

Abstract
Human leukocyte antigen (HLA)-B27-positive subjects are uncommon in their ability to control infection with human immunodeficiency virus type 1 (HIV-1). However, late viral escape from a narrowly directed immunodominant Gag-specific CD8+ T-lymphocyte (CTL) response has been linked to AIDS progression in these individuals. Identifying the mechanism of the immune-mediated control may provide critical insights into HIV-1 vaccine development. Here, we illustrate that the CTL escape mutation R264K in the HLA-B27-restricted KK10 epitope in the capsid resulted in a significant defect in viral replication in vitro. The R264K variant was impaired in generating late reverse transcription products, indicating that replication was blocked at a postentry step. Notably, the R264K mutation was associated in vivo with the development of a rare secondary mutation, S173A, which restored viral replication in vitro. Furthermore, infectivity of the R264K variant was rescued by the addition of cyclosporine A or infection of a cyclophilin A-deficient cell line. These data demonstrate a severe functional defect imposed by the R264K mutation during an early step in viral replication that is likely due to the inability of this variant to replicate efficiently in the presence of normal levels of cyclophilin A. We conclude that the impact of the R264K substitution on capsid structure constrains viral escape and enables long-term maintenance of the dominant CTL response against B27-KK10, providing an explanation for the protective effect of HLA-B27 during HIV infection.
Accumulating evidence supports a role for CD8+ cytotoxic T-lymphocyte (CTL) responses in the control of infections by human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus (SIV). The initial reduction of primary HIV-1 and SIV viremia correlates with the appearance of virus-specific CTL in the acute phase (49, 65), and depletion of these cells during chronic SIV infection results in an inability to control SIV (67). Furthermore, it is well established that particular major histocompatibility complex class I alleles are associated with slower disease progression following infection by these persistent lentiviruses. The most notable of these alleles include human leukocyte antigen (HLA)-B57 and HLA-B27 in HIV-1 infection (44, 55) and Mamu-A01 and Mamu-B17 in SIV infection of rhesus macaques (56, 77). While the mechanism of this control is not clearly understood, some studies suggest that it is related to the targeting of unique CD8+ T-cell epitopes. In support of this model, acute-phase immune responses in HLA-B57-positive (HLA-B57+) and HLA-B27+ subjects are dominated by HLA-B57- and HLA-B27-restricted CD8+ T-cell responses (7, 68), and HLA-B27 narrowly restricts an immunodominant CD8+ T-cell response in Gag (39, 58, 73). Similarly, in rhesus macaques expressing Mamu-A01, CTL responses against the Gag CM9 and Tat SL8 epitopes predominate during acute infection regardless of the expression of other class I alleles (6, 61).
Numerous studies have investigated correlations between the magnitude and breadth of CD8+ T-cell responses and control of HIV-1 by assessing CTL responses using the gamma interferon enzyme-linked immunospot assay (24, 28, 47, 53, 59). Despite these efforts, few consistent associations have come to light, with the exception of several reports linking Gag-specific CD8+ T-cell responses with lower viral loads and/or higher CD4+ T-cell counts in both clade B and clade C infections (18, 24, 36, 53, 59, 79). Other experiments indicate the enhanced ability of Gag-specific CTL clones to kill infected target cells and suppress viral replication (66, 75). Therefore, growing data suggest that Gag-specific CTL responses may be major contributors to the immune control of HIV-1. One explanation for this association is the critical role that the highly conserved p24 capsid protein of Gag plays during viral replication. Uncoating of the p24 core particle during viral entry appears to be a finely tuned process (23, 27) that may involve interactions with numerous host factors. In particular, efficient HIV-1 infection typically requires the binding of the host protein cyclophilin A (CypA) to the CypA binding loop of the N-terminal domain of p24 (14, 33, 35). Therefore, the requirement for p24 to perform multiple highly regulated steps during the viral life cycle may make it an extremely susceptible target for CD8+ T-cell responses.
A key factor limiting the success of CTL responses is the propensity for HIV-1 to mutate targeted epitopes to evade recognition (41, 64). Mutational escape from CD8+ T-cell responses is well defined in many viral infections (3, 70), and studies of both HIV-1 and SIV illustrate that CTL escape is a hallmark of both acute and chronic infection (4, 6, 43, 60). However, while CTL escape mutations enable the evasion of host immune responses and have been associated with the loss of HIV-1 control (12, 25, 41), the requirement to preserve the basic structure and function of viral proteins may limit the accommodation of these sequence changes. Indeed, several reports documented that some CTL escape mutations in HIV-1 and SIV diminish viral replication (31, 50, 52, 54) and may even revert upon transmission to an HLA-mismatched host (5, 31, 50, 51). This has been illustrated in SIV infection where a CTL escape mutation in the immunodominant Mamu-A01-CM9 epitope in p27 reduced viral replication capacity (32, 62, 78), presumably by interfering with the process of capsid formation, and more recently for two HLA-B57-associated escape mutations in HIV-1 p24 (22, 52). Together, these studies suggest that some immune-driven mutations disrupt functionally important regions of the virus and that a high barrier for CTL escape may thereby contribute to containment. Since many current HIV-1 vaccine strategies aim to sustain durable low viral loads as an alternative to seeking sterilizing protection, inducing immune responses that are capable of driving the virus into a less fit state may represent an important goal of a successful vaccine (8).
HLA-B27+ individuals infected with HIV-1 clade B mount an immunodominant CTL response targeting the KK10 epitope (KRWIILGLNK263-272) in p24 Gag (10, 41, 58). Viral escape from this conserved epitope typically arises late in infection and is associated with progression to AIDS (25, 41, 46). Here, we demonstrate that the predominant CTL escape mutation R264K in KK10 dramatically compromised in vitro viral replication capacity, highlighting the vulnerability of this conserved region of HIV-1 capsid to host immune pressures. Notably, replication of the mutated virus was restored to wild-type (WT) levels by the incorporation of a rare upstream in vivo compensatory mutation, S173A, by the modulation of CypA binding in the presence of cyclosporine A (CsA), and in a CypA knockout cell line, suggesting an impact of these mutations on the structure of HIV-1 capsid. These data provide a mechanistic explanation for the late escape observed in the KK10 epitope that may underlie the protective effect of HLA-B27.
MATERIALS AND METHODS
Patients.
Twenty-six HIV-positive subjects expressing HLA-B27 were examined. Partial sequences from some of the subjects were reported previously (SW, 007, 025, and 777) (38, 46). Subjects 17630, 11504, and 18030 were enrolled in the San Francisco City Clinic Cohort (17, 74). Subject M101 was enrolled in the Multicenter AIDS Cohort Study (45), and subjects H39, H1007, H178, H95, and H1006 were enrolled in a study by the Department for Internal Medicine III with the Institute for Clinical Immunology, Erlangen, Germany. The remaining subjects were enrolled in a study in Boston through the Fenway Community Health Center or Massachusetts General Hospital. Informed consent was obtained from all patients to conduct these investigations in accordance with guidelines of the appropriate local institutional ethics committees. Viral loads in copies per milliliter are also indicated where available.
Viral sequencing.
Genomic DNA was isolated from peripheral blood mononuclear cells (PBMC) either immediately or after stimulation with phytohemagglutinin for 48 h using the Puregene kit (Gentra Systems) and stored at −20°C. For nested PCR amplification of p24, the first-round PCR primer pair was CCCTTCAGACAGGATCAG and CCACATTTCCAACAGCCC, and the second-round primer pair was GCACAGCAAGCAGCAGCT and GTGCCCTTCTTTGCCACA. For PCR amplification of p17 plus p24, the primers were previously described (38). PCR products were gel purified and ligated into a TOPO sequencing vector (Invitrogen). Purified plasmid DNA was then sequenced bidirectionally on an ABI 377 sequencer. Sequences were aligned and analyzed using Bioedit (T. Hall, North Carolina State University) and Sequencer A3.1.1.1 (Applied Biosystems). For subjects from the Boston cohorts, Gag sequences were obtained as previously described (4, 51). Subtype B viral sequences were confirmed using BLAST software, available from the Los Alamos HIV Sequence Database (http://hiv-web.lanl.gov/).
Variant NL4-3 constructs.
HIV-1 strain NL4-3 was modified to express one or more mutations in p24 using the GeneTailor site-directed mutagenesis system (Invitrogen). A SacI-SbfI fragment (residues 491 to 2844) was isolated from pNL4-3 and ligated into pUC19. Mutagenesis was then performed using 5′ oligonucleotide primers R264K-F (CCAGTAGGAGAAATCTATAAAAAATGGATAATCCTG [nucleotide {nt} 1593]), R264K-R (TTTTATAGATTTCTCCTACTGGGATAGGTGG [nt] 1549), L268M-F (TCTATAAAAGATGGATAATCATGGGATTAAA [nt 1601]), L268M-R (GATTATCCATCTTTTATAGATTTCTCCTAC [nt 1561]), L268M on R264K-F (TCTATAAAAAATGGATAATCATGGGATTAAA [nt 1601]), L268M on R264K-R (GATTATCCATTTTTTATAGATTTCTCCTAC [nt 1561]), S173A-F (GAAGTAATACCCATGTTTGCAGCAT TATCA [nt 1317]), S173A-R (AAACATGGGTATTACTTCTGGGCTGAAAG [nt 1277]), A237T-F (AAGGGGAAGTGACATAACAGGAACTACTAGTACC [nt 1515]), and A237T-R (TATGTCACTTCCCCTTGGTTCTCTCAT [nt 1471]). Mutated codons are underlined, and primer positions are numbered according to those of NL4-3 (GenBank accession number AF324493). Mutated SacI-SbfI fragments were then isolated from pUC19 and cloned back into pNL4-3. The complete HIV-1 coding region of the variant proviruses was sequenced using previously reported primers (9) on an ABI 3730 XL DNA analyzer. Escherichia coli One Shot Stbl3 cells were used to propagate full-length proviral plasmids, and stocks were prepared using a QIAprep Spin Miniprep kit or HiSpeed Plasmid Midi kit (QIAGEN).
Cells.
HEK293T cells were cultured at 37°C in a 5% CO2 humidified incubator in Dulbecco's modified Eagle medium (Gibco) supplemented with 10% fetal bovine serum (Atlantic Biologicals), penicillin (50 IU/ml), and streptomycin (50 μg/ml). CEM-GXR cells (16), Jurkat cells (JKT), and PPIA−/− Jurkat cells (JKT CypA−/−), which do not express CypA (15), were grown in R10+ medium (RPMI 1640 [Sigma] supplemented with 10% fetal bovine serum, 2 mM l-glutamine, penicillin [50 IU/ml], and streptomycin [50 μg/ml]) at 37°C and 5% CO2. PBMC were separated from whole blood using Histopaque-1077 (Sigma) and then stimulated with a CD3:CD8-bispecific monoclonal antibody (72) at a concentration of 0.5 μg/ml in R10+ medium containing 50 U/ml human interleukin-2 (Murex) (R10-50) for 7 days prior to infection.
Viral stocks.
Viral stocks were generated by transfection of HEK293T cells with 5 μg of plasmid DNA in antibiotic-free Dulbecco's modified Eagle medium using Lipofectamine 2000 (Invitrogen). Pseudotyping with vesicular stomatitis virus glycoprotein G (VSV-g) was performed by cotransfection with 300 ng of pHEF-VSVG (20). Supernatants were harvested 48 h after transfection, and frozen aliquots were stored at −80°C. The capsid concentration of the viral stocks was quantified by p24 enzyme-linked immunosorbent assay (ELISA). Initial titers and relative infectivity were determined by flow cytometry using CEM-GXR cells.
p24 ELISA.
Extracellular p24 was measured using the Alliance HIV-1 p24 ELISA kit (Perkin-Elmer). Cell-free supernatants from infected cultures were harvested at the indicated times and stored at −80°C prior to analysis.
Viral infectivity assays.
One million CEM-GXR cells were inoculated with 100 μl of viral stocks for WT virus or the respective variants. The percentage of infected cells was measured using fluorescence-activated cell sorter (FACS) analysis to determine green fluorescent protein (GFP) expression at 48 h postinfection. Infectivity was determined as the percentage of infected cells per ng p24 of input virus. Values were normalized to those for the WT.
Viral replication assays.
One million CEM-GXR cells were pelleted and resuspended with WT or variant virus at a multiplicity of infection of 0.0015 in a total volume of 3 ml R10+ medium. Aliquots (500 μl) of the culture were harvested at the indicated times, and the volume was replaced with fresh R10+ medium. The proportion of GFP-expressing cells was determined by FACS analysis and normalized to the values observed at day 2 in order to calculate viral spread. For assays not using the GFP reporter cells, viral replication was determined by measuring the concentration of p24 in the supernatant of infected cultures. In this case, 1 million CEM-GXR, JKT, or JKT CypA−/− cells were infected with virus (100 ng p24) in a total volume of 600 μl R10+ medium, incubated for 5 h at 37°C, washed three times in phosphate-buffered saline, and resuspended in 2 ml of R10+ medium. At the indicated times, 200 μl supernatant was removed and replaced with fresh R10+ medium. For the analysis of primary cells, 2 million PBMC from healthy donors were stimulated for 1 week and then infected with 10 ng p24 equivalent of viruses in a volume of 200 μl R10-50 medium at 37°C for 7 h. Cells were then pelleted, resuspended, and plated at 1 million cells/ml in R10-50 medium. Two hundred fifty microliters of the culture was harvested at the indicated times, the volume was replaced with fresh R10-50 medium, cells were pelleted, and the supernatant was collected.
Flow cytometry.
CEM-GXR cells were fixed in phosphate-buffered saline containing 2% paraformaldehyde, and GFP expression was determined using FACS analysis. A signal 10-fold above the median fluorescence index of uninfected cells was considered to be positive, excluding greater than 99.95% of uninfected cells. FACS analysis was done using a FACSCalibur flow cytometer (Becton Dickenson) and FlowJo software (TreeStar). A minimum of 25,000 cells were analyzed for each sample.
Real-time PCR.
Quantitative PCR was completed according to previously reported procedures (14), with the following modifications. Two million CEM-GXR cells were infected with DNase I-treated VSV-g-pseudotyped virus (400 ng p24 equivalent in 600 μl R10+ medium) for 1 h, washed, and then resuspended in 2.5 ml of R10+ medium. Approximately 250,000 cells were harvested at the indicated times, and the volume was replaced by fresh R10+ medium. DNA was isolated using the DNeasy tissue kit (QIAGEN) and analyzed for late full-length DNA products of viral reverse transcriptase (primers J1 F [5′-ACAAGCTAGTACCAGTTGAGCCAGATAAG-3′] and J2 R [5′-GCCGTGCGCGCTTCAGCAAGC-3′]) (14) and for total DNA with primers specific for the actin gene (Act176F [5′-GTGACAGCAGTCGGTTGGAG-3′] and Act176R [5′-AGGACTGGGCCATTCTCCTT-3′]) by real-time PCR using Full Velocity SYBR Green 2× mix (Stratagene). Late reverse transcription products were normalized to the actin signal and are reported as 2(ΔΔCT) for each sample.
Capsid stability assay.
The stability of the viral cores was tested as previously described (27). Briefly, supernatants from transfected HEK293T cells were filtered to remove cellular debris, and HIV-1 particles were concentrated by ultracentrifugation (120,000 × g for 3 h at 4°C) through a cushion of 20% (wt/vol) sucrose in STE buffer (10 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA). Viral pellets were resuspended in 200 μl of STE buffer, and the concentrated virions were subjected to ultracentrifugation (100,000 × g for 16 h at 4°C) through a layer of 1% Triton X-100 into a linear sucrose density gradient (10 ml of STE buffer containing 30 to 70% sucrose). Fractions (1 ml) were collected from the top of the gradient and analyzed for capsid content by p24 ELISA. Samples of purified cores (100 μl of pooled fractions 7, 8, and 9) were diluted in STE buffer (1 ml) and incubated at 37°C for various times. Following incubation, samples were centrifuged at 100,000 × g (Beckman TLA-55 rotor at 45,000 rpm) for 20 min at 4°C. The supernatants were removed, the pellets were dissolved in sodium dodecyl sulfate sample buffer, and the capsid content in both fractions was quantified by ELISA. The extent of core disassembly was determined as the percentage of capsid in the supernatant versus the total quantity of capsid in the reaction (supernatant plus pellet).
CsA experiments.
Single-cycle infectivity was determined by the inoculation of CEM-GXR reporter cells with VSV-g-pseudotyped virus in the presence or absence of CsA. Cells were preincubated with the indicated amounts of CsA for 30 min, followed by inoculation with the WT or viral variants. Viral inocula were normalized to the amount of p24 determined by ELISA. Infected cells were quantified using FACS analysis to measure GFP expression at 48 h postinfection.
Statistical analysis.
All analyses were performed using Prism 4.0 (Graph Pad). HLA-associated sequence polymorphisms at the population level were calculated using Fisher's exact test.
Nucleotide sequence accession numbers.
The sequence data determined in this study were submitted to GenBank and assigned accession nos. EU113054 to EU113180 and EU183198 to EU183205.
RESULTS
Kinetics of viral escape in the B27-KK10 CTL epitope.
CTL escape in the B27-KK10 epitope is typified by the development of two mutations in a predictable order. The first mutation occurs at position 6 (L268M) of the epitope early after infection, followed by a second mutation at position 2 (R264K) that is documented to arise late in the course of disease (41, 46). The L268M mutation is hypothesized to represent a prerequisite compensatory mutation for R264K due to its minimal impact on HLA binding and T-cell recognition (38, 41, 46), while only the R264K mutation at the P2 anchor position has been shown to abrogate the binding of the KK10 epitope to the HLA-B27 molecule (11, 38, 41). To clarify the kinetics of these mutations, four HLA-B27+ subjects were identified early after infection, and virus was sequenced longitudinally. Figure 1A illustrates that subjects AC160 and AC88 developed the L268M mutation within the first 1 to 3 years of infection, while in subjects 777 and 007, the rare L268M mutation was already present at the earliest time points available. These results support the early evolution of the L268M escape mutation (46), but we cannot exclude the possibility that M268 was transmitted in the two cases. Follow-up samples from patients 777 and 007 revealed the development of R264K (Fig. 1A). Cross-sectional sequencing of the gag gene from an additional 22 chronic HLA-B27+ subjects showed that 19 of 26 subjects (73%) had developed mutations within KK10, including 16 subjects with L268M and 13 subjects with R264K (Fig. 1B). In each case, the predominant R264K mutation represented a single-nucleotide substitution from AGA-to-AAA. As previously described (46), the development of the R264K mutation was associated with L268M, except in one case (CRO206U), where it was associated with an L268I mutation. In addition to R264K, the alternative P2 mutations R264T, R264G, and R264Q have been described to occur at much lower frequencies and have also been associated with viral escape in KK10 (11, 13, 25, 38, 46, 63). In our study, two subjects developed the alternative R264T mutation, which has been shown to bind poorly to HLA-B27 and exhibit reduced presentation to CTL (25, 38). To verify these findings at the population level, 100 full viral genomes from HLA-typed subjects were examined. Mutations at R264 and L268 were found to be strongly associated with the expression of HLA-B27 (P < 0.0005 and P < 0.0005, respectively) (B. Li et al., unpublished data). Examination of the clinical impact of these escape mutations revealed that subjects bearing WT or L268M variants of KK10 had viral loads of 64,584 copies/ml on average, versus 356,294 copies/ml in subjects where viruses displayed mutations at residue R264 (Fig. 1B). In addition, the longitudinal follow-up of subject 007 revealed a greater-than-1.5-log increase in viral loads following the development of R264K. These data support previous studies assessing escape from the immunodominant response against the B27-KK10 epitope, indicating sequential viral evolution in KK10 and a correlation between the development of R264K and clinical progression (25, 41, 46).
FIG. 1.

CTL escape in B27-KK10 is associated with multiple mutations. Viral sequences of Gag p24 in subjects expressing HLA-B27 are aligned to consensus clade B and NL4-3. Areas in gray highlight amino acid position 173 and the KK10 epitope (amino acids 263 to 272). The CypA binding domain is shown in the consensus clade B sequence (underlined), and mixed amino acids are indicated by lowercase letters. ND indicates “not determined.” cl indicates sequence derived from a single full-length clone representative of the quasispecies. Where available, viral loads are indicated. (A) Longitudinal viral sequencing in four HLA-B27+ subjects illustrates that the L268M mutation arises within the first 1 to 3 years after infection and prior to the R264K mutation. Sample time points are indicated as days postpresentation (pp). Fractions in parentheses indicate the fractions of clones representing displayed sequence. (B) Cross-sectional sequencing of 26 chronically infected B27+ subjects illustrates the propensity for viral escape through the R264K and L268M mutations. The S173A mutation is strongly associated with the R264K mutation. Numbers in parentheses indicate the number of clones sequenced for which the consensus sequence is provided.
Escape mutation R264K substantially impacts viral replication in vitro.
To examine the impact of escape mutations in the KK10 epitope on viral replication, variant NL4-3 viruses (Table 1) containing the R264K (RK) or L268M (LM) mutations as well as a double mutant encoding R264K and L268M (RKLM) were constructed, and infectious viruses were produced by the transfection of HEK293T cells. To evaluate viral infectivity and replication, the percentage of infected cells was measured over time using a CEM-based GFP reporter T-cell line, CEM-GXR, as previously described (16). As illustrated in Fig. 2A, single-cycle infectivity of the LM variant was equivalent to WT NL4-3, but the RK and RKLM viruses exhibited profound defects, with percentages of HIV-infected cells reduced to only 4% and 5% of WT levels, respectively. Replication of these viruses was next assessed in a 7-day assay, which confirmed the ability of the LM mutant to spread at a rate similar to that of the WT virus, while the RK and RKLM variants failed to sustain viral replication above that of cell division (Fig. 2B). These data suggest that the R264K mutation induced a substantial defect in viral infectivity and cell-to-cell spread that was not compensated for by the adjacent L268M substitution, as was postulated previously (46).
TABLE 1.
Viral constructs and characteristics
| Virus | Sequence of KK10 epitope (amino acids 263-272) | Mutation(s) | Replicative capacitya | Influence of CypA on replicationb | Influence of CsA on replicationc |
|---|---|---|---|---|---|
| WT | KRWIILGLNK | ++ | ↑ | ↓ | |
| RK | -K-------- | R264K | − | ↓ | ↑ |
| LM | -----M---- | L268M | ++ | ND | ND |
| RKLM | -K---M---- | R264K, L268M | − | ND | ↑ |
| SA | ---------- | S173A | + | ND | ND |
| SARK | -K-------- | S173A, R264K | + | ND | ND |
| SARKLM | -K---M---- | S173A, R264K, L268M | + | ↑ | ↓ |
| A237T on RKLM | -K---M---- | A237T, R264K, L268M | + | ND | ↓ |
Measured as viral spread in CEM GXR cell culture. ++, replication at WT level or more than the WT level; +, replication 50 to 100% of the WT level; −, replication <10% of the WT level.
Measured as p24 production in JKT cells compared to that in JKT CypA−/− cells. ND, not determined; ↑, increased replication; ↓, decreased replication.
Measured as infectivity of CEM GXR cells in the presence of 0.5 μM CsA.
FIG. 2.

Escape mutation R264K substantially impacts viral replication in vitro. (A) CEM-GXR cells were infected with WT or variant viruses (100 μl of viral stocks), and the percentage of infected GFP-positive (GFP+) cells/ng p24 was determined at 48 h by flow cytometry. Values were normalized to WT values (black). The RK (red) and RKLM (orange) variants exhibited substantial deficiencies in infectivity, while only minor differences were observed for LM (blue), SA (purple), and SARKLM (green). Results shown are representative of three or more independent experiments. (B) CEM-GXR cells were infected (multiplicity of infection of 0.0015), and viral spread was measured over 7 days using flow cytometry to quantify GFP+ cells. The percentage of infected cells was normalized to the value at day 2 (0.15%). Again, RK (red) and RKLM (orange) exhibited deficiencies in replication, while SARKLM virus (green) replicated efficiently. Results shown are representative of three independent experiments. (C) The replicative defect of the RK variant was confirmed using primary cells. PBMC were inoculated with virus (10 ng p24), and viral spread was measured by p24 ELISA over 16 days. WT (black) and SARKLM (green) replicated similarly, while the RK variant (red) generated less p24 at all times tested. Results shown are representative of three independent experiments using different donors.
Viral escape in B27-KK10 is associated with an upstream putative compensatory mutation.
Compensatory mutations that restore the replicative defects of some CTL escape mutations in Gag have been previously identified in HIV-1 and SIV (32, 62). Therefore, sequences from HLA-B27+ subjects were examined for polymorphisms other than L268M that might be associated with R264K and would explain the ability of R264K to arise in vivo despite this significant replicative defect. Reexamination of longitudinal sequence data for subjects 777 and 007 supports the simultaneous development of R264K with a serine-to-alanine mutation at residue 173 (S173A) (Fig. 1A). Similarly, in the cross-sectional analysis, 12 of 13 HLA-B27+ subjects exhibiting R264K also displayed the S173A mutation (Fig. 1B). Residue S173 is well conserved in subtype B viruses from the Los Alamos National Laboratory HIV Sequence Database (www.hiv.lanl.gov), with only 10 of 203 (5%) full-length Gag sequences harboring S173A and 22 of 203 (11%) sequences harboring an alternative S173T mutation (data not shown). At the population level, we have observed that mutations at residue S173 were also associated with the expression of HLA-B27 (P < 0.05) (Li et al., unpublished). Notably, despite occupying linearly distant positions on alpha-helices II and VII, R264 and S173 lie in close proximity to one another on the same planar surface of the folded p24 molecule (Fig. 3). Taken together, the observed fitness defect of the RK and RKLM variants, and the nearly simultaneous development of S173A with R264K, suggested that the S173A mutation might represent a compensatory mutation for R264K.
FIG. 3.
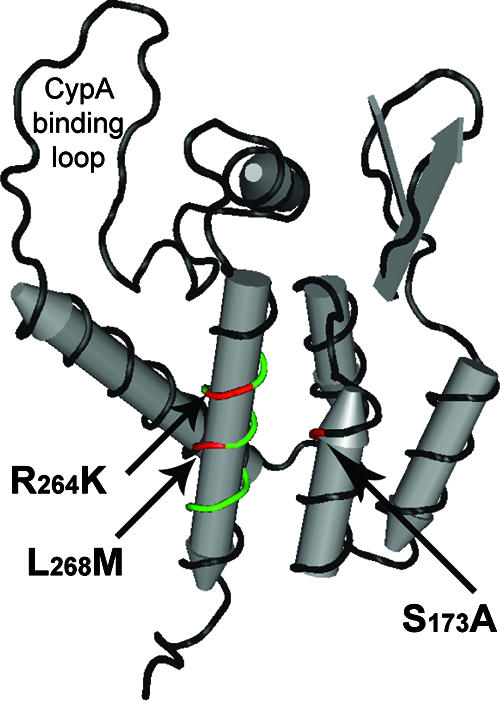
Location of KK10-associated mutations in the N-terminal domain of p24 capsid. The mutations associated with viral escape in B27-KK10 were mapped onto the structural model of the N-terminal domain of p24 capsid using Cn3D (National Center for Biotechnology Information). R264K and L268M (red) are located inside the KK10 epitope (green) on helix VII, while the S173A mutation (red) is located on helix II on the same planar surface of the folded p24 molecule as R264K and L268M.
The S173A mutation compensates for the replication defect of R264K.
To examine whether the S173A substitution functioned as a compensatory mutation to rescue viral replication of the RK mutant, NL4-3 variants encoding S173A alone (SA) and in combination with R264K (SARK) or R264K/L268M (SARKLM) were generated by site-directed mutagenesis (Table 1), and viral particles were produced by transfection of HEK293T cells. Following inoculation of CEM-GXR cells, the SA variant displayed a modest reduction in relative infectivity (73% of WT) (Fig. 2A). However, when S173A was combined with the R264K or R264K/L268M mutation (SARK or SARKLM variants), it substantially rescued the infectivity of these escape mutant viruses to 68% and 88% of WT levels, respectively (Fig. 2A and data not shown). Similarly, both the SARK and SARKLM viruses were observed to spread efficiently in a 7-day replication assay, illustrating significant recovery from the R264K fitness defect (Fig. 2B and data not shown). These results demonstrate that the upstream mutation S173A functions as an effective compensatory mutation for R264K, explaining their close temporal relationship (Fig. 1).
Replication of the RK variant is defective in primary cells.
To ensure that the replication phenotypes that we observed were not limited to the CEM-GXR cell line, viral variants were also evaluated using primary PBMC. Stimulated PBMC from two HIV-negative donors were infected with WT, RK, and SARKLM viruses using viral inocula normalized by p24 ELISA, and supernatant p24 values were monitored for 16 days (Fig. 2C). A 2-log reduction in p24 production was observed for the RK mutant compared to the WT, while the SARKLM variant displayed a p24 concentration in the supernatant that was similar to that of the WT. Therefore, the defective phenotype of RK and the compensatory function of S173A were confirmed using primary cells.
The RK variant is impaired at the step of viral reverse transcription.
To examine the mechanism behind the defect of RK replication, we first analyzed p24 production, protein release, and p55 proteolytic processing using HEK293T cells transfected with plasmids expressing the WT and RK, LM, RKLM, and SARKLM variants. Since the results of these assays were similar for all variants, the RK mutant did not appear to be impaired during late stages of the viral replication cycle (data not shown). We next examined the ability of the R264K mutation to disrupt viral replication during the entry step by measuring the generation of viral reverse transcription products, which depend on the initiation of capsid uncoating (23, 27). VSV-g-pseudotyped WT, RK, and SARKLM viruses, normalized to the amount of p24 by ELISA, were used to infect CEM-GXR cells, and quantitative PCR was performed using primers specific for a late viral reverse transcription product (14). These experiments revealed that while the WT and SARKLM viruses yielded similar amounts of viral DNA at each time point, the RK virus was defective in generating late reverse transcription products (Fig. 4A). Similar results were obtained when we assessed the formation of 2-long terminal repeat circles as a measurement of integration (data not shown). The reduced ability to efficiently generate reverse transcription products indicates that the RK variant is severely impaired at an early step of the viral replication cycle.
FIG. 4.

The RK variant is defective in the production of late viral reverse transcripts without affecting capsid-uncoating kinetics. (A) CEM-GXR cells were infected with VSV-g-pseudotyped WT, RK, and SARKLM viruses (400 ng p24). Cells were harvested at 3, 12, 24, and 48 h postinfection and analyzed for late reverse transcription (RT) products using real-time PCR, and values were normalized to the host cell actin gene. Late RT products were readily generated by WT (black) and SARKLM (green) within 12 h and then increased through 48 h, while the levels of reverse transcription products for RK (red) were significantly reduced at all times tested. Results are representative of two independent experiments. (B) The stabilities of WT (black) and RK (red) capsids were examined using a previously described assay (27). Purified cores were incubated in STE buffer at 37°C for the times shown. Following incubation, the samples were subjected to ultracentrifugation. The extent of disassembly was determined as the percentage of the total capsid protein in the reaction mixture present in the supernatant. In this in vitro assay, both viruses disassemble at the same rate. The results shown are mean values of duplicate determinations and are representative for two independent experiments.
The RK variant is not impaired due to altered stability of capsid.
Studies have indicated that the optimal stability of the incoming HIV-1 capsid is crucial for efficient viral infectivity, since mutations that hyper- or hypostabilize the core particle can disrupt the finely tuned process of uncoating and initiation of reverse transcription (23, 27). Therefore, a kinetic assay of core disassembly was used to analyze the impact of the R264K mutation on capsid stability (27). Purified cores of the WT and the RK variant were incubated for up to 2 h at 37°C and then subjected to ultracentrifugation. The quantity of intact cores pelleting at these conditions, and the amount of solubilized disassembled capsid in the supernatant, was determined by p24 ELISA. In this assay, WT and RK cores exhibited similar disassembly kinetics (Fig. 4B), indicating that the substantial viral replicative defect incurred by the R264K mutation is likely not due to the generation of a defective core particle that might prevent proper capsid uncoating. This interpretation is additionally supported by the observation that core particles with the R264K mutation were not impaired in their ability to saturate restriction of owl monkey TRIM-Cyp or rhesus macaque Trim5α (Data not shown) using previously reported protocols (26).
Infectivity of RK is restored through treatment of cells with CsA.
Efficient HIV-1 replication is normally dependent upon the binding of the host protein CypA to the viral capsid through an exposed CypA binding loop (14, 33, 35). The underlying mechanism of this dependency on CypA remains unclear, although it has been suggested to be related to the regulation of capsid stability (33, 37), uncoating (35), or impairment of recognition of capsid by host restriction factors (71). Since the R264K mutation did not appear to influence capsid uncoating, it remained possible that the postentry defect observed for the RK mutant might be related to an alteration in the interactions with CypA. To test this hypothesis, we infected CEM-GXR cells in the presence of CsA, a drug that binds to CypA and competes with HIV-1 capsid for this interaction (29). As expected based on previous reports (14, 29, 69), treatment of cells with 0.5 μM CsA impaired the infectivity of WT virus by more than twofold (Fig. 5A). Notably, a similar decline for the SARKLM variant was observed, indicating that both of these viruses required CypA interaction for optimal infectivity. In contrast, the infectivities of the RK and RKLM variants were increased by 5.7- and 3.4-fold, respectively, in the presence of CsA. Therefore, while the WT and the SARKLM variant displayed a normal CypA-dependent replication phenotype, CypA had a negative effect on the RK and RKLM variants, rendering them CsA dependent. The increase in infectivity of the RK variant in the presence of CsA verifies that this virus is not defective per se but rather that it exhibits a postentry block that is dependent upon host cellular factors and appears to be overcome by blocking CypA binding. Further support for this interpretation was sought by combining the RKLM variant with an additional mutation, A237T (residue A105T in p24 nomenclature) (Table 1). This substitution has recently been shown to rescue the replication of two in vitro-defined viral variants (T186A and A224E), which also exhibit a CsA-dependent phenotype (76). Similar to S173A, the A237T mutation was able to restore the infectivity of RKLM in the absence of CsA (Fig. 5A), while the infectivity of this virus in the presence of CsA was reduced by more than twofold.
FIG. 5.

RK infectivity is restored in the absence of CypA. (A) CEM-GXR cells were infected with VSV-g-pseudotyped WT, RK, RKLM, and SARKLM viruses (400 ng p24) in the presence or absence of 0.5 μM CsA. The percentage of GFP+ cells was determined by flow cytometry at 48 h postinfection and is shown for each variant. Infectivities of WT (black) and SARKLM (green) were reduced by 2.2- and 2.5-fold, respectively, in the presence of 0.5 μM CsA, while the percentages of infected cells for RK (red) and RKLM (orange) increased by 5.7- and 3.4-fold, respectively, in the presence of drug. The introduction of A237T increased the infectivity of RKLM (gray) by 4.2-fold in the absence of CsA. In the presence of the drug, the infectivity of this strain was reduced by threefold. Results displayed are mean values for duplicate cultures and are representative of three independent experiments. To further evaluate the role of CypA, 1 million CEM-GXR (B), JKT (C), or JKT CypA−/− (D) cells were infected with 100 ng p24 equivalent of the WT (black), RK (red), or SARKLM (green). The p24 concentration in the supernatant at the indicated days postinfection was determined by ELISA. Cultures with WT and SARKLM viruses generated similar amounts of p24 in CEM-GXR and JKT cells. In the JKT CypA−/− lines, p24 production was reduced by 1 log, and peak p24 levels were delayed by 4 days. RK virus failed to increase p24 values in CEM-GXR cells. Compared to the WT and SARKLM, p24 production by RK in JKT cells was reduced by 2.5 logs, while all three viruses displayed similar p24 values for JKT Cyp−/− cells. Results shown in B, C, and D are representative of three independent experiments.
Infectivity of RK is restored in a CypA knockout cell line.
The pharmacological mechanism of action of the immunosuppressive drug CsA involves initial binding to CypA to form a complex that then inhibits calcineurin, leading to the reduced transcription of various cytokine genes as part of the signal transduction pathway for T-cell activation (30). To exclude additional effects of CsA on cellular homeostasis, which might alter the course of the viral replicative cycle, we compared viral replication in JKT cells and CEM-GXR (GXR) cells with JKT CypA−/− cells (15). Here, the replication levels of the WT and the RK and SARKLM variants were compared by p24 production. The WT and SARKLM viruses readily produced similar amounts of p24 on CEM-GXR and JKT cells (Fig. 5B and C). In the JKT CypA−/− lines, as expected (15), p24 production was reduced by 1 log, and peak p24 levels were delayed by 4 days (Fig. 5D). In contrast, the RK virus failed to replicate efficiently in CEM-GXR and JKT cells, although it did replicate significantly better on the JKT cells, which contain lower levels of CypA (2). Furthermore, in the JKT CypA−/− cells, p24 production by the RK variant was actually enhanced 60-fold compared to the parent JKT cells (Fig. 5D). Testing of the RKLM variant in separate experiments indicated that this variant exhibited a phenotype that was identical to that of the RK variant (data not shown). These results confirm that a relevant portion of the replicative defect of the RK variant can be attributed to a detrimental effect of CypA, rendering this viral variant CsA resistant and dependent. This phenotype is rescued with the compensatory mutation S173A, as the SARKLM variant replicates in a CypA-dependent and CsA-sensitive manner with kinetics similar to those of WT virus.
DISCUSSION
In HLA-B27+ subjects, multiple reports now support a causal link between viral escape from the immunodominant KK10-specific CD8+ T-cell response late in infection and progression to AIDS (25, 41). Here, we illustrate that the development of the predominant CTL escape mutation R264K in the KK10 epitope results in a severe defect in viral replication and is strongly linked to the simultaneous development of the upstream compensatory mutation S173A. The L268M substitution has been hypothesized to compensate for the defect in replicative capacity imposed by R264K, but in our assays, only the combination of R264K with S173A efficiently allowed the outgrowth of mutant viruses that are able to escape from the KK10-specific CTL response. We show that a virus encoding the L268M mutation is not significantly defective in replication, and recent data suggest that the LM variant represents an early CTL escape mutation that does not inhibit recognition by all CTL clones (51a), suggesting that L268M functions primarily to partially evade early CTL pressure in B27+ individuals. In vitro experiments revealed that the R264K defect was associated with a failure to generate late reverse transcription products, indicating a block in replication at an early postentry step. Notably, infection of the RK variant was restored by blocking capsid binding to the host protein CypA using the drug CsA, and p24 production by the RK variant was less severely reduced in a JKT T-cell line and in PBMC than in a CEM-based T-cell line that expresses constitutively higher levels of CypA (2). Furthermore, replication levels of RK and the WT were similar in JKT cells deficient for CypA. Together, these data support a model that structural constraints on capsid residue R264 hinder the ability of HIV-1 to escape from the immunodominant KK10-directed CTL response, thereby enabling the maintenance of strong immune pressure that leads to long-term viral suppression (41).
The role of CypA in promoting HIV-1 infection remains elusive (15, 42, 69). It is believed to function by helping to maintain proper capsid conformation (35), by orchestrating the uncoating process through altering capsid stability (33), or by blocking the recognition of capsid by an unidentified antiviral host restriction factor (71). The observation that the infectivity of the RK and RKLM variants was substantially improved by the treatment of cells with CsA suggests that the R264K mutation alters this key interaction between capsid and cellular CypA. Notably, three other mutations in the N-terminal domain of p24, A224E, G226D, and T186A, that show similar CypA-mediated phenotypes have been identified (1, 76). The T186A mutation is located in helix III, while the A224E and G226D mutations are situated in the CypA binding loop. The CsA dependency of viruses containing R264K or these other mutations suggests a structural component to this phenotype. This is also supported by the observation that the A237T mutation, previously described by Yang et al. to rescue the impaired infectivity of the T186A and A224E mutants (76), was able to compensate for the poor infectivity of RKLM (Fig. 5A). Therefore, additional mutations, although rare, may restore viral replication of the RK variant. In this context, it should be noted that a previous study by Nietfeld et al. did not observe an impact of the R264K mutation on viral replication when introduced into HIV-1LAI (57). This may be due to amino acid differences between LAI and NL4-3 capsids, which will require further investigation.
The substantial impact of R264K on in vitro viral replication, and the requirement for compensation by S173A, likely explains the typical late escape from the KK10-specific CTL responses (41, 46). In addition, the partial replicative defect that we and others (19) have observed in vitro for the SA variant may diminish the frequency of this variant in the viral population. Examination of sequences from the Los Alamos National Laboratory HIV Sequence Database illustrates the relative absence of both R264K and S173A, with S173A accompanied by R264K and L268M in 6 out of 10 occurrences. Interestingly, S173A was never observed in viral sequences harboring any of the alternative CTL escape mutations at P2 of the KK10 epitope (R264T, R264G, or R264Q) (Fig. 1B) (A. Kelleher, personal communication), indicating that S173A solely compensates for the fitness defect imposed by the predominant R264K escape mutation. Otherwise, S173A was accompanied by a rare I256T substitution on the same planar face as R264 or by the H219Q substitution known to associate with CypA independence (21, 35). Therefore, the impaired replicative capacities of viruses harboring R264K or S173A alone are likely to diminish their chance to arise simultaneously to facilitate escape within KK10.
Although both HLA-B57 and HLA-B27 are associated with the control of HIV-1 infection, Gao et al. recently illustrated that these alleles differentially influence progression to AIDS (34). Here, the protective activity of HLA-B57 was observed relatively early following infection, resulting in a significantly slower decline to CD4+ T-cells count below 200 cells/mm3. The protective effect mediated by HLA-B27, on the other hand, was associated primarily with the later event of delaying disease progression to an AIDS-defining illness after the CD4+ T-cell count has declined. Those authors hypothesized that this discrepancy may be due to differences in immune pressure exerted by the immunodominant CD8+ T-cell responses restricted by these alleles and the costs to viral fitness associated with their respective CTL escape mutations (34, 50). Our data suggest that the difficulty in achieving viral escape due to fitness constraints enables the long-term maintenance of immune pressure against KK10 well into chronic infection, when viral escape has already occurred within many dominant CD8 epitopes.
The immunodominant targeting and viral escape from the B27-KK10 and B57-TW10 epitopes in HIV-1 p24 are strikingly similar to what has been observed in rhesus macaques for the immunodominant Mamu-A*01-CM9 epitope in SIV p27 that has also been associated with immune control (12). Viral escape in the CM9 epitope has been shown to diminish SIV fitness (31, 62, 78), presumably by disrupting the process of capsid assembly and maturation, and also required the development of multiple compensatory mutations in vivo (32, 62). Similarly, Matano et al. also observed that the control of SIVmac239 following vaccination of rhesus macaques was associated with the development of a single CTL escape mutation in capsid that likewise impaired viral replication (54). Each of these CD8+ T-cell responses targets highly conserved regions of the N-terminal domain of capsid and therefore may highlight a uniquely vulnerable target for vaccine design or novel drug therapy (8, 40). In particular, vaccines that are capable of inducing potent CTL responses against epitopes in this region that are restricted by other more common HLA alleles but that lack the immunodominance of B27-KK10 or B57-TW10 (10) might be able to drive viral evolution in a manner similar to that of B27-KK10. A recent study by Kiepiela et al. demonstrated that Gag-specific responses are strongly associated with lower viremia in a large cohort of HIV-1 clade C-infected individuals from South Africa (48). In addition, Sacha et al. indicated that Gag-specific CD8+ T cells are able to recognize infected lymphocytes as early as 2 h postinfection, before viral protein synthesis, and likely through the processing and presentation of capsid molecules of the incoming virion (66). Taken together, these data suggest that the structural conservation of the Gag protein, particularly the p24/p27 capsid, as well as the kinetics of epitope presentation to T cells may render Gag a highly susceptible target for effective CD8+ T-cell responses.
In conclusion, our data demonstrate that the CTL escape mutation R264K in the B27-KK10 epitope dramatically reduces HIV-1 replicative capacity. Furthermore, this escape variant imparts a rare CsA-dependent phenotype upon the virus, suggesting that the delicately balanced equilibrium of capsid interactions with host cellular factors essential to early postentry events has been disrupted. Thus, we propose that the long-term control of HIV-1 associated with HLA-B27, which has been linked to the immunodominant CTL response targeting the KK10 epitope, is maintained in vivo as a result of the difficulty of mutating this highly conserved region of the virus. Our findings further strengthen the notion that the HIV-1 capsid, particularly the region containing the KK10 epitope, might represent an attractive target for vaccine design and that the impact of CTL escape mutations on viral replication requires further consideration. Examination of immunodominant epitopes of HIV-1 that are similarly constrained may help to identify other critical targets for vaccine-induced immune responses.
Acknowledgments
Plasmids pNL4-3, from M. Martin, and pHEF-VSV-g, from L. J. Chang, were obtained via the NIH AIDS Research and Reference Reagent Program. The Jurkat PPIA−/− cells were obtained from the NIH AIDS Research and Reference Reagent Program (J. Luban). We thank all individuals who enabled this study by donating blood.
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other supporting agencies.
This study was supported by the National Institutes of Health grants R01-AI054178 (T.M.A.), R21-AI067078 (T.M.A.), and R01-AI050423 (C.A.) and the Acute Infection Early Disease Research Program (U01 AI052403). The National Centre in HIV Epidemiology and Clinical Research is supported by the Commonwealth Department of Health and Ageing through the Australian National Council on AIDS, Hepatitis C, and Related Diseases. A.D.K. is supported by a program grant from the Australian NHMRC.
Footnotes
Published ahead of print on 5 September 2007.
REFERENCES
- 1.Aberham, C., S. Weber, and W. Phares. 1996. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J. Virol. 70:3536-3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerson, B., O. Rey, J. Canon, and P. Krogstad. 1998. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. J. Virol. 72:303-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aebischer, T., D. Moskophidis, U. H. Rohrer, R. M. Zinkernagel, and H. Hengartner. 1991. In vitro selection of lymphocytic choriomeningitis virus escape mutants by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 88:11047-11051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen, T. M., M. Altfeld, S. C. Geer, E. T. Kalife, C. Moore, K. M. O'Sullivan, I. Desouza, M. E. Feeney, R. L. Eldridge, E. L. Maier, D. E. Kaufmann, M. P. Lahaie, L. Reyor, G. Tanzi, M. N. Johnston, C. Brander, R. Draenert, J. K. Rockstroh, H. Jessen, E. S. Rosenberg, S. A. Mallal, and B. D. Walker. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239-13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen, T. M., M. Altfeld, X. G. Yu, K. M. O'Sullivan, M. Lichterfeld, S. Le Gall, M. John, B. R. Mothe, P. K. Lee, E. T. Kalife, D. E. Cohen, K. A. Freedberg, D. A. Strick, M. N. Johnston, A. Sette, E. S. Rosenberg, S. A. Mallal, P. J. Goulder, C. Brander, and B. D. Walker. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 78:7069-7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386-390. [DOI] [PubMed] [Google Scholar]
- 7.Altfeld, M., M. M. Addo, E. S. Rosenberg, F. M. Hecht, P. K. Lee, M. Vogel, X. G. Yu, R. Draenert, M. N. Johnston, D. Strick, T. M. Allen, M. E. Feeney, J. O. Kahn, R. P. Sekaly, J. A. Levy, J. K. Rockstroh, P. J. Goulder, and B. D. Walker. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17:2581-2591. [DOI] [PubMed] [Google Scholar]
- 8.Altfeld, M., and T. M. Allen. 2006. Hitting HIV where it hurts: an alternative approach to HIV vaccine design. Trends Immunol. 27:504-510. [DOI] [PubMed] [Google Scholar]
- 9.Altfeld, M., T. M. Allen, X. G. Yu, M. N. Johnston, D. Agrawal, B. T. Korber, D. C. Montefiori, D. H. O'Connor, B. T. Davis, P. K. Lee, E. L. Maier, J. Harlow, P. J. Goulder, C. Brander, E. S. Rosenberg, and B. D. Walker. 2002. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature 420:434-439. [DOI] [PubMed] [Google Scholar]
- 10.Altfeld, M., E. T. Kalife, Y. Qi, H. Streeck, M. Lichterfeld, M. N. Johnston, N. Burgett, M. E. Swartz, A. Yang, G. Alter, X. G. Yu, A. Meier, J. K. Rockstroh, T. M. Allen, H. Jessen, E. S. Rosenberg, M. Carrington, and B. D. Walker. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 3:e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ammaranond, P., J. Zaunders, C. Satchell, D. van Bockel, D. A. Cooper, and A. D. Kelleher. 2005. A new variant cytotoxic T lymphocyte escape mutation in HLA-B27-positive individuals infected with HIV type 1. AIDS Res. Hum. Retrovir. 21:395-397. [DOI] [PubMed] [Google Scholar]
- 12.Barouch, D. H., J. Kunstman, M. J. Kuroda, J. E. Schmitz, S. Santra, F. W. Peyerl, G. R. Krivulka, K. Beaudry, M. A. Lifton, D. A. Gorgone, D. C. Montefiori, M. G. Lewis, S. M. Wolinsky, and N. L. Letvin. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415:335-339. [DOI] [PubMed] [Google Scholar]
- 13.Betts, M. R., B. Exley, D. A. Price, A. Bansal, Z. T. Camacho, V. Teaberry, S. M. West, D. R. Ambrozak, G. Tomaras, M. Roederer, J. M. Kilby, J. Tartaglia, R. Belshe, F. Gao, D. C. Douek, K. J. Weinhold, R. A. Koup, P. Goepfert, and G. Ferrari. 2005. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc. Natl. Acad. Sci. USA 102:4512-4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braaten, D., E. K. Franke, and J. Luban. 1996. Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIVCPZGAB but not group O HIV-1 or other primate immunodeficiency viruses. J. Virol. 70:4220-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braaten, D., and J. Luban. 2001. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 20:1300-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockman, M. A., G. O. Tanzi, B. D. Walker, and T. M. Allen. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131:134-142. [DOI] [PubMed] [Google Scholar]
- 17.Buchbinder, S. P., M. H. Katz, N. A. Hessol, P. M. O'Malley, and S. D. Holmberg. 1994. Long-term HIV-1 infection without immunologic progression. AIDS 8:1123-1128. [DOI] [PubMed] [Google Scholar]
- 18.Buseyne, F., J. Le Chenadec, B. Corre, F. Porrot, M. Burgard, C. Rouzioux, S. Blanche, M. J. Mayaux, and Y. Riviere. 2002. Inverse correlation between memory Gag-specific cytotoxic T lymphocytes and viral replication in human immunodeficiency virus-infected children. J. Infect. Dis. 186:1589-1596. [DOI] [PubMed] [Google Scholar]
- 19.Cartier, C., P. Sivard, C. Tranchat, D. Decimo, C. Desgranges, and V. Boyer. 1999. Identification of three major phosphorylation sites within HIV-1 capsid. Role of phosphorylation during the early steps of infection. J. Biol. Chem. 274:19434-19440. [DOI] [PubMed] [Google Scholar]
- 20.Chang, L. J., V. Urlacher, T. Isakuma, Y. Cui, and J. Zucali. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene. Ther. 6:715-728. [DOI] [PubMed] [Google Scholar]
- 21.Chatterji, U., M. D. Bobardt, R. Stanfield, R. G. Ptak, L. A. Pallansch, P. A. Ward, M. J. Jones, C. A. Stoddart, P. Scalfaro, J. M. Dumont, K. Besseghir, B. Rosenwirth, and P. A. Gallay. 2005. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in Owl monkey cells. J. Biol. Chem. 280:40293-40300. [DOI] [PubMed] [Google Scholar]
- 22.Crawford, H., J. G. Prado, A. Leslie, S. Hué, I. Honeyborne, S. Reddy, M. van der Stok, Z. Mncube, C. Brander, C. Rousseau, J. I. Mullins, R. Kaslow, P. Goepfert, S. Allen, E. Hunter, J. Mulenga, P. Kiepiela, B. D. Walker, and P. J. R. Goulder. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346-8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dismuke, D. J., and C. Aiken. 2006. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J. Virol. 80:3712-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards, B. H., A. Bansal, S. Sabbaj, J. Bakari, M. J. Mulligan, and P. A. Goepfert. 2002. Magnitude of functional CD8+ T-cell responses to the Gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J. Virol. 76:2298-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feeney, M. E., Y. Tang, K. A. Roosevelt, A. J. Leslie, K. McIntosh, N. Karthas, B. D. Walker, and P. J. Goulder. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927-8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forshey, B. M., J. Shi, and C. Aiken. 2005. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J. Virol. 79:869-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forshey, B. M., U. von Schwedler, W. I. Sundquist, and C. Aiken. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667-5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frahm, N., B. T. Korber, C. M. Adams, J. J. Szinger, R. Draenert, M. M. Addo, M. E. Feeney, K. Yusim, K. Sango, N. V. Brown, D. SenGupta, A. Piechocka-Trocha, T. Simonis, F. M. Marincola, A. G. Wurcel, D. R. Stone, C. J. Russell, P. Adolf, D. Cohen, T. Roach, A. StJohn, A. Khatri, K. Davis, J. Mullins, P. J. Goulder, B. D. Walker, and C. Brander. 2004. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J. Virol. 78:2187-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franke, E. K., and J. Luban. 1996. Inhibition of HIV-1 replication by cyclosporine A or related compounds correlates with the ability to disrupt the Gag-cyclophilin A interaction. Virology 222:279-282. [DOI] [PubMed] [Google Scholar]
- 30.Friedman, J., and I. Weissman. 1991. Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: one in the presence and one in the absence of CsA. Cell 66:799-806. [DOI] [PubMed] [Google Scholar]
- 31.Friedrich, T. C., E. J. Dodds, L. J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D. T. Evans, R. C. Desrosiers, B. R. Mothe, J. Sidney, A. Sette, K. Kunstman, S. Wolinsky, M. Piatak, J. Lifson, A. L. Hughes, N. Wilson, D. H. O'Connor, and D. I. Watkins. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10:275-281. [DOI] [PubMed] [Google Scholar]
- 32.Friedrich, T. C., C. A. Frye, L. J. Yant, D. H. O'Connor, N. A. Kriewaldt, M. Benson, L. Vojnov, E. J. Dodds, C. Cullen, R. Rudersdorf, A. L. Hughes, N. Wilson, and D. I. Watkins. 2004. Extraepitopic compensatory substitutions partially restore fitness to simian immunodeficiency virus variants that escape from an immunodominant cytotoxic-T-lymphocyte response. J. Virol. 78:2581-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gamble, T. R., F. F. Vajdos, S. Yoo, D. K. Worthylake, M. Houseweart, W. I. Sundquist, and C. P. Hill. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285-1294. [DOI] [PubMed] [Google Scholar]
- 34.Gao, X., A. Bashirova, A. K. Iversen, J. Phair, J. J. Goedert, S. Buchbinder, K. Hoots, D. Vlahov, M. Altfeld, S. J. O'Brien, and M. Carrington. 2005. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat. Med. 11:1290-1292. [DOI] [PubMed] [Google Scholar]
- 35.Gatanaga, H., D. Das, Y. Suzuki, D. D. Yeh, K. A. Hussain, A. K. Ghosh, and H. Mitsuya. 2006. Altered HIV-1 Gag protein interactions with cyclophilin A (CypA) on the acquisition of H219Q and H219P substitutions in the CypA binding loop. J. Biol. Chem. 281:1241-1250. [DOI] [PubMed] [Google Scholar]
- 36.Geldmacher, C., J. R. Currier, E. Herrmann, A. Haule, E. Kuta, F. McCutchan, L. Njovu, S. Geis, O. Hoffmann, L. Maboko, C. Williamson, D. Birx, A. Meyerhans, J. Cox, and M. Hoelscher. 2007. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 81:2440-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gitti, R. K., B. M. Lee, J. Walker, M. F. Summers, S. Yoo, and W. I. Sundquist. 1996. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273:231-235. [DOI] [PubMed] [Google Scholar]
- 38.Goulder, P. J., C. Brander, Y. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. Korber, and B. D. Walker. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412:334-338. [DOI] [PubMed] [Google Scholar]
- 39.Goulder, P. J., A. K. Sewell, D. G. Lalloo, D. A. Price, J. A. Whelan, J. Evans, G. P. Taylor, G. Luzzi, P. Giangrande, R. E. Phillips, and A. J. McMichael. 1997. Patterns of immunodominance in HIV-1-specific cytotoxic T lymphocyte responses in two human histocompatibility leukocyte antigens (HLA)—identical siblings with HLA-A*0201 are influenced by epitope mutation. J. Exp. Med. 185:1423-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goulder, P. J., and D. I. Watkins. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat. Rev. Immunol. 4:630-640. [DOI] [PubMed] [Google Scholar]
- 41.Goulder, P. J. R., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland-Jones. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212-217. [DOI] [PubMed] [Google Scholar]
- 42.Hatziioannou, T., D. Perez-Caballero, S. Cowan, and P. D. Bieniasz. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 79:176-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones, N. A., X. Wei, D. R. Flower, M. Wong, F. Michor, M. S. Saag, B. H. Hahn, M. A. Nowak, G. M. Shaw, and P. Borrow. 2004. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 200:1243-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaslow, R. A., M. Carrington, R. Apple, L. Park, A. Munoz, A. J. Saah, J. J. Goedert, C. Winkler, S. J. O'Brien, C. Rinaldo, R. Detels, W. Blattner, J. Phair, H. Erlich, and D. L. Mann. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405-411. [DOI] [PubMed] [Google Scholar]
- 45.Kaslow, R. A., D. G. Ostrow, R. Detels, J. P. Phair, B. F. Polk, and C. R. Rinaldo, Jr. 1987. The Multicenter AIDS Cohort Study: rationale, organization, and selected characteristics of the participants. Am. J. Epidemiol. 126:310-318. [DOI] [PubMed] [Google Scholar]
- 46.Kelleher, A. D., C. Long, E. C. Holmes, R. L. Allen, J. Wilson, C. Conlon, C. Workman, S. Shaunak, K. Olson, P. Goulder, C. Brander, G. Ogg, J. S. Sullivan, W. Dyer, I. Jones, A. J. McMichael, S. Rowland-Jones, and R. E. Phillips. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769-775. [DOI] [PubMed] [Google Scholar]
- 48.Kiepiela, P., K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne, E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, K. Bishop, M. van der Stok, K. Nair, N. Khan, H. Crawford, R. Payne, A. Leslie, J. Prado, A. Prendergast, J. Frater, N. McCarthy, C. Brander, G. H. Learn, D. Nickle, C. Rousseau, H. Coovadia, J. I. Mullins, D. Heckerman, B. D. Walker, and P. Goulder. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46-53. [DOI] [PubMed] [Google Scholar]
- 49.Koup, R. A., J. T. Safrit, Y. Cao, C. A. Andrews, G. McLeod, W. Borkowsky, C. Farthing, and D. D. Ho. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650-4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. St. John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282-289. [DOI] [PubMed] [Google Scholar]
- 51.Li, B., A. D. Gladden, M. Altfeld, J. M. Kaldor, D. A. Cooper, A. D. Kelleher, and T. M. Allen. 2007. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J. Virol. 81:193-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51a.Lichterfeld, M., D. G. Kavanagh, K. L. Williams, B. Moza, S. K. Mui, T. Miura, R. Sivamurthy, R. Allgaier, F. Pereyra, A. Trocha, M. Feeney, R. T. Gandhi, E. S. Rosenberg, M. Altfeld, T. M. Allen, R. Allen, B. D. Walker, E. J. Sundberg, and X. G. Yu. A viral CTL escape mutation leading to immunoglobulin-like transcript 4 (ILT4)-mediated functional inhibition of myelomonocytic cells. J. Exp. Med., in press. [DOI] [PMC free article] [PubMed]
- 52.Martinez-Picado, J., J. G. Prado, E. E. Fry, K. Pfafferott, A. Leslie, S. Chetty, C. Thobakgale, I. Honeyborne, H. Crawford, P. Matthews, T. Pillay, C. Rousseau, J. I. Mullins, C. Brander, B. D. Walker, D. I. Stuart, P. Kiepiela, and P. Goulder. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masemola, A., T. Mashishi, G. Khoury, P. Mohube, P. Mokgotho, E. Vardas, M. Colvin, L. Zijenah, D. Katzenstein, R. Musonda, S. Allen, N. Kumwenda, T. Taha, G. Gray, J. McIntyre, S. A. Karim, H. W. Sheppard, and C. M. Gray. 2004. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J. Virol. 78:3233-3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matano, T., M. Kobayashi, H. Igarashi, A. Takeda, H. Nakamura, M. Kano, C. Sugimoto, K. Mori, A. Iida, T. Hirata, M. Hasegawa, T. Yuasa, M. Miyazawa, Y. Takahashi, M. Yasunami, A. Kimura, D. H. O'Connor, D. I. Watkins, and Y. Nagai. 2004. Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J. Exp. Med. 199:1709-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Migueles, S. A., M. S. Sabbaghian, W. L. Shupert, M. P. Bettinotti, F. M. Marincola, L. Martino, C. W. Hallahan, S. M. Selig, D. Schwartz, J. Sullivan, and M. Connors. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. USA 97:2709-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mothé, B. R., J. Weinfurter, C. Wang, W. Rehrauer, N. Wilson, T. M. Allen, D. B. Allison, and D. I. Watkins. 2003. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 77:2736-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nietfeld, W., M. Bauer, M. Fevrier, R. Maier, B. Holzwarth, R. Frank, B. Maier, Y. Riviere, and A. Meyerhans. 1995. Sequence constraints and recognition by CTL of an HLA-B27-restricted HIV-1 gag epitope. J. Immunol. 154:2189-2197. [PubMed] [Google Scholar]
- 58.Nixon, D. F., A. R. Townsend, J. G. Elvin, C. R. Rizza, J. Gallwey, and A. J. McMichael. 1988. HIV-1 gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature 336:484-487. [DOI] [PubMed] [Google Scholar]
- 59.Novitsky, V., P. Gilbert, T. Peter, M. F. McLane, S. Gaolekwe, N. Rybak, I. Thior, T. Ndung'u, R. Marlink, T. H. Lee, and M. Essex. 2003. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J. Virol. 77:882-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O'Connor, D. H., T. M. Allen, T. U. Vogel, P. Jing, I. P. DeSouza, E. Doods, E. Dunphy, C. Melsaether, B. Mothe, H. Horton, A. L. Hughes, and D. I. Watkins. 2002. Acute phase CTL escape is a hallmark of SIV infection. Nat. Med. 8:493-499. [DOI] [PubMed] [Google Scholar]
- 61.O'Connor, D. H., B. R. Mothe, J. T. Weinfurter, S. Fuenger, W. M. Rehrauer, P. Jing, R. R. Rudersdorf, M. E. Liebl, K. Krebs, J. Vasquez, E. Dodds, J. Loffredo, S. Martin, A. B. McDermott, T. M. Allen, C. Wang, G. G. Doxiadis, D. C. Montefiori, A. Hughes, D. R. Burton, D. B. Allison, S. M. Wolinsky, R. Bontrop, L. J. Picker, and D. I. Watkins. 2003. Major histocompatibility complex class I alleles associated with slow simian immunodeficiency virus disease progression bind epitopes recognized by dominant acute-phase cytotoxic-T-lymphocyte responses. J. Virol. 77:9029-9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peyerl, F. W., H. S. Bazick, M. H. Newberg, D. H. Barouch, J. Sodroski, and N. L. Letvin. 2004. Fitness costs limit viral escape from cytotoxic T lymphocytes at a structurally constrained epitope. J. Virol. 78:13901-13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pillay, T., H. T. Zhang, J. W. Drijfhout, N. Robinson, H. Brown, M. Khan, J. Moodley, M. Adhikari, K. Pfafferott, M. E. Feeney, A. St. John, E. C. Holmes, H. M. Coovadia, P. Klenerman, P. J. Goulder, and R. E. Phillips. 2005. Unique acquisition of cytotoxic T-lymphocyte escape mutants in infant human immunodeficiency virus type 1 infection. J. Virol. 79:12100-12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Price, D. A., P. J. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94:1890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reimann, K. A., K. Tenner-Racz, P. Racz, D. C. Montefiori, Y. Yasutomi, W. Lin, B. J. Ransil, and N. L. Letvin. 1994. Immunopathogenic events in acute infection of rhesus monkeys with simian immunodeficiency virus of macaques. J. Virol. 68:2362-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sacha, J. B., C. Chung, E. G. Rakasz, S. P. Spencer, A. K. Jonas, A. T. Bean, W. Lee, B. J. Burwitz, J. J. Stephany, J. T. Loffredo, D. B. Allison, S. Adnan, A. Hoji, N. A. Wilson, T. C. Friedrich, J. D. Lifson, O. O. Yang, and D. I. Watkins. 2007. Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J. Immunol. 178:2746-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmitz, J. E., M. J. Kuroda, S. Santra, V. G. Sasseville, M. A. Simon, M. A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B. J. Scallon, J. Ghrayeb, M. A. Forman, D. C. Montefiori, E. P. Rieber, N. L. Letvin, and K. A. Reimann. 1999. Control of viremia in simian immunodeficiency virus infection by CD8(+) lymphocytes. Science 283:857-860. [DOI] [PubMed] [Google Scholar]
- 68.Streeck, H., M. Lichterfeld, G. Alter, A. Meier, N. Teigen, B. Yassine-Diab, H. K. Sidhu, S. Little, A. Kelleher, J. P. Routy, E. S. Rosenberg, R. P. Sekaly, B. D. Walker, and M. Altfeld. 2007. Recognition of a defined region within p24 Gag by CD8+ T cells during primary human immunodeficiency virus type 1 infection in individuals expressing protective HLA class I alleles. J. Virol. 81:7725-7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thali, M., A. Bukovsky, E. Kondo, B. Rosenwirth, C. T. Walsh, J. Sodroski, and H. G. Gottlinger. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature 372:363-365. [DOI] [PubMed] [Google Scholar]
- 70.Timm, J., G. M. Lauer, D. G. Kavanagh, I. Sheridan, A. Y. Kim, M. Lucas, T. Pillay, K. Ouchi, L. L. Reyor, J. Schulze zur Wiesch, R. T. Gandhi, R. T. Chung, N. Bhardwaj, P. Klenerman, B. D. Walker, and T. M. Allen. 2004. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 200:1593-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Towers, G. J., T. Hatziioannou, S. Cowan, S. P. Goff, J. Luban, and P. D. Bieniasz. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138-1143. [DOI] [PubMed] [Google Scholar]
- 72.Wilson, C. C., J. T. Wong, D. D. Girard, D. P. Merrill, M. Dynan, D. D. An, S. A. Kalams, R. P. Johnson, M. S. Hirsch, R. T. D'Aquila, and B. D. Walker. 1995. Ex vivo expansion of CD4 lymphocytes from human immunodeficiency virus type 1-infected persons in the presence of combination antiretroviral agents. J. Infect. Dis. 172:88-96. [DOI] [PubMed] [Google Scholar]
- 73.Wilson, J. D., G. S. Ogg, R. L. Allen, C. Davis, S. Shaunak, J. Downie, W. Dyer, C. Workman, S. Sullivan, A. J. McMichael, and S. L. Rowland-Jones. 2000. Direct visualization of HIV-1-specific cytotoxic T lymphocytes during primary infection. AIDS 14:225-233. [DOI] [PubMed] [Google Scholar]
- 74.Winkelstein, W., Jr., M. Samuel, N. S. Padian, and J. A. Wiley. 1987. Selected sexual practices of San Francisco heterosexual men and risk of infection by the human immunodeficiency virus. JAMA 257:1470-1471. [DOI] [PubMed] [Google Scholar]
- 75.Yang, O. O., S. A. Kalams, M. Rosenzweig, A. Trocha, N. Jones, M. Koziel, B. D. Walker, and R. P. Johnson. 1996. Efficient lysis of human immunodeficiency virus type 1-infected cells by cytotoxic T lymphocytes. J. Virol. 70:5799-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang, R., and C. Aiken. 2007. A mutation in alpha helix 3 of CA renders human immunodeficiency virus type 1 cyclosporin A resistant and dependent: rescue by a second-site substitution in a distal region of CA. J. Virol. 81:3749-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yant, L. J., T. C. Friedrich, R. C. Johnson, G. E. May, N. J. Maness, A. M. Enz, J. D. Lifson, D. H. O'Connor, M. Carrington, and D. I. Watkins. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 Replication. J. Virol. 80:5074-5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yeh, W. W., E. M. Cale, P. Jaru-Ampornpan, C. I. Lord, F. W. Peyerl, and N. L. Letvin. 2006. Compensatory substitutions restore normal core assembly in simian immunodeficiency virus isolates with Gag epitope cytotoxic T-lymphocyte escape mutations. J. Virol. 80:8168-8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuniga, R., A. Lucchetti, P. Galvan, S. Sanchez, C. Sanchez, A. Hernandez, H. Sanchez, N. Frahm, C. H. Linde, H. S. Hewitt, W. Hildebrand, M. Altfeld, T. M. Allen, B. D. Walker, B. T. Korber, T. Leitner, J. Sanchez, and C. Brander. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80:3122-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]