

Abstract
Three proteins encoded by murine cytomegalovirus (MCMV)— gp34, encoded by m04 (m04/gp34), gp48, encoded by m06 (m06/gp48), and gp40, encoded by m152 (m152/gp40)—act together to powerfully impact the ability of primed cytotoxic CD8 T lymphocytes (CTL) to kill virus-infected cells. Of these three, the impact of m152/gp40 on CTL lysis appears greater than would be expected based on its impact on cell surface major histocompatibility complex (MHC) class I. In addition to MHC class I, m152/gp40 also downregulates the RAE-1 family of NKG2D ligands, which can provide costimulation for CD8 T cells. We hypothesized that m152/gp40 may impact CTL lysis so profoundly because it inhibits both antigen presentation and NKG2D-mediated costimulation. We therefore tested the extent to which m152/gp40's ability to inhibit CTL lysis of MCMV-infected cells could be accounted for by its inhibition of NKG2D signaling. As was predictable from the results reported in the literature, NKG2D ligands were not detected by NKG2D tetramer staining of cells infected with wild-type MCMV, whereas those infected with MCMV lacking m152/gp40 displayed measurable levels of the NKG2D ligand. To determine whether NKG2D signaling contributed to the ability of CTL to lyse these cells, we used a blocking anti-NKG2D antibody. Blocking NKG2D signaling did affect the killing of MCMV-infected cells for some epitopes. However, for all epitopes, the impact of m152/gp40 on CTL lysis was much greater than the impact of inhibition of NKG2D signaling. We conclude that the downregulation of NKG2D ligands by MCMV makes only a small contribution to the impact of m152/gp40 on CTL lysis and only for a small subset of CTL.
All cytomegaloviruses (CMVs) interfere with the major histocompatibility complex (MHC) class I pathway of antigen presentation. Although the biological ramifications of this interference remain unclear, the variety of molecular mechanisms employed by different CMVs to attack MHC class I is impressive. Murine CMV (MCMV) contains three viral genes whose products interfere with MHC class I: m04/gp34, m06/gp48, and m152/gp40 (37). This interference has been demonstrated by their ability to impair the lysis of infected targets by cytotoxic T-lymphocytes (CTL) (17, 31).
m06/gp48 binds MHC class I and diverts it to lysosomes for destruction (33). m152/gp40 causes nascent MHC class I to be retained in the endoplasmic reticulum cis-Golgi intermediate compartment (6, 34, 38). m04/gp34 binds some MHC class I and escorts it to the cell surface; in different circumstances, m04/gp34 can either increase or decrease susceptibility to CTL lysis (17, 21, 30, 31). Both m06/gp48 and m152/gp40 reduce the cell surface expression of MHC class I, whereas m04/gp34 either has no impact on cell surface MHC class I or slightly elevates the levels (22, 31, 35). We recently assessed the relative impacts of the three MHC class I immune evasion genes on the ability of CTL specific for 15 H-2b-restricted epitopes to lyse infected cells (31). This study revealed that the three genes act powerfully together to impair CTL lysis of infected cells. We used a panel of mutant viruses generated by Wagner et al. (35) in which m04, m06, and m152 are deleted either alone or in combination. This revealed that m06/gp48 and m152/gp40 made the major contribution to this inhibition, with a contribution from m04/gp34 being needed only for certain epitopes. Thus, the two genes that downregulate cell surface MHC class I also had the greatest impact on CTL lysis.
However, the degree of cell surface MHC class I downregulation did not correlate precisely with the inhibition of CTL lysis. This was most marked in the case of m152/gp40, which had only a moderate impact on the endoplasmic reticulum export of nascent MHC class I and on cell surface MHC class I levels but had a profound impact on CTL lysis. The fact that m152/gp40's impact on CTL lysis was greater than would be expected from its impact on cell surface MHC class I was also noted by Wagner et al. in the original description of their panel of immunoevasin-deficient viruses. These authors postulated that the discrepancy might be explained by a recently uncovered second function of m152/gp40, namely, its ability to prevent the expression of the RAE-1 family of ligands for NKG2D (23, 26).
NKG2D is an activating receptor found on natural killer (NK) cells and on antigen-experienced CD8 T cells. Most NKG2D ligands are not constitutively expressed but are induced by transformation or stress, notably by viral infection (1, 12). In the mouse, there are three groups of known NKG2D ligands, the RAE-1 family of molecules, H-60, and MULT-1 (5, 8, 9). These molecules are major targets of CMV immune evasion: four MCMV genes (m138, m145, m152, and m155) are involved in preventing their expression on infected cells (16, 23-25, 27). The downregulation of the individual NKG2D ligands by each of these MCMV genes affects the ability of NK cells to control MCMV infection (16, 23-27).
m152/gp40 prevents the expression of the RAE-1 family of ligands for NKG2D (23). Thus, m152/gp40 has at least two targets: MHC class I and RAE-1 molecules. Although it is best characterized as a major activating receptor on NK cells, NKG2D does also function as a costimulatory molecule on CD8 T cells (19). In fact, for two human CMV (HCMV)-specific CTL clones, costimulation through NKG2D was necessary to enable the lysis of infected targets once the HCMV immune evasion genes had downregulated MHC class I (14). We therefore wondered whether the apparently disproportionate impact of m152/gp40 on CTL lysis could be explained by its ability to inhibit RAE-1 expression, depriving the CTL of NKG2D-mediated costimulation.
The purpose of the present study was to test the hypothesis that NKG2D inhibition contributes to the profound impact of m152/gp40 on the ability of CD8 T cells to lyse MCMV-infected targets. Although all MCMV-specific CD8 T cells come to express NKG2D over time, we found that costimulation by the NKG2D ligand RAE-1 played only a small role in promoting CTL lysis and only for a subset of epitope specificities. For this minority of specificities, the inhibition of NKG2D signaling did contribute to m152/gp40's impact on CTL lysis. Contrary to our initial hypothesis, overall, targeting of NKG2D does not play a major role in m152/gp40's impact on CD8 T-cell killing.
MATERIALS AND METHODS
Cells.
IC-21 cells (a simian virus 40-transformed macrophage cell line from C57BL/6 mice [4], a gift from Ann Campbell, Eastern Virginia Medical School) were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/liter glucose, 1.5 g/liter sodium bicarbonate, and antibiotics. K42 cells (simian virus 40-transformed H-2b fibroblasts, a gift from Marek Michalak, University of Alberta), C57BL/6 mouse embryo fibroblasts (MEFs) (isolated from C57BL/6 mice), BALB/c MEFs (isolated from BALB/c mice), L929 cells (ATCC), and B16-FL cells (murine melanoma tumor cell line producing FLT-3 ligand [11], a gift from Glen Dranoff, Harvard Medical School) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and antibiotics. The L929 supernatant, a source of macrophage colony-stimulating factor, was harvested from L929 cells grown for 10 days after reaching confluence. Primary bone marrow macrophages (BMMΦ) were isolated by the procedure described by Bouwer et al. (2). Briefly, bone marrow was cultured on non-tissue-culture-treated petri dishes in Dulbecco's modified Eagle's medium with 10% FBS, 30% macrophage colony-stimulating factor from L929 supernatant, and antibiotics. Lymphokine-activated killer (LAK) cells were prepared by incubating C57BL/6 mouse splenocytes for 4 days in RPMI complete medium containing 1 μg/ml interleukin-2.
Viruses.
Wild-type bacterial artificial chromosome-derived MCMV strain MW97.01 (36) Δm04, Δm06, Δm152 Δm04+m06, Δm04+m152, Δm06+m152, and Δm04+m06+m152 (35) mutant viruses were grown in C57BL/6 MEFs and then purified by being pelleted over a 15% sucrose cushion (3). Each virus stock was titered without centrifugal enhancement on BALB/3T3 cells. The mean of the results of three virus titrations was used to calculate titers for use in these assays.
T-cell lines.
Female C57BL/6 or BALB/c mice were purchased from NCI Fredrick (Baltimore, MD) or The Jackson Laboratory (Bar Harbor, ME) and infected with either 1 × 106 or 5 × 106 PFU of MCMV. Spleens were harvested from mice that had been infected at least 11 weeks previously. As a source of dendritic cell-enriched splenocytes to stimulate CTL lines, we used spleens from mice that had been injected 14 days previously with the Flt-3 ligand-secreting B16-FL tumor cells. Splenocytes from the B16-FL-cell-injected mice were gamma irradiated, pulsed with peptide at 10−8 M, and cultured with splenocytes from MCMV-infected mice in RPMI supplemented with 10% FBS for 3 days, after which 10 U/ml recombinant interleukin-2 (eBioscience) was added. After 10 days, the percentage of CD8 T cells responding to the stimulating peptide epitopes was assessed by intracellular cytokine staining (ICS), and the cells used in 51Cr release assays.
Antibodies and tetramers.
Anti-gB and anti-gH were a gift from Lambert Loh (28, 29, 32). Anti-NKG2D (MI-6) (19) and anti-pp89 (7) were purified on protein A and G (Sigma-Aldrich) columns and conjugated to fluorescein isothiocyanate (Molecular Probes), phycoerythrin, or allophycocyanin (Cyanotech) according to published protocols (15). Anti-gamma interferon (IFN-γ) (XMG1.2) and anti-CD8 (53-6.7) were purchased from eBioscience. Anti-MULT-1 (237104), anti-RAE-1pan-specific (186107), and anti-H60 (205326) were purchased from R&D Systems and used according to the manufacturer's instructions. Staining with NKG2D tetramers was done as previously described (9). Db-M45 tetramers, generated as previously described (13), and M38, m139, and m141 tetramers, a kind gift from Sophie Sierro and Paul Klenerman (University of Oxford, Oxford, United Kingdom) were coupled to streptavidin-phycoerythrin.
FACS analysis.
IC-21 cells were infected overnight with the panel of mutant viruses at a multiplicity of infection of 20 in the presence of 0.3 mg/ml phosphonoacetic acid (PAA) (Sigma-Aldrich). PAA is used to limit cytopathic effect by blocking late gene expression. We have previously reported that PAA does not alter immune evasion gene function in macrophages (30). For intranuclear staining, cells were stained with tetramer specific for anti-NKG2D ligands, fixed with CytoFix/CytoPerm (BD Bioscience), and then permeabilized by incubation for 5 min with 0.1% Triton X-100 (Sigma-Aldrich) in phosphate-buffered saline. The cells were then stained for 30 min with anti-pp89 in the presence of 0.1% Triton X-100. The cells were washed three times in 0.1% Triton X-100 and then washed one time in fluorescence-activated cell sorter (FACS) buffer before analysis. ICS was used to demonstrate the antigen specificity. For ICS, CD8 T-cell lines were incubated with their appropriate peptide at 1 μM in the presence of brefeldin A (Golgi-plug; BD Bioscience) for 6 h, stained with anti-CD8 and anti-NKG2D, fixed with CytoFix/CytoPerm (BD Bioscience), permeabilized with PermWash (BD Bioscience), and stained with anti-IFN-γ. All cells were analyzed by using a FACSCalibur flow cytometer (BD Bioscience) in conjunction with Cell Quest (BD Bioscience). All further analysis was performed using FlowJo software (Treestar).
Assay for cell-mediated cytotoxicity.
Amounts of 104 MEF IC-21, K42, or BMMΦ target cells per well were plated in 96-well plates, infected with the indicated viruses at a multiplicity of infection of 20, and labeled with 100 μCi 51Cr (NEN) in the presence of 0.3 mg/ml of PAA for 12 h. For peptide-pulsed targets, 51Cr-labeled cells were incubated with 1 μM peptide for 1 h at 37°C and then washed three times. Effector T cells were incubated with the appropriate concentration of anti-NKG2D (MI-6) or isotype control immunoglobulin G (IgG) for 1 h, and then the effector cells were added at the indicated effector-to-target (E/T) ratios in the presence of blocking antibody or control antibody. The cells were incubated for 6 h, and the supernatants were harvested and assayed with a Topcount scintillation counter (Packard Instruments). The background 51Cr release was determined by incubating targets with medium alone, and the total 51Cr release by lysing targets with medium containing 1% Nonidet P-40 (USB). The percent specific lysis was calculated as follows: (experimental counts per minute [cpm] − background cpm)/(total cpm − background cpm).
Statistics.
The statistical significance was determined by using Student's t test. A paired two-tailed t test was used, and all comparisons were determined to be of equal variance.
RESULTS
NKG2D is expressed on MCMV-specific CD8 T cells in vivo and in vitro.
According to the literature, NKG2D is expressed on murine CD8 T cells within several days of activation (10). Because CMV-specific memory CD8 T cells have an unusual phenotype and usually fail to express the costimulatory molecules CD28 and CD27, we first assessed whether MCMV-specific CD8 T cells express NKG2D. C57BL/6 mice were infected with MCMV and then sacrificed at various times postinfection. MCMV-specific responses were assessed by ICS for IFN-γ, and NKG2D expression determined by costaining with the NKG2D-specific antibody (Fig. 1A). Figure 1B shows that the rate of acquisition of NKG2D differed for different epitope-specific responses. CD8 T cells specific for m139 and m141 were mostly NKG2D positive from day 7 postinfection, whereas it took 30 days before the majority of CD8 T cells specific for M38 and M45 expressed NKG2D. However, most antigen-specific CD8 T cells continued to express NKG2D at 123 days postinfection.
FIG. 1.

Expression of NKG2D by MCMV-specific CD8 T cells. (A) Splenocytes from five mice infected 7 days previously were stimulated with the indicated peptides for ICS. The left panels show CD8 and NKG2D staining from a representative animal and from a naïve control. The numbers indicate the percentage of lymphocytes that express CD8, NKG2D, or both. The middle panels are gated on CD8+ cells, and the numbers show the IFN-γ response to the indicated peptides from a representative infected mouse. The numbers indicate the percentage of lymphocytes that express CD8, and the numbers in the upper quadrant indicate the CD8 T cells that are secreting IFN-γ. The right panels show the number of IFN-γ-positive cells stained with NKG2D. (B) Percent CD8 T cells specific for the indicated peptides that expressed NKG2D on the indicated days postinfection. The means ± standard deviations of the results for 5 mice per group are shown. (C) Polyclonal T-cell lines were generated against the indicated peptide epitopes and analyzed by FACS 10 days later. Antigen-specific cells were identified by tetramer staining (top panel), with the numbers representing the CD8 T cells that stained positive or negative for each antigen-specific tetramer. NKG2D expression on tetramer-positive cells is shown below, as indicated by the numbers for each FACS plot.
The phenomenon of m152/gp40's disproportionate impact on CTL lysis, which we wanted to address in these experiments, was described by using in vitro CTL lines. To confirm that NKG2D expression was maintained upon culture, we generated short-term polyclonal CTL lines specific for four epitopes and assessed their NKG2D expression. Figure 1C shows that the majority of cells in these lines continued to express NKG2D.
Impact of m04/gp34, m06/gp48, and m152/gp40 on expression of NKG2D ligands in infected macrophages.
As described above, m152/gp40 has been shown to downregulate the RAE-1 family of NKG2D ligands. Neither m04/gp34 nor m06/gp48 has been reported to have any impact on NKG2D ligands. However, the previous reports of the downregulation of NKG2D ligands by m152/gp40 have all been carried out in infected fibroblasts. Because our recent characterization of the impact of immune evasion genes on CTL lysis was performed using the macrophage cell line IC-21, we wanted to confirm that m152/gp40 impacted RAE-1 expression in this cell line. Staining with NKG2D tetramers demonstrated that IC-21 cells constitutively express NKG2D ligands (Fig. 2). Upon infection with MCMV, the expression was downregulated, and this downregulation was not relieved by infection with viruses lacking m04 or m06. However, infection with MCMV lacking m152 allowed the expression of NKG2D at almost the same level as in uninfected cells. Three other MCMV genes, m145, m155, and m138, are known to downregulate one or both of the NKG2D ligands MULT-1 and H60 (16, 24, 25, 27). The fact that, in IC-21 cells, infection with a virus lacking m152 restored NKG2D ligand expression to a level similar to that observed in uninfected cells suggested that RAE-1 molecules are the predominant NKG2D ligand expressed by IC-21 cells. This was confirmed by staining both infected and uninfected cells with antibodies against MULT-1, H60, and RAE-1 antibodies (data not shown).
FIG. 2.

m152 interferes with expression of NKG2D ligands on infected IC-21 macrophages. IC-21 cells were infected with the indicated viruses, stained with NKG2D tetramers, and analyzed by FACS. For each infected cell type, NKG2D-tetramer staining of uninfected cells is shown in gray for comparison.
Impact of NKG2D blockade on the ability of CTL to lyse MCMV-infected cells.
In order to assess the extent to which m152/gp40's ability to inhibit CTL lysis can be attributed to its downregulation of NKG2D ligands, we used the blocking anti-NKG2D antibody (MI-6) to inhibit CTL lysis. We first determined the concentration of anti-NKG2D that could completely inhibit NKG2D signaling by titrating anti-NKG2D in a 51Cr release assay using LAK cells against two cell lines that constitutively express NKG2D ligands: IC-21 cells and K42 cells. Figure 3 shows that maximal inhibition of lysis was achieved using an antibody concentration of 50 μg/ml. We note that this concentration did not completely inhibit LAK lysis of IC-21 cells, presumably because other non-NKG2D-mediated LAK receptors are involved in the killing of that cell line (19). We concluded that anti-NKG2D at a concentration of 50 μg/ml could be used to block NKG2D signaling.
FIG. 3.

Determining the anti-NKG2D antibody concentration needed to completely inhibit NKG2D signaling. Two cell lines, IC-21 macrophages and K42 fibroblasts, constitutively expressed NKG2D ligands. (A) To identify the concentration of anti-NKG2D antibody needed to completely inhibit NK cells lysis, LAK cells were incubated with the targets in the presence of the indicated concentration of antibody at an E/T ratio of 20:1 for a 51Cr release assay. (B) E/T ratio titration of LAK cells with IC-21 cells and K42 targets at 50 μg/ml of anti-NKG2D antibody or control.
We then tested the impact of NKG2D blockade on the ability of MCMV-specific CTL to lyse IC-21 cells infected with the panel of immunoevasin-deficient MCMV viruses created by Wagner et al. (35). Short-term polyclonal CTL lines were tested in 51Cr release assays against targets incubated with anti-NKG2D or rat IgG at 50 μg/ml. The results of a typical assay are shown in Fig. 4A. We tested CTL lines against eight different epitope specificities, and each epitope specificity was tested at least three times. To integrate the data from multiple assays, the results for each assay were normalized, with the lysis of each of the mutants, with or without anti-NKG2D, expressed as a percentage of the lysis in cells infected with Δm04+m06+m152 in the presence of rat IgG. An E/T ratio that was below the plateau of maximum killing was used for the calculation. The mean and standard error of multiple assays were then calculated.
FIG. 4.

The impact of the inhibition of NKG2D on CD8 T-cell killing in the presence and absence of m152. (A) Representative 51Cr release assay in the presence of 50 μg/ml of anti-NKG2D. (B) CTL lysis in the presence of anti-NKG2D (open bars) or control IgG (solid bars). +m152 and −m152 indicate the expression of m152 in the viruses used in each group. At least three 51Cr assays were completed for each epitope, and the results normalized by expressing the lysis of each data point as a percentage of the lysis of Δm4 m6 m152 with control IgG. The means ± standard errors of the means of the normalized results from at least three assays per group using an E/T ratio that was off the maximal plateau are shown. A significant difference (P < 0.05) between lysis in the presence and absence of NKG2D blockade is indicated by an asterisk. (C) Direct comparison of the impacts of m152/gp40 and NKG2D blockade on cytolysis by M97-specific T cells. The data shown in panel B are regraphed to compare virus pairs that differ only by the absence or presence of m152 with and without NKG2D blockade. Error bars show the means ± standard errors of the means. (D) Comparison of the effects of m152/gp40 or m06/gp48 alone in the presence of anti-NKG2D. The data shown in panel B are regraphed to compare the impacts of m152/gp40 and m06/gp48 when NKG2D inhibition is playing no role. Error bars show the means ± standard errors of the means.
Figure 4B shows the results of these normalized assays for eight CTL specificities. In the cases where NKG2D blockade resulted in reduced CTL lysis, significant reduction of lysis by the addition of NKG2D is shown as indicated in the figure legend. The impact of NKG2D blockade was most clearly seen for M97-specific CTL. Importantly, NKG2D blockade reduced the lysis of targets infected with viruses that lacked m152 but not of targets infected with viruses in which m152 was present. This result is consistent with the above observation that IC-21 cells infected with a virus containing m152 do not express ligands capable of engaging NKG2D.
Several other H2-Kb CTL specificities, M38, m139, and m141, also showed a significant impact of NKG2D blockade in some but not all viruses lacking m152. However, we saw no impact of NKG2D blockade on any of the H2-Db epitope specificities we tested. For the majority of epitope-specific CTL tested, NKG2D blockade in the absence of m152/gp40 did not cause a significant reduction of target lysis.
Contribution of NKG2D blockade to m152/gp40's impact on CTL lysis.
For those CTL that were affected by NKG2D blockade, we were interested in determining how much of m152/gp40's impact could be attributed to its impact on NKG2D signaling. Figure 4C shows the normalized results from Fig. 4B, redisplayed in order to contrast the impact of m152/gp40 with the impact of NKG2D blockade. Even for the specificities that were most impacted by NKG2D blockade, such as M97, the addition of m152/gp40 caused a greater reduction of target cell lysis than was achieved by NKG2D blockade alone (e.g., for M97, 85% lysis by the Δm04+m06+m152 mutant virus in the presence of anti-NKG2D compared with 25% lysis by the Δm04 m06 mutant virus). We conclude that NKG2D blockade contributes only modestly to m152/gp40's impact on CTL lysis and then only for a minority of epitopes.
This investigation was prompted in part by the observation that m06/gp48 has a greater impact on cell surface MHC class I levels than m152/gp40 but generally has less impact on CTL lysis. In order to compare the impact of m152/gp40 and m06/gp48 on CTL lysis with m152/gp40's impact on NKG2D signaling taken out of the picture, we graphed the lysis of cells infected with Δm04+m06 (m152/gp40 acting alone) and Δm04+m152 (m06/gp48 acting alone) mutant viruses, both in the presence of anti-NKG2D antibody. Figure 4D shows that, even when NKG2D signaling was blocked, m152/gp40 generally had a greater impact on CTL lysis than m06/gp48. We conclude that m152/gp40's disproportionate impact on CTL lysis cannot be explained solely by its ability to inhibit NKG2D ligand costimulation.
Impact of NKG2D on CTL lysis with different mouse strains and cell types.
We considered the possibility that NKG2D signaling might play a greater role in CD8 T-cell lysis of primary rather than transformed cells or, possibly, of different cell types. To investigate this, we generated bone marrow macrophages (BMMΦ) and primary MEFs from C57BL/6 mice for use as target cells in the 51Cr release assays. CD8 T-cell lines were generated against M38 and M97, and 51Cr release assays were performed in the presence of blocking anti-NKG2D antibody or rat IgG as described above. Figure 5A shows that blocking NKG2D resulted in some inhibition of lysis by M97-specific CTL in MEF and BMMΦ targets and by M38-specific CTL in MEFs. However, as was seen with the IC-21 cells, the impact of NKG2D blockade was modest and made only a small contribution to the ability of m152/gp40 to affect CTL killing.
FIG. 5.
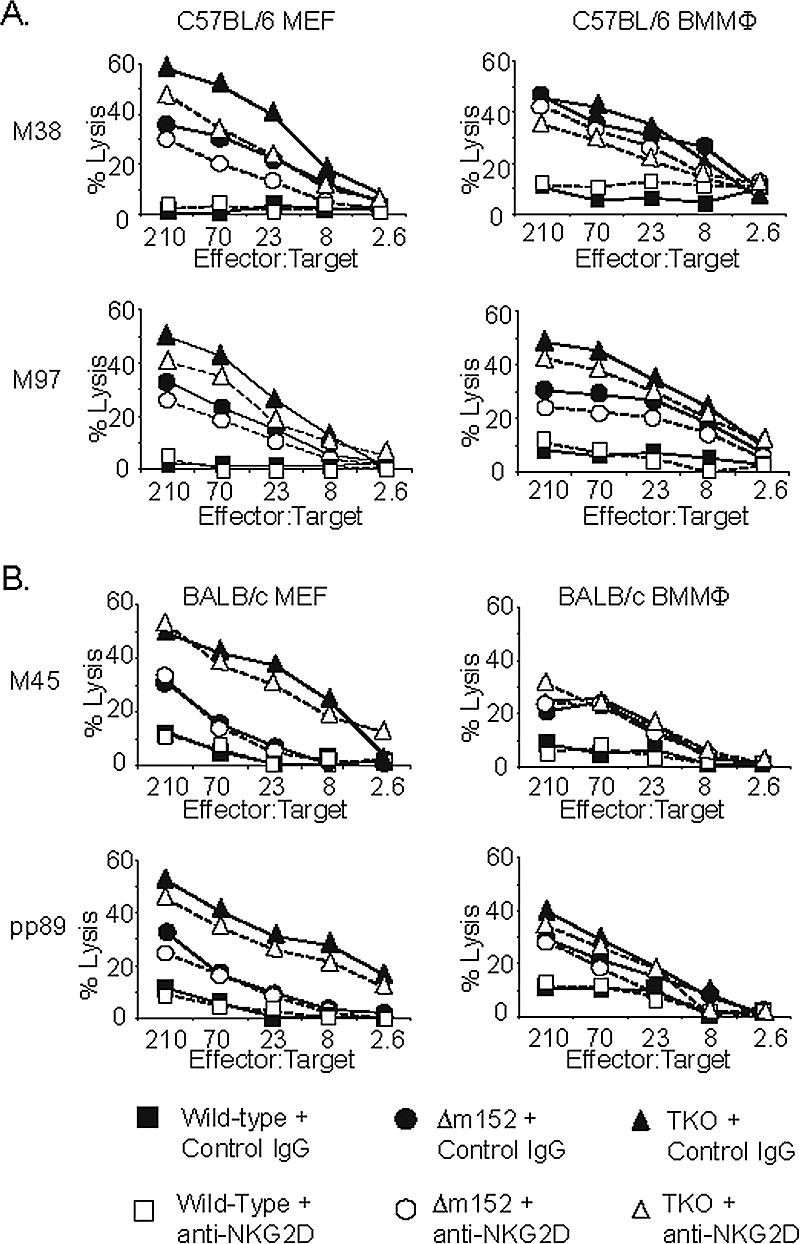
The effect of NKG2D costimulation on CD8 T-cell killing in different cell types and mouse strains. (A) 51Cr release assay using M38- and M97-specific T-cell lines and C57BL/6 MEF and primary BMMΦ targets, infected as indicated in the presence of 50 μg/ml of anti-NKG2D (open symbol) or control IgG antibody (closed symbol). (B) CD8 T-cell lines specific for pp89 and M45 were generated from BALB/c mice and used in 51Cr release assays with BALB/c MEF and primary BMMΦ targets with anti-NKG2D or control antibody as described for panel A.
To test the impact of NKG2D blockade in a different mouse strain, we generated BMMΦ cells and MEFs from BALB/c mice. Flow cytometry staining for the expression of NKG2D ligands on BALB/c MEFs and BMMΦ cells revealed that H60 was downregulated by MCMV infection, but as expected, the downregulation was independent of m152/gp40 (data not shown). CD8 T-cell lines were generated against H-2d-restricted M45 and pp89 peptides, and 51Cr release assays performed as described above (Fig. 5B). In this instance, we saw no impact on killing in the presence of blocking anti-NKG2D antibody. We conclude that NKG2D signaling has a small but perceptible impact on killing of MCMV-infected macrophages or fibroblasts from C57BL/6 mice but no impact on killing of infected primary cells generated from BALB/c mice.
The impact of NKG2D blockade is not determined by peptide-MHC density.
Finally, we were intrigued by the finding that CTL specific for different MCMV epitopes differed in their susceptibility to NKG2D blockade (Fig. 4). We thought that costimulation via NKG2D might be required only when the target cell presents a low-avidity peptide target for the T-cell receptor (TCR). We postulated that those MCMV-specific CTL that were affected by NKG2D might be specific for epitopes that are poorly presented by infected cells, resulting in a suboptimal number of peptide-MHC complexes at the cell surface. To test this idea, we performed a CTL assay in the presence or absence of anti-NKG2D using IC-21 targets incubated with a range of peptide concentrations. If NKG2D costimulation contributes to CD8 T-cell activation when the peptide-MHC density is low, we expected that anti-NKG2D would inhibit lysis at lower peptide concentrations. However, Fig. 6 shows that anti-NKG2D did not inhibit CTL lysis at any concentration of peptide on uninfected IC-21 cells for any of the epitopes tested.
FIG. 6.

The effect of peptide density on anti-NKG2D inhibition of CTL killing. IC-21 targets, incubated with peptides as indicated, were tested for lysis by antigen-specific CD8 T cells in the presence of anti-NKG2D or IgG control.
DISCUSSION
The fact that m152/gp40's efficacy in inhibiting CD8 T-cell function seems greater than its impact on MHC class I levels has been noted by several investigators (18, 20, 31, 35). A simple explanation for this discrepancy seemed to be offered by the finding that, in addition to targeting MHC class I, m152/gp40 could also target the RAE-1 family of NKG2D ligands. Since activated CD8 T cells express NKG2D, inhibition of NKG2D's costimulatory activity seemed a likely explanation for m152/gp40's disproportionate impact on CTL lysis. This explanation was rendered even more plausible by the clear demonstration of the importance of NKG2D costimulation in enabling the lysis of HCMV-infected targets by two pp65-specific CTL clones (14). However, the data presented here do not support this hypothesis. NKG2D inhibition did impact lysis by CTL specific for four epitopes, M97, M38, m141, and m139. Notably, this inhibition was only seen for targets infected with viruses lacking m152, confirming that m152/gp40 effectively inhibits the majority of NKG2D ligand expression in the IC-21 cells used in this assay. However, NKG2D inhibition had no impact on CTL lysis by polyclonal CTL lines specific for many of the MCMV epitopes tested (Fig. 4B). For the epitope specificities for which NKG2D blockade did inhibit lysis, NKG2D blockade had a smaller impact on target lysis than did adding m152 to the genes expressed by MCMV (Fig. 4C). When we directly compared the efficacies of m152/gp40 and m06/gp48 at inhibiting lysis in the presence of NKG2D blockade, it was evident that m152/gp40's impact on CTL lysis was greater than that of m06/gp48 (Fig. 4D), despite the fact that m06/gp48 has a greater impact on cell surface MHC class I levels (31, 35). m152/gp40's disproportionate impact must therefore have another explanation. One possibility would be that, acting at the peptide-loading complex, m152/gp40 has some level of peptide cargo discrimination, perhaps more effectively impacting higher-affinity epitopes that are likely to become immunodominant. Another possibility would be that m152/gp40 has yet further cellular targets that impact CTL efficacy. It is becoming apparent that some MCMV immune evasion genes do indeed have multiple cellular targets: for example, m152/gp40 targets MHC class I and RAE-1. The mechanism of m152/gp40's disproportionate impact thus requires further investigation.
It was surprising to note that NKG2D inhibition impacted CTL lysis by T-cell lines specific for only a subset of epitopes. The reason for this selectivity remains unclear. NKG2D seems to play a costimulatory role when TCR stimulation might be suboptimal, such as in tumor or autoimmune epitopes. The initial observation that NKG2D provided necessary costimulation for HCMV-infected targets seemed in keeping with this idea, since HCMV profoundly downregulates MHC class I. Hence, we suspected that the MCMV epitopes affected might be those that presented lower-avidity ligands for CTL, either because they were present at low epitope density on the surface of infected targets or because they presented low-affinity ligands for the TCR. We have not directly assessed the affinity of peptide-MHC for the TCR. However, we note that the epitopes that were affected by NKG2D were not obviously of low affinity for the TCR, as assessed by peptide titration (compare Fig. 4 and 6). Furthermore, peptide titration on IC-21 cells in the presence or absence of NKG2D blockade provided no evidence for the hypothesis that NKG2D costimulation would prove crucial when peptide MHC density is limiting, at least for the target cells used in this assay. Hence, the reason for NKG2D's affecting only a minority of epitopes remains unclear.
Finally, our studies of NKG2D expression on MCMV-specific CD8 T cells revealed an unexpected feature. NKG2D was expressed on the majority of memory CD8 T cells by 3 weeks postinfection. However, the rate at which CD8 T cells specific for different epitopes acquired NKG2D was quite different: m139- and m141-specific CD8 T cells were almost entirely NKG2D positive by day 7 postinfection, whereas the total population of CD8 T cells specific for M38 and M45 took much longer to become positive. This difference does not correlate with continued activation during the chronic phase of infection. Responses to both M38 and m139 undergo pronounced “memory inflation,” whereas responses to both M45 and m141 contract severely in the chronic phase. m139 and m141 are both members of the same gene family and appear to play an important role in macrophage tropism, whereas M45 is involved in endothelial cell tropism, and the role of M38 is not known. Hence, one possible explanation for these epitope-specific differences could be the types of target cells that express greater quantities of the antigens in vivo. This speculation obviously requires further investigation. We do note, however, that the slow rate of acquisition of NKG2D positivity by M45- and M38-specific CD8 T cells is the exception to the rapid acquisition of NKG2D that has been more commonly described in the literature.
The importance of NKG2D in CMV biology can be deduced from the number of genes that both MCMV and HCMV devote to downregulating its ligands. Inhibition of NKG2D signaling has a major impact on the efficacy of murine NK cells, resulting in significantly higher virus titers in the first few days of infection. Our data suggest that NKG2D signaling in NK cells is likely the major focus of the viral genes that inhibit NKG2D ligands and that targeting NKG2D on CD8 T cells has only a minor impact on CTL function.
Acknowledgments
This research was support by the National Institutes of Health (grants AI47206A and AI50099A to A.B.H.), an American Heart Association fellowship (0215188Z to A.K.P.), and a National Eye Institute training grant (grant ACAEI0071 to A.K.P.).
Footnotes
Published ahead of print on 12 September 2007.
REFERENCES
- 1.Bauer, S., V. Groh, J. Wu, A. Steinle, J. H. Phillips, L. L. Lanier, and T. Spies. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727-729. [DOI] [PubMed] [Google Scholar]
- 2.Bouwer, H. G., M. S. Seaman, J. Forman, and D. J. Hinrichs. 1997. MHC class Ib-restricted cells contribute to antilisterial immunity: evidence for Qa-1b as a key restricting element for Listeria-specific CTLs. J. Immunol. 159:2795-2801. [PubMed] [Google Scholar]
- 3.Brune, W., H. Hengel, and U. H. Koszinowski. 2005. A mouse model for cytomegalovirus infection, unit 19.7. In J. E. Coligan et al. (ed.), Current protocols in immunology, vol. 4. John Wiley & Sons, Hoboken, NJ. [DOI] [PubMed]
- 4.Cavanaugh, V. J., R. M. Stenberg, T. L. Staley, H. W. Virgin IV, M. R. MacDonald, S. Paetzold, H. E. Farrell, W. D. Rawlinson, and A. E. Campbell. 1996. Murine cytomegalovirus with a deletion of genes spanning HindIII-J and -I displays altered cell and tissue tropism. J. Virol. 70:1365-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerwenka, A., A. B. Bakker, T. McClanahan, J. Wagner, J. Wu, J. H. Phillips, and L. L. Lanier. 2000. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 12:721-727. [DOI] [PubMed] [Google Scholar]
- 6.del Val, M., H. Hengel, H. Hacker, U. Hartlaub, T. Ruppert, P. Lucin, and U. H. Koszinowski. 1992. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J. Exp. Med. 176:729-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Del Val, M., H. Volkmer, J. B. Rothbard, S. Jonjic, M. Messerle, J. Schickedanz, M. J. Reddehase, and U. H. Koszinowski. 1988. Molecular basis for cytolytic T-lymphocyte recognition of the murine cytomegalovirus immediate-early protein pp89. J. Virol. 62:3965-3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diefenbach, A., J. K. Hsia, M. Y. Hsiung, and D. H. Raulet. 2003. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur. J. Immunol. 33:381-391. [DOI] [PubMed] [Google Scholar]
- 9.Diefenbach, A., A. M. Jamieson, S. D. Liu, N. Shastri, and D. H. Raulet. 2000. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1:119-126. [DOI] [PubMed] [Google Scholar]
- 10.Diefenbach, A., E. Tomasello, M. Lucas, A. M. Jamieson, J. K. Hsia, E. Vivier, and D. H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142-1149. [DOI] [PubMed] [Google Scholar]
- 11.Driessen, C., R. A. Bryant, A. M. Lennon-Dumenil, J. A. Villadangos, P. W. Bryant, G. P. Shi, H. A. Chapman, and H. L. Ploegh. 1999. Cathepsin S controls the trafficking and maturation of MHC class II molecules in dendritic cells. J. Cell Biol. 147:775-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasser, S., S. Orsulic, E. J. Brown, and D. H. Raulet. 2005. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436:1186-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gold, M. C., M. W. Munks, M. Wagner, C. W. McMahon, A. Kelly, D. G. Kavanagh, M. K. Slifka, U. H. Koszinowski, D. H. Raulet, and A. B. Hill. 2004. Murine cytomegalovirus interference with antigen presentation has little effect on the size or the effector memory phenotype of the CD8 T cell response. J. Immunol. 172:6944-6953. [DOI] [PubMed] [Google Scholar]
- 14.Groh, V., R. Rhinehart, J. Randolph-Habecker, M. S. Topp, S. R. Riddell, and T. Spies. 2001. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2:255-260. [DOI] [PubMed] [Google Scholar]
- 15.Hardy, R. 1986. Purification and coupling of fluorescent proteins for use in flow cytometry, 4th ed. Blackwell Scientific Publications, Boston, MA.
- 16.Hasan, M., A. Krmpotic, Z. Ruzsics, I. Bubic, T. Lenac, A. Halenius, A. Loewendorf, M. Messerle, H. Hengel, S. Jonjic, and U. H. Koszinowski. 2005. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 79:2920-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holtappels, R., D. Gillert-Marien, D. Thomas, J. Podlech, P. Deegen, S. Herter, S. A. Oehrlein-Karpi, D. Strand, M. Wagner, and M. J. Reddehase. 2006. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 80:7613-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holtappels, R., J. Podlech, M.-F. Pahl-Seibert, M. Julch, D. Thomas, C. O. Simon, M. Wagner, and M. J. Reddehase. 2004. Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J. Exp. Med. 199:131-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamieson, A. M., A. Diefenbach, C. W. McMahon, N. Xiong, J. R. Carlyle, and D. H. Raulet. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 17:19-29. [DOI] [PubMed] [Google Scholar]
- 20.Kavanagh, D. G., M. C. Gold, M. Wagner, U. H. Koszinowski, and A. B. Hill. 2001. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194:967-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kavanagh, D. G., U. H. Koszinowski, and A. B. Hill. 2001. The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-Golgi compartment. J. Immunol. 167:3894-3902. [DOI] [PubMed] [Google Scholar]
- 22.Kleijnen, M. F., J. B. Huppa, P. Lucin, S. Mukherjee, H. Farrell, A. E. Campbell, U. H. Koszinowski, A. B. Hill, and H. L. Ploegh. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16:685-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krmpotic, A., D. H. Busch, I. Bubic, F. Gebhardt, H. Hengel, M. Hasan, A. A. Scalzo, U. H. Koszinowski, and S. Jonjic. 2002. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3:529-535. [DOI] [PubMed] [Google Scholar]
- 24.Krmpotic, A., M. Hasan, A. Loewendorf, T. Saulig, A. Halenius, T. Lenac, B. Polic, I. Bubic, A. Kriegeskorte, E. Pernjak-Pugel, M. Messerle, H. Hengel, D. H. Busch, U. H. Koszinowski, and S. Jonjic. 2005. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 201:211-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lenac, T., M. Budt, J. Arapovic, M. Hasan, A. Zimmermann, H. Simic, A. Krmpotic, M. Messerle, Z. Ruzsics, U. H. Koszinowski, H. Hengel, and S. Jonjic. 2006. The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J. Exp. Med. 203:1843-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lodoen, M., K. Ogasawara, J. A. Hamerman, H. Arase, J. P. Houchins, E. S. Mocarski, and L. L. Lanier. 2003. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 197:1245-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodoen, M. B., G. Abenes, S. Umamoto, J. P. Houchins, F. Liu, and L. L. Lanier. 2004. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 200:1075-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loh, L. C., N. Balachandran, and L. F. Qualtiere. 1988. Characterization of a major virion envelope glycoprotein complex of murine cytomegalovirus and its immunological cross-reactivity with human cytomegalovirus. Virology 166:206-216. [DOI] [PubMed] [Google Scholar]
- 29.Loh, L. C., and L. F. Qualtiere. 1988. A neutralizing monoclonal antibody recognizes an 87K envelope glycoprotein on the murine cytomegalovirus virion. Virology 162:498-502. [DOI] [PubMed] [Google Scholar]
- 30.LoPiccolo, D. M., M. C. Gold, D. G. Kavanagh, M. Wagner, U. H. Koszinowski, and A. B. Hill. 2003. Effective inhibition of Kb- and Db-restricted antigen presentation in primary macrophages by murine cytomegalovirus. J. Virol. 77:301-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinto, A. K., M. W. Munks, U. H. Koszinowski, and A. B. Hill. 2006. Coordinated function of murine cytomegalovirus genes completely inhibits CTL lysis. J. Immunol. 177:3225-3234. [DOI] [PubMed] [Google Scholar]
- 32.Rapp, M., P. Lucin, M. Messerle, L. C. Loh, and U. H. Koszinowski. 1994. Expression of the murine cytomegalovirus glycoprotein H by recombinant vaccinia virus. J. Gen. Virol. 75:183-188. [DOI] [PubMed] [Google Scholar]
- 33.Reusch, U., W. Muranyi, P. Lucin, H. G. Burgert, H. Hengel, and U. H. Koszinowski. 1999. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18:1081-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thale, R., U. Szepan, H. Hengel, G. Geginat, P. Lucin, and U. H. Koszinowski. 1995. Identification of the mouse cytomegalovirus genomic region affecting major histocompatibility complex class I molecule transport. J. Virol. 69:6098-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner, M., A. Gutermann, J. Podlech, M. J. Reddehase, and U. H. Koszinowski. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 196:805-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner, M., S. Jonjic, U. H. Koszinowski, and M. Messerle. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056-7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yewdell, J. W., and A. B. Hill. 2002. Viral interference with antigen presentation. Nat. Immunol. 3:1019-1025. [DOI] [PubMed] [Google Scholar]
- 38.Ziegler, H., R. Thale, P. Lucin, W. Muranyi, T. Flohr, H. Hengel, H. Farrell, W. Rawlinson, and U. H. Koszinowski. 1997. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6:57-66. [DOI] [PubMed] [Google Scholar]