 1% ammonium molybdate (pH 5.5). Isolation of RNA from the peak fraction containing NV was performed using the QIAamp viral RNA mini kit (QIAGEN) following the manufacturer's instructions.
RNA transfection.
Cells were plated into 48-well plates at a density of 5 × 104 cells per well. After incubation overnight at 37°C, cells were washed twice with DMEM containing 2% FBS, and 250 ng of RNA per well was transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. In some experiments, Lipofectamine 2000 was used in combination with Magnetofection technology (OZ Biosciences). Briefly, after a 15-min incubation of RNA with Lipofectamine 2000, 1 μl of CombiMag transfection reagent was added to the solution per μg of RNA to be transfected. After a 15-min incubation at room temperature, the mixture was added to each well and cell culture plates were incubated on the magnetic plate for 20 min at 37°C. After a 3-h incubation at 37°C, cells were washed twice with DMEM-2% FBS and incubated in complete media at 37°C for the indicated periods.
Detection of NV proteins in transfected cells by IF analysis.
At various hours posttransfection (hpt), cells were rinsed once with 0.1 M PBS and fixed in 4% paraformaldehyde (PF) for 30 min at room temperature. After incubation of cells in 0.5% Triton X-100-0.1 M PBS for 15 min at room temperature for permeabilization, cells were blocked at 37°C for 2 h in 0.1 M PBS-1% bovine serum albumin (BSA). Primary antibodies were added to the wells at the appropriate dilution in 0.1 M PBS-1% BSA (1:1,000 for rabbit anti-VPg and 1:5,000 for MAbs NV3901 and NV8812) and incubated overnight at 4°C. Cells were washed in 0.1 M PBS and incubated for 2 h at room temperature in a 1:1,000 dilution of the corresponding secondary antibody conjugated to Alexa Fluor 594 or 488. Nuclei were stained with 300 nM DAPI (4′,6′-diamidino-2-phenylindole) for 15 min at room temperature. Following the final wash step, immunofluorescence (IF) was detected using an Olympus IX-70 inverted-system microscope.
Proteinase K treatment of NV RNA.
Pretreatment of RNA with proteinase K was carried out by incubation of purified RNA in 0.1 M NaCl, 10 mM Tris (pH 8.0), 1 mM EDTA, 0.5% sodium dodecyl sulfate, and 200 μg/ml of proteinase K for 45 min at 55°C followed by precipitation with ethanol. As a control, equivalent amounts of NV RNA were also treated as described above but with the omission of proteinase K.
Northern blotting.
RNA samples were loaded onto a denaturing agarose gel for Northern blotting, using the NorthernMax kit (Ambion). Membranes were probed with a negative-sense 32P-labeled RNA probe complementary to nucleotides (nt) 5929 to 6808 of the NV genome. The RNA probe was prepared by in vitro transcription using the Promega Riboprobe in vitro transcription systems, in the presence of 100 μCi of [α-32P]UTP (20 mCi/ml). Genomic and subgenomic RNA controls included RNA transcripts derived from in vitro transcription from the previously described plasmids containing genomic and subgenomic sequences under a T7 promoter (2).
qRT-PCR assay.
Two qRT-PCR assays targeted to ORF1 (nt 4641 to 4715) and ORF2 (nt 5412 to 5522) genomic regions were developed. Standard curves or absolute RNA quantification were included in every assay and were generated by using RNA transcripts produced by in vitro transcription of a cDNA that contained both ORF1- and ORF2-targeted regions (nt 4487 to 5671). Primers and probes used to amplify both regions have been previously described (2). Normalization versus glyceraldehyde-3-phosphate dehydrogenase (GAPDH) titers was performed using the TaqMan GAPDH control reagents (human) (Applied Biosystems). Standard curves included five dilutions and three replicate wells for each dilution. All samples were quantified in at least duplicate wells. Reactions were performed using the TaqMan One-Step RT-PCR master mix reagent kit (Applied Biosystems) in an Applied Biosystems 7500 real-time PCR system. The thermal protocol consisted of 30 min at 48°C, followed by 10 min at 95°C and 45 cycles of 15 s at 95°C and 1 min at 60°C.
Analysis of intracellular RNA.
Total cellular RNA was isolated from 1 × 105 cells using the RNeasy mini kit (QIAGEN). Poly(A)+ RNA was further purified from total cellular RNA using the MicroPoly(A) Purist kit (Ambion) and analyzed by Northern blotting and qRT-PCR using the same set of primers and probes described above.
Detection of viral RNA, proteins, and viral particles in the culture supernatant of transfected cells.
At different hours posttransfection (hpt), the culture supernatants were harvested and clarified at 16,000 × g for 15 min at 4°C. NV capsid protein in the supernatant was detected using an ELISA specific for VP1 as previously described (3). For quantification of encapsidated NV RNA, 70 μl of culture supernatant was treated with 20 μg/ml of RNase A for 30 min at 37°C. Viral RNA was purified using the QIAamp viral RNA mini kit (QIAGEN) and subjected to qRT-PCR for ORF1 NV-specific primers. Viral particles were detected by electron microscopy after negative staining with 1% ammonium molybdate (pH 5.5), after concentration of the samples through a 40% sucrose cushion for 3 h at 124,000 × g.
Binding assays.
Forty-eight hours before performing the binding assay, Huh-7 cells grown on poly-d-lysine-coated 48-well plates were transfected with a plasmid encoding the FUT2 enzyme or the empty vector using Lipofectamine 2000. Both recombinant NV VLPs produced in the baculovirus expression system (43) and wild-type virus isolated from infected human stools were used in binding assays. Cells were washed twice in cold PBS-1% BSA and incubated with 100 μl of 5 μg/ml of VLPs or different doses of purified virus diluted in cold PBS-1% BSA for 1 h at 4°C with gentle agitation. Unbound VLPs or virus were removed by washing three times in cold PBS-1% BSA and two times in cold PBS. To detect VLPs bound to cell monolayers, cells were fixed with 4% PF for 30 min at room temperature and processed for IF as described above, but omitting the permeabilization step, using the MAb NV8812 against VP1 protein. The amount of bound wild-type virus was quantified by qRT-PCR. After the final washing steps, total RNA from each well was isolated using the RNeasy mini kit (QIAGEN) and analyzed by qRT-PCR with NV ORF2 and GAPDH primers. The input virus was measured by adding equivalent amounts of virus to wells that had been incubated with PBS-1% BSA alone just before purifying the RNA. Experiments were performed in duplicate wells, and the percentage of bound virus relative to the input virus was calculated after determining the ORF2/GAPDH ratio for each well. The ability of specific MAbs against VP1 to block binding was examined by preincubating the sample in PBS-1% BSA with serial 10-fold dilutions of ascites fluids for 2 h at room temperature as previously described (43), prior to performing the binding assay as described above.
Infection of cultures.
Forty-eight hours after transfection of Huh-7 cells grown on 48-well plates with a plasmid encoding the FUT2 enzyme or the empty vector using Lipofectamine 2000, cells were washed twice with serum-free media. The inocula containing wild-type NV isolated from stool diluted in serum-free media was left on the cells for 3 h at 37°C and was replaced with fresh media containing 10% FBS. Cells were fixed with PF at 24 h postinfection (hpi), and viral replication was analyzed by IF using the MAb NV8812 against VP1 protein.
Statistics.
All statistical analyses were conducted using the Student's t test in SigmaPlot 10.0. Error bars represent standard deviation, and statistical significance was defined as P < 0.01.
RESULTS
Isolation of NV RNA from human stool samples.
Stools from two volunteers containing high titers of NV were used as a source of wild-type NV RNA. Virus was purified from 5-g aliquots of stool by CsCl gradient centrifugation, and each fraction was suspended in a final volume of 100 μl of milli-Q water. The presence of VP1 antigen and NV RNA in each fraction was analyzed by ELISA (Fig. 1A) and qRT-PCR (Fig. 1B). The peak viral antigen and nucleic acid corresponded to fractions with CsCl densities of 1.37 to 1.39 g/ml, which is consistent with previous reports for noroviruses (22). The presence of viral particles in these fractions was confirmed by electron microscopy (Fig. 1C). NV RNA was purified from 15 μl of the peak fraction (fraction 8), and the RNA integrity was assessed by Northern blotting before transfection (Fig. 1D), showing a major band of 7.7 kb corresponding to the size of NV genomic RNA.
FIG. 1.

Purification of NV from stool samples from infected volunteers by CsCl gradient centrifugation. (A) Detection of NV capsid protein VP1 by ELISA in each fraction. (B) Quantification of NV RNA in each fraction by qRT-PCR. (C) Electron micrograph of NV particles after CsCl gradient purification (density, 1.37 g/ml). Scale bar, 100 nm. (D) Northern blot analysis of viral RNA purified from fraction 8 (F8) prior to transfection. In vitro-transcribed positive-sense genomic (G) and subgenomic (S) RNAs were used as positive controls.
Transfection of wild-type NV RNA into CaCo-2 and Huh-7 cells leads to expression of viral antigens.
Since it is believed that the NV RNA 5′ end is covalently linked to a VPg protein, proteinase K was not used to extract the RNA from NV particles purified from stool. NV wild-type RNA was purified from CsCl fractions using the QIAamp viral RNA mini kit (QIAGEN), which is based on the binding properties of nucleic acids to a silica gel-based membrane, in the presence of carrier RNA. Purified RNA used for transfection had a concentration of 70 ng/μl, containing between 1 × 108 and 1 × 109 NV RNA molecules/μl. When 250 ng of purified viral RNA was used to transfect cells of the human intestinal cell line CaCo-2 and human hepatoma cell line Huh-7 grown on 48-well plates, expression of both nonstructural and structural proteins was observed in both cell lines at 48 h after transfection, using antiserum against VPg and the MAb NV3901 against VP1 protein, respectively (Fig. 2). Although the number of positive cells was low in all cases, Huh-7 cells showed the highest ratio of transfected cells/copy of transfected NV RNA. Around 100 to 150 positive cells could be detected after transfecting 5 × 108 NV RNA molecules in 1 × 105 cells. Other cell lines such as Int407 and HEK293T cells also supported NV protein expression after RNA transfection, but the ratios of transfected cells per copy of transfected NV RNA were lower than that for Huh-7 cells (data not shown). Since capsid proteins are translated from subgenomic RNA, detection of both structural and nonstructural proteins indicated that transfected NV RNA was able to replicate and that synthesis of subgenomic RNA occurred.
FIG. 2.
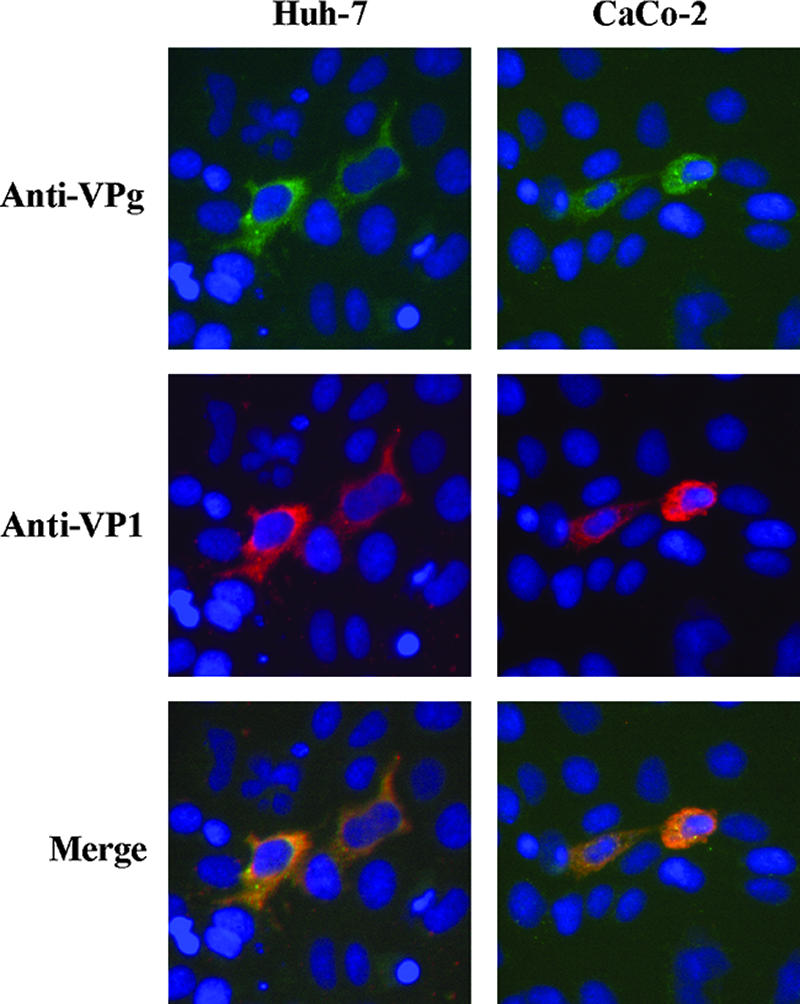
Detection of NV protein expression in Huh-7 and CaCo-2 RNA-transfected cells at 48 hpt. Immunofluorescent staining for nonstructural proteins was performed using antiserum against the VPg protein. Capsid protein VP1 was stained with the MAb NV3901. Nuclei were counterstained with DAPI.
Expression of viral RNA is likely dependent on VPg linkage to genomic RNA.
To further analyze the infectious properties of wild-type NV RNA, RNA isolated from stool was treated with or without proteinase K. Prior to transfection, integrity of RNA was confirmed by Northern blotting (Fig. 3) and qRT-PCR. As measured by qRT-PCR with primers targeted to the ORF2 region, the titers of NV RNA in the absence and presence of proteinase K treatment were similar [(2.05 ± 0.01) × 106 and (3.54 ± 0.12) × 106 NV RNA copies/ng of RNA, respectively]. The number of positive cells after transfection of equal amounts of RNA in Huh-7 cells was evaluated by IF staining using antibodies against VPg and MAb NV8812 at 24 h after transfection with 250 ng of RNA in Huh-7 cells grown on 48-well plates. While in the absence of proteinase K treatment 105 cells expressed VP1 protein, proteinase K treatment completely abolished viral expression, suggesting that NV viral protein expression is dependent on an RNA-protein interaction.
FIG. 3.
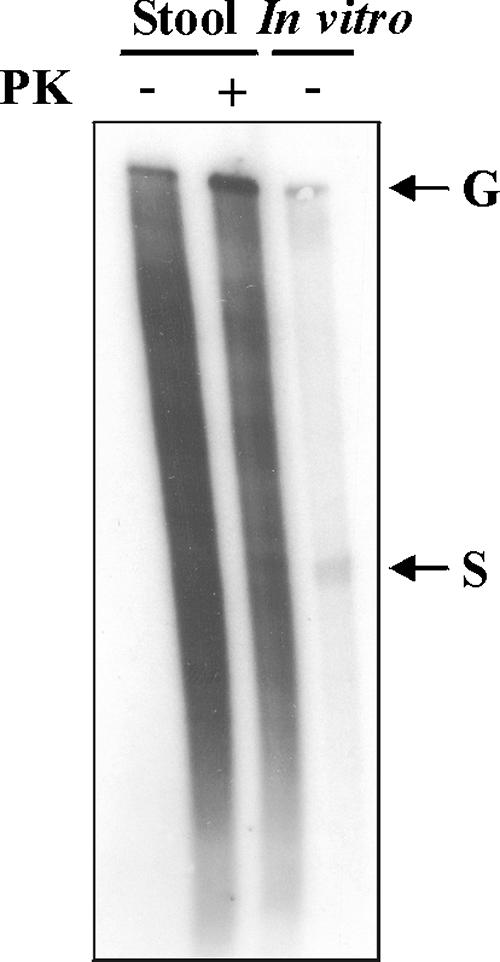
Northern blot analysis of equal amounts of NV RNA isolated from stool after treatment in the presence (+) or absence (−) of proteinase K (PK). In vitro-transcribed positive-sense genomic (G) and subgenomic (S) RNAs without PK treatment were used as positive controls.
NV RNA transfection leads to a single cycle of viral replication.
A kinetic analysis of NV protein expression in Huh-7 cells showed that although clusters containing four to six positive cells were frequently observed by 2 days posttransfection (dpt), a significant increase in the percentage of positive cells over time was not detected, and morphological changes were observed by 4 dpt (Fig. 4A). Morphological changes observed in NV-expressing cells are analyzed in more detail at higher magnification in Fig. 4B. Between 2 and 3 dpt, NV-transfected cells started to detach from the plate surface, and by 5 dpt, the vast majority of NV-transfected cells showed dramatic cytopathic effects that resulted in cell lysis. Altogether, these results suggest that cell-to-cell spread of virus resulting in infection of the complete cell monolayer did not occur. Next, to elucidate whether a complete cycle of viral replication occurred, we tested whether different steps of the viral replication cycle, such as viral RNA replication and assembly and release of progeny virus to the media, could be observed.
FIG. 4.

Time course analysis of NV capsid protein expression by IF staining in transfected Huh-7 cells. (A) NV RNA-transfected Huh-7 cells at 2 and 4 dpt. Magnification, ×40. Arrows indicate clusters of four to six transfected cells, and arrowheads indicate lysed cells. (B) Analysis of changes in Huh-7 cell morphology over time after NV RNA transfection. Magnification, ×200.
Poly(A)-containing RNA produced in Huh-7-transfected cells at different times posttransfection was examined by Northern blot analysis, using an antisense RNA probe specific for the NV ORF2 region and qRT-PCR using primers targeting regions either in ORF1 or ORF2. While ORF2 primers are able to amplify both genomic and subgenomic RNAs, ORF1 primers will only amplify genomic RNA. By Northern blotting, a band corresponding to positive-sense subgenomic RNA was detected starting at 24 hpt. A clear band corresponding to genomic RNA could not be observed, probably due to the low number of NV-transfected cells and low sensitivity of Northern blots, especially to detect high-molecular-weight bands (Fig. 5A). By qRT-PCR, however, an increase in RNA templates amplified by both ORF1 and ORF2 primers began to be observed at 24 hpt and was more obvious at 48 hpt (Fig. 5B). Although at 0 hpt, the titer using ORF2 primers was approximately 3 times lower than the titer obtained with ORF1 primers, probably due to different primer sensitivities, increases of 9-fold and 31-fold were observed from 0 to 48 hpt for ORF1 and ORF2 primers, respectively, indicating that both genomic and subgenomic RNAs were being synthesized. Levels of GAPDH mRNA did not change. The higher increase (fold) detected with ORF2 primers indicated that subgenomic RNA was being synthesized and confirmed the results obtained by Northern blotting. The larger amounts of subgenomic RNA molecules compared to genomic RNA molecules are expected, since similar results are seen during replication of other positive-strand RNA viruses that use subgenomic RNA to synthesize large amounts of capsid proteins, such as togavirus (38) or astrovirus (33).
FIG. 5.

Analysis of NV RNA replication within transfected Huh-7 cells. (A) Northern blot analysis of poly(A)-purified RNA from transfected cells using a negative-strand RNA probe targeting the ORF2 region at different hours posttransfection (hpt). In vitro-transcribed positive-sense genomic (G) and subgenomic (S) RNAs were used as positive controls. Nonspecific bands corresponding to 28S and 18S ribosomal RNAs are indicated. (B) Levels of NV RNA in poly(A)-purified RNA from transfected cells were measured by qRT-PCR using primers against ORF1 and ORF2 regions and GAPDH.
To determine whether viral particles were generated and released from cells, the presence of viral RNA and VP1 protein in the culture supernatant was confirmed by qRT-PCR and ELISA, respectively (Fig. 6A). To rule out the detection of input or free viral RNA, culture supernatants were treated with RNase A prior to extraction of RNA. Overall, a 3-log10 increase in viral RNA was detected from 0 h to 96 hpt, reaching a titer of (1.4 ± 0.4) × 107 NV RNA copies/ml. A correlation was observed in the detection of viral protein by ELISA and RNA titer, and intact virions were detected by electron microscopy in samples concentrated by pelleting through a sucrose cushion (Fig. 6B). Taking into account the number of positive cells detected by IF at 24 hpt in each transfected well, it was possible to estimate that at 96 hpt, each NV-transfected cell had released approximately (2.8 ± 0.8) × 104 genome-containing virions. Finally, biophysical properties of released virions were examined by subjecting the concentrated culture supernatants to isopycnic CsCl gradient centrifugation. The presence of encapsidated viral RNA in each CsCl fraction was analyzed by qRT-PCR. Prior to RNA extraction, each fraction was treated with RNase A to avoid the detection of free viral RNA. A peak of NV RNA was detected at a density of 1.37 g/ml (Fig. 6C), which is similar to that of wild-type virions isolated from human stool samples. Studies to analyze whether RNA extracted from released virus was also infectious were not possible due to the insufficient amount of NV RNA isolated from these particles. While a minimum concentration of 5 × 107 NV RNA copies/μl of purified stool sample was required to detect positive transfected cells by IF, the amount of virus particles released into the supernatant after RNA transfection was not higher than 1 × 104 NV RNA copies/μl.
FIG. 6.

Detection of NV particles in the supernatant of transfected Huh-7 cells. (A) NV RNA and VP1 protein in the supernatant of transfected cultures over time were measured by qRT-PCR using ORF1 primers and ELISA, respectively. (B) Electron micrograph of viral particles released into the supernatant after a concentration step by sucrose cushion. Scale bar, 50 nm. (C) Analysis of biophysical properties of released particles by CsCl density gradient. The titer of NV RNA in each CsCl fraction was measured by qRT-PCR using ORF1 primers.
Taken together, our results indicate that NV RNA isolated from infected stools is infectious and can produce virions when transfected into Huh-7 cells. However, since a significant increase in the percentage of positive cells was not observed over time by IF, NV RNA transfection most likely leads to a single cycle of viral replication, and released virions are not able to infect new cells. Failure to infect new Huh-7 cultures using either wild-type virus isolated from stool or virus produced by NV RNA transfection confirmed these observations (data not shown).
Overexpression of the human FUT2 gene in Huh-7 cells enhances virus binding but does not lead to viral infection.
Since expression of certain H antigens is required for susceptibility to NV infection, we wanted to analyze whether Huh-7 cells express histo-blood group antigens on the cell surface. A phenotyping assay for the expression of some of the main carbohydrate antigens previously shown to be involved in NV binding (H type 1, H type 2, and Leb) was performed on Huh-7 cells. Differentiated CaCo-2 cells (D-CaCo-2) were used as a positive control. Confluent cell monolayers fixed with PF were tested for histo-blood group antigen expression on the cell surface by IF, omitting the permeabilization step. The results indicated that while D-CaCo-2 cells express high levels of H type 1, H type 2, and Leb carbohydrates (Fig. 7, A to C), only a low percentage of Huh-7 cells express levels of Leb antigen detected by IF staining (Fig. 7D to F).
FIG. 7.

Histo-blood group antigen phenotyping by IF assay on differentiated CaCo-2 cells (D-CaCo-2) (A to C), Huh-7 cells (D to F), and Huh-7 cells overexpressing the human FUT2 gene (48 hpt) (G to I). Antibodies against the H type 1, H type 2, and Leb carbohydrates were used to detect carbohydrates.
In case the expression level of histo-blood group antigens on Huh-7 cells was not sufficient to support NV binding and entry, and to enhance expression of the putative NV receptor, Huh-7 cells were transfected with a plasmid encoding the human secretor blood group α(1,2) fucosyltransferase (FUT2). Transient expression of FUT2 in these cells resulted in a significant increase in Leb antigen expression at 48 hpt, but we did not observe an increase in the levels of H type 1 or H type 2, at least not up to levels detectable by IF with the available antibodies (Fig. 7G to I).
Binding assays using recombinant NV VLPs and NV purified from stool were performed with Huh-7 cells transfected with either the FUT2 plasmid or pcDNAI empty vector as a negative control and showed that FUT2 transient expression significantly enhanced NV and VLP binding. IF analysis using MAb NV8812 showed that the VP1 protein of VLPs could be detected on the surface of cells (Fig. 8A). Table 1 shows the percentage of bound virus after incubating FUT2- and pcDNAI-transfected cultures with three different doses of wild-type NV virus purified from stool. Bound virus was quantified by qRT-PCR and normalized versus the number of cells with measurements of GAPDH mRNA levels. While less than 1% of the input virus bound to Huh-7 cells transfected with the empty vector, significantly more virus (up to 16.6%) bound to Huh-7 cells transfected with the FUT2 gene.
FIG. 8.

Analysis of virus binding and viral replication in Huh-7 cells overexpressing FUT2. (A) Effect of FUT2 transient expression on NV binding to Huh-7 cells. Huh-7 cells were transfected with a plasmid carrying the human FUT2 gene or the empty vector (pcDNAI), and 48 hpt, binding assays were performed. Binding of recombinant NV VLPs (5 μg/ml) to Huh-7 cells was detected by IF using the MAb NV8812. (B) Effect of MAbs against NV VP1 on the binding of wild-type NV to FUT2-expressing Huh-7 cells. Purified NV from stool was incubated in three serial 10-fold dilutions of ascites fluids from MAbs NV8812 and NV3901, starting at a 10−3 dilution, for 2 h at room temperature before performing the binding assay. A dose of 2 × 107 NV RNA copies/1 × 105 cells was used. Bound virus was measured by qRT-PCR and normalized versus the number of cells by measurements of GAPDH mRNA levels. Results are expressed as percentage of the control (absence of MAb). The data plotted represent the means of duplicate wells. (C) Quantification of the NV virus yield in the supernatant of cultures cotransfected with NV RNA plus pcDNAI and NV RNA plus FUT2 by qRT-PCR at 96 hpt. The data plotted represent the means ± standard deviation of triplicate wells. (D) Infection of Huh-7 cells transfected with the empty vector (pcDNAI) or the FUT2 plasmid, and detection of VP1 protein by IF at 24 hpi using MAb NV8812. DAPI was used for nuclear counterstaining.
TABLE 1.
Percentage of NV bound to Huh-7 cells transfected with pcDNAI or the FUT2 gene
| Amt of input virus (NV RNA copies/1 × 105 cells) | % of virus bound
|
|
|---|---|---|
| Empty vector (pcDNAI) | FUT2 | |
| 1.4 × 107 | 0.58 ± 0.08 | 10.99 ± 0.48 |
| 7.0 × 107 | 0.97 ± 0.13 | 9.91 ± 1.00 |
| 3.4 × 108 | 0.93 ± 0.08 | 16.64 ± 3.44 |
Due to the limited amounts of NV that can be isolated from stool samples, it was not possible to analyze whether NV binding to FUT2-expressing Huh-7 was saturable. However, specificity of the virus binding was examined by preincubating the virus with serial dilutions of ascites fluids with MAbs NV8812 and NV3901. MAb NV8812 recognizes a conformational epitope in the P2 protruding domain of VP1 and was previously shown to block binding of NV VLPs to D-CaCo-2 cells (43), as well as to inhibit hemagglutination of red blood cells by NV VLPs (21). MAb NV3901, a GI-cross-reactive MAb that recognizes an epitope within the C-terminal P1 subdomain of VP1 (35) and does not have any effect on NV VLP binding to D-CaCo-2 cells, was used as a negative control. While MAb NV3901 had no effect on NV binding, MAb NV8812 was able to block NV binding in a dose-dependent manner (Fig. 8B). These results confirm that attachment of both NV VLPs and wild-type virus to cells requires expression of FUT2 and is mediated by recognition of a histo-blood group H-related antigen by some residues of the P2 protruding domain of VP1 protein.
Cotransfection of NV wild-type RNA with FUT2 expression plasmid does not enhance released virus spreading to neighboring cells.
Since FUT2 overexpression increased the amount of NV binding, we next asked whether cells overexpressing FUT2 would support a productive NV infection following NV RNA transfection or NV infection. To examine whether transfection of NV RNA in FUT2-expressing cells would result in a productive NV infection of the cell monolayer with virus spreading from initially transfected cells to naïve cells, NV RNA from stool was purified using the QIAamp viral RNA mini kit (QIAGEN), adding either pcDNAI or FUT2 plasmids instead of carrier RNA to lysis buffer AVL, and transfected into Huh-7 cells. Ninety-six hours after transfection of samples in triplicate wells, IF staining for Leb carbohydrate was performed in one well to monitor efficiency of FUT2 expression, and IF staining for NV capsid protein was performed in the remaining two wells to count the number of NV-transfected cells per well. NV RNA in the media of each well was quantified by qRT-PCR using NV primers targeted to ORF1 region, and virus yields were compared between NV RNA- pcDNAI- and NV RNA-FUT2-transfected wells. No statistically significant differences were detected either in the number of VP1-positive cells counted by IF (data not shown) nor in the virus yield measured in the supernatant by qRT-PCR (Fig. 8C), indicating that overexpression of FUT2 did not result in virus spreading through the cell culture. Although differences in virus yield were not statistically significant (P < 0.01), the NV titer detected in the supernatant of NV RNA-FUT2-transfected cells was 0.2 log lower than in NV RNA-pcDNAI-transfected cells (Fig. 8C). Since overexpression of FUT2 induced an increase in virus binding to cells, virus yield measured in the supernatant of NV RNA-FUT2-transfected cells may have been lower due to the binding of a portion of the released viruses to the remaining uninfected cells on the monolayer.
In an alternative approach, 48 h after transfection of Huh-7 cells with the FUT2 plasmid or the empty vector, cultures were infected with wild-type NV virions isolated from stool, using a multiplicity of infection of 7 × 108 NV RNA copies/1 × 105 cells. Results of IF staining using the MAb NV8812 against VP1 protein performed at 24 hpi showed that bound virus was only detected on Huh-7 cells that expressed FUT2 (Fig. 8D). No expression of nonstructural proteins was detected (data not shown). Although VP1 of bound virus could still be detected 72 hpi, no viral replication occurred. These results indicated that although FUT2 expression increased binding, cells were not permissive to infection, either due to the lack of a factor required during viral entry and/or uncoating or due to the induction of a cellular response that would inhibit replication.
Inactivation of RIG-I does not affect NV RNA replication.
Recent studies have suggested that cellular innate immune responses play an important role in the control of norovirus replication (7, 44). To examine whether NV RNA replication could be enhanced by suppressing innate immunity, we compared the abilities of the Huh-7 and Huh-7.5.1 cell lines to replicate and produce NV. Huh-7.5.1 cells were derived from the parental Huh-7 cells that harbored a subgenomic hepatitis C virus (HCV) replicon, after curing them by prolonged treatment with alpha interferon (IFN-α). These cells are deficient in virus-activated signaling of IFN-β synthesis through the intracellular retinoic acid-inducible gene I (RIG-I) pathway and are highly permissive for HCV replication (4, 41). To determine whether there were quantitative differences in replication efficiencies between Huh-7 and Huh-7.5.1 cells, equal amounts of NV RNA were transfected in both cell lines in parallel, and kinetics of viral replication were compared after quantifying the released virus in the supernatant at different times posttransfection. The numbers of positive cells by IF at 24 hpt were similar for both cell lines, indicating that RNA transfection efficiencies were similar for both cell lines. NV particle release into the supernatant of transfected Huh-7 cells was slightly delayed compared to production by Huh-7.5.1 cells (Fig. 9), but the differences were not statistically significant. Similar levels of intracellular synthesized subgenomic RNA were detected by Northern blotting between both cell lines (data not shown). These results demonstrate that NV RNA replication efficiencies are similar between Huh-7 and Huh-7.5.1 cells and suggest that an inactivating mutation in RIG-I does not have any effect on NV RNA replication and that the RIG-I signaling pathway does not account for the block to NV infection in Huh-7 cells.
FIG. 9.

Kinetics of NV infection in Huh-7 and Huh-7.5.1 cells after RNA transfection. Equal amounts of purified NV RNA were used to transfect Huh-7 and Huh-7.5.1 cells. Released virus in the supernatant was measured by qRT-PCR at the indicated times posttransfection. The data plotted represent the means ± standard deviation of triplicate wells.
DISCUSSION
The absence of a robust cell culture model for NV infection has limited the study of the NV life cycle, development of effective antivirals and vaccines, and determination of correlates of protection for infection. Here we report for the first time that NV RNA isolated from stool from human volunteers is fully infectious in cultured human cells. Transfection of NV RNA into human hepatoma cells leads to expression of viral antigens and viral replication, with release of viral particles into the medium. These results provide useful information for understanding the block(s) in NV replication in cell culture, since they demonstrate that once NV RNA is introduced into Huh-7 cells, viral replication and viral progeny release occur unhindered. Thus, a lack of a host factor involved in intracellular expression and replication of the viral genome does not seem to be the cause of restriction for virus replication.
Since Huh-7 cells were isolated from a human hepatocarcinoma (34), the high susceptibility of these cells to NV replication initially was unexpected. However, our results are consistent with data published by Chang et al. (7), which show that Huh-7 cells can harbor a NV RNA replicon, suggesting that these cells are highly susceptible to NV replication. Not only have Huh-7 cells been very successful in allowing replication of other fastidious gastrointestinal viruses such as HCV (29, 42, 45, 46) and hepatitis A virus (28), but they have also been shown to support growth of many respiratory RNA viruses (11).
Since translation and infectivity of RNA purified from infected cells or virions of other caliciviruses are drastically reduced by treatment of RNA with proteinase K (5, 8), we examined the effect of this enzyme on wild-type NV RNA. Prior treatment of the RNA with proteinase K completely abolished protein expression after transfection, suggesting that the covalent linkage of RNA to a protein, likely VPg, plays an important role in infectivity and expression of the NV genome, similar to animal caliciviruses (8, 15, 17, 39). Our attempts to recover viral particles by transfecting cells with capped in vitro-transcribed RNA from a full-length NV genomic cDNA have been unsuccessful (data not shown). Although norovirus genome replication and packaging by mammalian cells have been recently demonstrated using different recombinant vaccinia virus expression systems (2, 25), the low efficiency of these systems has not allowed the visualization of recovered virus particles with calicivirus morphology. Since neither of these reverse genetics systems has determined whether VPg was covalently attached to the 5′ end of generated genomic and subgenomic RNAs, the low efficiency of these systems may be explained in part by the fact that a high percentage of the transcripts produced by T7 RNA polymerase in vaccinia virus-infected cells are not linked to a VPg protein but to a cap structure (12).
Demonstrating that human NV RNA is infectious when transfected into cells potentially has important implications. First, although the amount of NV RNA that can be isolated from human stools is limited, the ability to monitor wild-type NV RNA expression and replication in human cells can be used to test the effect of antivirals on NV replication and will help to provide more insights into the NV life cycle, such as the characterization of the RNA replication sites within the cell or the molecular basis of cell death after infection, and proteins involved in these key pathways may become new targets for development of antivirals. Second, since most current methods to detect noroviruses are based on detection of viral nucleic acid by RT-PCR, it is not possible to know whether detected virus is truly infectious or not and whether it represents a potential risk for virus transmission. Until a robust cell culture system is widely available to grow human noroviruses, testing of infectivity by RNA transfection may provide additional information about the infectious capacity of a potentially contaminated sample. However, methods to increase NV RNA isolation from different kinds of samples and transfection efficiency will be required, as currently large amounts of viral RNA are needed to detect replication. Since a certain level of denaturation of the RNA-linked VPg protein may occur with the use of guanidine salts in the RNA purification process, and this could partially account for the low number of positive cells after transfection, the use of other RNA extraction methods might improve the efficiency of the RNA infectivity assays.
Previous work showed that transfection of cells with fucosyltransferases can enable recombinant NV VLP binding. This was achieved with Chinese hamster ovary (CHO) cells, which normally do not express H antigen carbohydrates and show little binding to VLPs. When these cells were transfected with the rat homologue of the human FUT2 gene, they expressed H type 1 and bound NV VLPs (31). Our current work demonstrates that the lack of carbohydrate expression does not explain the block to NV replication. For example, CaCo-2 cells are a human intestinal cell line derived from a secretor-positive individual of blood type O and these cells express the H antigen (1) and show the best binding to NV VLPs (43). Although these cells can replicate wild-type NV RNA and express capsid proteins after transfection, infection does not spread cell to cell over time. Our results indicate that if receptor binding, internalization, and uncoating steps are circumvented, these cells can fully replicate the viral RNA. Unfortunately, due to the low transfection efficiency observed with this cell line, we were unable to confirm the presence of released virions from RNA-transfected CaCo-2 cells, and the possibility that these cells block viral replication at the assembly and/or release step still exists.
In contrast, our results using Huh-7 cells show that although FUT2 expression is a prerequisite for NV attachment and significantly enhances binding of wild-type virus as well as VLPs, it is not sufficient to lead to a productive infection and other factors are required. Although previous work had suggested that the block in viral replication in cell culture occurs at postbinding steps (20), our data rule out the presence of blocks at the level of RNA expression, replication, viral assembly, and progeny release.
Several possibilities can be hypothesized to explain why NV cannot spread to new cells: either cells lack some factor(s) required for viral entry and/or uncoating, or cellular antiviral responses inhibit viral replication. Based on data obtained using the NV replicon developed in Huh-7 cells, Chang et al. (7) suggested that innate immunity may have a role in the control of human norovirus replication, as reported for other animal members of the Caliciviridae family such as the murine norovirus (44) or porcine enteric calicivirus (6), since the NV replicon did not block the ability of Huh-7 cells to produce IFN in response to Sendai virus infection. In addition, expression of the NV replicon RNA was significantly reduced in the presence of exogenous IFN-α (7). Thus, it is possible that replication of wild-type NV RNA transfected into cells could activate the innate immune response, making other cells in the culture resistant to infection. However, whether a cellular innate immune response is activated in cells transfected with wild-type NV RNA or cells harboring the NV replicon remains to be determined.
A recent report describing a reverse genetics system for murine noroviruses using a recombinant fowlpox virus expressing T7 RNA polymerase (9) showed no differences in virus recovery when using Huh-7.5.1 cells or Vero cells, which are defective for IFN production, suggesting that the interferon system does not play a role in the restriction of virus recovery.
Our results show that NV replication is not enhanced in Huh-7.5.1 cells, which contain an inactivating mutation in RIG-I, suggesting that RIG-I pathway activation is not suppressing NV replication in Huh-7 cells. These results are not unexpected, since RIG-I mediates antiviral responses by recognition of single-stranded RNA bearing 5′ phosphates (18, 37), and it is believed that NV genomic and subgenomic RNAs are covalently linked to VPg. RIG-I and MDA-5 (melanoma differentiation-associated gene 5) are similar helicase proteins that induce type I IFN responses through the same signaling pathway, but RNA viruses are differentially recognized by RIG-I and MDA-5. While RIG-I is essential for the production of IFN in response to paramyxoviruses, influenza virus, and Japanese encephalitis virus, MDA-5 is critical for picornavirus detection (13, 26). Whether or not activation of cellular innate immune responses in NV RNA-transfected Huh-7 cells takes place through the MDA-5 signaling pathway will be a focus of our future studies.
If cellular antiviral responses do not account for the complete block of NV replication in cell culture, it is reasonable to think that cells may lack a cellular factor required for viral entry and/or uncoating. Besides interaction with H antigens, NV infection could require an interaction with a coreceptor that triggers internalization signals. Although our results cannot rule out the possibility that overexpression of FUT2 results in an expression level of carbohydrates sufficient to enhance binding of NV but not at the proper location on the cell surface or at the optimal density to allow successful viral entry, our observations lead us to predict that studies to identify other factors such as a coreceptor for internalization or a missing maturation/activation step that elicits uncoating are worthwhile. Understanding the molecular basis of the block(s) of NV infection in vitro should ultimately lead to the development of new cell culture systems to study NV replication and pathogenesis.
Acknowledgments
We are grateful to S. Makino for providing the Huh-7 cells, Francis V. Chisari for providing the Huh-7.5.1 cells, and John B. Lowe for providing the vector carrying the human FUT2 gene. We also thank Richard E. Lloyd, Tyler M. Sharp, and Margarita Lay for critical reading of the manuscript and excellent suggestions.
This work was funded by the National Institutes of Health (P01 AI 57788, N01 AI 25465, and M01 RR-000188), and the Fulbright Scholar Program (FMECD2004/46139439).
Footnotes
Published ahead of print on 12 September 2007.
REFERENCES
- 1.Amano, J., and M. Oshima. 1999. Expression of the H type 1 blood group antigen during enterocytic differentiation of Caco-2 cells. J. Biol. Chem. 274:21209-21216. [DOI] [PubMed] [Google Scholar]
- 2.Asanaka, M., R. L. Atmar, V. Ruvolo, S. E. Crawford, F. H. Neill, and M. K. Estes. 2005. Replication and packaging of Norwalk virus RNA in cultured mammalian cells. Proc. Natl. Acad. Sci. USA 102:10327-10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertolotti-Ciarlet, A., L. J. White, R. Chen, B. V. Venkataram Prasad, and M. K. Estes. 2002. Structural requirements for the assembly of Norwalk virus-like particles. J. Virol. 76:4044-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burroughs, J. N., and F. Brown. 1978. Presence of a covalently linked protein on calicivirus RNA. J. Gen. Virol. 41:443-446. [DOI] [PubMed] [Google Scholar]
- 6.Chang, K. O., S. V. Sosnovtsev, G. Belliot, Y. Kim, L. J. Saif, and K. Y. Green. 2004. Bile acids are essential for porcine enteric calicivirus replication in association with down-regulation of signal transducer and activator of transcription 1. Proc. Natl. Acad. Sci. USA 101:8733-8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, K. O., S. V. Sosnovtsev, G. Belliot, A. D. King, and K. Y. Green. 2006. Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 353:463-473. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhry, Y., A. Nayak, M. E. Bordeleau, J. Tanaka, J. Pelletier, G. J. Belsham, L. O. Roberts, and I. G. Goodfellow. 2006. Caliciviruses differ in their functional requirements for eIF4F components. J. Biol. Chem. 281:25315-25325. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhry, Y., M. A. Skinner, and I. G. Goodfellow. 2007. Recovery of genetically defined murine norovirus in tissue culture by using a fowlpox virus expressing T7 RNA polymerase. J. Gen. Virol. 88:2091-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheetham, S., M. Souza, T. Meulia, S. Grimes, M. G. Han, and L. J. Saif. 2006. Pathogenesis of a genogroup II human norovirus in gnotobiotic pigs. J. Virol. 80:10372-10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freymuth, F., A. Vabret, F. Rozenberg, J. Dina, J. Petitjean, S. Gouarin, L. Legrand, S. Corbet, J. Brouard, and P. Lebon. 2005. Replication of respiratory viruses, particularly influenza virus, rhinovirus, and coronavirus in HuH7 hepatocarcinoma cell line. J. Med. Virol. 77:295-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuerst, T. R., and B. Moss. 1989. Structure and stability of mRNA synthesized by vaccinia virus-encoded bacteriophage T7 RNA polymerase in mammalian cells. Importance of the 5′ untranslated leader. J. Mol. Biol. 2:333-348. [DOI] [PubMed] [Google Scholar]
- 13.Gitlin, L., W. Barchet, S. Gilfillan, M. Cella, B. Beutler, R. A. Flavell, M. S. Diamond, and M. Colonna. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 103:8459-8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glass, P. J., L. J. White, J. M. Ball, I. LeParc-Goffart, M. E. Hardy, and M. K. Estes. 2000. Norwalk virus open reading frame 3 encodes a minor structural protein. J. Virol. 74:6581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodfellow, I., Y. Chaudhry, I. Gioldasi, A. Gerondopoulos, A. Natoni, L. Labrie, J. F. Laliberte, and L. Roberts. 2005. Calicivirus translation initiation requires an interaction between VPg and eIF4E. EMBO Rep. 6:968-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy, M. E., T. N. Tanaka, N. Kitamoto, L. J. White, J. M. Ball, X. Jiang, and M. K. Estes. 1996. Antigenic mapping of the recombinant Norwalk virus capsid protein using monoclonal antibodies. Virology 217:252-261. [DOI] [PubMed] [Google Scholar]
- 17.Herbert, T. P., I. Brierley, and T. D. Brown. 1997. Identification of a protein linked to the genomic and subgenomic mRNAs of feline calicivirus and its role in translation. J. Gen. Virol. 78:1033-1040. [DOI] [PubMed] [Google Scholar]
- 18.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-triphosphate RNA is the ligand for RIG-I. Science 314:994-997. [DOI] [PubMed] [Google Scholar]
- 19.Hutson, A. M., F. Airaud, J. LePendu, M. K. Estes, and R. L. Atmar. 2005. Norwalk virus infection associates with secretor status genotyped from sera. J. Med. Virol. 77:116-120. [DOI] [PubMed] [Google Scholar]
- 20.Hutson, A. M., R. L. Atmar, and M. K. Estes. 2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol. 12:279-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutson, A. M., R. L. Atmar, D. M. Marcus, and M. K. Estes. 2003. Norwalk virus-like particle hemagglutination by binding to H histo-blood group antigens. J. Virol. 77:405-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang, X., D. Y. Graham, K. Wang, and M. K. Estes. 1990. Norwalk virus genome cloning and characterization. Science 250:1580-1583. [DOI] [PubMed] [Google Scholar]
- 23.Jiang, X., M. Wang, D. Y. Graham, and M. K. Estes. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66:6527-6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang, X., M. Wang, K. Wang, and M. K. Estes. 1993. Sequence and genomic organization of Norwalk virus. Virology 195:51-61. [DOI] [PubMed] [Google Scholar]
- 25.Katayama, K., G. S. Hansman, T. Oka, S. Ogawa, and N. Takeda. 2006. Investigation of norovirus replication in a human cell line. Arch. Virol. 151:1291-1308. [DOI] [PubMed] [Google Scholar]
- 26.Kato, H., O. Takeuchi, S. Sato, M. Yoneyama, M. Yamamoto, K. Matsui, S. Uematsu, A. Jung, T. Kawai, K. J. Ishii, O. Yamaguchi, K. Otsu, T. Tsujimura, C. S. Koh, C. Reis e Sousa, Y. Matsuura, T. Fujita, and S. Akira. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101-105. [DOI] [PubMed] [Google Scholar]
- 27.Kelly, R. J., S. Rouquier, D. Giorgi, G. G. Lennon, and J. B. Lowe. 1995. Sequence and expression of a candidate for the human secretor blood group α(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J. Biol. Chem. 270:4640-4649. [DOI] [PubMed] [Google Scholar]
- 28.Konduru, K., and G. G. Kaplan. 2007. Stable growth of wild-type hepatitis A virus in cell culture. J. Virol. 80:1352-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 30.Lindesmith, L., C. Moe, S. Marionneau, N. Ruvoen, X. Jiang, L. Lindblad, P. Stewart, J. LePendu, and R. Baric. 2003. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9:548-553. [DOI] [PubMed] [Google Scholar]
- 31.Marionneau, S., F. Airaud, N. V. Bovin, J. LePendu, and N. Ruvoen-Clouet. 2005. Influence of the combined ABO, FUT2, and FUT3 polymorphism on susceptibility to Norwalk virus attachment. J. Infect. Dis. 192:1071-1077. [DOI] [PubMed] [Google Scholar]
- 32.Marionneau, S., N. Ruvoen, B. Le Moullac-Vaidye, M. Clement, A. Cailleau-Thomas, G. Ruiz-Palacios, P. Huang, X. Jiang, and J. LePendu. 2002. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 122:1967-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monroe, S. S., S. E. Stine, L. Gorelkin, J. E. Herrmann, N. R. Blacklow, and R. I. Glass. 1991. Temporal synthesis of proteins and RNAs during human astrovirus infection of cultured cells. J. Virol. 65:641-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 35.Parker, T. D., N. Kitamoto, T. Tanaka, A. M. Hutson, and M. K. Estes. 2005. Identification of genogroup I and genogroup II broadly reactive epitopes on the norovirus capsid. J. Virol. 79:7402-7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parrino, T. A., D. S. Schreiber, J. S. Trier, A. Z. Kapikian, and N. R. Blacklow. 1977. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med. 297:86-89. [DOI] [PubMed] [Google Scholar]
- 37.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 38.Schlesinger, S., and M. J. Schlesinger. 2001. Togaviridae: the viruses and their replication, p. 895-916. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott, Williams and Wilkins, Philadelphia, PA.
- 39.Sosnovtsev, S., and K. Y. Green. 1995. RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VpG for infectivity. Virology 210:383-390. [DOI] [PubMed] [Google Scholar]
- 40.Straub, T. M., K. H. zu Bentrup, P. Orosz-Coghlan, A. Dohnalkova, B. K. Mayer, R. A. Bartholomew, C. O. Valdez, C. J. Bruckner-Lea, C. P. Gerba, M. Abbaszadegan, and C. A. Nickerson. 2007. In vitro cell culture infectivity assay for human noroviruses. Emerg. Infect. Dis. 13:396-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumpter, R., Jr., Y.-M. Loo, E. Foy, K. Li, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White, L. J., J. M. Ball, M. E. Hardy, T. N. Tanaka, N. Kitamoto, and M. K. Estes. 1996. Attachment and entry of recombinant Norwalk virus capsids to cultured human and animal cell lines. J. Virol. 70:6589-6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wobus, C. E., S. M. Karst, L. B. Thackray, K. O. Chang, S. V. Sosnovtsev, G. Belliot, A. Krug, J. M. Mackenzie, K. Y. Green, and H. W. Virgin. 2004. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2:e432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi, M., R. A. Villanueva, D. L. Thomas, T. Wakita, and S. M. Lemon. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. USA 103:2310-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]