

Abstract
Vaccinia virus (VACV) has been attracting attention recently not only as a vector for various vaccines but also as an immunization tool against smallpox because of its potential use as a bioterrorism agent. It has become evident that in spite of a long history of studies of VACV, its tissue pathogenesis remains to be fully understood. Here, we investigated the pathogenesis of VACV and its interactions with human immunodeficiency virus type 1 (HIV-1) in the context of human lymphoid tissues. We found that ex vivo-cultured tonsillar tissue supports productive infection by the New York City Board of Health strain, the VACV strain of the Dryvax vaccine. VACV readily infected both T and non-T (B) lymphocytes and depleted cells of both of these subsets equally over a 12-day period postinfection. Among T lymphocytes, CD8+ cells are preferentially depleted in accordance with their preferential infection: the probability that a CD8+ T cell will be productively infected is almost six times higher than for a CD4+ T cell. T cells expressing CCR5 and the activation markers CD25, CD38, and HLA-DR are other major targets for infection by VACV in lymphoid tissue. As a consequence, VACV predominantly inhibits the replication of the R5SF162 phenotype of HIV-1 in coinfected tissues, as R5-tropic HIV-1 requires activated CCR5+ CD4+ cells for productive infection. Human lymphoid tissue infected ex vivo by VACV can be used to investigate interactions of VACV with other viruses, in particular HIV-1, and to evaluate various VACV vectors for the purpose of recombinant vaccine development.
The global eradication of smallpox declared by the World Health Organization in 1980 eliminated the need for poxvirus immunization and reduced interest in vaccinia virus (VACV) biology (24). The threat of smallpox virus being used as a biological weapon, however, has prompted the reintroduction of the vaccination program as a preventive measure against acts of bioterrorism and produced a resurgence in scientific attention to VACV. VACV, like variola virus, the causal agent of smallpox, belongs to the genus Orthopoxvirus. Vaccination with VACV confers cross-protection against smallpox disease, and several strains of VACV have been developed for this purpose. These include the replication-deficient modified virus Ankara (MVA) (10) and the New York City Board of Health (NYCBH) strain. Such strains have been developed to avoid complications in immunocompromised people. Indeed, progressive vaccinia (vaccinia necrosum) has been described in human immunodeficiency virus type 1 (HIV-1)-infected people (29). Similarly, it was reported that AIDS patients receiving a recombinant vaccinia virus-based anti-HIV-1 vaccine developed “wide necrosis” at the sites of subcutaneous and/or intramuscular injections (19, 27).
VACV infects cells of a wide variety of types, including T cells, monocytes/macrophages, NK cells, and B cells (36). Despite a long history of related studies, the components of the viral entry/fusion complex which control VACV attachment to and entry into target cells remain incompletely characterized. Identification of the VACV receptor responsible for virus binding and entry into cells is complicated by the existence of several distinct forms of viral particles which may enter cells by different mechanisms (25, 34). Recently, it has been demonstrated that VACV-mediated tyrosine phosphorylation of CC chemokine receptor 5 (CCR5) occurs at a postentry step of VACV infection which is followed by the downregulation of CCR5 from the surface of VACV-infected T cells. These events play a critical role in VACV replication (28). CCR5 also serves as a coreceptor for HIV-1 variants responsible for primary infection, suggesting a potential interaction between VACV and HIV-1.
Here, we studied VACV infection and its interaction with HIV-1 in human tonsillar tissues coinfected ex vivo with VACV and HIV-1. Smallpox virus is most commonly acquired from inhalation of infectious aerosols (after which the virus attaches to the nasopharyngeal and respiratory mucosa) and then is transferred to local lymphoid tissue, where replication occurs. Here, we used a system of ex vivo lymphoid tissue culture to study VACV pathogenesis and Orthopoxvirus-HIV interactions which may take place in individuals coinfected with variola virus and HIV.
Using this infection model, we demonstrate that VACV preferentially depletes activated T cells of the CCR5+ CD4+ phenotype, thus dramatically suppressing replication of the R5 strain of HIV-1.
MATERIALS AND METHODS
Tissue culture and viral infection.
Human tonsils surgically removed during routine tonsillectomy were received within 5 h of excision and were dissected into 2- to 3-mm3 blocks. Tissue blocks were placed onto collagen sponge gels in culture medium at the air-liquid interface and infected the next day, as described earlier (12, 16). MVA and NYCBH strains were obtained from ATCC (Manassas, VA) and used in VACV infections. Five microliters of viral stock containing approximately 1 × 106 genome equivalents of VACV was used to infect each block. A total of 27 tissue blocks from each donor were used for each tested condition. We evaluated VACV replication using calibrated real-time PCR. For HIV-1 infection, 5 μl of clarified HIV-1-containing medium (approximately 0.5 ng of p24 per block of a prototypic R5 HIV variant, SF162 [R5SF162], or of a prototypic X4 HIV variant, LAI.04 [X4LAI.04]) was applied to the top of each tissue block. We measured HIV-1 replication as the production of p24 core antigen released into the pooled medium bathing all 27 blocks, using an HIV-1 p24 enzyme-linked immunosorbent assay (ELISA; Perkin-Elmer, Wellesley, MA). In the coinfection experiments, tissues were inoculated with VACV and then infected with HIV-1 within 3 h. Experiments were repeated n times, each time with tissue from a different donor; n is indicated in the text and figure legends. UV inactivation of VACV (250 nm) was performed on a UV Stratalinker 2400 (Stratagene, La Jolla, CA) at 5,000 J/m2.
Quantification of VACV DNA with calibrated real-time PCR.
We extracted viral DNA from virus-containing culture supernatants using the QIAamp kit (QIAGEN Gmbh, Hilden, Germany) according to the instructions of the manufacturer. The number of viral genome copies (VACV load) was determined in a quantitative calibrated real-time PCR with an ABI Prism 7000 sequence detector (PE Applied Biosystems, Foster City, CA). The specific primer sets used to detect VACV were the following: VACV forward, 5′-CCGTCCAGTCTGAACATCAATC-3′; VACV reverse, 5′-ACAAATAGAAAAGTGTTGTAAACGCAA-3′. The probe (5′-FAM-CCAACCTAAATAGAACTTCAT-3′-MGBNFQ) was covalently linked to the reporter dye 6-carboxy-fluorescein (FAM) at the 5′ end and to a minor groove binder and a nonfluorescent quencher (MGBNFQ) at the 3′ end (26). The reaction mixture contained TaqMan PCR master mix, each primer at 300 nM, and 5 μl of DNA. Following activation of AmpliTaq Gold at 95°C for 10 min, 40 cycles of amplification (denaturation step, 95°C for 15 s; annealing-extension step, 60°C for 1 min) were performed. Data were analyzed using sequence detection software (Applied Biosystems). We performed VACV DNA quantification using a commercially available reference standard (Advanced Biotechnologies, Columbia, MD) and normalized the results using a synthetic DNA calibrator molecule (106 copies per sample) that was added to the samples before the extraction step; this procedure allowed us to control for intersample extraction efficiency and to monitor PCR artifacts (4). Primers and the calibrator probe were not cross-reactive with the VACV sequences.
Flow cytometry.
Flow cytometry was performed on cells mechanically isolated from control and infected tissue blocks on day 12 after infection. Lymphocytes, identified according to their light-scattering properties, were analyzed for expression of lymphocyte markers. We stained cells for surface markers, including CD3, CD4, CD8, CD25, CD38, CD69, CD95, HLA-DR, CD45RA, CD62L, CCR5, and CXCR4, using anti-CD3-phycoerythrin (PE)-Cy7 or PE-Alexa 610, anti-CD4-PE-Cy5.5, or allophycocyanin (APC)-Cy7, anti-CD8-APC or Pacific blue, anti-CD25-APC, anti-CD38-APC-Cy5.5, anti-CD69-PE-Cy5.5, anti-HLA-DR-peridinin chlorophyll protein-Cy5.5, anti-CD45RA-PE-Cy7, anti-CD62L-APC-Cy7 (Caltag Laboratories, Burlingame, CA), or anti-CCR5-APC or APC-Cy7 and anti-CXCR4-PE or APC (Becton-Dickinson, Pharmingen, San Jose, CA). To identify productively infected cells, we stained cells for surface markers, washed them two times, fixed and permeabilized them with Fix&Perm (Caltag Laboratories), and then stained them with an anti-p24-PE-labeled antibody (KC57; Beckman-Coulter, Miami, FL) and a fluorescein isothiocyanate-labeled polyclonal antibody to cell-associated vaccinia virus antigens recognizing early and late VACV proteins (U.S. Biological, Swampscott, MA). To quantify cell depletion, we added Caltag counting beads (Caltag Laboratories) to each tube prior to cell surface staining. For each condition, 15,000 beads were acquired and then the number of cells was normalized according to the weight of the 27 blocks taken on the day of the staining and expressed as a percentage of the uninfected tissue. Normalization based on the weight of the blocks at day zero was also performed and gave similar results. Data were acquired on a BD LSRII instrument using DIVA software version 3.0 and analyzed with FlowJo software (Tree Star, San Carlos, CA).
Multiplexed fluorescent microsphere array of human cytokines.
We determined the levels of CCL3 (macrophage inflammatory protein 1α), CCL4 (macrophage inflammatory protein 1β), CCL5 (RANTES), and CXCL12 (SDF-1β) in culture medium using multiplex bead array assays on a Luminex 100 platform. All the antibodies and cytokine standards were purchased from R&D Systems (Minneapolis, MN). Individual Luminex bead sets were coupled to cytokine-specific capture antibodies according to the manufacturer's instructions. Conjugated beads were washed and kept at 4°C until use. Biotinylated polyclonal antibodies were used at twice the concentrations recommended for a classical ELISA procedure. All the assay procedures were performed in phosphate-buffered saline supplemented with 1% normal mouse serum, 1% normal goat serum, and 20 mM Tris-HCl (pH 7.4) filtered through a 0.2-μm-pore-size membrane. Fifty microliters of each sample was combined in a well with 50 μl of a mix containing 1,200 beads per set and incubated overnight at 4°C in a Millipore multiscreen plate. We then aspirated the liquid by using a Millipore vacuum manifold and washed the plates twice with 200 μl of the assay buffer. The beads were then resuspended in 50 μl of solution containing biotinylated polyclonal antibodies and incubated at room temperature for 20 min. The plates were washed twice with phosphate-buffered saline, the beads were resuspended in 50 μl of assay buffer, and 50 μl per well of a 16-μg/ml solution of streptavidin-PE (Molecular Probes, Carlsbad, CA) was added. The plates were read on a Luminex 100 platform. A total of 61 beads was collected for each bead set.
Statistical analysis.
We have normalized the final data in order to compare results obtained in different experiments. For each experiment, we compared infected and control tissues obtained from an individual donor in replicates of 27 tissue blocks for each data point. To average the results of different experiments and to analyze them statistically, we normalized the data as percentages of controls and determined the means ± standard errors of the means (SEM). We used Student's paired t test to evaluate the significance of the differences between various data sets. Statistical analysis of p24 ELISA data was performed with Deltasoft software (version 3.0; BioMetallics), with which we combined data from three dilutions and calculated the weighted interpolated p24 concentration and SE. Data analysis of the median of the fluorescence intensity recorded for 61 beads of each bead set for the Luminex assays was performed with Bioplex Manager software (version 3.0; Bio-Rad) using a 5P regression algorithm.
RESULTS
VACV infection of human lymphoid tissue.
The NYCBH strain of VACV, but not MVA, replicated efficiently in human tonsillar tissue ex vivo (Fig. 1A). Viral replication became detectable in tissues of all the tested donors at day 3 after virus inoculation and continued to increase until the end of the experiment at day 12 after infection. The choice of day 12 as the end point is based on the standard protocol for this system in order to avoid the possible effect of tissue deterioration on viral replication (12). The viral concentration in culture supernatants reached, on average, (4.8 ± 1.3) × 1010 genome equivalents per milliliter (range, 1.27 × 109 to 1.64 × 1011), corresponding to approximately a 3-log amplification compared with the inoculum. Each block of tissue produced on average around 1.6 × 1010 viral genome copies into the culture medium over the period of 12 days. The wide interassay variability of virus yield produced by ex vivo-infected lymphoid tissues from various donors has been previously documented for replication of human herpesvirus 6, human herpesvirus 7, measles virus, and HIV-1 (12, 14, 17, 18, 22).
FIG. 1.

Replication of VACV in human lymphoid tissue ex vivo. Tissue blocks (27 from each human tonsil) were infected with either NYCBH or MVA. Each of the 27 tissue blocks from a single donor was inoculated with 5 μl of viral stock containing approximately 1 × 106 genome equivalents. The culture medium was changed every 3 days, and VACV replication was monitored in these medium samples by means of real-time PCR (A) or flow cytometry (B and C). A. Typical replication kinetics of NYCBH and MVA are presented. The inset represents the average kinetics of replication of NYCBH (n = 13); results are shown as means ± SEM. Each point represents the measurement of medium pooled from three wells, each of which contained nine tissue blocks. To merge the data from different experiments, we normalized the amount of VACV DNA for each experiment as a percentage of the maximum production of VACV over the 12-day period. B. Expression of VACV antigen by lymphocytes as detected with flow cytometry. Density plots of lymphocytes, isolated from VACV-infected (right panel) and matched uninfected tissue (left panel), indicate preferential staining of CD3− cells for cell-associated vaccinia virus antigen on day 12 postinfection. One representative experiment (out of 11) is shown. C. Frequencies of VACV-infected CD3−, CD3+, CD3/CD4+, and CD3/CD8+ cells detected at day 12 postinfection. Cells were stained at day 12 for CD3, CD4, CD8, and cell-associated vaccinia virus antigens. Data are presented as means ± SEM (error bars) of VACV-infected cells from 11 donors.
To determine the type(s) of cells supporting VACV replication, we prepared a suspension of single cells of tonsillar tissue by mechanical dissociation and stained it for CD3, CD4, CD8, and cell-associated vaccinia virus antigens. We then analyzed stained cells using flow cytometry. Positive staining for VACV reflected viral infection rather than adsorption of free virus, since no significant increase in the number of vaccinia-positive cells was observed when cells were incubated with UV-inactivated VACV at a range mimicking the amount of replicating VACV (data not shown).
Figure 1B shows VACV-infected CD3+ and CD3− lymphocytes. The latter subset consisted of 90% B cells (9). In eight tissue samples inoculated with VACV, on average 2% ± 0.3% of CD3+ cells and 3.4% ± 0.2% of CD3− cells were productively infected at day 12 postinfection, reflecting a higher rate of infection of non-T cells (P = 0.0003). Among T cells, 1.8% ± 0.3% of CD4+ cells and 2.6% ± 0.4% of CD8+ cells (n = 8; P = 0.02) were productively infected on day 12 postinfection, showing a preferential infection of CD8 T cells (Fig. 1C). To define B cells more precisely, we stained lymphocytes for CD19. In these experiments, 3.2% ± 0.5% of CD19+ cells were infected by VACV (n = 4), confirming the above-presented conclusion that VACV infects B cells at a rate not significantly different from that for CD3− cells.
Thus, both real-time PCR and flow cytometric data demonstrated productive VACV infection of human lymphoid tissue ex vivo.
VACV preferentially depletes CD4+ CCR5+ T cells in human tonsillar tissues.
To further identify productively infected cells and investigate preferential depletion of various tissue lymphocytes by VACV, we stained tissue-isolated cells for CD3, CD4, CD8, CXCR4, CCR5, and cell-associated vaccinia virus antigens. We evaluated the cytopathicity of VACV for different cell subsets by comparing the number of cells in infected tissues to that in matched uninfected tissues. To account for size differences in tissue blocks, we normalized the data by the weight of the tissue (Fig. 2A).
FIG. 2.

Preferential depletion of the CD4+ CCR5+ cell subset in VACV-inoculated ex vivo-cultured human lymphoid tissues. Tissue blocks (27 from each individual human tonsil donor) were infected with NYCBH. At day 12 postinfection, cells were stained for CD3, CD4, or CD8. Data are means ± SEM (error bars) of the numbers of lymphocyte subsets normalized by the weight of the tissue blocks and by the numbers of corresponding cells in matched uninfected control tissues. A. Depletion of CD3−, CD3+, CD3+/CD4+, and CD3+/CD8+ cells by VACV in human lymphoid tissue ex vivo (n = 12). B. Depletion of CCR5+ versus CCR5− cells within the CD3+ versus the CD3− cell subsets (n = 9). C. Depletion of CCR5+ versus CCR5− cells within the CD3+/CD4+ versus the CD3+/CD8+ cell subsets (n = 9).
Kinetic studies showed that cell depletion was insignificant at day 6 postinfection. It became significant at day 9 of culture when VACV depleted 15.6% ± 1.7% of lymphocytes (14.5% ± 1.9% and 18% ± 4.8% of CD3+ and CD3− cells, respectively; n = 3).
At day 12 postinfection, VACV depleted 29.8% ± 8% of lymphocytes in infected tissues and, similarly to day 9 results, we found no significant difference between VACV-induced depletion of T and non-T cells (n = 10; P = 0.086) (Fig. 1C). Among T cells, a significant preferential depletion of CD8+ over CD4+ T-cell populations was observed (41% ± 6.8% versus 29.8% ± 8.3%; n = 10; P = 0.0019). We next evaluated VACV-induced depletion among cells within the CXCR4+ and CCR5+ lymphocyte subsets. Among CD3+ cells, VACV depleted 47.8% ± 9.7% of CCR5+and 31.6% ± 9.9% of CCR5− cells, demonstrating a preferential depletion of CCR5+ subsets (n = 7; P = 0.0059). This preferential loss of CCR5+ subsets was observed among both CD4+ and CD8+ T cells: within the CD4+ T-cell population, 46.2% ± 11% of CCR5+ cells were depleted compared with 27.8% ± 11.7% of CCR5− cells (n = 7; P = 0.0066), and among CD8+ T lymphocytes, 53.8 ± 7.9% of the CCR5+ cells were depleted versus 36.6% ± 9.6% of CCR5− cells (n = 7; P = 0.0038).
No preferential depletion of CCR5+ cells over CCR5− cells was detected among CD3− lymphocytes (24.8% ± 18.8% versus 30.6% ± 12.5% of CCR5, respectively; n = 7; P = 0.52), nor was any preferential depletion observed between CD4/CXCR4+ and CD4/CXCR4− T cells.
VACV preferentially depletes activated T cells in human tonsillar tissues.
Since VACV downregulates CCR5 (28) and CCR5 expression is linked to cell activation in human lymphoid tissue (21), we investigated the relationship between VACV-induced T-cell depletion and the cellular activation status. For this purpose, we defined as “activated T cells” any CD3+ cells expressing at least one of the following markers: CD25, CD38, CD69, and HLA-DR. As shown in Fig. 3A, VACV depleted 65.1% ± 6.9% of CD3+/CD25+ cells, 65.2% ± 7.5% of CD3+/CD38+ cells, 46.1% ± 6.5% of CD3+/CD69+ cells, and 51.9% ± 9.1% of CD3+/HLA-DR+ cells, while the corresponding activation marker-negative lymphocytes were depleted by only 22.4% ± 9.3% (P = 0.0017), 22.5% ± 6.7% (P = 0.0032), 13.8% ± 10.1% (P = 0.0128), and 21.6% ± 9.4% (P = 0.0222) (n = 7).
FIG. 3.
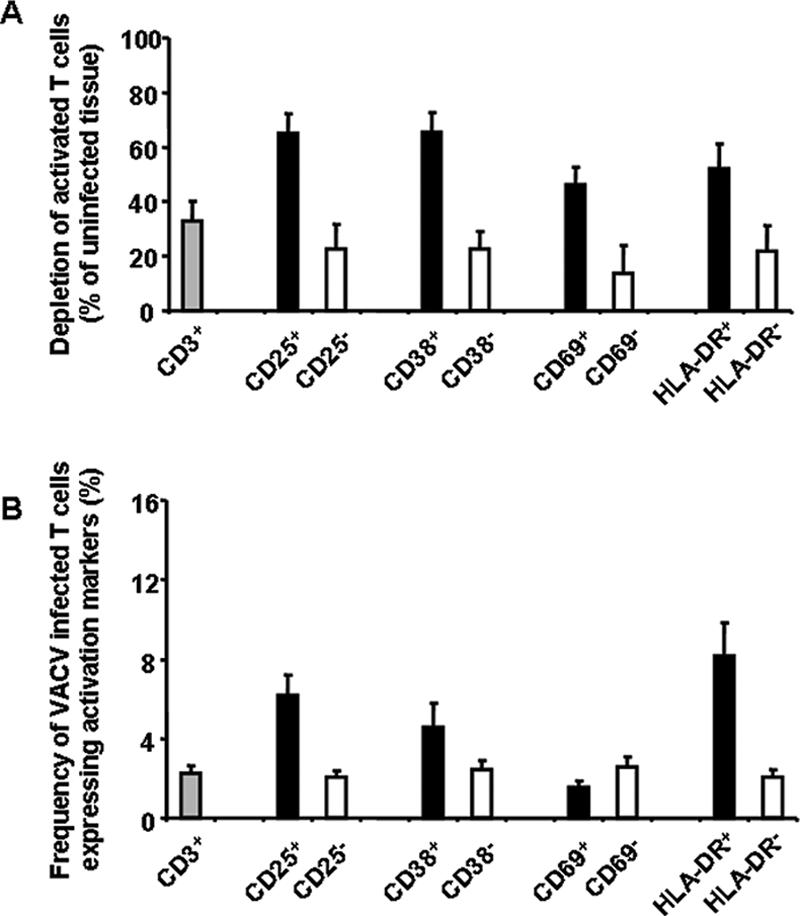
Preferential depletion of activated T cells in human lymphoid tissues inoculated ex vivo with VACV. Tissue blocks (27 from each individual human tonsil donor) were infected with NYCBH. At day 12 postinfection, cells were stained for the following activation markers: CD3, CD25, CD38, CD69, and HLA-DR. Data are means ± SEM (error bars) of the numbers of lymphocytes positive or negative for any of the activation markers listed above normalized by the weight of the tissue blocks and by the numbers of corresponding cells in matched uninfected control tissues. A. Depletion of activated CD3+ cells by VACV in human lymphoid tissue ex vivo (n = 12). B. Fraction of VACV-infected activated T cells detected at day 12 postinfection (n = 7).
We then analyzed the levels of infection of activated T cells by VACV at day 12 postinfection. VACV infected 6.2% ± 1% of CD3+/CD25+ cells, 4.6% ± 1.2% of CD3+/CD38+ cells, 1.6% ± 0.3% of CD3+/CD69+ cells, and 8.2% ± 1.6% of CD3+/HLA-DR+ cells, while the infection rates of the corresponding activation marker-negative lymphocytes were close to the infection rate of all CD3+ cells, i.e., 2.3% ± 0.4% (n = 7; P = 0.002, P = 0.050, P = 0.079, and P = 0.008, respectively). Altogether, these results show that VACV preferentially infects and depletes activated T cells in lymphoid tissue ex vivo.
Interactions between VACV and HIV-1.
Since VACV preferentially depletes CCR5+ cells, we compared the effects of this virus on tissue infection with HIV-1 variants that differ in their coreceptor usage. We used a prototypic R5 HIV-1 variant, R5SF162, and a prototypic X4 HIV-1 variant, X4LAI.04. To study the interactions between VACV and HIV-1 in human lymphoid tissue, we coinfected tissue blocks first with VACV and then within 3 h with HIV-1. HIV-1 replication in coinfected tissues was compared with that in matched singly HIV-1-infected tissues (Fig. 4A).
FIG. 4.

Interactions of VACV and HIV-1 in human lymphoid tissues coinfected ex vivo. Tissue blocks were coinfected with VACV and either R5SF-162 or X4LAI.04 or were infected singly with one of these isolates. HIV-1 replication was evaluated based on measurement of the p24 core antigen. VACV replication was evaluated based on measurement of VACV genome equivalents in the culture medium by using real-time PCR. A. Typical kinetics of HIV-1 replication in tissue blocks infected singly with HIV-1 or coinfected with VACV. Each point represents a measurement of the pooled medium bathing 27 blocks. B. Average HIV-1 replication in tissues coinfected with VACV and HIV-1, relative to that in matched tissues infected singly with HIV-1. Data are means ± SEM (error bars) of experiments with tissues from 13 donors infected ex vivo with R5SF-162 and from 6 donors infected ex vivo with X4LAI.04. C. Average VACV replication in tissues coinfected with VACV and HIV-1, relative to that in matched tissues infected singly with VACV. Data are means ± SEM (error bars) of experiments with tissues from 13 donors infected ex vivo with R5SF-162 and from 6 donors infected ex vivo with X4LAI.04.
Replication of HIV-1 became evident at day 6 postinoculation and increased thereafter, to reach maximum levels that varied among different donors from 2 to 88 ng and from 2 to 100 ng of p24 per milliliter of culture medium for R5SF162 and X4LAI.04, respectively. Despite this variability, X4LAI.04 and R5SF162 HIV-1 variants replicated to approximately the same levels within the tissue from an individual donor: the cumulative production of p24 per milliliter of culture medium was on average 30 ± 4 ng and 24 ± 3 ng for R5SF162 and X4LAI.04, respectively (n = 13 and n = 6). VACV inhibited replication of R5SF162 by 75.2% ± 3.7% (n = 13; P = 0.0005) relative to matched tissues infected by R5SF162 alone (Fig. 4B). Consistent with the inhibition of p24 release, we observed a significant reduction in the number of HIV-1-positive CD4 T cells by using flow cytometry (data not shown). Replication of X4LAI.04 was inhibited in VACV-coinfected tissues by 25.1% ± 7% compared with matched singly X4LAI.04-infected tissues (Fig. 4B) (n = 6; P = 0.06). Inactivated VACV and MVA did not suppress R5SF-162 replication, indicating that VACV-induced inhibition of HIV-1 required VACV replication (data not shown).
We next evaluated the effect of HIV-1 on replication of VACV in coinfected tissues. As described above, replication of VACV was measured by means of a calibrated real-time PCR. The levels of VACV replication in R5SF162- and in X4LAI.04-coinfected tissues were 110.3% ± 24.1% (n = 13; P = 0.68) and 68.7% ± 19.6% (n = 5; P = 0.19), respectively, of the levels detected in tissues singly infected with VACV alone (Fig. 4C). Despite the fact that VACV was inhibited in four out of five VACV/X4LAI.04-coinfected tissue samples, the difference was not statistically significant.
VACV infection does not modulate HIV-1 replication by upregulation of CCL3, CCL4, CCL5, or CXCL12 in lymphoid tissue.
Several viruses have been shown to inhibit R5 HIV-1 by upregulating the CCR5-binding chemokines CCL3, CCL4, and CCL5 (14, 15) and to inhibit X4 HIV-1 by upregulating CXCL12 (35). To investigate whether a similar mechanism was responsible for VACV-induced HIV-1 inhibition, we analyzed the levels of these chemokines in infected tissues. Using the multiplex bead technology, we measured the levels of CCL3, CCL4, CCL5, and CXCL12 released by tissue blocks into the culture medium between days 3 and 12 of culture. Chemokine and cytokine concentrations in the media from tissues infected with VACV or coinfected with VACV plus R5SF162 or VACV plus X4LAI.04 were compared with those in uninfected or singly infected matched tissues. No significant change in the levels of CCL3, CCL4, CCL5, and CXCL12 was detected. In media from tissues infected with VACV, the concentrations of CCL3, CCL4, CCL5, and CXCL12 were, respectively, 93.8% ± 4.4%, 98% ± 5.3%, 104% ± 9.4%, and 109.4% ± 9% of the concentrations measured in control medium bathing matched uninfected tissue blocks (0.2 < P < 0.71; n = 6).
DISCUSSION
In recent years, interest in VACV has increased enormously, largely because of its use for immunization against the potential bioterrorism agent variola virus and its use as a vector for various vaccines (2, 3, 11, 24, 30). Although there is a long history of VACV infection studies, most experimental data have come from studies performed on cell lines or cultures of isolated primary cells infected in vitro.
To understand VACV pathogenesis in the context of human tissues with a rich repertoire of cells that preserve structural and functional relations, more adequate in vivo-like experimental systems are needed. Ex vivo-cultured tonsillar tissue, as used here, is one such experimental system. It harbors cells of different types that retain the expression of CCR5, CXCR4, and other surface molecules necessary for pathogen entry, requires no exogenous activation to support viral replication (13, 15, 17), and allows analysis of the effects of VACV on tissues of the immune system.
In this study, we focused on the role of the chemokine receptor CCR5 in VACV replication, since it has recently been reported (28) that phosphorylation of this molecule plays a critical role in VACV infection of isolated T cells and since the same molecule is involved in HIV-1 entry, indicating possible interactions between VACV and HIV-1.
Using real-time PCR and flow cytometry, we showed that ex vivo lymphoid tissues support productive infection by NYBCH, the VACV strain of Dryvax. We found that VACV readily infected both T and non-T lymphocytes, the latter more efficiently. This observation is in agreement with earlier studies that demonstrated the presence of VACV-infected B and T cells among peripheral blood lymphocytes (1, 31). Among T lymphocytes, the number of infected CD4+ cells was approximately 30% lower than that of infected CD8+ cells. Since in tonsillar tissues the latter are, on average, four times less abundant than the former, the probability of a CD8+ T cell being productively infected is almost six times higher than that of a CD4+ T cell. The predominance of VACV-positive CD8+ T cells over CD4+ T cells at day 12 postinoculation could be the result of selective cell depletion. The same may be true regarding the predominance of VACV-positive B over T cells. However, in spite of the difference in infection, the levels of depletion of T and B cells over 12 days of infection were similar. These results suggest that infected B cells survive longer than infected T cells. This longer survival may be explained by the fact that, in contrast to T cells, infection of B cells is abortive because of the lack of transcription of late viral genes (8).
Both CD4+ and CD8+ T cells can be further divided into subtypes according to their expression of the chemokine receptors CCR5 and/or CXCR4, which also serve as HIV-1 coreceptors. We found that among T cells, depletion preferentially occurs in cells expressing CCR5. Our analysis might, however, underestimate the depletion of these cells, since it is possible that they are expressing CCR5 at levels that are below the limit of detection but sufficient for VACV replication, leading to their depletion. These cells would be then counted as CCR5− in our flow cytometric assay. A similar phenomenon has been already suggested for lamina propria lymphocytes in the case of HIV-1 infection (23).
We showed that VACV-infected tissues were depleted of activated cells expressing at least one of the following activation markers: CD25, CD38, CD69, and HLA-DR. It seems that this reflects preferential cell death rather than downregulation of these molecules, since it is unlikely that VACV downregulates all of these functionally distinct activation markers. Also, activated cells may express an extracellular VACV receptor (8) that makes them a preferential target for VACV infection. CCR5 is mainly expressed on activated T cells. Therefore, the great decrease in the number of activated cells also strongly suggests a preferential depletion of CCR5-expressing cells rather than CCR5 downregulation.
Among B cells, in contrast to T cells, no preferential depletion of CCR5+ over CCR5− cells was observed. The lack of discrimination in depletion of CCR5+ and CCR5− B cells may be related to the lack of CCR5 activity in these cells (20). Indeed, since VACV infection requires activation of CCR5-controlled signaling, the absence of functional CCR5 would explain why VACV infection is abortive in B but not in T cells.
To study VACV-HIV-1 interactions, human lymphoid tissue ex vivo was coinfected with VACV and either a prototypic R5 HIV-1 variant, R5SF162, or a prototypic X4 HIV-1 variant, X4LAI.04. While VACV significantly inhibited R5SF162 replication, the replication of X4LAI.04 was inhibited only mildly and nonsignificantly. VACV preferentially depletes activated cells. Since activated cells are mainly CCR5-expressing cells and constitute the main producer of HIV-1, it is conceivable that HIV-1 inhibition is related to VACV-induced depletion of this specific cell subset. Moreover, VACV induces a strong depletion of CD4+ CCR5+ T cells, thus additionally inhibiting R5 HIV-1 replication. In contrast, depletion of CD4+ CXCR4+ T cells was mild and, accordingly, the replication of X4 HIV-1 was inhibited mildly. Inactivated VACV did not deplete T cells and did not inhibit the replication of either R5 or X4 HIV-1, indicating that replication of VACV is necessary for the observed inhibition of HIV-1 infection. Conversely, no significant effect of R5SF162 was found on VACV replication, while X4LAI.04 decreased VACV replication by more than 30% in tissues from four out of five donors.
Although depletion of target cells seems to be a plausible explanation for HIV-1 suppression, we investigated whether upregulation of chemokines interfering with HIV-1 coreceptor binding can contribute to this phenomenon, as has been demonstrated for several other viruses (15, 35). We found that the average concentrations of CC chemokines (CCL3, -4, and -5) were similar in the culture media from R5SF162-VACV-coinfected tissues and from matched tissues singly infected with R5SF162. Likewise, in medium from X4LAI.04-VACV coinfected tissues, the average concentration of CXCL12 was similar to that in matched singly X4LAI.04-infected tissues, indicating that upregulation of these chemokines was not the cause of HIV-1 inhibition.
X4LAI.04 upregulates the above-mentioned CC chemokines in infected tissues, as previously described (14, 15, 35). This upregulation of CCR5-specific ligands may explain the decrease of VACV replication observed in X4LAI.04-VACV coinfection, since VACV replication in T cells involves tyrosine phosphorylation of CCR5 signaling pathways. Consistent with this idea, we found that 100 μM CCL5 strongly inhibited VACV replication in ex vivo lymphoid tissue (data not shown). It is interesting that VACV evolved to secrete a CC chemokine inhibitor (vCCI), a soluble factor which binds natural ligands of CCR5 (7, 33). It is well established that this protein protects the virus from the cellular immune response (32) by inhibiting leukocyte trafficking (5, 6). In addition, our results suggest that vCCI may also directly contribute to the increase of viral production by preventing CCR5 natural ligands from inhibiting VACV replication.
In summary, we have described a novel experimental approach to study the replication and pathogenesis of VACV in human lymphoid tissues. Since VACV replicates efficiently ex vivo in tonsillar tissue, this model can be used to evaluate various VACV vectors for recombinant vaccine development. Using this system, we have demonstrated that VACV preferentially infects non-T cells. However, the major depletion occurs in activated T cells, the principal site of productive HIV-1 infection. As the CCR5+ T cells are preferentially depleted at the same time, replication of R5-tropic HIV-1 is particularly suppressed in tissues coinfected by VACV and HIV-1.
Acknowledgments
We are grateful to Bernard Moss for helpful comments and M. Santi and the entire staff of the Department of Anatomic Pathology of Children's National Medical Center for their generous assistance in obtaining human tonsillar tissues.
This research was supported, in part, by the Intramural Research Program of the NIH National Institute of Child Health and Human Development and by funds from the Department of Microbiology, Immunology and Tropical Medicine at the George Washington University Medical Center.
Footnotes
Published ahead of print on 5 September 2007.
REFERENCES
- 1.Alonso, J. M., J. Rodriguez, E. Vinuela, G. Kroemer, and C. Martinez. 1991. Highly efficient expression of proteins encoded by recombinant vaccinia virus in lymphocytes. Scand. J. Immunol. 34:619-626. [DOI] [PubMed] [Google Scholar]
- 2.Amara, R. R., F. Villinger, J. D. Altman, S. L. Lydy, S. P. O'Neil, S. I. Staprans, D. C. Montefiori, Y. Xu, J. G. Herndon, L. S. Wyatt, M. A. Candido, N. L. Kozyr, P. L. Earl, J. M. Smith, H. L. Ma, B. D. Grimm, M. L. Hulsey, J. Miller, H. M. McClure, J. M. McNicholl, B. Moss, and H. L. Robinson. 2001. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292:69-74. [DOI] [PubMed] [Google Scholar]
- 3.Breman, J. G., and D. A. Henderson. 2002. Diagnosis and management of smallpox. N. Engl. J. Med. 346:1300-1308. [DOI] [PubMed] [Google Scholar]
- 4.Broccolo, F., G. Locatelli, L. Sarmati, S. Piergiovanni, F. Veglia, M. Andreoni, S. Butto, B. Ensoli, P. Lusso, and M. S. Malnati. 2002. Calibrated real-time PCR assay for quantitation of human herpesvirus 8 DNA in biological fluids. J. Clin. Microbiol. 40:4652-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bursill, C. A., J. L. Cash, K. M. Channon, and D. R. Greaves. 2006. Membrane-bound CC chemokine inhibitor 35K provides localized inhibition of CC chemokine activity in vitro and in vivo. J. Immunol. 177:5567-5573. [DOI] [PubMed] [Google Scholar]
- 6.Bursill, C. A., R. P. Choudhury, Z. Ali, D. R. Greaves, and K. M. Channon. 2004. Broad-spectrum CC-chemokine blockade by gene transfer inhibits macrophage recruitment and atherosclerotic plaque formation in apolipoprotein E-knockout mice. Circulation 110:2460-2466. [DOI] [PubMed] [Google Scholar]
- 7.Carfi, A., C. A. Smith, P. J. Smolak, J. McGrew, and D. C. Wiley. 1999. Structure of a soluble secreted chemokine inhibitor vCCI (p35) from cowpox virus. Proc. Natl. Acad. Sci. USA 96:12379-12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chahroudi, A., R. Chavan, N. Kozyr, E. K. Waller, G. Silvestri, and M. B. Feinberg. 2005. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J. Virol. 79:10397-10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Condack, C., J. C. Grivel, P. Devaux, L. Margolis, and R. Cattaneo. 2007. Measles virus vaccine attenuation: suboptimal infection of lymphatic tissue and tropism alteration. J. Infect. Dis. 196:541-549. [DOI] [PubMed] [Google Scholar]
- 10.Drexler, I., C. Staib, and G. Sutter. 2004. Modified vaccinia virus Ankara as antigen delivery system: how can we best use its potential? Curr. Opin. Biotechnol. 15:506-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earl, P. L., J. L. Americo, L. S. Wyatt, L. A. Eller, J. C. Whitbeck, G. H. Cohen, R. J. Eisenberg, C. J. Hartmann, D. L. Jackson, D. A. Kulesh, M. J. Martinez, D. M. Miller, E. M. Mucker, J. D. Shamblin, S. H. Zwiers, J. W. Huggins, P. B. Jahrling, and B. Moss. 2004. Immunogenicity of a highly attenuated MVA smallpox vaccine and protection against monkeypox. Nature 428:182-185. [DOI] [PubMed] [Google Scholar]
- 12.Glushakova, S., B. Baibakov, L. B. Margolis, and J. Zimmerberg. 1995. Infection of human tonsil histocultures: a model for HIV pathogenesis. Nat. Med. 1:1320-1322. [DOI] [PubMed] [Google Scholar]
- 13.Glushakova, S., J. C. Grivel, W. Fitzgerald, A. Sylwester, J. Zimmerberg, and L. B. Margolis. 1998. Evidence for the HIV-1 phenotype switch as a causal factor in acquired immunodeficiency. Nat. Med. 4:346-349. [DOI] [PubMed] [Google Scholar]
- 14.Grivel, J. C., M. Garcia, W. J. Moss, and L. B. Margolis. 2005. Inhibition of HIV-1 replication in human lymphoid tissues ex vivo by measles virus. J. Infect. Dis. 192:71-78. [DOI] [PubMed] [Google Scholar]
- 15.Grivel, J. C., Y. Ito, G. Faga, F. Santoro, F. Shaheen, M. S. Malnati, W. Fitzgerald, P. Lusso, and L. Margolis. 2001. Suppression of CCR5- but not CXCR4-tropic HIV-1 in lymphoid tissue by human herpesvirus 6. Nat. Med. 7:1232-1235. [DOI] [PubMed] [Google Scholar]
- 16.Grivel, J. C., N. Malkevitch, and L. Margolis. 2000. Human immunodeficiency virus type 1 induces apoptosis in CD4+ but not in CD8+ T cells in ex vivo-infected human lymphoid tissue. J. Virol. 74:8077-8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grivel, J. C., and L. B. Margolis. 1999. CCR5- and CXCR4-tropic HIV-1 are equally cytopathic for their T-cell targets in human lymphoid tissue. Nat. Med. 5:344-346. [DOI] [PubMed] [Google Scholar]
- 18.Grivel, J. C., F. Santoro, S. Chen, G. Faga, M. S. Malnati, Y. Ito, L. Margolis, and P. Lusso. 2003. Pathogenic effects of human herpesvirus 6 in human lymphoid tissue ex vivo. J. Virol. 77:8280-8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillaume, J. C., P. Saiag, J. Wechsler, M. C. Lescs, and J. C. Roujeau. 1991. Vaccinia from recombinant virus expressing HIV genes. Lancet 337:1034-1035. [DOI] [PubMed] [Google Scholar]
- 20.Honczarenko, M., Y. Le, A. M. Glodek, M. Majka, J. J. Campbell, M. Z. Ratajczak, and L. E. Silberstein. 2002. CCR5-binding chemokines modulate CXCL12 (SDF-1)-induced responses of progenitor B cells in human bone marrow through heterologous desensitization of the CXCR4 chemokine receptor. Blood 100:2321-2329. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson, I., J. C. Grivel, S. S. Chen, A. Karlsson, J. Albert, E. M. Fenyo, and L. B. Margolis. 2005. Differential pathogenesis of primary CCR5-using human immunodeficiency virus type 1 isolates in ex vivo human lymphoid tissue. J. Virol. 79:11151-11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lisco, A., J. C. Grivel, A. Biancotto, C. Vanpouille, F. Origgi, M. S. Malnati, D. Schols, P. Lusso, and L. B. Margolis. 2007. Viral interactions in human lymphoid tissue: human herpesvirus 7 suppresses the replication of CCR5-tropic human immunodeficiency virus type 1 via CD4 modulation. J. Virol. 81:708-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattapallil, J. J., D. C. Douek, B. Hill, Y. Nishimura, M. Martin, and M. Roederer. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093-1097. [DOI] [PubMed] [Google Scholar]
- 24.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott-Raven Publishers, Philadelphia, PA.
- 25.Moss, B. 2006. Poxvirus entry and membrane fusion. Virology 344:48-54. [DOI] [PubMed] [Google Scholar]
- 26.Olson, V. A., T. Laue, M. T. Laker, I. V. Babkin, C. Drosten, S. N. Shchelkunov, M. Niedrig, I. K. Damon, and H. Meyer. 2004. Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J. Clin. Microbiol. 42:1940-1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picard, O., J. Lebas, J. C. Imbert, P. Bigel, and D. Zagury. 1991. Complication of intramuscular/subcutaneous immune therapy in severely immune-compromised individuals. J. Acquir. Immune Defic. Syndr. 4:641-643. [PubMed] [Google Scholar]
- 28.Rahbar, R., T. T. Murooka, A. A. Hinek, C. L. Galligan, A. Sassano, C. Yu, K. Srivastava, L. C. Platanias, and E. N. Fish. 2006. Vaccinia virus activation of CCR5 invokes tyrosine phosphorylation signaling events that support virus replication. J. Virol. 80:7245-7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Redfield, R. R., D. C. Wright, W. D. James, T. S. Jones, C. Brown, and D. S. Burke. 1987. Disseminated vaccinia in a military recruit with human immunodeficiency virus (HIV) disease. N. Engl. J. Med. 316:673-676. [DOI] [PubMed] [Google Scholar]
- 30.Robinson, H. L. 2003. Prime boost vaccines power up in people. Nat. Med. 9:642-643. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez-Puig, J. M., L. Sanchez, G. Roy, and R. Blasco. 2004. Susceptibility of different leukocyte cell types to vaccinia virus infection. Virol. J. 1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21:377-423. [DOI] [PubMed] [Google Scholar]
- 33.Smith, C. A., T. D. Smith, P. J. Smolak, D. Friend, H. Hagen, M. Gerhart, L. Park, D. J. Pickup, D. Torrance, K. Mohler, K. Schooley, and R. G. Goodwin. 1997. Poxvirus genomes encode a secreted, soluble protein that preferentially inhibits beta chemokine activity yet lacks sequence homology to known chemokine receptors. Virology 236:316-327. [DOI] [PubMed] [Google Scholar]
- 34.Vanderplasschen, A., M. Hollinshead, and G. L. Smith. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79:877-887. [DOI] [PubMed] [Google Scholar]
- 35.Xiang, J., S. L. George, S. Wunschmann, Q. Chang, D. Klinzman, and J. T. Stapleton. 2004. Inhibition of HIV-1 replication by GB virus C infection through increases in RANTES, MIP-1α, MIP-1β, and SDF-1. Lancet 363:2040-2046. [DOI] [PubMed] [Google Scholar]
- 36.Yu, Q., B. Jones, N. Hu, H. Chang, S. Ahmad, J. Liu, M. Parrington, and M. Ostrowski. 2006. Comparative analysis of tropism between canarypox (ALVAC) and vaccinia viruses reveals a more restricted and preferential tropism of ALVAC for human cells of the monocytic lineage. Vaccine 24:6376-6391. [DOI] [PubMed] [Google Scholar]