

Abstract
Certain histocompatibility leukocyte antigen (HLA) alleles are associated with improved clinical outcomes for individuals infected with human immunodeficiency virus type 1 (HIV-1), but the mechanisms for their effects remain undefined. An early CD8+ T-cell escape mutation in the dominant HLA-B57-restricted Gag epitope TW10 (TSTLQEQIGW) has been shown to impair HIV-1 replication capacity in vitro. We demonstrate here that this T242N substitution in the capsid protein is associated with upstream mutations at residues H219, I223, and M228 in the cyclophilin A (CypA)-binding loop in B57+ individuals with progressive disease. In an independent cohort of epidemiologically linked transmission pairs, the presence of these substitutions in viruses encoding T242N was associated with significantly higher plasma viremia in donors, further suggesting that these secondary mutations compensated for the replication defect of T242N. Using NL4-3 constructs, we illustrate the ability of these CypA loop changes to partially restore replication of the T242N variant in vitro. Notably, these mutations also enhanced viral resistance to the drug cyclosporine A, indicating a reduced dependence of the compensated virus on CypA that is normally essential for optimal infectivity. Therefore, mutations in TW10 allow HIV-1 to evade a dominant early CD8+ T-cell response, but the benefits of escape are offset by a defect in capsid function. These data suggest that TW10 escape variants undergo a postentry block that is partially overcome by changes in the CypA-binding loop and identify a mechanism for an HIV-1 fitness defect that may contribute to the slower disease progression associated with HLA-B57.
CD8+ cytotoxic T-lymphocyte (CTL) responses represent an important antiviral defense mechanism against human immunodeficiency virus type 1 (HIV-1). Major histocompatibility complex class I alleles, such as HLA-B57 and HLA-B27 in humans (27, 37, 38) or Mamu-A01 and Mamu-B17 in rhesus macaques (16, 54, 66), present particular viral peptides to CD8+ T cells and are associated with slower progression to AIDS in humans or monkeys infected with HIV-1 or simian immunodeficiency virus (SIV). However, the basis for the link between certain HLA alleles and enhanced viral control remains unclear. Understanding the viral and cellular factors that mediate control in these cases may enable the design of HIV-1 vaccines capable of exploiting these mechanisms.
HLA-allele-specific viral mutations can be identified within circulating strains of HIV-1, providing strong support for immune-mediated selective pressure on the virus in vivo (51). Notably, some CTL-targeted epitopes mutate rapidly during acute HIV-1 or SIV infection, while others remain invariant (6, 18, 37, 53, 57). Such differences in sequence evolution may be due to variation in CTL efficacy or may reflect deleterious effects of particular escape mutations on the virus. Recent reports for HIV-1 and SIV illustrate that CTL escape mutations can impact viral replication (30, 46, 55, 56), revert upon transmission to a new host (5, 17, 29, 44, 45), or require the development of numerous compensatory mutations (30, 41, 44, 55, 56). Each of these observations implies a negative impact of immune escape on the replicative capacity of the virus. In addition, Matano et al. (48) observed that induction of CTL escape mutations in the SIV capsid protein was associated with control of the highly pathogenic SIVmac239 in rhesus macaques. These studies highlight the ability of some HLA alleles to direct CTL pressure against highly conserved regions of the virus and reveal an important mechanism by which to control HIV-1, requiring the virus either to endure an effective CTL response or to develop mutations that negatively impact its ability to replicate (8).
Of the HLA alleles associated with improved clinical outcomes following HIV-1 infection in population-based studies, HLA-B57 and HLA-B5801 confer the greatest advantage (7, 40, 50, 52). These alleles present multiple epitopes within the HIV-1 capsid protein p24 to CTLs (7, 10), and viral escape has been described for three of these epitopes, TW10 (44), ISW9 (26), and KF11 (24). Notably, mutations in the TW10 epitope have been observed to revert following transmission to an HLA-mismatched host (44), providing in vivo evidence for a viral fitness cost incurred by these escape mutations. Recently, Martinez-Picado et al. (46) confirmed this hypothesis by using an in vitro culture system, although the mechanisms responsible for the viral defect were not examined.
CTL escape in the B57-TW10 epitope in the capsid occurs primarily at residue T242 and is associated with a putative upstream compensatory mutation in the adjacent cyclophilin A (CypA)-binding loop (44). Binding of the capsid to target cell protein CypA is critical for efficient viral replication (19, 20, 39), likely through alteration of capsid stability (31, 36), regulation of the viral uncoating process (33), or prevention of host restriction factor recognition (62). These observations prompted us to examine whether secondary capsid mutations associated with CTL escape in TW10 altered this critical viral protein-host protein interaction, thus compromising the ability of HIV-1 to replicate efficiently. We demonstrate here the accumulation of specific amino acid substitutions in the CypA-binding loop in HLA-B57+ individuals with progressive disease and provide evidence that transmission of these mutations is associated with higher viral loads in the recipient. Furthermore, we describe the impact of both the CTL escape mutations in the B57-TW10 epitope and the secondary sequence changes in the CypA-binding loop on viral replicative capacity in vitro. Finally, we show that these mutations in the capsid affect the sensitivity of virus to the CypA-binding drug cyclosporine A (CsA), suggesting a functional mechanism to explain the observed fitness defect of T242N.
MATERIALS AND METHODS
Patient data.
Clinical data and viral sequences depicted in Fig. 1 were described previously (49, 50) (accession numbers AV986516 to AV986733). Patients were classified as progressors or nonprogressors by Migueles et al. (49, 50) based on longitudinal clinical data not shown here. Subjects in the nonprogressor group had been infected for a median of 16 years and had maintained normal CD4 T-cell counts and low or undetectable viral loads during this time, and therefore, they were not exhibiting any signs of progression over an extended period of follow-up. Progressors were studied for a median of 8 years after diagnosis of HIV infection and were antiretroviral naïve at the time of study. The predominant clonal plasma viral sequence was used for this analysis, except in the case of nonprogressors with undetectable plasma viremia (viral loads of less than 50 copies/ml), from whom only clonal proviral DNA sequences were available. Minor populations in patient 5 (Pt5) and Pt14 contained mutations in the TW10 epitope and are shown in lowercase font. Pt12, Pt25, and Pt34 were identified as heterozygous for the CCR5Δ32 mutation (49). Data shown in Fig. 2 were obtained from a cohort of 88 epidemiologically linked subtype C heterosexual transmission pairs enrolled in a discordant couple cohort in Lusaka, Zambia. Details of this cohort have been described previously (25, 63). The set point viral load in donors was determined by repeated longitudinal testing to ensure values were stable within 0.3 logs. For recipients, the viral load was measured at the first seropositive visit (range of 3 to 6 months posttransmission).
FIG. 1.

Escape in the B57-TW10 epitope is associated with multiple potential compensatory mutations. (a) The predominant HIV-1 Gag sequences from HLA-B57+ progressors and nonprogressors are shown, aligned to the consensus subtype B Gag sequence from the LANL HIV database. Patient classification, viral loads (in copies/milliliter), CD4 counts (in cells/microliter), and Gag sequences (accession numbers AV986516 to AV986733) are from Migueles et al. (49). Minor clonal populations are shown in lowercase for Pt5 and Pt12 (asterisks). The HLA-B57-restricted CD8 epitope TW10 (TSTLQEQIGW) is highlighted (boxed), and the CypA-binding domain is indicated (underlined). The predominant TW10 escape mutation, T242N, is shaded. Polymorphisms at residues H219, I223, M228, and G248 (boldface) were more frequent in HLA-B57+ progressors than nonprogressors in this cohort, as well as in subtype B sequences from the LANL HIV database that contained the T242N substitution. Statistical calculations were determined using Fisher's exact test, and P values are shown beneath each residue. (b) A structural diagram of the HIV-1 capsid is shown, with residues associated with TW10 escape highlighted. The TW10 epitope is shown (green), and polymorphic residues are identified (red). The escape mutation T242N and residues G248A and N252H are located within or adjacent to alpha helix VI. Residues H219Q, I223V, and M228I are located within the CypA-binding loop.
FIG. 2.

Acquisition of mutations in the CypA-binding loop correlates with higher plasma viremia following transmission of TW10 escape variants. Viral sequence polymorphism data from 22 transmission pairs from a discordant couple cohort in Lusaka, Zambia, were analyzed in which a donor infected with HIV-1 subtype C transmitted the T242N escape mutation in the presence of zero, one, two, or three or more potential compensating mutations (i.e., H219Q, I223V, M228I, and G248A). (a) In the donors, the median plasma viral load was significantly higher when one or more of these additional mutations was present (P = 0.03). Furthermore, a correlation was observed for donors between viral load and number of mutations (r = 0.34), but this failed to reach significance (P = 0.13). (b) A similar correlation also was observed for recipients between viral load and the number of mutations in this region (r = 0.29), but again this failed to reach statistical significance (P = 0.19). Viral loads were not significantly different with the addition of one or more mutations to the recipients (P = 0.56). (c) When data from donors and recipients were combined for analysis, the median viral load was significantly higher when T242N escape occurred in the presence of one or more of the identified mutations (P = 0.03). A correlation was observed between viral load and the number of mutations (r = 0.34), which reached significance with this larger number of samples (P = 0.03).
NL4-3 constructs.
HIV-1 strain NL4-3 (2) was modified using unique restriction sites SpeI (nucleotide [nt] 1504) and SbfI (nt 2838). Inserts were generated by PCR using primers SpeI-T242N (5′-GGA ACT ACT AGT AAC CTT CAG GAA C-3′; nt 1501), SpeI-T242S (5′-GGA ACT ACT AGT TCC CTT CAG GAA C-3′; nt 1501), SpeI-T242A (5′-GGA ACT ACT AGT GCC CTT CAG GAA C-3′; nt 150), SpeI-T242W (5′-GGA ACT ACT AGT TGG CTT CAG GAA C-3′; nt 1501), SpeI-T242N-G248A (5′-ACT ACT AGT AAC CTT CAG GAA CAA ATA GCA TGG ATC-3′; nt 1501), and SbfI-Rev1 (5′-GGC AGC ACT ATA GGC TGT ACT GTC C-3′; nt 2839). Mutated residues are underlined, and positions listed are according to the NL4-3 sequence (GenBank accession no. AF324493). Mutations upstream of TW10 were introduced using sites SphI (nt 1443) and SbfI (nt 2838). Inserts were generated by PCR using phosphorylated primer SphI-H219Q (5′-Phos-CAA GCA GGG CCT ATT GCA CC-3′; nt 1444) or SphI-H219Q-I223V-M228I (5′-Phos-CAA GCA GGG CCT GTT GCA CCA GGC CAG ATT AGA G-3′; nt 1477) and the 3′ primer SbfI-Rev1. Enzymes were purchased from New England Biolabs. Mutants were verified by sequencing using published primers (9), Escherichia coli STBL3 cells (Invitrogen) were used to propagate full-length proviral plasmids, and stocks were prepared using a DNA Midi kit (QIAGEN).
Viral stocks.
Viruses were generated by transfection of HEK293T cells using Lipofectamine 2000 (Invitrogen). Plasmid pHEF-VSV-g (22) was used to pseudotype viral particles. Supernatant was harvested at 48 h posttransfection, and aliquots were stored at −80°C. Stock concentrations were quantified by p24 enzyme-linked immunosorbent assay (ELISA), and titers were determined by infection of CEM-GXR cells.
Cells.
The green fluorescent protein (GFP) reporter T-cell line CEM-GXR was described previously (21). Cells were maintained in RPMI 1640 medium (Sigma-Aldrich) containing 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% (vol/vol) bovine calf serum (R10; Atlantic Biologicals). Geneticin (G418; Gibco-BRL) at 500 μg/ml was added for selection. Jurkat-GFP cells were generated by transfection with the plasmid pLTR-GFP (35). A stable clone was selected using 500 μg/ml Geneticin, tested for GFP induction, and used for subsequent experiments. For primary cell assays, peripheral blood mononuclear cells (PBMCs) were isolated from healthy HIV-negative donors at Massachusetts General Hospital by gradient centrifugation using Ficoll-Histopaque (Sigma-Aldrich) and were stimulated for 3 days using phytohemaglutinin (PHA; Murex Pharmaceuticals) in R10 medium containing 50 U/ml interleukin-2 (R10-50).
Virus inoculation and culture.
Cells (1 × 106) were infected at the indicated multiplicity of infection (MOI), incubated for 3 h at 37°C, washed, and plated at 0.5 × 105 cells/ml in R10. A portion of the culture was harvested at the indicated times, and cells were prepared for fluorescence activated cell sorter (FACS) analysis. Cell density was monitored, and cultures were maintained at 0.5 × 105 to 2 × 105 cells/ml. Single-cycle infectivity was determined by inoculation of CEM-GXR or Jurkat-GFP reporter cells with vesicular stomatitis virus g (VSV-g)-pseudotyped virus in the presence or absence of CsA (Sigma-Aldrich). Cells were preincubated with either 0.5 or 2.5 μM of CsA for 30 min, followed by inoculation with the indicated variant (MOI = 0.1). Infected cells were quantified using FACS analysis to measure GFP expression at 48 h postinfection. For primary cell assays, day-3 PHA-stimulated PBMCs were inoculated (MOI = 0.001) in the absence or presence of 0.5 μM CsA, washed, and then resuspended in R10-50 medium containing no drug or CsA. Supernatants were collected at the indicated times for analysis of viral replication by p24 ELISA, and the volume was replaced with R10-50 medium containing no drug or 0.5 μM CsA.
Flow cytometry.
For flow cytometry, cells were fixed in phosphate-buffered saline containing 2% paraformaldehyde, and GFP expression was determined using FACS analysis. A signal 10-fold above the mean fluorescent intensity of uninfected cells was considered positive. In preliminary experiments, intracellular p24 was measured using the KC57-fluorescein isothiocyanate antibody (Beckman-Coulter). Data were collected using a FACSCalibur flow cytometer (Becton Dickinson) and analyzed using FlowJo software (TreeStar, Inc.).
p24 ELISA.
Extracellular p24 was measured using the Alliance HIV-1 p24 ELISA kit (Perkin-Elmer) according to the manufacturer's instructions. Cell-free supernatants from infected cultures were harvested and stored at −80°C prior to quantification.
Amplification and sequencing of the Gag gene from HIV-1 subtype C transmission pairs.
Epidemiologically linked transmission pairs were identified as described previously during follow-up of a discordant couple cohort at the Zambia Emory HIV Research Project (25, 63). Couples were counseled and provided condoms at each 3-month return visit. Plasma was collected from both partners during each visit and was tested to detect transmission of HIV-1. gag gene sequences were amplified and sequenced using methods and primers described previously (43, 44).
Statistical analysis.
Sequence polymorphisms were compared using Fisher's exact test. Median plasma viral loads in the transmission studies whose results are shown in Fig. 2 were analyzed using the Mann-Whitney test, and correlations with the number of compensatory substitutions were determined using the Spearman rank order test. Calculations of mean viral slope were determined in Excel (Microsoft) using the LOGEST function and were converted to natural logs for presentation in Fig. 4c and Table 1. Variation in mean virus infectivity and replication slopes was evaluated using Student's t test. All statistical analyses were performed using Prism 4.0 (GraphPad).
FIG. 4.
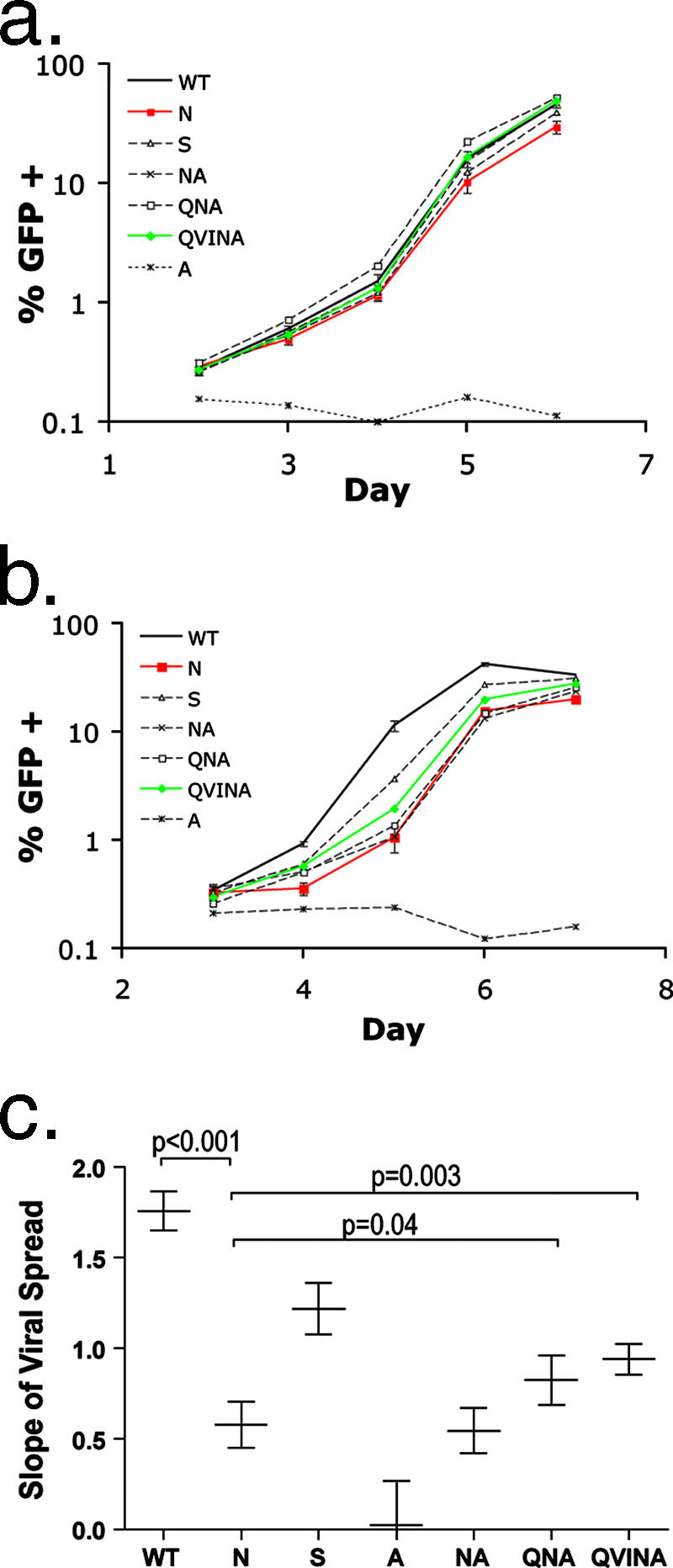
Escape in TW10 reduces HIV-1 replication capacity. (a and b) GFP reporter cells were inoculated in quadruplicate cultures with WT or TW10 variant N, S, A, NA, QNA, or QVINA (MOI = 0.0025), and the percentage of GFP+ cells was determined by FACS analysis to measure viral spread. Mean values ± standard deviations are shown for the WT, N, and QVINA. The A variant did not replicate efficiently and was included in these experiments as a negative control. (a) In CEM-GXR cells, no substantial differences in replication were seen between the WT (black line) and any of the TW10-associated mutants. (b) In contrast, in Jurkat-GFP cells a significant difference in replication was observed between the WT (black line) and the N variant (red line), as indicated by a 10-fold-greater proportion of GFP+ cells in WT-infected cultures at day 5. Incorporation of mutations in the CypA-binding loop enhanced the spread of the N mutant, as seen for the QVINA variant (green line). (c) The natural log slope of cell-to-cell spread in Jurkat-GFP cells was determined for each variant as described in Materials and Methods. Mean values ± standard deviations are shown. Results were compared using Student's t test, and the slope of the WT virus was found to be significantly greater than those of each of the variants (P < 0.001). Furthermore, the QNA and QVINA variants displayed significantly higher slopes than the N mutant (P = 0.039 and 0.003, respectively).
TABLE 1.
Replication capacity (slopea) of TW10 variants
| Virus variant | Result for cell type:
|
|||||
|---|---|---|---|---|---|---|
| CEM-GXR
|
Jurkat-GFP
|
|||||
| Replication capacity | Significance (P value) compared to virusb:
|
Replication capacity | Significance (P value) compared to virusb:
|
|||
| WT | N | WT | N | |||
| WT | 1.64 ± 0.11 | 1.76 ± 0.11 | ||||
| N | 1.51 ± 0.08 | NS | 0.58 ± 0.13 | <0.001 | ||
| S | 1.57 ± 0.07 | NS | NS | 1.22 ± 0.14 | <0.001 | <0.001 |
| A | 0.07 ± 0.11 | <0.001 | <0.001 | 0.02 ± 0.24 | <0.001 | <0.001 |
| NA | 1.65 ± 0.04 | NS | NS | 0.54 ± 0.12 | <0.001 | NS |
| QNA | 1.73 ± 0.04 | NS | NS | 0.82 ± 0.14 | <0.001 | 0.039 |
| QVINA | 1.72 ± 0.07 | NS | NS | 0.94 ± 0.09 | <0.001 | 0.003 |
The mean slope was determined by best-fit analysis to an exponential curve (± standard deviations).
P values were calculated by Student's t test. NS, not significant.
RESULTS
Viral escape in the B57-TW10 epitope is associated with multiple capsid mutations.
Following HIV-1 infection, the earliest peptide sequence targeted by CTL in HLA-B57/B58+ individuals typically is the TW10 epitope (TSTLQEQIGW240-249) in the capsid (7, 10). Viral escape from this dominant response most often occurs through a Thr-to-Asn alteration at position 3 of the epitope (residue T242N in Gag p55) (44). Two putative compensatory mutations have been associated with T242N, namely, a Gly-to-Ala (G248A) change at position 9 within the epitope and an upstream His-to-Gln (H219Q) mutation within the CypA-binding loop (44). Because viral escape may require multiple compensatory mutations (30, 55, 56, 67), we first investigated whether additional sequence changes were associated with T242N by using data from studies by Migueles et al. (49, 50) that identified variations in Gag CTL epitopes in B57+ individuals with progressive disease. Note that in contrast to plasma viral sequences obtained for the viremic individuals, data for subjects with an undetectable viral load (<50 copies/ml) were derived from cellular proviral DNA using a limiting dilution technique. In sequences containing T242N, residues H219, I223, M228, and N252 also were commonly altered from the residues of consensus HIV-1 subtype B (Fig. 1a). Statistical analysis indicated that mutations H219Q, I223V, T242N, G248A, and N252H were significantly more frequent in B57+ progressors than in B57+ nonprogressors from this cohort (P < 0.05). Further comparison of these polymorphisms to the Los Alamos National Laboratory (LANL) HIV sequence database (http://hiv-web.lanl.gov) produced similar results, with H219Q, I223V, M228I, and G248A appearing significantly more frequently in sequences that contained Asn at residue 242 (P < 0.05). In this latter analysis, however, residue 252 was seen to be highly variable in HIV-1 subtype B sequences (approximately 58% N, 20% S, and 18% H at this position), and H252 was not significantly associated with a T242N substitution (P = 0.37).
The location of each residue in a model of the HIV-1 p24 N-terminal domain structure is displayed in Fig. 1b and indicates that these polymorphisms cluster near capsid helix VI (T242N, G248A, and N252H) or within the CypA-binding loop (H219Q, I223V, and M228I). This loop region does not contain putative HLA-B57 epitopes based on binding motif analysis (15), although a subdominant HLA-B7-restricted epitope does lie in this area of the capsid (4, 59). Since identical T242N-associated polymorphisms in the CypA loop region were observed in B7+ and B7-negative individuals in this cohort (data not shown), mutations in this area are unlikely to be related to immune pressure mediated by HLA-B7. Furthermore, the proximity of these mutations to TW10 and to each other in the capsid structure suggested an association with TW10 rather than other B57 epitopes in Gag (49). Thus, escape in the TW10 epitope in the capsid appeared to be associated with two structurally distinct clusters of mutations.
Examination of clinical data from the subjects without disease progression indicates that development of the T242N mutation was associated with a modest increase in HIV-1 plasma viremia (<2,000 copies/ml in 4/4 subjects) (Fig. 1a). In this subgroup, viremia appeared to be contained in most patients despite escape from the dominant early TW10-directed immune response. For individuals with progressive disease, higher plasma viremia (>10,000 copies/ml) occurred frequently in conjunction with the development of more than one mutation in the CypA-binding loop (7/10 subjects) (Fig. 1a).
Transmission of CypA-binding loop mutations in conjunction with T242N is associated with higher plasma viral loads in donors and recipients.
To further assess the in vivo relevance of these potential compensatory substitutions, gag gene-sequencing data from transmission events for 88 discordant couples in Lusaka, Zambia, were analyzed. Twenty-two instances were identified in which a donor infected with HIV-1 subtype C had transmitted a virus containing the T242N escape mutation to his or her partner. We subdivided these cases based on sequence data and assessed plasma viral loads in the donor and the recipient when T242N was transmitted in the presence of potential compensating mutations (i.e., H219Q, I223V, M228I, or G248A). No evidence of T242N reversion was observed in the recipients at this time point, despite the fact that only two individuals expressed HLA alleles that potentially could target this epitope (one B5703+ and one B5801+). In agreement with the results shown in Fig. 1a, we observed that the median plasma viral load for donors having one or more of these additional mutations was significantly higher than that for donors harboring the T242N mutation alone (P = 0.03) (Fig. 2a). A correlation also was observed for the donors between viral load and the number of mutations (r = 0.34), although this did not reach statistical significance (P = 0.13). A trend, although not significant, similarly was observed for recipients (Fig. 2b) between viral load and the number of mutations in this region (r = 0.29). Although median viral loads appeared to increase in recipients with the addition of these associated mutations, the difference between individuals with zero additional changes and those encoding one or more of these mutations was not statistically significant (P = 0.56). This may be attributable to a lack of statistical power as a result of the number of subjects available for the study and the presence of a potential outlier individual with T242N alone who displayed a very high viral load (232,129 copies/ml) at the first seropositive visit, indicating that some plasma samples may have been collected prior to the set point. When data from the donors and recipients were combined, we observed that the median viral load was significantly higher when the T242N escape mutation occurred in the presence of at least one of the identified mutations (P = 0.03) (Fig. 2c). Similarly, a correlation was observed between viral load and the number of mutations (r = 0.34) that reached significance (P = 0.03). These results using an independent patient cohort, in particular the data obtained from the donor group, strengthen the observation that mutations in the CypA-binding loop are associated with increases in viremia following escape in TW10 and provide additional evidence that the accumulation of multiple substitutions in this region likely functions to compensate for a defect of T242N.
Escape mutations associated with TW10 do not diminish virion production.
To assess the functional significance of mutations associated with escape in the TW10 epitope, we modified HIV-1 strain NL4-3 (2) to encode T242N and the other identified changes. The viral variants used for our studies are shown in Fig. 3a. Note that the NL4-3 strain contains His at residue 252, which was not altered in our constructs. T242N (N) and T242S (S) are the most common clinically observed variations at this residue, while T242A (A) is rarely seen and T242W (W) was never observed at this position in the LANL database. Therefore, Ala and Trp were chosen to represent more dramatic substitutions at this site. To generate infectious viral stocks, we transfected HEK293T cells with the relevant proviral DNA constructs. In light of a recent report indicating that the fitness cost associated with escape in the SIV p11C, C-M epitope resulted from a significant defect in capsid assembly that limited particle production (67), we examined p24 release by these variants by using ELISA. No significant differences in p24 values were observed at 24 h posttransfection for the N, S, NA, QNA, or QVINA variant using this assay (Fig. 3b). In contrast, replacement of T242 with the atypical nonpolar residue Ala or Trp severely compromised the ability to release p24 (Fig. 3b). Similar results were seen at 48 h posttransfection (data not shown). Thus, while selection of alternative amino acids at residue T242 appeared to be constrained, the common naturally arising variants did not substantially impact p24 particle production. These data indicate that none of the clinically observed substitutions resulted in gross defects in virion production in this in vitro system.
FIG. 3.

TW10-associated mutations in NL4-3. (a) Variants were aligned to the consensus clade B sequence and were constructed by PCR mutagenesis using the HIV-1 molecular clone NL4-3 as described in Materials and Methods. (b) HEK293T cells were transfected in triplicate with 5 μg of proviral plasmid DNA for each variant as described in Materials and Methods. Culture supernatants were collected at 24 h, and particle production was assessed by p24 ELISA. Values shown are the means ± standard deviations for each variant. No significant differences were observed between WT NL4-3 and the N, S, NA, QNA, or QVINA mutant strain. Replacement of T242 with A or W resulted in a severe defect in virion production.
Escape mutation T242N reduces HIV-1 replication capacity.
It has been reported that the T242N mutation confers an in vitro fitness defect on HIV-1 (46). However, more than 20 days of passage in coculture were required to observe outgrowth of wild-type (WT) virus, and the impact of this substitution on the MT4 T-cell line used in these assays was subtle (46). We therefore wished to examine the effect of TW10 escape and associated mutations on viral replication using other T-cell lines. First, we utilized a CEM-derived GFP reporter cell (CEM-GXR) assay that we developed to quantify viral infection and cell-to-cell spread using flow cytometry (21). CEM-GXR cells were inoculated in quadruplicate with virus (MOI = 0.0025), and infection was verified by measuring GFP expression at 48 h (day 2) (Fig. 4a). Because any replicating virus eventually will saturate the culture in this in vitro assay, we assessed the spread of viral variants during the exponential-growth phase, routinely using only GFP values between 0.5 and 10%. The slope of cell-to-cell spread therefore was determined between days 3 and 5 by best-fit analysis to an exponential curve (Table 1). We detected no significant impact of either the T242N or T242S substitution on viral replication compared to replication of WT NL4-3 in these cells during a 7-day culture. These results are similar to those reported for MT4 cells and suggest that additional time in culture or coculture assays is necessary to observe growth differences between these viral strains using CEM cells. Preliminary analyses of viral replication in other human T-cell lines, however, indicated a reduction in growth for the N variant when we infected Jurkat T cells (data not shown). Therefore, we constructed a GFP-expressing Jurkat reporter cell line, and parallel experiments were completed with CEM-GXR and Jurkat-GFP cells. In quadruplicate assays with Jurkat-GFP cells, each of the variant viruses replicated less well than the WT (Fig. 4b), with the number of GFP+ cells present following infection with the N variant reduced approximately 10-fold compared to that for the WT at day 5 postinfection (Fig. 4b). As for CEM-GXR cells, we calculated the mean slopes of viral spread for these variants in Jurkat cells and observed that the T242N mutant replicated significantly less well than WT NL4-3 (0.58 ± 0.13 and 1.76 ± 0.11, respectively; P < 0.001) (Fig. 4c and Table 1). Partial recovery of this defect was observed for variants encoding the associated mutations, as indicated by statistically significant increases in the mean slopes of viral spread for the QNA (0.82 ± 0.14; P = 0.039) and QVINA (0.94 ± 0.09; P = 0.003) variants, respectively. Notably, all of the variant viruses replicated significantly less well than WT NL4-3 in Jurkat cells. These results indicate that escape in TW10 reduced HIV-1 replication in vitro, which partially could be compensated for by additional changes within the CypA-binding loop of the capsid.
Impact of TW10 escape mutations in primary cells.
The observation that mutation T242N reverts upon transmission to a non-B57+ individual (44) provides strong evidence to support an in vivo fitness defect for TW10 escape variants; however, the in vitro data to support this model have utilized only immortalized T-cell lines (46). To assess the impact of TW10 escape and compensation in a more relevant system, we evaluated viral growth in PBMC cultures. PHA-stimulated PBMCs were inoculated with the indicated viral variant at an MOI of 0.001, and supernatants were collected for analysis of soluble p24 using ELISA. We detected significantly less p24 accumulation for the N variant at days 5 and 7 postinfection than for the WT (P = 0.031 and 0.004, respectively), while replication of the NA variant was significantly reduced on day 7 (P = 0.041) (Fig. 5a). The addition of compensatory mutations in the CypA-binding loop restored p24 values for the QVINA variant, which were not significantly different from WT levels at either day 5 or 7. These results are consistent with our in vitro data acquired using immortalized Jurkat T cells, demonstrating that the N and NA variants display subtle replicative defects that can be corrected by additional alterations in the CypA-binding loop.
FIG. 5.
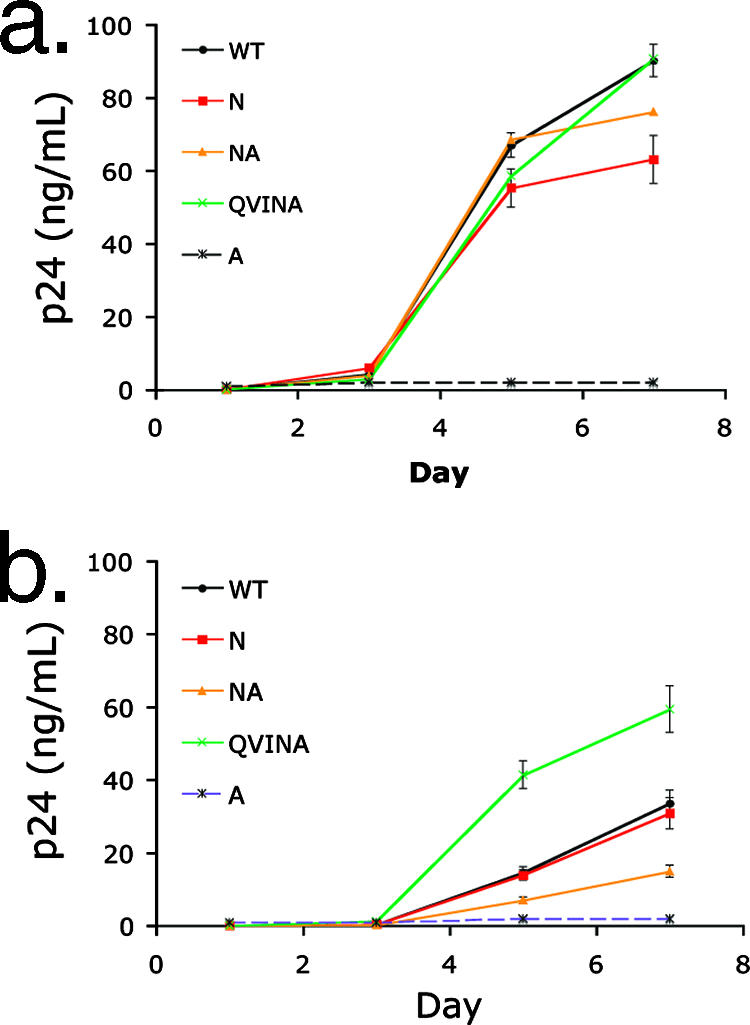
Replication of TW10 variants in primary cells. PHA-stimulated PBMCs from a healthy HIV-negative donor were inoculated with TW10 variants at an MOI of 0.001 in the absence (a) or presence (b) of 0.5 μM CsA. Replication of each virus was determined by ELISA to measure p24 in the culture supernatant at the indicated day postinfection, and results are displayed as the mean values of triplicate samples ± standard deviations. (a) In the absence of drug, a reduction in p24 accumulation compared to that of the WT was observed for the N variant (red line) at days 5 and 7 (black line) (P = 0.031 and 0.004, respectively), and replication of the NA variant (orange line) also was reduced on day 7 (P = 0.041). (b) Addition of CsA resulted in a decline in viral replication for all variants. The NA variant (orange line) appeared to be more sensitive to this drug than the WT, while the QVINA strain (green line) was less sensitive to CsA during this 7-day assay.
T242N-associated mutations modulate viral sensitivity to CsA.
Optimal HIV-1 infection requires the finely tuned interaction between capsid and target cell CypA (19, 39, 60, 68). Certain T-cell lines, such as CEM, which express two- to fourfold-higher levels of CypA protein per cell, have been shown to support replication of HIV-1 capsid mutants with a decreased ability to bind to this host protein (1). To address the relationship between CypA and replication of the TW10 escape variants directly, we examined replication of these viral variants in the presence of CsA, a competitive inhibitor of HIV-1 capsid binding to CypA (28). PHA-stimulated PBMCs were inoculated at an MOI of 0.001 in the presence of 0.5 μM CsA, and supernatant p24 was measured. As expected, the WT virus yielded less p24 production over the course of a 7-day infection in the presence of CsA (Fig. 5b) than in the absence of drug (Fig. 5a). In addition, we observed that the NA variant displayed significantly less p24 accumulation at days 5 and 7 postinfection than the WT in the presence of drug (P = 0.002 and 0.001, respectively), while the N strain was as sensitive to CsA as the WT in these assays (Fig. 5b). Notably, the QVINA variant appeared less sensitive to CsA in primary cells, as indicated by significantly higher p24 values, than the WT at days 5 and 7 postinfection (P < 0.001 and 0.004, respectively).
To further address the role of CypA, CEM-GXR or Jurkat-GFP cells were inoculated with VSV-g envelope-pseudotyped viruses (MOI = 0.1) in the presence or absence of CsA, and GFP expression was measured at 48 h. In CEM-GXR cells, the WT, N, S, and NA viruses were similarly sensitive to this drug in a dose-dependent manner, exhibiting a threefold reduction in single-round infectivity in the presence of 2.5 μM CsA (Fig. 6a) that indicated a normal dependence of these viruses on CypA for infection (19, 39). However, the QNA and QVINA variants were significantly less sensitive to the effects of 0.5 or 2.5 μM CsA than the WT in these assays (P = 0.001). In Jurkat-GFP cells, infection by the N, S, and NA variants was more sensitive than that of the WT to 0.5 μM CsA (P < 0.05 for each), while the QNA and QVINA variants were minimally affected by 2.5 μM of the drug (P < 0.001) (Fig. 6b).
FIG. 6.

TW10 mutations alter viral sensitivity to CsA. GFP reporter cells were inoculated with VSV-g-pseudotyped WT, N, S, NA, QNA, or QVINA virus (MOI = 0.1) in triplicate in the presence of either 0.5 or 2.5 μM CsA. The proportion of GFP+ cells was measured at 48 h, and values were normalized to those of the no-drug control. Mean values ± standard deviations are shown, and significant differences were determined using Student's t test (*, P < 0.05; **, P < 0.01; ***, P < 0.005). (a) No significant differences were seen between the WT, N, S, and NA variants in CEM-GXR cells, as each appeared to be equally sensitive to both 0.5 and 2.5 μM concentrations of CsA. Infection by the QNA and QVINA variants in the presence of both CsA concentrations was significantly higher than that of the WT (P < 0.01 in all cases), indicating that these variants were resistant to the effects of the drug. (b) In Jurkat-GFP cells, the N, S, and NA variants were significantly more sensitive than WT virus to 0.5 μM CsA (P < 0.001, P < 0.05, and P < 0.001, respectively), while the QVINA variant was significantly more resistant to the drug (P < 0.001). At 2.5 μM CsA, the N mutant was again more sensitive than the WT (P < 0.001), while both the QNA and QVINA variants appeared to be resistant to the drug (P < 0.001 for each).
Taken together, these data indicate that CTL escape mutations in the TW10 epitope generate a capsid molecule that was more dependent on CypA to maintain optimal infectivity. This may account for the observed replication defect in Jurkat T cells, but not CEM-derived T cells, and the decrease in viral replication observed for these variants in primary cells. In turn, mutations that develop within the CypA-binding loop appeared to further alter capsid function, allowing compensation for the replication defect but also altering the state of the capsid to that of an unusual CypA-independent phenotype.
DISCUSSION
Our results indicate that viral escape in the HLA-B57-restricted TW10 epitope is associated with a series of mutations within the CypA-binding loop region of the capsid. We observed that the viruses of B57+ individuals with progressive disease were more likely to encode these substitutions than those of B57+ nonprogressors. In addition, using independent data from transmission studies, we show that TW10 escape variants containing these additional mutations were associated with higher plasma viremia in the donors, while a trend toward higher viral loads was observed in the recipients. Together, these data suggest that mutations in the CypA-binding loop can serve to compensate for an in vivo replication defect associated with TW10 escape. Using in vitro assays, the T242N escape mutation reduced the HIV-1 replication capacity of NL4-3. The in vitro defect correlated with an enhanced sensitivity to the drug CsA, indicating that the T242N mutation hindered capsid interactions with the host cell protein CypA that may result in an early postentry defect in viral replication. Notably, this defect was apparent when Jurkat T cells and PBMCs were used, but it was not observed with CEM-derived cells during a 7-day assay. This cell-line-dependent phenotype will require further analysis but suggests that cellular CypA levels may dictate the impact of these mutations on replication. This is particularly relevant in light of a recent report that polymorphisms in the CypA gene are associated with a rapid disease course in HIV-1 infection (11). Therefore, we propose that while the T242N mutation enables evasion from the dominant HLA-B57-mediated CTL response elicited during primary infection (44), the in vivo benefits of this substitution are significantly diminished by its impact on capsid function. Clinically associated mutations H219Q, I223V, and M228I in the CypA-binding loop enhanced viral replication in Jurkat T cells and PBMCs, and the fact that these compensatory mutations acted to reduce viral sensitivity to CsA provides further evidence for the central role of capsid-CypA interactions in this case. Together, these data demonstrate that the replication defect observed following escape at T242N likely is due to an alteration of Gag function at an early postentry stage during viral infection and that this defect can be partially compensated for by specific mutations within the CypA-binding loop that can modulate the critical capsid interaction with CypA.
The TW10-specific CTL response typically is early and dominant in B57+ subjects (10), and the T242N mutation develops quickly following infection (44). Therefore, a possible explanation of long-term control of HIV-1 is the inability of virus to escape at T242N in some B57+ individuals, resulting in a viral load of <50 copies/ml. Recent evidence using sensitive methods to sequence plasma viral RNA in aviremic B57+ individuals, however, revealed a prevalence of T242N similar to that of viremic subjects (reference 13 and data not shown). Our results and recent data from Martinez-Picado et al. (46) indicate that mutations in this region of the capsid result in a replication defect, and we now demonstrate that the impact of T242N on replication can be relieved through acquisition of compensatory mutations. Thus, a revised model of control is that HLA-B57 commonly selects a less fit viral variant containing T242N and that compensation for this capsid defect results in higher viremia and progressive disease. This model is consistent with the work of Gao et al. (32), demonstrating that the protective effect of HLA-B57 occurs at early stages of disease and accumulating data indicating the importance of compensatory mutations coincident with or subsequent to CTL epitope escape (24, 30, 41, 67). In addition to the potential for unknown compensatory mutations, control of HIV-1 likely is due to CTL pressure against multiple epitopes, which may be the case for HLA-B57 in particular, for which numerous CTL responses against Gag and other proteins have been described. Therefore, simple correlation of viral sequence evolution with clinical data may be problematic and may explain the relatively weak associations we observed between substitutions H219Q, I223V, and M228I and viral load measurements.
Efficient replication of HIV-1 typically requires binding to host cell CypA (1, 19, 20, 39, 68). The precise effects of this interaction are not well understood, but it is likely that CypA alters capsid stability (31, 36), regulates viral uncoating (33), and/or prevents host restriction factor recognition (62). Therefore, the variable phenotype observed between Jurkat and CEM T cells may be due to differences in the expression of CypA or another protein related to these processes. CEM cells have been described to express two- to fourfold-higher levels of CypA protein than Jurkat cells, and CypA expression correlated with the ability of T-cell lines to support replication of HIV-1 strains with defects in CypA binding (1). Gag residues H219 and I223 in the CypA-binding loop have been shown to interact with CypA (3), and therefore, TW10-associated changes in the loop region may directly affect binding to CypA. Our results are consistent with those of recent reports demonstrating that the H219Q mutation alters capsid interactions with CypA, as well as data indicating that the QVI mutations diminished viral sensitivity to owl monkey Trim5-CypA and CsA (23, 33). Together, these data suggest that modest differences in CypA interactions by TW10 escape variants result in a postentry capsid defect in primary cells and Jurkat T cells that can be enhanced by the addition of CsA. The impact of these mutations may be masked in CEM cells due to their increased CypA expression. Though subtle in single-round assays, any defect is likely to be amplified by additional rounds of cell-to-cell spread during in vitro culture or in vivo pathogenesis, accounting for the reversion of the T242N mutation observed in vivo.
A detailed understanding of the structural constraints on HIV-1 capsid is essential to explain the development of escape and compensatory mutations for TW10 and other epitopes. Compensation for the T242N defect appears to be possible through alteration of the natural interaction between the capsid and CypA, as indicated by increased resistance to the drug CsA. The in vivo association of TW10 escape with H219Q, I223V, and M228I mutations within the CypA-binding loop strongly suggests that escape from CTL pressure altered this critical interaction between the capsid and CypA. It is striking that the H219Q, I223V, and M228I mutations are present in rare but naturally occurring HIV-1 group M viruses that also replicate in a CypA-independent manner (23) and that most group O strains encode comparable sequence polymorphisms at these residues and also display a reduced dependence on CypA (19). Since a minority of individuals were observed to progress in the absence of compensatory changes in the CypA-binding loop, other unknown mutations in the capsid are likely to be able to compensate for viral fitness following TW10 escape. We have not completed a comprehensive sequence analysis of p24 from HLA-B57+ individuals to identify additional mutations that may result in better compensation for T242N or perhaps cause a more pronounced primary replication defect in association with escape. This point may be relevant to the clinical data illustrated in Fig. 2a, since donors with a single compensatory change appeared to have a higher viral load than those with two or more identified changes. Therefore, it is possible that other combinations of AA substitutions in the capsid will be uncovered that affect the T242N phenotype. In particular, many clinically derived sequences contain Asp at residue 252, while all of our NL4-3 variants encoded His at this site. Further analysis of capsid function will be necessary to evaluate these possibilities. Regardless of these limitations, our data indicate that mutations that effectively reduce capsid dependence on CypA may function to compensate for T242N. Upon binding to CypA, alpha helices I, II, IV, and VI in the amino-terminal domain show significant movement (3, 31). Therefore, alternative mutations in any of these regions could impact CypA binding, capsid stability, and early postentry steps in viral infection (58, 64, 65) and thus also function to compensate for TW10 escape. Together, the in vivo and in vitro results suggest that compensation for the T242N mutation is an important step during disease progression in most B57+ individuals.
Reversion of certain CTL escape mutations upon transmission to a new host is a well-characterized phenomenon (5, 29, 43, 44, 61), and several CTL escape mutations now have been shown to occur at a substantial cost to viral replicative capacity (29, 46, 67). The important role that viral fitness plays in control of HIV-1 also is supported by reversion of transmitted drug resistance mutations in the absence of highly active antiretroviral therapy and observations of inherent intra- and intersubtype differences in viral replication capacity (12, 14). A comprehensive approach to determine the extent to which viral escape and reversion occur during HIV-1 infection may help to elucidate the role of particular CD8+ T-cell responses in immune control. Furthermore, achieving a deeper understanding of the impact of these mutations on viral replication capacity should help to identify the most sensitive regions of this highly variable virus. Such data may assist in explaining the association observed between Gag-specific CTL responses and control of HIV-1 viremia in both subtype B and subtype C infections (34, 42, 47).
In summary, HLA-B57/B58 has been associated with enhanced control of HIV-1, but the mechanisms for its effect remained unknown. Early CTL pressure on the dominant B57-restricted epitope TW10 in HIV-1 capsid is known to select for a viral escape variant that displays reduced in vitro replication capacity (46). For B57+ individuals with progressive disease, we observed that TW10 escape was associated with additional changes in the CypA-binding loop of the capsid. Prior to and following transmission, escape variants that contained these mutations were associated with higher viremia. We provide in vitro evidence that escape and compensatory mutations alter capsid function by modulating CypA interactions that normally are critical in the viral life cycle. TW10 escape variants displayed enhanced sensitivity to the CypA-binding drug CsA, while viruses encoding mutations in the CypA-binding loop exhibited decreased susceptibility to this drug. Together, these data illustrate a mechanism that explains the viral fitness defect observed for the B57-mediated TW10 escape mutation and a pathway of compensation that may be relevant to disease progression. Therefore, selective pressure by the adaptive immune response on this region in Gag may contribute to the ability of HLA-B57 to control HIV-1 infection. A detailed understanding of the limits of sequence variation by measuring the impact of immune escape mutations on viral fitness may help to guide vaccine strategies or therapeutics aimed at driving HIV-1 evolution toward a less fit state.
Acknowledgments
We thank Stephen Migueles and Mark Connors for viral sequence data and discussions related to their clinical data; Philip Goulder for communication of unpublished data; Jacques Corbeil for the plasmid pLTR-GFP; and the physicians, staff, and volunteers at the Zambia Emory HIV Research Project (ZEHRP), Lusaka, Zambia. Plasmids pNL4-3 (M. Martin) and pHEF-VSV-g (L. J. Chang) were obtained from the NIH AIDS Research and Reference Reagents Program.
This study was supported by the Howard Hughes Medical Institute (B.D.W.) and National Institutes of Health grants R01AI054178 and R21AI067078 (T.M.A.), R01A164030 (E.H.), and R01AI028568 (B.D.W.). B.D.W. is the recipient of a Distinguished Clinical Scientist Award from the Doris Duke Charitable Foundation.
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the Howard Hughes Medical Institute or the National Institutes of Health.
Footnotes
Published ahead of print on 29 August 2007.
REFERENCES
- 1.Ackerson, B., O. Rey, J. Canon, and P. Krogstad. 1998. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. J. Virol. 72:303-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and M. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal, P. K. 2004. Cis/trans isomerization in HIV-1 capsid protein catalyzed by cyclophilin A: insights from computational and theoretical studies. Proteins 56:449-463. [DOI] [PubMed] [Google Scholar]
- 4.Allen, T. M., M. Altfeld, S. C. Geer, E. T. Kalife, C. Moore, M. O'Sullivan, K. I. Desouza, M. E. Feeney, R. L. Eldridge, E. L. Maier, D. E. Kaufmann, M. P. Lahaie, L. Reyor, G. Tanzi, M. N. Johnston, C. Brander, R. Draenert, J. K. Rockstroh, H. Jessen, E. S. Rosenberg, S. A. Mallal, and B. D. Walker. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239-13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen, T. M., M. Altfeld, X. G. Yu, K. M. O'Sullivan, M. Lichterfeld, S. Le Gall, M. John, B. R. Mothe, P. K. Lee, E. T. Kalife, D. E. Cohen, K. A. Freedberg, D. A. Strick, M. N. Johnston, A. Sette, E. S. Rosenberg, S. A. Mallal, P. J. Goulder, C. Brander, and B. D. Walker. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 78:7069-7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386-390. [DOI] [PubMed] [Google Scholar]
- 7.Altfeld, M., M. M. Addo, E. S. Rosenberg, F. M. Hecht, P. K. Lee, M. Vogel, X. G. Yu, R. Draenert, M. N. Johnston, D. Strick, T. M. Allen, M. E. Feeney, J. O. Kahn, R. P. Sekaly, J. A. Levy, J. K. Rockstroh, P. J. Goulder, and B. D. Walker. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17:2581-2591. [DOI] [PubMed] [Google Scholar]
- 8.Altfeld, M., and T. M. Allen. 2006. Hitting HIV where it hurts: an alternative approach to HIV vaccine design. Trends Immunol. 27:504-510. [DOI] [PubMed] [Google Scholar]
- 9.Altfeld, M., T. M. Allen, X. G. Yu, M. N. Johnston, D. Agrawal, B. T. Korber, D. C. Montefiori, D. H. O'Connor, B. T. Davis, P. K. Lee, E. L. Maier, J. Harlow, P. J. Goulder, C. Brander, E. S. Rosenberg, and B. D. Walker. 2002. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature 420:434-439. [DOI] [PubMed] [Google Scholar]
- 10.Altfeld, M., E. T. Kalife, Y. Qi, H. Streeck, M. Lichterfeld, M. N. Johnston, N. Burgett, M. E. Swartz, A. Yang, G. Alter, X. G. Yu, A. Meier, J. K. Rockstroh, T. M. Allen, H. Jessen, E. S. Rosenberg, M. Carrington, and B. D. Walker. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8+ T cell response against HIV-1. PLoS Med. 3:e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An, P., L. H. Wang, H. Hutcheson-Dilks, G. Nelson, S. Donfield, J. J. Goedert, C. R. Rinaldo, S. Buchbinder, G. D. Kirk, J. O'Brien, and C. A. Winkler. 2007. Regulatory polymorphisms in the cyclophilin A gene, PPIA, accelerate progression to AIDS. PLoS Pathogens 3:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ariën, K. K., A. Abraha, M. E. Quinones-Mateu, L. Kestens, G. Vanham, and E. J. Arts. 2005. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J. Virol. 79:8979-8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailey, J. R., H. Zhang, B. W. Wegweiser, H. C. Yang, L. Herrera, A. Ahonkhai, T. M. Williams, R. F. Siliciano, and J. N. Blankson. 2007. Evolution of HIV-1 in an HLA-B*57-positive patient during virologic escape. J. Infect. Dis. 196:50-55. [DOI] [PubMed] [Google Scholar]
- 14.Ball, S. C., A. Abraha, K. R. Collins, A. J. Marozsan, H. Baird, M. E. Quinones-Mateu, A. Penn-Nicholson, M. Murray, N. Richard, M. Lobritz, P. A. Zimmerman, T. Kawamura, A. Blauvelt, and E. J. Arts. 2003. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J. Virol. 77:1021-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber, L. D., L. Percival, K. L. Arnett, J. E. Gumperz, L. Chen, and P. Parham. 1997. Polymorphism in the alpha(1) helix of the HLA-B heavy chain can have an overriding influence on peptide-binding specificity. J. Immunol. 158:1660-1669. [PubMed] [Google Scholar]
- 16.Barouch, D. H., J. Kunstman, J. Glowczwskie, K. J. Kunstman, M. A. Egan, F. W. Peyerl, S. Santra, M. J. Kuroda, J. E. Schmitz, K. Beaudry, G. R. Krivulka, M. A. Lifton, D. A. Gorgone, S. M. Wolinsky, and N. L. Letvin. 2003. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J. Virol. 77:7367-7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barouch, D. H., J. Powers, D. M. Truitt, M. G. Kishko, J. C. Arthur, F. W. Peyerl, M. J. Kuroda, D. A. Gorgone, M. A. Lifton, C. I. Lord, V. M. Hirsch, D. C. Montefiori, A. Carville, K. G. Mansfield, K. J. Kunstman, S. M. Wolinsky, and N. L. Letvin. 2005. Dynamic immune responses maintain cytotoxic T lymphocyte epitope mutations in transmitted simian immunodeficiency virus variants. Nat. Immunol. 6:247-252. [DOI] [PubMed] [Google Scholar]
- 18.Borrow, P., H. Lewicki, X. P. Wei, M. S. Horwitz, N. Peffer, H. Meyers, J. A. Nelson, J. E. Gairin, B. H. Hahn, M. B. A. Oldstone, and G. M. Shaw. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205-211. [DOI] [PubMed] [Google Scholar]
- 19.Braaten, D., E. K. Franke, and J. Luban. 1996. Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J. Virol. 70:4220-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braaten, D., and J. Luban. 2001. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 20:1300-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brockman, M. A., G. O. Tanzi, B. D. Walker, and T. M. Allen. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131:134-142. [DOI] [PubMed] [Google Scholar]
- 22.Chang, L. J., V. Urlacher, T. Isakuma, Y. Cui, and J. Zucali. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 6:715-728. [DOI] [PubMed] [Google Scholar]
- 23.Chatterji, U., M. D. Bobardt, R. Stanfield, R. G. Ptak, L. A. Pallansch, P. A. Ward, M. J. Jones, C. A. Stoddart, P. Scalfaro, J. M. Dumont, K. Besseghir, B. Rosenwirth, and P. A. Gallay. 2005. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in owl monkey cells. J. Biol. Chem. 280:40293-40300. [DOI] [PubMed] [Google Scholar]
- 24.Crawford, H., J. G. Prado, A. Leslie, S. Hue, I. Honeyborne, S. Reddy, M. van der Stok, Z. Mncube, C. Brander, C. Rousseau, J. I. Mullins, R. Kaslow, P. Goepfert, S. Allen, E. Hunter, J. Mulenga, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus infection. J. Virol. 81:8346-8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derdeyn, C. A., J. M. Decker, F. Bibollet-Ruche, J. L. Mokili, M. Muldoon, S. A. Denham, M. L. Heil, F. Kasolo, R. Musonda, B. H. Hahn, G. M. Shaw, B. T. Korber, S. Allen, and E. Hunter. 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303:2019-2022. [DOI] [PubMed] [Google Scholar]
- 26.Draenert, R., S. Le Gall, K. J. Pfafferott, A. J. Leslie, P. Chetty, C. Brander, E. C. Holmes, S. C. Chang, M. E. Feeney, M. M. Addo, L. Ruiz, D. Ramduth, P. Jeena, M. Altfeld, S. Thomas, Y. Tang, C. L. Verrill, C. Dixon, J. G. Prado, P. Kiepiela, J. Martinez-Picado, B. D. Walker, and P. J. Goulder. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199:905-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feeney, M. E., Y. Tang, K. A. Roosevelt, A. J. Leslie, K. McIntosh, N. Karthas, B. D. Walker, and P. J. Goulder. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927-8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franke, E. K., and J. Luban. 1996. Inhibition of HIV-1 replication by cyclosporine A or related compounds correlates with the ability to disrupt the Gag-cyclophilin A interaction. Virology 222:279-282. [DOI] [PubMed] [Google Scholar]
- 29.Friedrich, T. C., E. J. Dodds, L. J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D. T. Evans, R. C. Desrosiers, B. R. Mothe, J. Sidney, A. Sette, K. Kunstman, S. Wolinsky, M. Piatak, J. Lifson, A. L. Hughes, N. Wilson, D. H. O'Connor, and D. I. Watkins. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10:275-281. [DOI] [PubMed] [Google Scholar]
- 30.Friedrich, T. C., C. A. Frye, L. J. Yant, D. H. O'Connor, N. A. Kriewaldt, M. Benson, L. Vojnov, E. J. Dodds, C. Cullen, R. Rudersdorf, A. L. Hughes, N. Wilson, and D. I. Watkins. 2004. Extraepitopic compensatory substitutions partially restore fitness to simian immunodeficiency virus variants that escape from an immunodominant cytotoxic-T-lymphocyte response. J. Virol. 78:2581-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gamble, T. R., F. F. Vajdos, S. Yoo, D. K. Worthylake, M. Houseweart, W. I. Sundquist, and C. P. Hill. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285-1294. [DOI] [PubMed] [Google Scholar]
- 32.Gao, X., A. Bashirova, A. K. Iversen, J. Phair, J. J. Goedert, S. Buchbinder, K. Hoots, D. Vlahov, M. Altfeld, S. J. O'Brien, and M. Carrington. 2005. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat. Med. 11:1290-1292. [DOI] [PubMed] [Google Scholar]
- 33.Gatanaga, H., D. Das, Y. Suzuki, D. D. Yeh, K. A. Hussain, A. K. Ghosh, and H. Mitsuya. 2006. Altered HIV-1 Gag protein interactions with cyclophilin A (CypA) on the acquisition of H219Q and H219P substitutions in the CypA binding loop. J. Biol. Chem. 281:1241-1250. [DOI] [PubMed] [Google Scholar]
- 34.Geldmacher, C., J. R. Currier, E. Herrmann, A. Haule, E. Kuta, F. McCutchan, L. Njovu, S. Geis, O. Hoffmann, L. Maboko, C. Williamson, D. Birx, A. Meyerhans, J. Cox, and M. Hoelscher. 2007. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 81:2440-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gervaix, A., D. West, L. M. Leoni, D. D. Richman, F. Wong-Staal, and J. Corbeil. 1997. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. USA 94:4653-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gitti, R. K., B. M. Lee, J. Walker, M. F. Summers, S. Yoo, and W. I. Sundquist. 1996. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273:231-235. [DOI] [PubMed] [Google Scholar]
- 37.Goulder, P., D. Price, M. Nowak, S. Rowland-Jones, R. Phillips, and A. McMichael. 1997. Co-evolution of human immunodeficiency virus and cytotoxic T-lymphocyte responses. Immunol. Rev. 159:17-29. [DOI] [PubMed] [Google Scholar]
- 38.Goulder, P. J., C. Brander, Y. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. Korber, and B. D. Walker. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412:334-338. [DOI] [PubMed] [Google Scholar]
- 39.Hatziioannou, T., D. Perez-Caballero, S. Cowan, and P. D. Bieniasz. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 79:176-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaslow, R. A., M. Carrington, R. Apple, L. Park, A. Munoz, A. J. Saah, J. J. Goedert, C. Winkler, S. J. O'Brien, C. Rinaldo, R. Detels, W. Blattner, J. Phair, H. Erlich, and D. L. Mann. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405-411. [DOI] [PubMed] [Google Scholar]
- 41.Kelleher, A. D., C. Long, E. C. Holmes, R. L. Allen, J. Wilson, C. Conlon, C. Workman, S. Shaunak, K. Olson, P. Goulder, C. Brander, G. Ogg, J. S. Sullivan, W. Dyer, I. Jones, A. J. McMichael, S. Rowland-Jones, and R. E. Phillips. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiepiela, P., K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne, E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, K. Bishop, M. van der Stok, K. Nair, N. Khan, H. Crawford, R. Payne, A. Leslie, J. Prado, A. Prendergast, J. Frater, N. McCarthy, C. Brander, G. H. Learn, D. Nickle, C. Rousseau, H. Coovadia, J. I. Mullins, D. Heckerman, B. D. Walker, and P. Goulder. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46-53. [DOI] [PubMed] [Google Scholar]
- 43.Leslie, A., D. Kavanagh, I. Honeyborne, K. Pfafferott, C. Edwards, T. Pillay, L. Hilton, C. Thobakgale, D. Ramduth, R. Draenert, S. Le Gall, G. Luzzi, A. Edwards, C. Brander, A. K. Sewell, S. Moore, J. Mullins, C. Moore, S. Mallal, N. Bhardwaj, K. Yusim, R. Phillips, P. Klenerman, B. Korber, P. Kiepiela, B. Walker, and P. Goulder. 2005. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J. Exp. Med. 201:891-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. St. John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282-289. [DOI] [PubMed] [Google Scholar]
- 45.Li, B., A. D. Gladden, M. Altfeld, J. M. Kaldor, D. A. Cooper, A. D. Kelleher, and T. M. Allen. 2007. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J. Virol. 81:193-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Picado, J., J. G. Prado, E. E. Fry, K. Pfafferott, A. Leslie, S. Chetty, C. Thobakgale, I. Honeyborne, H. Crawford, P. Matthews, T. Pillay, C. Rousseau, J. I. Mullins, C. Brander, B. D. Walker, D. I. Stuart, P. Kiepiela, and P. Goulder. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masemola, A., T. Mashishi, G. Khoury, P. Mohube, P. Mokgotho, E. Vardas, M. Colvin, L. Zijenah, D. Katzenstein, R. Musonda, S. Allen, N. Kumwenda, T. Taha, G. Gray, J. McIntyre, S. A. Karim, H. W. Sheppard, and C. M. Gray. 2004. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J. Virol. 78:3233-3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matano, T., M. Kobayashi, H. Igarashi, A. Takeda, H. Nakamura, M. Kano, C. Sugimoto, K. Mori, A. Iida, T. Hirata, M. Hasegawa, T. Yuasa, M. Miyazawa, Y. Takahashi, M. Yasunami, A. Kimura, D. H. O'Connor, D. I. Watkins, and Y. Nagai. 2004. Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J. Exp. Med. 199:1709-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Migueles, S. A., A. C. Laborico, H. Imamichi, W. L. Shupert, C. Royce, M. McLaughlin, L. Ehler, J. Metcalf, S. Liu, C. W. Hallahan, and M. Connors. 2003. The differential ability of HLA B*5701+ long-term nonprogressors and progressors to restrict human immunodeficiency virus replication is not caused by loss of recognition of autologous viral gag sequences. J. Virol. 77:6889-6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Migueles, S. A., M. S. Sabbaghian, W. L. Shupert, M. P. Bettinotti, F. M. Marincola, L. Martino, C. W. Hallahan, S. M. Selig, D. Schwartz, J. Sullivan, and M. Connors. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. USA 97:2709-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439-1443. [DOI] [PubMed] [Google Scholar]
- 52.O'Brien, S. J., X. Gao, and M. Carrington. 2001. HLA and AIDS: a cautionary tale. Trends Mol. Med. 7:379-381. [DOI] [PubMed] [Google Scholar]
- 53.O'Connor, D. H., T. M. Allen, T. U. Vogel, P. Jing, I. P. DeSouza, E. Doods, E. Dunphy, C. Melsaether, B. Mothe, H. Horton, A. L. Hughes, and D. I. Watkins. 2002. Acute phase CTL escape is a hallmark of SIV infection. Nat. Med. 8:493-499. [DOI] [PubMed] [Google Scholar]
- 54.O'Connor, D. H., B. R. Mothe, J. T. Weinfurter, S. Fuenger, W. M. Rehrauer, P. Jing, R. R. Rudersdorf, M. E. Liebl, K. Krebs, J. Vasquez, E. Dodds, J. Loffredo, S. Martin, A. B. McDermott, T. M. Allen, C. Wang, G. G. Doxiadis, D. C. Montefiori, A. Hughes, D. R. Burton, D. B. Allison, S. M. Wolinsky, R. Bontrop, L. J. Picker, and D. I. Watkins. 2003. Major histocompatibility complex class I alleles associated with slow simian immunodeficiency virus disease progression bind epitopes recognized by dominant acute-phase cytotoxic-T-lymphocyte responses. J. Virol. 77:9029-9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peyerl, F. W., D. H. Barouch, W. W. Yeh, H. S. Bazick, J. Kunstman, K. J. Kunstman, S. M. Wolinsky, and N. L. Letvin. 2003. Simian-human immunodeficiency virus escape from cytotoxic T-lymphocyte recognition at a structurally constrained epitope. J. Virol. 77:12572-12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peyerl, F. W., H. S. Bazick, M. H. Newberg, D. H. Barouch, J. Sodroski, and N. L. Letvin. 2004. Fitness costs limit viral escape from cytotoxic T lymphocytes at a structurally constrained epitope. J. Virol. 78:13901-13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phillips, R. E., S. Rowland-Jones, D. F. Nixon, F. M. Gotch, J. P. Edwards, A. O. Ogunlesi, J. G. Elvin, J. A. Rothbard, C. R. Bangham, C. R. Rizza, and A. J. McMichael. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453-459. [DOI] [PubMed] [Google Scholar]
- 58.Reicin, A. S., A. Ohagen, L. Yin, S. Hoglund, and S. P. Goff. 1996. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J. Virol. 70:8645-8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sipsas, N. V., S. A. Kalams, A. Trocha, S. He, W. A. Blattner, B. D. Walker, and R. P. Johnson. 1997. Identification of type-specific cytotoxic T lymphocyte responses to homologous viral proteins in laboratory workers accidentally infected with HIV-1. J. Clin. Investig. 99:752-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sokolskaja, E., D. M. Sayah, and J. Luban. 2004. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 78:12800-12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Timm, J., G. M. Lauer, D. G. Kavanagh, I. Sheridan, A. Y. Kim, M. Lucas, T. Pillay, K. Ouchi, L. L. Reyor, J. Schulze zur Wiesch, R. T. Gandhi, R. T. Chung, N. Bhardwaj, P. Klenerman, B. D. Walker, and T. M. Allen. 2004. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 200:1593-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Towers, G. J., T. Hatziioannou, S. Cowan, S. P. Goff, J. Luban, and P. D. Bieniasz. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138-1143. [DOI] [PubMed] [Google Scholar]
- 63.Trask, S. A., C. A. Derdeyn, U. Fideli, Y. Chen, S. Meleth, F. Kasolo, R. Musonda, E. Hunter, F. Gao, S. Allen, and B. H. Hahn. 2002. Molecular epidemiology of human immunodeficiency virus type 1 transmission in a heterosexual cohort of discordant couples in Zambia. J. Virol. 76:397-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trono, D., M. B. Feinberg, and D. Baltimore. 1989. HIV-1 Gag mutants can dominantly interfere with the replication of the wild-type virus. Cell 59:113-120. [DOI] [PubMed] [Google Scholar]
- 65.Wang, C.-T., and E. Barklis. 1993. Assembly, processing, and infectivity of human immunodeficiency virus type 1 Gag mutants. J. Virol. 67:4264-4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yant, L. J., T. C. Friedrich, R. C. Johnson, G. E. May, N. J. Maness, A. M. Enz, J. D. Lifson, H. O'Connor, D. M. Carrington, and D. I. Watkins. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80:5074-5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yeh, W. W., E. M. Cale, P. Jaru-Ampornpan, C. I. Lord, F. W. Peyerl, and N. L. Letvin. 2006. Compensatory substitutions restore normal core assembly in simian immunodeficiency virus isolates with Gag epitope cytotoxic T-lymphocyte escape mutations. J. Virol. 80:8168-8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin, L., D. Braaten, and J. Luban. 1998. Human immunodeficiency virus type 1 replication is modulated by host cyclophilin A expression levels. J. Virol. 72:6430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]