

Abstract
Herpes simplex virus (HSV)-specific T cells are essential for viral clearance. However, T cells do not prevent HSV latent infection or reactivation, suggesting that HSV has the potential to modulate T-cell function. T-cell receptor (TCR) stimulation is a potent and specific means of activating T cells. To investigate how HSV affects T-cell function, we have analyzed how HSV affects TCR-stimulated intracellular signaling and cytokine synthesis in mock-infected and HSV-infected T cells. Mock-infected T cells stimulated through the TCR synthesized a broad range of cytokines that included the proinflammatory cytokines tumor necrosis factor alpha, gamma interferon, and interleukin-2. In contrast, HSV-infected T cells stimulated through the TCR selectively synthesized interleukin-10, a cytokine that suppresses cellular immunity and favors viral replication. To achieve selective interleukin-10 synthesis, HSV differentially affected TCR signaling pathways. HSV inhibited TCR-stimulated formation of the linker for activation of the T-cell signaling complex, and HSV inhibited TCR-stimulated NF-κB activation. At the same time, HSV activated the p38 and JNK mitogen-activated protein kinases as well as the downstream transcription factors ATF-2 and c-Jun. HSV did not inhibit TCR-stimulated activation of STAT3, a transcription factor involved in interleukin-10 synthesis. The activation of p38 was required for interleukin-10 synthesis in HSV-infected T cells. The ability of HSV to differentially target intracellular signaling pathways and transform an activating stimulus into an immunosuppressive response represents a novel strategy for pathogen-mediated immune modulation. Selective, TCR-stimulated interleukin-10 synthesis may play an important role in HSV pathogenesis.
The herpes simplex viruses (HSV-1 and HSV-2) are prevalent human pathogens that cause substantial global morbidity (28). Primary HSV infection progresses to persistent latency in the sensory neural ganglia, and viral reactivation produces painful, recurrent oral and genital mucocutaneous lesions in one-third of those infected (55). Serious complications of HSV infection include stromal keratitis, a leading cause of corneal blindness (64), neonatal encephalitis (12), and an increased risk for human immunodeficiency virus transmission (7).
HSV-specific T cells are a critical component of the adaptive immune response generated by HSV infection. Adoptive transfer experiments have demonstrated that HSV-specific cytotoxic T cells are required to resolve the epithelial manifestations of HSV infection (3, 59). However, T cells do not prevent viral latency, reactivation, or transmission (54). In part, this may be due to HSV-encoded mechanisms that modulate the immune response. For example, HSV has been reported to inhibit the type I interferon response, impair complement, and interfere with neutralizing antibody (14, 26, 41, 47). HSV has also been reported to modulate T-cell function. HSV-infected cells can evade T-cell recognition by interfering with antigen presentation (15), and HSV-infected cells can resist T-cell-induced apoptosis (25). We have previously shown that infection of T cells with HSV inhibits T-cell receptor (TCR)-stimulated effector functions (60, 61).
TCR stimulation triggers a sequential cascade of protein phosphorylations and translocations that propagate the TCR signal from the plasma membrane to the T-cell nucleus. Formation of the membrane-anchored linker for activation of T cells (LAT) signaling complex, a proximal event in TCR signal propagation, is an essential link between the membrane and cytoplasmic TCR signaling machinery (39). Following TCR stimulation, LAT is phosphorylated at four critical C-terminal tyrosine residues by ZAP-70. Binding of phosphorylated LAT to phospholipase C γ1 (PLCγ1) activates inositol trisphosphate and diacylglycerol, resulting in calcium mobilization (13). Binding of phosphorylated LAT to growth factor receptor-binding protein 2 (Grb2) and Grb2-related adaptor downstream of Shc (GADS) activates the three major mitogen-activated protein kinase (MAPK) pathways: extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 kinase (p38) (38, 74). When completely activated, the MAPKs activate a network of transcription factors that can up-regulate both proinflammatory and immunosuppressive cytokines.
Cytokines can be divided into two groups, T helper 1 (Th1) and Th2. Th1 and Th2 cytokines have opposing functions (46). Th1 cytokines (e.g., gamma interferon [IFN-γ], tumor necrosis factor alpha [TNF-α], and interleukin-2 [IL-2]) induce major histocompatibility complex molecules and activate T cells. Alternatively, Th2 cytokines (e.g., IL-4, IL-5, IL-6, and IL-10) activate B cells and stimulate antibody development. The Th1 cytokine IL-2 has been shown to suppress Th2 cytokine development, whereas IL-10, a Th2 cytokine, has been shown to suppress Th1 development. We have previously reported that HSV inhibits TCR-stimulated synthesis of Th1 cytokines (61). A Th1 environment favors recognition and removal of virus-infected cells, and HSV may gain a growth advantage by inhibiting TCR-stimulated Th1 cytokines. As a possible mechanism to explain inhibition of Th1 cytokine synthesis, we have reported that HSV inhibits TCR-stimulated phosphorylation of LAT at C-terminal tyrosine residues (60). Interestingly, mice homozygous for mutations in the equivalent C-terminal tyrosine residues of LAT have altered T-cell development characterized by reduced levels of Th1 cytokines and increased levels of Th2 cytokines, such as IL-10 (1, 51). IL-10 is a particularly attractive candidate for pathogens to affect, as it can dramatically inhibit the proinflammatory environment. For example, IL-10-deficient mice develop chronic enterocolitis as a result of excessive immune responses to intestinal bacterial antigens (35).
In this report, we demonstrate that HSV inhibits formation of the LAT signaling complex by inhibiting TCR-stimulated binding of LAT to Grb2, GADS, and PLCγ1. In addition, we demonstrate that HSV selectively activates the p38 and JNK pathways. The net result of LAT inhibition and p38 activation is isolated IL-10 synthesis in HSV-infected T cells stimulated through the TCR. As IL-10 has been shown to suppress the T-cell response and favor viral replication (45), elucidation of HSV-induced TCR signal remodeling may improve management of HSV pathogenesis. In a broader sense, we believe that this process represents a previously unreported form of pathogen-mediated immune modulation.
MATERIALS AND METHODS
Cells, virus, and infections.
Peripheral blood mononuclear cells (PBMC) were isolated from the whole blood of healthy adult volunteers or from Pall leukocyte filters obtained from the Puget Sound Blood Center using Ficoll-Hypaque (Amersham Pharmacia) density centrifugation. PBMC were incubated for 2 h at 37°C, and nonadherent cells were used for subsequent steps. Naive CD4+ T cells were isolated from nonadherent PBMC using a magnetic-activated cell sorter (MACS) CD4+ T-cell isolation kit following the manufacturer's instructions (Miltenyi Biotec). The Jurkat immortalized T-cell clone E6-1, purchased from the American Type Culture Collection, was maintained between 2 × 105 and 1 × 106 cells/ml in RPMI 1640 supplemented with 4 mM HEPES, 3 mM l-glutamine, 10% fetal calf serum. Human fibroblasts, obtained from human foreskin samples, were used from passage 5 to 12 and maintained in Dulbecco's modified Eagle's medium, 10% fetal calf serum, 50 U/ml penicillin, and 50 mg/ml streptomycin. Vero cells were obtained from ATCC and grown in the same medium as fibroblasts. Cells were screened regularly for mycoplasma by the Biologics Production Facility at the Fred Hutchinson Cancer Research Center.
The wild-type virus HSV-1 (strain F; obtained from J. A. Blaho) was propagated and titers were determined on Vero cells as previously described (60). Fibroblasts were split 24 to 48 h prior to HSV infection. At the time of infection, fibroblasts were 100% confluent in individual six-well plates. All infections and subsequent steps were done in the presence of 25 μM acyclovir. These conditions prevent viral replication but permit transfer of HSV proteins between the HSV-infected monolayers and the T cells (60).
In all of the experiments described in this paper, PBMC or T cells were infected with HSV by coincubating them with mock-infected or HSV-infected fibroblasts to permit efficient cell-to-cell transmission of virus. T cells were not directly infected with HSV. Fibroblasts were first infected with HSV at a multiplicity of infection of 10 infectious viral particles per cell for 6 h. Fibroblasts were then washed twice, and 1 to 2 million PBMC or T cells were added to each well for 16 h. PBMC or T cells were then removed from the monolayers by gentle aspiration and counted before continuing with TCR stimulation and subsequent analysis for cytokine production or intracellular signaling events. In experiments that investigated the effects of p38 inhibition on TCR-stimulated production of IL-10, PBMC were preincubated with 10 μM SB203580 (Sigma) or dimethyl sulfoxide (0.1%) as carrier control, and SB203580 was included throughout the coincubation with HSV-infected fibroblasts and subsequent TCR stimulation.
Cytokine detection.
PBMC or T cells were transferred from mock-infected or HSV-infected fibroblast monolayers to control wells or wells coated with 10 μg/ml immobilized anti-CD3 antibody (clone OKT3; University of Washington Pharmacy). CD3 is a TCR-associated molecule required for TCR signal transduction. We have determined that HSV affects TCR signaling whether we stimulate with immobilized anti-CD3, cross-linked anti-CD3, or cross-linked anti-CD3 plus costimulation with anti-CD28 (data not shown). PBMC or T cells were incubated for 48 h, and the medium was removed. Cell-free supernatants were submitted to the Cytokine Analysis Shared Resources Laboratory at the Fred Hutchinson Cancer Research Center, and an enzyme-linked immunosorbent assay (ELISA) was used to determine cytokine concentrations in triplicate wells.
LAT immunoprecipitation and Western blotting.
Jurkat T cells were removed from mock-infected or HSV-infected fibroblasts, washed in serum-free medium, and rested at 37°C for 1 h. Cells were stimulated through the TCR with anti-CD3 antibody at 10 μg/ml for 2 min at 37°C. Cells were then washed in ice-cold buffer containing protease and phosphatase inhibitors (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonide, 1 μg/ml N-tosyl-l-phenylalanine chloromethyl ketone, and 1 μg/ml N-tosyl-lysine chloromethyl ketone), pelleted, and resuspended in lysis buffer (same buffer plus 1% Brij-97 detergent σ. Lysates were incubated for 1 h with rotation at 4°C and pelleted at 10,000 × g for 5 min at 4°C. Lysate supernatants (250 μg each) were immunoprecipitated with the following antibodies: 5 μg anti-GADS (Upstate), 10 μg anti-Grb2 (Santa Cruz), and 2 μg anti-PLCγ1 (Cell Signaling). Lysates were incubated for 1 h on ice, and 50 μl of protein G-coupled magnetic MicroBeads (Miltenyi) was added, following the manufacturer's protocol. Lysates were run through μMACS columns, washed with lysis buffer, and eluted with 1.3× sodium dodecyl sulfate loading buffer (Invitrogen) preheated to 95°C. Samples were boiled for 5 min, loaded on 4 to 12% bis-Tris-sodium dodecyl sulfate-polyacrylamide gels (Invitrogen), transferred to nitrocellulose membranes, and blocked with phosphate-buffered saline (PBS)-5% milk. Membranes were probed with anti-phosphotyrosine (clone 4G10; Upstate Biotechnology), anti-phospho-LAT (no. 3581; Cell Signaling Technology), anti-LAT (clone 2E9; Upstate), or with the immunoprecipitation antibodies described above for 16 h at 4°C in PBS-5% milk. Horseradish peroxidase-conjugated secondary antibody (Cell Signaling) was added for 1 h at 25°C. Signals were developed with SuperSignal chemiluminescent substrate (Pierce) and detected using autoradiographic film.
MAPK and transcription factor phosphorylation analysis by flow cytometry.
Jurkat T cells were removed from mock-infected or HSV-infected fibroblasts and incubated on ice for 10 min. Cells were incubated with anti-CD3 antibody (clone OKT3) and anti-CD28 antibody (clone L293; BD Biosciences) at 10 μg/ml and cross-linked with goat anti-mouse antibody (Jackson Immunoresearch) at 17 μg/ml for 15 min on ice. Cells were TCR stimulated by incubating at 37°C for 10 min to detect phosphorylation of p38, JNK, ERK, ATF-2, and NF-κB or for 20 min to detect phosphorylation of c-Jun, STAT1, and STAT3. Cells were fixed by adding fresh 2% methanol-free formaldehyde (Polysciences, Inc.) and incubation for 10 min at 37°C. Cells were permeabilized with drop-wise addition of ice-cold methanol while vortexing and incubated for 20 min on ice. Cells were washed twice in 0.8% bovine serum albumin in PBS by centrifugation at 400 × g for 4 min. Rabbit antibodies against the phospho-specific forms of p38 (catalog no. 9215; 1:25 dilution), JNK (no. 9251; 1:25), ERK (no. 9101; 1:50), ATF-2 (no. 5112; 1:50), c-Jun (no. 9261; 1:100), NF-κB (no. 3033; 1:50), STAT1-Tyr (no. 9167; 1:100), STAT1-Ser (no. 9177; 1:50), and STAT3 (no. 9145; 1:25) (all from Cell Signaling Technology) were added for 30 min at 25°C. Cells were washed, and donkey anti-rabbit, allophycocyanin-conjugated secondary antibody (no. 711-136-152; Jackson Immunoresearch) was added at a dilution of 1:50 for 30 min at 25°C. Cells were washed, resuspended in wash buffer, and analyzed on a FACSCalibur flow cytometer (BD Biosciences).
RESULTS
HSV-infected T cells stimulated through the TCR selectively synthesize IL-10.
HSV has been shown to infect primary T cells and immortalized Jurkat cells, a human T-cell line that is frequently used to study TCR signal transduction (4, 21). We have reported that entry of HSV into T cells, either by direct infection or by contact with HSV-infected cell monolayers, reduced LAT phosphorylation and inhibited Th1 cytokine synthesis (60, 61). To examine how HSV affected T-cell-derived Th2 cytokine production, human PBMC or CD4-enriched T cells from PBMC were incubated with mock-infected or HSV-1-infected human fibroblasts to allow sufficient cell-to-cell viral transmission. As described previously, we confirmed that T cells were infected with HSV after coincubating them with HSV-infected fibroblasts by using a viral construct that expresses green fluorescent protein (60). Mock-infected T cells and HSV-infected T cells were removed and either left untreated or specifically stimulated through the TCR for an additional 48 h, the time of maximal cytokine secretion. Cell supernatants were then analyzed for the following secreted cytokines: TNF-α and IL-2 (Th1), granulocyte-macrophage colony-stimulating factor (GM-CSF; Th1/Th2), and the Th2 cytokines IL-4, IL-5, IL-6, and IL-10. We have previously reported that HSV inhibits TCR-stimulated synthesis of the Th1 cytokine IFN-γ (61). As a control to confirm that the cytokines detected were not derived from the HSV-infected fibroblasts, we detected background levels of each cytokine when PBMC or T cells were not added (data not shown).
Prior to TCR stimulation, the coincubation of PBMC with HSV-infected fibroblasts did not significantly affect synthesis of any of the cytokines tested. In mock-infected PBMC stimulated through the TCR, there was a statistically significant increase in TNF-α, GM-CSF, IL-4, IL-5, and IL-10 (Fig. 1), first versus third bars) (P < 0.05, Student's t test). Compared to mock-infected PBMC stimulated through the TCR, HSV-infected PBMC stimulated through the TCR had a significant reduction in synthesis of TNF-α, GM-CSF, IL-4, IL-5, and IL-6 (Fig. 1, third versus fourth bars) (P < 0.05). In contrast, HSV did not inhibit TCR-stimulated synthesis of IL-10. As with the other cytokines tested, HSV infection alone did not stimulate IL-10 synthesis. To further assess the effects of HSV on TCR-stimulated cytokine synthesis and confirm the results seen with IL-10, we isolated T cells from PBMC using CD4-coupled magnetic beads. Infection of CD4+ T cells with HSV significantly inhibited TCR-stimulated synthesis of IL-2, a Th1 cytokine. However, infection of CD4+ T cells with HSV did not inhibit TCR-stimulated synthesis of IL-10 (Fig. 2). These findings suggest that HSV infection of resting human primary T cells results in the selective TCR-stimulated synthesis of IL-10.
FIG. 1.

HSV-infected human PBMC selectively synthesize IL-10 in response to TCR stimulation. Human PBMC were incubated with mock-infected or HSV-infected human fibroblasts, as described in Materials and Methods. PBMC were removed by aspiration and transferred to plates that were untreated or coated with immobilized anti-CD3 antibody to specifically stimulate T cells through the TCR. Forty-eight hours later, supernatant was analyzed for cytokine production in triplicate using an ELISA format. The results from five separate experiments for TNF-α, GM-CSF, IL-4, IL-5, and IL-6 and from eight separate experiments for IL-10 are depicted as mean cytokine levels (left column) and median cytokine levels (right column). *, P < 0.05, Student's t test.
FIG. 2.

HSV-infected human T cells selectively synthesize IL-10 in response to TCR stimulation. CD4+ T cells were isolated from human PBMC using CD4-coupled magnetic beads. T cells were incubated with mock-infected or HSV-infected fibroblasts as described in Materials and Methods, removed, and stimulated through the TCR. Secreted IL-2 and IL-10 were measured using an ELISA. The results from four separate experiments for IL-2 and IL-10 are depicted as mean cytokine levels (left column) and median cytokine levels (right column). *, P < 0.05, Student's t test.
HSV inhibits LAT-mediated TCR signal transduction in T cells.
Our previous findings demonstrated that HSV infection of T cells reduced phosphorylation of LAT at tyrosine residues involved in TCR signal transduction without affecting the total levels of LAT (60). However, the reduced phosphorylation pattern of LAT was incomplete, and it was not known how downstream binding events were affected. In an effort to understand how HSV facilitates selective synthesis of IL-10, we examined how HSV affected binding of LAT to Grb2, GADS, and PLCγ1 2 min after TCR stimulation, as maximal binding of LAT to Grb2, GADS, and PLCγ1 in mock-infected T cells was observed 2 min after TCR stimulation. In lysates from mock-infected T cells stimulated through the TCR and immunoprecipitated with LAT, we were able to detect PLCγ1 but not Grb2 or GADS (data not shown). Therefore, we immunoprecipitated lysates with Grb2, GADS, and PLCγ1. Using this approach, we detected equivalent amounts of Grb2, GADS, and PLCγ1 in lysates from mock-infected and HSV-infected Jurkat T cells (Fig. 3A to C, bottom panels, respectively). Immunoprecipitated lysates were probed for LAT using an anti-phospho-tyrosine antibody (P-Tyr) or with an antibody that recognizes LAT phosphorylated at tyrosine residue 191 (P-LAT). While P-LAT was used to confirm the correct size of the doublet recognized by P-Tyr, P-Tyr is a superior detection reagent, as it can recognize LAT phosphorylated at all four C-terminal tyrosine residues. The characteristic 36- and 38-kDa LAT doublet present in mock-infected cells stimulated through the TCR (column 3) was absent in immunoprecipitated lysates from HSV-infected T cells (Fig. 3A to C, column 5). A higher-molecular-weight band was observed in HSV lysates immunoprecipitated with Grb2 and PLCγ1. For this reason, we performed a further probe with total LAT antibody, which confirmed that this band did not represent a hyperphosphorylated form of LAT (Fig. 3A and C). These results suggest that an HSV-induced reduction in LAT phosphorylation is sufficient to disrupt LAT-dependent TCR signal transduction by inhibiting TCR-stimulated binding of LAT to Grb2, GADS, and PLCγ1.
FIG. 3.
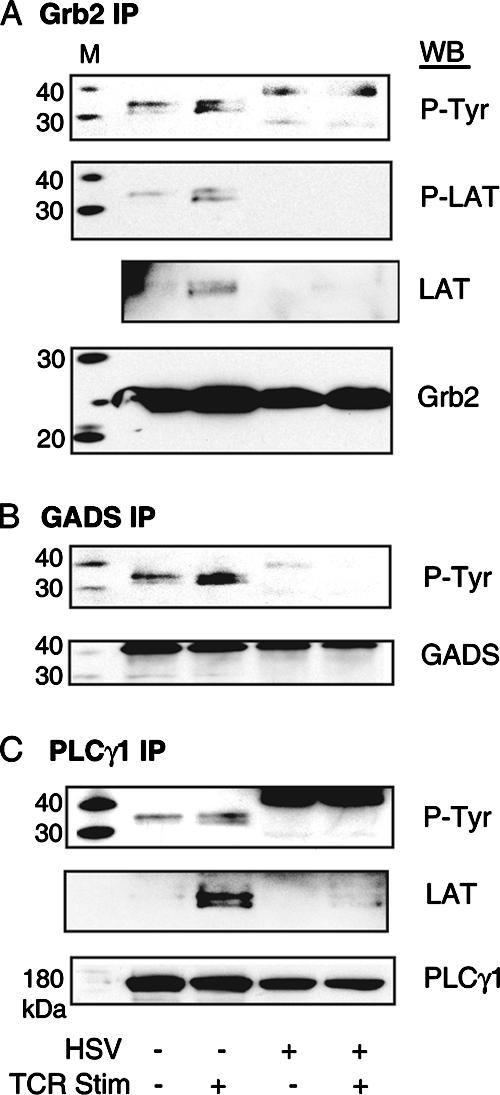
HSV inhibits binding of LAT to Grb2, GADS, and PLCγ1 in TCR-stimulated T cells. Jurkat T cells were incubated with mock-infected fibroblasts or HSV-infected fibroblasts. T cells were removed and either unstimulated or TCR stimulated. T-cell lysates were immunoprecipitated (IP) with antibodies against Grb2 (A), GADS (B), and PLCγ1 (C), run on 4 to 12% acrylamide denaturing gels with molecular mass standards (M), transferred to nitrocellulose, and blotted (WB) with antibodies against phospho-tyrosine (P-Tyr), phospho-LAT (P-LAT), or total LAT (LAT). Equivalent levels of protein from each immunoprecipitation were confirmed by blotting with Grb2, GADS, and PLCγ1 antibodies. The results are representative of three experiments each.
HSV induces phosphorylation of p38 and JNK in T cells.
Maximal activation of p38, JNK, and ERK by TCR stimulation involves phosphorylated LAT binding to Grb2 and GADS. An alternative pathway exists whereby TCR-activated ZAP-70 can directly activate the p38 pathway in a LAT-independent manner (57). We have reported that TCR-stimulated phosphorylation of ZAP-70 is not inhibited by HSV (60). We were therefore interested in investigating how HSV affects the p38 pathway in unstimulated and TCR-stimulated T cells. For these studies, we used antibodies against the phospho-specific forms of each MAPK to determine how HSV infection of Jurkat T cells affected MAPK activation at 10 min after TCR stimulation, the time of maximal phosphorylation in mock-infected T cells. As an initial control, we determined that compared to mock infection, HSV infection did not alter the total protein levels of each MAPK (data not shown). Compared to mock-infected T cells, HSV-infected T cells had increased phosphorylation levels of p38 and JNK but not ERK prior to TCR stimulation (Fig. 4). HSV inhibited TCR-stimulated phosphorylation of p38, JNK, and ERK, but only inhibition of ERK phosphorylation reached statistical significance (P < 0.05) (Fig. 4). To examine whether the phosphorylation of p38 and JNK that occurred in HSV-infected T cells was sufficient to activate these pathways, we subsequently analyzed how HSV affected the phosphorylation of downstream transcription factors.
FIG. 4.

HSV differentially affects MAPK activation in T cells. Jurkat T cells were incubated with mock- or HSV-infected fibroblasts and removed. T cells were TCR stimulated, fixed, and permeabilized. T cells were stained with phospho-specific antibodies against the MAPKs p38, JNK, and ERK. The histogram plots depicted are representative of three independent flow cytometry experiments each. Plots of the mean fluorescence intensities from these experiments are depicted in the bar graphs on the right. *, P < 0.05, Student's t test.
HSV induces phosphorylation of ATF-2 and c-Jun in T cells.
Activation of the JNK pathway has been shown to be primarily responsible for activation of the transcription factor c-Jun (9). Although activation of p38 and JNK can both activate ATF-2, p38 activation is primarily responsible for ATF-2 activation in T cells (19, 73). Prior to TCR stimulation, there was increased phosphorylation of ATF-2 and c-Jun in HSV-infected T cells relative to mock-infected T cells (Fig. 5). Following TCR stimulation for 10 min (ATF-2) or 20 min (c-Jun), the phosphorylation levels of ATF-2 and c-Jun were roughly equivalent in HSV-infected and mock-infected T cells. Unlike ATF-2 and c-Jun activation, activation of the transcription factor NF-κB is not dependent on activation of p38 or JNK. TCR-stimulated NF-κB activation involves IKK-induced degradation of the inhibitory protein IκB, a complex process that includes LAT-dependent activation of protein kinase C and calmodulin-dependent kinase (8, 23). In contrast to ATF-2 and c-Jun, HSV had no effect on phosphorylation of NF-κB in unstimulated T cells. Furthermore, HSV significantly inhibited TCR-stimulated phosphorylation of NF-κB (Fig. 5) (P < 0.05), most likely due to inhibition of LAT-mediated signal transduction. Taken together, these findings suggest that the phosphorylation of p38 and JNK by HSV is sufficient to activate ATF-2 and c-Jun, transcription factors that have both been implicated in IL-10 synthesis in T cells (37, 69).
FIG. 5.

HSV induces ATF-2 and c-Jun activation in T cells. Jurkat T cells were incubated with mock- or HSV-infected fibroblasts and removed. T cells were TCR stimulated, fixed, and permeabilized. T cells were stained with phospho-specific antibodies against the transcription factors ATF-2, c-Jun, and NF-κB. The histogram plots depicted are representative of three independent flow cytometry experiments each. Plots of the mean fluorescence intensities from these experiments are depicted in the bar graphs on the right. *, P < 0.05, Student's t test.
TCR stimulation induces phosphorylation of STAT3 in HSV-infected T cells.
Although HSV activated ATF-2 and c-Jun in T cells, selective IL-10 synthesis required TCR stimulation. TCR stimulation in HSV-infected T cells did not substantially enhance ATF-2 or c-Jun phosphorylation, suggesting that activation of additional transcription factors might be involved. Signal transducer and activator of transcription 3 (STAT3) is activated by TCR stimulation (16), and activated STAT3 has been implicated in IL-10 synthesis in T cells (30). We were therefore interested in determining how HSV affected STAT3 phosphorylation. STAT3 and STAT1 are both activated by IL-10 (70), and activated STAT1 has been shown to repress IL-10 expression (67). Furthermore, p38 activation has been reported to increase STAT1 serine phosphorylation (33). For these reasons, we also examined STAT1 phosphorylation.
While HSV infection of T cells moderately induced STAT1 phosphorylation at serine residue 727 and tyrosine residue 705, there was less HSV-induced phosphorylation of STAT3 (Fig. 6). At 20 min after TCR stimulation, HSV did not significantly inhibit phosphorylation of STAT1 or STAT3. When comparing phosphorylation levels between unstimulated and TCR-stimulated HSV-infected T cells, only STAT3 showed a statistically significant increase (P < 0.05) (Fig. 6). Taken together, these results suggest that STAT3 activation may play a role in the selective synthesis of IL-10 in HSV-infected T cells stimulated through the TCR.
FIG. 6.

TCR stimulation induces STAT3 phosphorylation in HSV-infected T cells. Jurkat T cells were incubated with mock- or HSV-infected fibroblasts and removed. T cells were TCR stimulated, fixed, and permeabilized. T cells were stained with phospho-specific antibodies against the transcription factors STAT1 (Ser727), STAT1 (Tyr705), and STAT3. The histogram plots depicted are representative of four independent flow cytometry experiments each. Plots of the mean fluorescence intensities from these experiments are depicted in the bar graphs on the right. *, P < 0.05, Student's t test.
Activation of the p38 pathway is required for TCR-stimulated IL-10 synthesis in HSV-infected T cells.
The p38 pathway has been implicated in the production of multiple Th2 cytokines, including IL-10 (32). However, it has not been reported whether TCR-stimulated IL-10 synthesis requires p38 activation in the context of viral infection. In HSV-infected T cells, we observe activation of p38 and activation of transcription factors downstream of p38. We were therefore interested in determining how inhibition of p38 activity affected TCR-stimulated IL-10 synthesis. In these experiments, PBMC were untreated or treated with a specific inhibitor of p38 kinase prior to HSV infection. As seen previously, HSV significantly inhibited TCR-stimulated synthesis of TNF-α but not IL-10 (Fig. 7). In the presence of the p38 inhibitor SB203580, there was significant inhibition of TCR-stimulated IL-10 synthesis but not TNF-α synthesis in both mock- and HSV-infected cells (Fig. 7) (P < 0.05). These results suggest that HSV-infected T cells require p38 activity to selectively synthesize IL-10 in response to TCR stimulation.
FIG. 7.
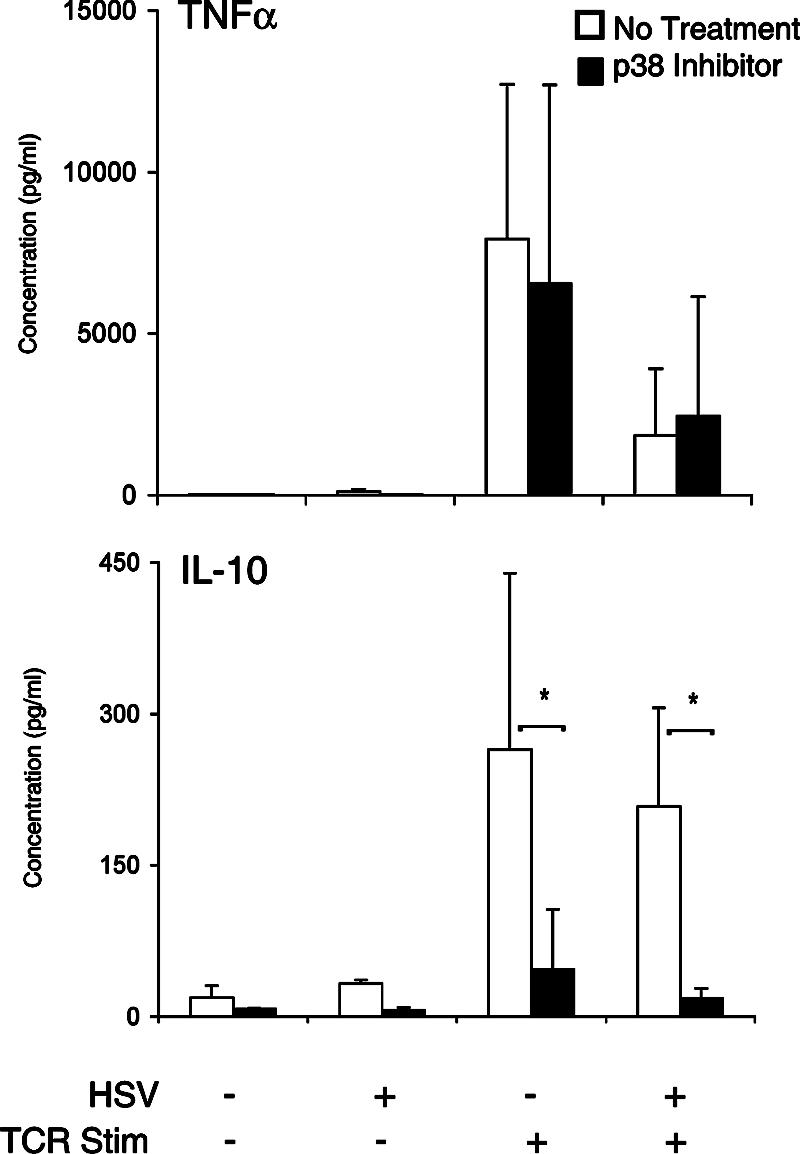
TCR-stimulated cytokine synthesis of IL-10 requires p38 in HSV-infected T cells. Fibroblast monolayers were infected with HSV as before. Human PBMC were preincubated with 10 μM SB203580, a p38-specific inhibitor, or with the control carrier. PBMC were added to fibroblasts, and SB203580 was maintained throughout the assay. Supernatants were removed, and TNF-α and IL-10 were detected using an ELISA. Data represent the mean cytokine levels from four independent experiments. *, P < 0.05, Student's t test.
DISCUSSION
The ability of IL-10 to potently suppress the cellular immune response and increase host susceptibility has made it a strategic target for intracellular human pathogens. The immunosuppressive ability of IL-10 was recently demonstrated in mice infected with cytomegalovirus (CMV), where IL-10 receptor blockade resulted in reduced viral load in the salivary glands (24). Increased IL-10 synthesis is thought to play an important role in persistent infections caused by viruses, bacteria, and parasites. Pathogens have been shown to induce IL-10 synthesis as well as encode their own IL-10 homologues (42). For example, the Herpesviridae family members CMV and Epstein-Barr virus (EBV) each encode functional viral homologues of IL-10 (56, 63). HSV does not encode an IL-10 homologue, nor does it directly induce significant levels of IL-10 in T cells. Instead, in an elegant example of convergent evolution, HSV has developed an alternative strategy. By differentially targeting intracellular signaling pathways, HSV transforms an activating stimulus into the isolated synthesis of an immunosuppressive cytokine that favors viral replication. Whereas other pathogens have been reported to directly induce cellular cytokines or encode cytokine homologues, the strategy employed by HSV represents a novel and sophisticated mechanism of immune modulation that involves an altered intracellular response to an extracellular stimulus.
To achieve selective TCR-stimulated IL-10 synthesis, HSV markedly remodels intracellular signal pathways in T cells by simultaneously inhibiting signaling through LAT and inducing signaling through p38 and JNK. Our evidence suggests that IL-10 synthesis requires p38 activity in HSV-infected T cells, a finding that is consistent with reports in uninfected T cells (11, 32). Although it is unclear how HSV activates p38 and JNK in T cells, our previous reported findings suggest that HSV does not directly activate ZAP-70 (60). HSV may induce activation of Rac1 and/or MEKK, as these upstream kinases are both required for p38 and JNK activation (27). The HSV genes UL48 and UL54 are required and sufficient to activate the p38 and JNK pathways in nonhuman fibroblasts (22, 71). In human T cells, UL48 and UL54, as well as Us3 or UL13 (HSV-encoded kinases), may be involved in p38 and JNK activation. Activation of the MAPKs is not specific to HSV. For example, other herpesviruses have been shown to phosphorylate p38 and JNK (EBV) or phosphorylate p38 and ERK (CMV) (22). However, inhibition of TCR signaling appears to be specific to HSV, as other herpesviruses (e.g., cytomegalovirus and varicella-zoster virus) do not affect TCR-stimulated effector functions (G. Zahariadis and K. R. Jerome, unpublished data).
IL-10 gene expression is a complex process that involves transcriptional and posttranscriptional regulation. In addition, regulation of IL-10 expression varies depending on the stimulus and the type of cell, i.e., T cells versus monocytes and macrophages (45). Although a comprehensive analysis of the transcription factors required for IL-10 expression in T cells is beyond the scope of this report, our results demonstrate that infection of T cells with HSV activates the p38 pathway. Furthermore, we demonstrate that p38 activation is required for IL-10 synthesis in HSV-infected T cells stimulated through the TCR. ATF-2 and c-Jun, transcription factors downstream of p38, have been implicated in IL-10 expression in T cells (37, 69). Furthermore, ATF-2 activation has been associated with increased IL-10 synthesis in the context of parasitic infection (6). Although HSV appears to maximally activate ATF-2 and c-Jun, IL-10 synthesis is only induced after TCR stimulation in HSV-infected T cells, suggesting that additional transcription factors may be involved. In contrast to ATF-2 and c-Jun, HSV infection alone does not activate STAT3. Furthermore, we have demonstrated that HSV does not inhibit TCR-stimulated phosphorylation of STAT3, suggesting that STAT3 may play a role in the TCR-stimulated synthesis of IL-10 in HSV-infected T cells. STAT3 activation has been associated with IL-10 synthesis in T cells (29, 30). STAT3 is phosphorylated by TCR stimulation on serine and tyrosine residues (16, 49). We focused on STAT3 tyrosine phosphorylation, as STAT3 serine phosphorylation requires ERK activation, which HSV inhibits in T cells. It is quite possible that additional transcription factors implicated in IL-10 expression, e.g., AP-1 (34) and Sp1 (65), may also play a role in the selective synthesis of IL-10 in HSV-infected T cells stimulated through the TCR.
We previously reported that a tyrosine phosphatase inhibitor restored TCR-stimulated calcium flux in HSV-infected T cells, suggesting that HSV-induced tyrosine phosphatase activity may contribute to reduced LAT phosphorylation (60). The ability of HSV to inhibit TCR-stimulated formation of the LAT signaling complex sheds new light on results seen in vivo in mutant LAT knock-in models. Mice homozygous for a mutation in a LAT C-terminal tyrosine residue have impaired T-cell development. These mice accumulated T cells that chronically secreted significant amounts of IL-4 and IL-10, and they eventually developed disease states related to an elevated Th2 environment (e.g., eosinophilia and immunoglobulinemia) (1). In similar experiments involving point mutations in three out of the four C-terminal LAT tyrosine residues, there was a block in T-cell maturation, and T cells accumulated in the spleens and lymph nodes of experimental mice. In this model, there were also large amounts of Th2 cytokines chronically produced (51). Infection of T cells with HSV effectively inhibits LAT-dependent TCR signaling, and the isolated IL-10 phenotype that results is reminiscent of the phenotype observed in the aforementioned LAT mutant mouse models. In LAT-deficient Jurkat T cells, an alternative pathway for TCR-stimulated activation of p38 by ZAP-70 has been reported (57). HSV does not inhibit TCR-stimulated ZAP-70 phosphorylation (60), and TCR-simulated p38 activation in HSV-infected T cells may involve ZAP-70 activation.
We contend that HSV induces dual and enzymatically opposed kinase and phosphatase activities in T cells to skew TCR stimulation toward selective IL-10 production. Intracellular signal remodeling likely requires an HSV protein(s) that can induce phosphatase activity to inhibit LAT and an additional HSV protein(s) that can induce kinase activity to facilitate p38- and JNK-mediated signaling. This added level of complexity could enhance regulation by limiting IL-10 production to sites of viral reactivation, such as the herpetic ulcer, an anatomic site where T cells may become infected and activated. A study of herpetic ulcers in vivo demonstrated increased levels of IL-10 as well as increased beta chemokines IL-1, IL-6, and IL-12 (44). In in vivo mouse models, the role of IL-10 in HSV infection appears to vary depending on the site of inoculation as well as the site of persistent infection studied. For example, significant levels of IFN-γ, IL-2, IL-6, and IL-10 proteins were detected in the trigeminal ganglion of ocular-inoculated mice (20), whereas T cells isolated from the cornea of ocular-inoculated mice with active herpetic stromal keratitis could be stimulated to synthesize IFN-γ and IL-2 but not IL-10 (50). In another model of herpetic stromal keratitis, mice pretreated with IL-10 prior to HSV ocular inoculation had significantly reduced corneal inflammation and the incidence of blindness was reduced by 60% (66). In contrast, a mouse model of herpetic encephalomyelitis has implicated HSV-induced increased Th2 cytokine synthesis, including IL-10, with increased disease severity (48).
Although the origin of IL-10 in HSV infection has not been determined, in vitro studies of HSV-infected cells have demonstrated that epithelial cells and antigen-presenting cells may be an important source of HSV-induced cytokines. One study demonstrated that immortalized mouse keratinocytes infected with HSV had increased levels of IL-10 mRNA (72). However, these investigators did not analyze IL-10 protein levels, and mRNA and protein levels do not always correlate with one another. For example, a study of HSV-infected dendritic cells (DC) reported increased IL-10 mRNA that did not translate into increased IL-10 protein secretion (17). Infection of human PBMC and DC with HSV induced IL-6 synthesis (17, 52, 53), and infection of human corneal epithelial cells with HSV induced IFN-α, IL-6, IL-8, and TNF-α synthesis (36). In this latter study, there was increased activation of p38, JNK, and NF-κB. NF-κB activation has also been implicated in IL-12 induced in HSV-infected macrophages (40) and in TNF-α induced in HSV-infected DC (43). In HSV-infected T cells, we have demonstrated a distinct pattern characterized by p38 and JNK activation and NF-κB inhibition. HSV-induced NF-κB inhibition may play a role in inhibition of Th1 cytokine synthesis, as NF-κB activation has been associated with increased Th1 cytokine synthesis (10). While epithelial cells and antigen-presenting cells may be a source of IL-10 in HSV infection, ours is the first report demonstrating that HSV-infected T cells may be an additional, important source of IL-10 produced during HSV infection.
A fascinating feature of the natural history of HSV infection and reactivation is that significant levels of virus can be shed in the absence of noticeable herpetic ulcers. In fact, asymptomatic shedding is believed to be the most common cause of horizontal and vertical HSV transmission in immunocompetent individuals (31). While this is clinically relevant, it may also indicate the potential for pervasive modulation of immune function by HSV. Like HSV, additional members of the Herpesviridae family (e.g., CMV, EBV, varicella-zoster virus, and human herpesvirus 6 [HHV-6]) are characterized by their ability to establish latent infection and reactivate despite eliciting innate and adaptive immune responses. The herpesviruses have large genomes, encoding 70 to 80 open reading frames. However, only a fraction of these genes are required for viral replication in vitro, suggesting that a substantial number of herpesvirus proteins may have immune modulation function. While the herpesviruses share many features, they have each evolved distinct mechanisms to cope with immune surveillance. This is particularly evident in studies that report differential induction and inhibition of cytokine synthesis between the different herpesvirus members. For example, one study reported that infection of PBMC with EBV induced IL-6 and inhibited TNF-α, whereas HSV had a minimal effect on either cytokine, and HHV-6 inhibited IL-6 and induced TNF-α (18). In other studies, infection of macrophages with HHV-6 inhibited IFN-γ-induced IL-12 synthesis but not TNF-α (62), and infection of monocytes with varicella-zoster virus induced selective IL-6 synthesis, a process that required activation of NF-κB (68).
By inhibiting LAT-mediated signaling while activating the p38 pathway, HSV inhibits antiviral Th1 cytokines and skews the TCR-stimulated T-cell response toward a Th2 environment dominated by IL-10. The selective synthesis of IL-10 that results from the remodeling of TCR signaling by HSV may facilitate viral latency and reactivation, and identification of the mechanisms involved in the remodeling of TCR signaling by HSV may improve the management of HSV disease. Other potential benefits of studying mechanisms to remodel TCR signaling include the specific and selective targeting of proinflammatory T-cell function that has been implicated in a variety of human disease states, such as cancer, transplant rejection, and autoimmunity (2, 5, 58).
Acknowledgments
We thank M. Aubert, S. Polyak, T. Spies, and V. Groh for discussion.
This work was supported by the Developmental Awards Program of the NIAID Sexually Transmitted Infections and Topical Microbicide Cooperative Research Centers at the University of Washington, grant U19 AI 31448 (D.D.S.), and NIH grant R56 AI 65956 (K.R.J.).
Footnotes
Published ahead of print on 5 September 2007.
REFERENCES
- 1.Aguado, E., S. Richelme, S. Nunez-Cruz, A. Miazek, A. M. Mura, M. Richelme, X. J. Guo, D. Sainty, H. T. He, B. Malissen, and M. Malissen. 2002. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science 296:2036-2040. [DOI] [PubMed] [Google Scholar]
- 2.Amrolia, P. J., S. D. Reid, L. Gao, B. Schultheis, G. Dotti, M. K. Brenner, J. V. Melo, J. M. Goldman, and H. J. Stauss. 2002. Allo-restricted cytotoxic t cells specific for human CD45 show potent antileukemic activity. Blood 12:12. [DOI] [PubMed] [Google Scholar]
- 3.Bonneau, R. H., and S. R. Jennings. 1989. Modulation of acute and latent herpes simplex virus infection in C57BL/6 mice by adoptive transfer of immune lymphocytes with cytolytic activity. J. Virol. 63:1480-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braun, R. W., H. K. Teute, H. Kirchner, and K. Munk. 1984. Replication of herpes simplex virus in human T lymphocytes: characterization of the viral target cell. J. Immunol. 132:914-919. [PubMed] [Google Scholar]
- 5.Burrows, S. R., R. Khanna, J. M. Burrows, and D. J. Moss. 1994. An alloresponse in humans is dominated by cytotoxic T lymphocytes (CTL) cross-reactive with a single Epstein-Barr virus CTL epitope: implications for graft-versus-host disease. J. Exp. Med. 179:1155-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carneiro-Santos, P., L. F. Alves-Oliveira, R. Correa-Oliveira, and P. Hagan. 2002. p38 mitogen-activated protein kinase influence on the production of IL-10 in human Schistosoma mansoni. Parasite Immunol. 24:493-497. [DOI] [PubMed] [Google Scholar]
- 7.Corey, L., A. Wald, C. L. Celum, and T. C. Quinn. 2004. The effects of herpes simplex virus-2 on HIV-1 acquisition and transmission: a review of two overlapping epidemics. J. Acquir. Immune Defic. Syndr. 35:435-445. [DOI] [PubMed] [Google Scholar]
- 8.Coudronniere, N., M. Villalba, N. Englund, and A. Altman. 2000. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc. Natl. Acad. Sci. USA 97:3394-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Derijard, B., M. Hibi, I. H. Wu, T. Barrett, B. Su, T. Deng, M. Karin, and R. J. Davis. 1994. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76:1025-1037. [DOI] [PubMed] [Google Scholar]
- 10.Dieckhoff, K., P. Graf, B. Beinhauer, C. Schwaerzler, J. M. Carballido, C. Neumann, K. Zachmann, and T. Jung. 2005. Deficient translocation of c-Rel is associated with impaired Th1 cytokine production in T cells from atopic dermatitis patients. Exp. Dermatol. 14:17-25. [DOI] [PubMed] [Google Scholar]
- 11.Dodeller, F., A. Skapenko, J. R. Kalden, P. E. Lipsky, and H. Schulze-Koops. 2005. The p38 mitogen-activated protein kinase regulates effector functions of primary human CD4 T cells. Eur. J. Immunol. 35:3631-3642. [DOI] [PubMed] [Google Scholar]
- 12.Elbers, J. M., A. Bitnun, S. E. Richardson, E. L. Ford-Jones, R. Tellier, R. M. Wald, M. Petric, H. Kolski, H. Heurter, and D. MacGregor. 2007. A 12-year prospective study of childhood herpes simplex encephalitis: is there a broader spectrum of disease? Pediatrics 119:e399-e407. [DOI] [PubMed] [Google Scholar]
- 13.Finco, T. S., T. Kadlecek, W. Zhang, L. E. Samelson, and A. Weiss. 1998. LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity 9:617-626. [DOI] [PubMed] [Google Scholar]
- 14.Friedman, H. M., G. H. Cohen, R. J. Eisenberg, C. A. Seidel, and D. B. Cines. 1984. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 309:633-635. [DOI] [PubMed] [Google Scholar]
- 15.Fruh, K., K. Ahn, H. Djaballah, P. Sempe, P. M. van Endert, R. Tampe, P. A. Peterson, and Y. Yang. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375:415-418. [DOI] [PubMed] [Google Scholar]
- 16.Gerwien, J., M. Nielsen, T. Labuda, M. H. Nissen, A. Svejgaard, C. Geisler, C. Ropke, and N. Odum. 1999. Cutting edge: TCR stimulation by antibody and bacterial superantigen induces STAT3 activation in human T cells. J. Immunol. 163:1742-1745. [PubMed] [Google Scholar]
- 17.Ghanekar, S., L. Zheng, A. Logar, J. Navratil, L. Borowski, P. Gupta, and C. Rinaldo. 1996. Cytokine expression by human peripheral blood dendritic cells stimulated in vitro with HIV-1 and herpes simplex virus. J. Immunol. 157:4028-4036. [PubMed] [Google Scholar]
- 18.Gosselin, J., L. Flamand, M. D'Addario, J. Hiscott, I. Stefanescu, D. V. Ablashi, R. C. Gallo, and J. Menezes. 1992. Modulatory effects of Epstein-Barr, herpes simplex, and human herpes-6 viral infections and coinfections on cytokine synthesis. A comparative study. J. Immunol. 149:181-187. [PubMed] [Google Scholar]
- 19.Gupta, S., D. Campbell, B. Derijard, and R. J. Davis. 1995. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267:389-393. [DOI] [PubMed] [Google Scholar]
- 20.Halford, W. P., B. M. Gebhardt, and D. J. Carr. 1996. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J. Immunol. 157:3542-3549. [PubMed] [Google Scholar]
- 21.Hammer, S. M., W. P. Carney, V. R. Iacoviello, B. R. Lowe, and M. S. Hirsch. 1982. Herpes simplex virus infection of human T-cell subpopulations. Infect. Immun. 38:795-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hargett, D., T. McLean, and S. L. Bachenheimer. 2005. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J. Virol. 79:8348-8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes, K., S. Edin, A. Antonsson, and T. Grundstrom. 2001. Calmodulin-dependent kinase II mediates T cell receptor/CD3- and phorbol ester-induced activation of IκB kinase. J. Biol. Chem. 276:36008-36013. [DOI] [PubMed] [Google Scholar]
- 24.Humphreys, I. R., C. de Trez, A. Kinkade, C. A. Benedict, M. Croft, and C. F. Ware. 2007. Cytomegalovirus exploits IL-10-mediated immune regulation in the salivary glands. J. Exp. Med. 204:1217-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jerome, K. R., Z. Chen, R. Lang, M. R. Torres, J. Hofmeister, S. Smith, R. Fox, C. J. Froelich, and L. Corey. 2001. HSV and glycoprotein J inhibit caspase activation and apoptosis induced by granzyme B or Fas. J. Immunol. 167:3928-3935. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, D. C., M. C. Frame, M. W. Ligas, A. M. Cross, and N. D. Stow. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 62:1347-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson, G. L., and R. Lapadat. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298:1911-1912. [DOI] [PubMed] [Google Scholar]
- 28.Johnson, R. E., A. J. Nahmias, L. S. Magder, F. K. Lee, C. A. Brooks, and C. B. Snowden. 1989. A seroepidemiologic survey of the prevalence of herpes simplex virus type 2 infection in the United States. N. Engl. J. Med. 321:7-12. [DOI] [PubMed] [Google Scholar]
- 29.Kasprzycka, M., M. Marzec, X. Liu, Q. Zhang, and M. A. Wasik. 2006. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc. Natl. Acad. Sci. USA 103:9964-9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinjyo, I., H. Inoue, S. Hamano, S. Fukuyama, T. Yoshimura, K. Koga, H. Takaki, K. Himeno, G. Takaesu, T. Kobayashi, and A. Yoshimura. 2006. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J. Exp. Med. 203:1021-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koelle, D. M., and A. Wald. 2000. Herpes simplex virus: the importance of asymptomatic shedding. J. Antimicrob. Chemother. 45(Suppl. T3):1-8. [DOI] [PubMed] [Google Scholar]
- 32.Koprak, S., M. J. Staruch, and F. J. Dumont. 1999. A specific inhibitor of the p38 mitogen activated protein kinase affects differentially the production of various cytokines by activated human T cells: dependence on CD28 signaling and preferential inhibition of IL-10 production. Cell. Immunol. 192:87-95. [DOI] [PubMed] [Google Scholar]
- 33.Kovarik, P., D. Stoiber, P. A. Eyers, R. Menghini, A. Neininger, M. Gaestel, P. Cohen, and T. Decker. 1999. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc. Natl. Acad. Sci. USA 96:13956-13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kremer, K. N., A. Kumar, and K. E. Hedin. 2007. Haplotype-independent costimulation of IL-10 secretion by SDF-1/CXCL12 proceeds via AP-1 binding to the human IL-10 promoter. J. Immunol. 178:1581-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75:263-274. [DOI] [PubMed] [Google Scholar]
- 36.Li, H., J. Zhang, A. Kumar, M. Zheng, S. S. Atherton, and F. S. Yu. 2006. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117:167-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, L. B., D. Y. Leung, M. J. Strand, and E. Goleva. 2007. ATF2 impairs glucocorticoid receptor-mediated transactivation in human CD8+ T cells. Blood 110:1570-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin, J., and A. Weiss. 2001. Identification of the minimal tyrosine residues required for linker for activation of T cell function. J. Biol. Chem. 276:29588-29595. [DOI] [PubMed] [Google Scholar]
- 39.Malissen, B., E. Aguado, and M. Malissen. 2005. Role of the LAT adaptor in T-cell development and Th2 differentiation. Adv. Immunol. 87:1-25. [DOI] [PubMed] [Google Scholar]
- 40.Malmgaard, L., S. R. Paludan, S. C. Mogensen, and S. Ellermann-Eriksen. 2000. Herpes simplex virus type 2 induces secretion of IL-12 by macrophages through a mechanism involving NF-κB. J. Gen. Virol. 81:3011-3020. [DOI] [PubMed] [Google Scholar]
- 41.Mao, H., and K. S. Rosenthal. 2002. An N-terminal arginine-rich cluster and a proline-alanine-threonine repeat region determine the cellular localization of the herpes simplex virus type 1 ICP34.5 protein and its ligand, protein phosphatase 1. J. Biol. Chem. 277:11423-11431. [DOI] [PubMed] [Google Scholar]
- 42.Mege, J. L., S. Meghari, A. Honstettre, C. Capo, and D. Raoult. 2006. The two faces of interleukin 10 in human infectious diseases. Lancet Infect. Dis. 6:557-569. [DOI] [PubMed] [Google Scholar]
- 43.Melchjorsen, J., J. Siren, I. Julkunen, S. R. Paludan, and S. Matikainen. 2006. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-κB and IRF-3. J. Gen. Virol. 87:1099-1108. [DOI] [PubMed] [Google Scholar]
- 44.Mikloska, Z., V. A. Danis, S. Adams, A. R. Lloyd, D. L. Adrian, and A. L. Cunningham. 1998. In vivo production of cytokines and beta (C-C) chemokines in human recurrent herpes simplex lesions: do herpes simplex virus-infected keratinocytes contribute to their production? J. Infect. Dis. 177:827-838. [DOI] [PubMed] [Google Scholar]
- 45.Moore, K. W., R. de Waal Malefyt, R. L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683-765. [DOI] [PubMed] [Google Scholar]
- 46.Mosmann, T. R., H. Cherwinski, M. W. Bond, M. A. Giedlin, and R. L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348-2357. [PubMed] [Google Scholar]
- 47.Murphy, J. A., R. J. Duerst, T. J. Smith, and L. A. Morrison. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 77:9337-9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakajima, H., M. Kobayashi, R. B. Pollard, and F. Suzuki. 2000. A pathogenic role of Th2 responses on the severity of encephalomyelitis induced in mice by herpes simplex virus type 2 infection. J. Neuroimmunol. 110:106-113. [DOI] [PubMed] [Google Scholar]
- 49.Ng, J., and D. Cantrell. 1997. STAT3 is a serine kinase target in T lymphocytes. Interleukin 2 and T cell antigen receptor signals converge upon serine 727. J. Biol. Chem. 272:24542-24549. [DOI] [PubMed] [Google Scholar]
- 50.Niemialtowski, M. G., and B. T. Rouse. 1992. Predominance of Th1 cells in ocular tissues during herpetic stromal keratitis. J. Immunol. 149:3035-3039. [PubMed] [Google Scholar]
- 51.Nunez-Cruz, S., E. Aguado, S. Richelme, B. Chetaille, A. M. Mura, M. Richelme, L. Pouyet, E. Jouvin-Marche, L. Xerri, B. Malissen, and M. Malissen. 2003. LAT regulates γδ T cell homeostasis and differentiation. Nat. Immunol. 4:999-1008. [DOI] [PubMed] [Google Scholar]
- 52.Paludan, S. R. 2001. Requirements for the induction of interleukin-6 by herpes simplex virus-infected leukocytes. J. Virol. 75:8008-8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peretti, S., A. Shaw, J. Blanchard, R. Bohm, G. Morrow, J. D. Lifson, A. Gettie, and M. Pope. 2005. Immunomodulatory effects of HSV-2 infection on immature macaque dendritic cells modify innate and adaptive responses. Blood 106:1305-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Posavad, C. M., D. M. Koelle, and L. Corey. 1998. Tipping the scales of herpes simplex virus reactivation: the important responses are local. Nat. Med. 4:381-382. [DOI] [PubMed] [Google Scholar]
- 55.Roizman, B. 1996. Herpes simplex virus, p. 2231. In B. Fields (ed.), Virology, 3rd ed., vol. 2. Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 56.Salek-Ardakani, S., J. R. Arrand, and M. Mackett. 2002. Epstein-Barr virus encoded interleukin-10 inhibits HLA-class I, ICAM-1, and B7 expression on human monocytes: implications for immune evasion by EBV. Virology 304:342-351. [DOI] [PubMed] [Google Scholar]
- 57.Salvador, J. M., P. R. Mittelstadt, T. Guszczynski, T. D. Copeland, H. Yamaguchi, E. Appella, A. J. Fornace, Jr., and J. D. Ashwell. 2005. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 6:390-395. [DOI] [PubMed] [Google Scholar]
- 58.Seki, N., A. D. Brooks, C. R. Carter, T. C. Back, E. M. Parsoneault, M. J. Smyth, R. H. Wiltrout, and T. J. Sayers. 2002. Tumor-specific CTL kill murine renal cancer cells using both perforin and Fas ligand-mediated lysis in vitro, but cause tumor regression in vivo in the absence of perforin. J. Immunol. 168:3484-3492. [DOI] [PubMed] [Google Scholar]
- 59.Sethi, K. K., Y. Omata, and K. E. Schneweis. 1983. Protection of mice from fatal herpes simplex virus type 1 infection by adoptive transfer of cloned virus-specific and H-2-restricted cytotoxic T lymphocytes. J. Gen. Virol. 64:443-447. [DOI] [PubMed] [Google Scholar]
- 60.Sloan, D. D., J. Y. Han, T. K. Sandifer, M. Stewart, A. J. Hinz, M. Yoon, D. C. Johnson, P. G. Spear, and K. R. Jerome. 2006. Inhibition of TCR signaling by herpes simplex virus. J. Immunol. 176:1825-1833. [DOI] [PubMed] [Google Scholar]
- 61.Sloan, D. D., G. Zahariadis, C. M. Posavad, N. T. Pate, S. J. Kussick, and K. R. Jerome. 2003. CTL are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J. Immunol. 171:6733-6741. [DOI] [PubMed] [Google Scholar]
- 62.Smith, A., F. Santoro, G. Di Lullo, L. Dagna, A. Verani, and P. Lusso. 2003. Selective suppression of IL-12 production by human herpesvirus 6. Blood 102:2877-2884. [DOI] [PubMed] [Google Scholar]
- 63.Spencer, J. V. 2007. The cytomegalovirus homolog of interleukin-10 requires phosphatidylinositol 3-kinase activity for inhibition of cytokine synthesis in monocytes. J. Virol. 81:2083-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Streilein, J. W., M. R. Dana, and B. R. Ksander. 1997. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol. Today 18:443-449. [DOI] [PubMed] [Google Scholar]
- 65.Tone, M., M. J. Powell, Y. Tone, S. A. Thompson, and H. Waldmann. 2000. IL-10 gene expression is controlled by the transcription factors Sp1 and Sp3. J. Immunol. 165:286-291. [DOI] [PubMed] [Google Scholar]
- 66.Tumpey, T. M., V. M. Elner, S. H. Chen, J. E. Oakes, and R. N. Lausch. 1994. Interleukin-10 treatment can suppress stromal keratitis induced by herpes simplex virus type 1. J. Immunol. 153:2258-2265. [PubMed] [Google Scholar]
- 67.VanDeusen, J. B., M. H. Shah, B. Becknell, B. W. Blaser, A. K. Ferketich, G. J. Nuovo, B. M. Ahmer, J. Durbin, and M. A. Caligiuri. 2006. STAT-1-mediated repression of monocyte interleukin-10 gene expression in vivo. Eur. J. Immunol. 36:623-630. [DOI] [PubMed] [Google Scholar]
- 68.Wang, J. P., E. A. Kurt-Jones, O. S. Shin, M. D. Manchak, M. J. Levin, and R. W. Finberg. 2005. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J. Virol. 79:12658-12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang, Z. Y., H. Sato, S. Kusam, S. Sehra, L. M. Toney, and A. L. Dent. 2005. Regulation of IL-10 gene expression in Th2 cells by Jun proteins. J. Immunol. 174:2098-2105. [DOI] [PubMed] [Google Scholar]
- 70.Wehinger, J., F. Gouilleux, B. Groner, J. Finke, R. Mertelsmann, and R. M. Weber-Nordt. 1996. IL-10 induces DNA binding activity of three STAT proteins (STAT1, STAT3, and Stat5) and their distinct combinatorial assembly in the promoters of selected genes. FEBS Lett. 394:365-370. [DOI] [PubMed] [Google Scholar]
- 71.Zachos, G., B. Clements, and J. Conner. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 274:5097-5103. [DOI] [PubMed] [Google Scholar]
- 72.Zak-Prelich, M., K. E. Halliday, C. Walker, C. M. Yates, M. Norval, and R. C. McKenzie. 2001. Infection of murine keratinocytes with herpes simplex virus type 1 induces the expression of interleukin-10, but not interleukin-1 alpha or tumour necrosis factor-alpha. Immunology 104:468-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang, J., K. V. Salojin, J. X. Gao, M. J. Cameron, I. Bergerot, and T. L. Delovitch. 1999. p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J. Immunol. 162:3819-3829. [PubMed] [Google Scholar]
- 74.Zhang, W., J. Sloan-Lancaster, J. Kitchen, R. P. Trible, and L. E. Samelson. 1998. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92:83-92. [DOI] [PubMed] [Google Scholar]