

Abstract
Covalently closed circular DNA (cccDNA) of hepatitis B virus (HBV) is formed by conversion of capsid-associated relaxed circular DNA (rcDNA) via unknown mechanisms and exists in the nucleus of the infected hepatocyte as a minichromosome that serves as the transcription template for viral RNAs. To study the molecular pathway of cccDNA formation and its regulation by viral and cellular factors, we have established a cell line that supports the replication of an envelope protein-deficient HBV genome in a tetracycline-inducible manner. Following induction of HBV replication, the cells accumulate higher levels of cccDNA as well as larger amounts of deproteinized rcDNA (DP-rcDNA) than cells that replicate wild-type HBV genomes. These results indicate that HBV envelope proteins negatively regulate cccDNA formation, and conversion of DP-rcDNA into cccDNA is a rate-limiting step of cccDNA formation in HepG2 cells. Detailed analyses reveal the following: (i) DP-rcDNA exists in both cytoplasm and nucleus; (ii) while nuclear DP-rcDNA is sensitive to DNase I digestion, a small fraction of cytoplasmic DP-rcDNA is DNase I resistant; (iii) both DNase I-sensitive and -resistant cytoplasmic DP-rcDNAs cosediment with capsids and can be immunoprecipitated with HBV core antibody; and (iv) a primer extension assay maps the 5′ end of the minus strand of DP-rcDNA at the authentic end of virion rcDNA. Hence, our results favor a hypothesis that the removal of viral polymerase protein covalently linked to the 5′ end of the minus-strand DNA occurs inside the capsid in the cytoplasm and most possibly via a reaction that cleaves the phosphodiester bond between the tyrosine of the polymerase and the 5′ phosphoryl group of minus-strand DNA.
Hepatitis B virus (HBV) causes transient and chronic infections of the liver. Transient infection frequently leads to acute hepatitis but, in rare cases, results in fatal, fulminant hepatitis. Chronic infection represents a major public health problem affecting an estimated 400 million individuals worldwide and carries a high risk for the development of chronic active hepatitis, cirrhosis, and primary hepatocellular carcinoma (20, 27).
HBV is the prototype virus of the Hepadnaviridae family and contains a relaxed circular partially double-stranded DNA (rcDNA; 3.2 kb in length) genome (43). A hallmark of HBV genomic DNA replication is protein-primed reverse transcription of an RNA intermediate called pregenomic RNA (pgRNA) (42, 49). The overall replication scheme of HBV is related to that of retroviruses but is mechanistically distinct (40). One of the most obvious differences is that the integration of viral genomic DNA into host cellular chromosomes is not an obligatory step in the HBV life cycle. Instead, upon the entry into hepatocytes, viral genomic DNA (rcDNA) in the nucleocapsid is transported into the nucleus and converted into an episomal covalently closed circular DNA (cccDNA), which serves as the template for the transcription of viral RNAs (3, 31, 40). As a key replication intermediate in HBV infection, a small and regulated number (approximately 10 to 50 copies per cell) of cccDNAs in the nuclei of infected hepatocytes are formed during the early phase of infection and stably maintained throughout persistent infection (30, 48, 53, 56). Therapeutic elimination of cccDNA with highly active viral DNA polymerase inhibitors has not been achieved and remains a major challenge to a cure for chronic HBV infections (19, 23, 26, 59). Further investigations into the molecular mechanism of cccDNA formation and maintenance are clearly warranted.
The molecular pathway of cccDNA formation from its rcDNA precursor residing in the cytoplasmic nucleocapsids would include as necessary steps viral capsid disassembly, transport of rcDNA into the nucleus, and conversion of rcDNA into cccDNA. Thus far, it is not yet known where in the cells capsid disassembly occurs and how rcDNA is delivered into the nucleus (17). Moreover, explanations of the conversion of rcDNA into cccDNA must account for the distinct structural features of rcDNA illustrated in Fig. 1. These include the following: (i) the minus strand contains a terminal redundant sequence and is longer than unit length, but the plus strand is incompletely synthesized; (ii) viral DNA polymerase is covalently linked to the 5′ terminus of minus-strand DNA as a result of protein-primed minus-strand DNA synthesis (9, 50); and (iii) the 5′ end of the plus-strand DNA is linked to a 5′ capped RNA oligonucleotide of 17 to 18 nucleotides (nt) in length as a result of RNA-primed plus-strand DNA synthesis (9, 29). It is quite obvious that for cccDNA formation, these obstructive terminal modifications must be removed, the gap in plus-strand DNA needs to be filled in, and both strands of DNA must be covalently ligated (Fig. 1). How these biochemical reactions are achieved remains to be resolved. While it is reasonable to envisage that general cellular DNA repair enzymes may catalyze the removal of RNA primer and the ligation of the two ends of both DNA strands, the removal of viral polymerase protein from the 5′ end of minus-strand DNA represents an unusual reaction. Biochemically, this deproteinization reaction can be achieved via either nucleolytic cleavage of the viral DNA close to the 5′ end by an endonuclease or hydrolysis of the phosphodiester bond between the tyrosine residue of polymerase and the 5′ phosphoryl group of minus-strand DNA. The role of cellular and/or viral proteins, such as DNA polymerase itself, in this reaction remains to be determined. Also, it is not yet known whether the deproteinization of rcDNA occurs before or after its translocation across the nuclear membrane (40).
FIG. 1.

Schematic illustration of rcDNA structural features and requirement for the conversion of rcDNA into cccDNA. Incompletely synthesized plus-strand DNA is indicated with dashed line. RNA primer for plus-strand DNA synthesis is shown as curved line, and sequence is shown in lowercase letters. Polymerase (pol) covalently linked to the 5′ end of minus-strand rcDNA is highlighted with a filled circle. Direct repeat (DR) sequences 1 and 2 are indicated.
A major obstacle to answering these questions is that HBV cccDNA formation in cultured cells is very inefficient, and isolation of intermediates that occur during cccDNA formation has been difficult to achieve. To overcome this problem, we have established a stable cell line that supports the replication of an envelope protein-deficient HBV genome in a tetracycline-inducible manner and demonstrated that the cells accumulate higher levels of cccDNA and, more strikingly, larger amounts of deproteinized rcDNA (DP-rcDNA) than cells that support the replication of the wild-type HBV genome. While this article was in preparation, Gao and Hu (8) reported a similar observation by a transient transfection assay with plasmids containing wild-type and envelope protein-deficient HBV genomes and convincingly demonstrated that the DP-rcDNA (named protein-free rcDNA by these authors) is derived from capsid-associated rcDNA upon removal of its covalently attached polymerase protein. In this report, we further extended these observations and provide evidence showing that DP-rcDNA is a functional precursor of cccDNA and that the removal of HBV genome-bound viral DNA polymerase occurs inside the viral capsid in the cytoplasm and most probably via a reaction that hydrolyzes the phosphodiester bond between the tyrosine of polymerase and the 5′ phosphoryl group of minus-strand DNA. The work reported herein provides insight to the molecular pathway of cccDNA formation.
MATERIALS AND METHODS
Plasmids.
To construct the tetracycline-inducible-HBV pgRNA expression plasmid pTREHBV, a DNA fragment containing HBV nt 2331 to 3182 and 1 to 1990 (GenBank accession number V01460) was retrieved from pCMVHBV (where CMV is cytomegalovirus) (6) by BspEI digestion and blunted with Klenow enzyme, followed by further digestion with PciI. The obtained 2.8-kb DNA fragment was then ligated to pTRE2 vector (Clontech, Mountain View, CA) which had been cut with HindIII, blunted, and digested with PciI, to yield plasmid pTRE2/HBV. Then, a PCR fragment containing the CMV-IE TATA box and HBV nt 1805 to 2618 was amplified from pCMVHBV by using the primers 5′-ATGGTACCCGGGTCGAGTAGGCGTGTACGGTGGGA-3′ and 5′-TCTCATTAACTGTGAGTGGGCCTA-3′. The PCR fragment was restricted with KpnI and PciI and ligated with pTRE2/HBV that had been digested by the same enzymes to yield plasmid pTREHBV. To eliminate the 5′ AUG codon of the precore open reading frame, a single nucleotide deletion (1816A) mutation was introduced into the HBV genome in pTREHBV by using a QuickChange kit (Stratagene, La Jolla, CA). The resulting plasmid was designated pTREHBVDE. To obtain HBV genome that is unable to produce three envelope proteins, two tandem stop codons were introduced into the coding region of the amino terminus of the small (S) envelope protein (217TTGTTG222 to 217TAGTAG222; mutations are underlined) in plasmid pTREHBVDE with a QuickChange kit to yield plasmid pTREHBVDES. The intended mutations in both plasmids were confirmed by sequence analysis. Plasmids pE and pS that express large (L) and middle (M)/S HBV envelope proteins, respectively, were described previously and kindly provided by Volker Bruss (University of Gottingen, Germany) (39).
Cell culture.
HepG2 cells were obtained from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM)-F12 medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The HepAD38 cell line was kindly provided by Christoph Seeger (Fox Chase Cancer Center, Philadelphia, PA) (18). The cell lines HepDE19 and HepDES19 were developed upon the principles of the HepAD38 cell line with some modifications. Briefly, the HepG2 cells were transfected with plasmid pTet-off (Clontech) that expresses the Tet-responsive transcriptional activator and plasmid pTREHBVDE or pTREHBVDES, in which HBV pgRNA expression is controlled by a CMV early promoter with a tetracycline-responsive element. Transfected HepG2 cells were selected with 500 μg/ml G418 in the presence of 1 μg/ml tetracycline. G418-resistant colonies were picked and expanded into cell lines. HBV replication was induced by culturing cells in tetracycline-free medium, and the levels of viral DNA replicative intermediates were determined by Southern blot hybridization. The cell lines with high levels of HBV replication were chosen and designated as HepDE19 and HepDES19. All three HBV-expressing stable cell lines were cultured in the same way as HepG2, except for the addition of G418 at 500 μg/ml. Where needed, tetracycline was routinely added at 1 μg/ml to suppress HBV pgRNA transcription (18). dstet5 is an LMH cell-derived cell line that supports the replication of an envelope protein-deficient duck hepatitis B virus (DHBV) genome in the tetracycline-inducible manner, and it was cultured as previously described (11).
Transient transfection assay.
HepDES19 cells were seeded into 35-mm diameter dishes at a density of 1.2 × 106 cells per dish and cultured in antibiotic-free complete DMEM-F12 medium. Six hours postseeding, each well was transfected with either plasmid pE (2 μg) and plasmid pS (2 μg) to express HBV envelope proteins (L, M, and S) or with 4 μg of vector plasmid (pUC19) with Lipofectamine 2000 (Invitrogen), according to the manufacturer's directions. Transfected cells were cultured in the absence of tetracycline. Cells and culture fluids were harvested at the indicated time points (see Fig. 4), and intracellular HBV core DNA and protein-free viral DNA were extracted and analyzed by Southern blot hybridization. The levels of HBV particles in culture medium were analyzed by particle assay as described below.
FIG. 4.
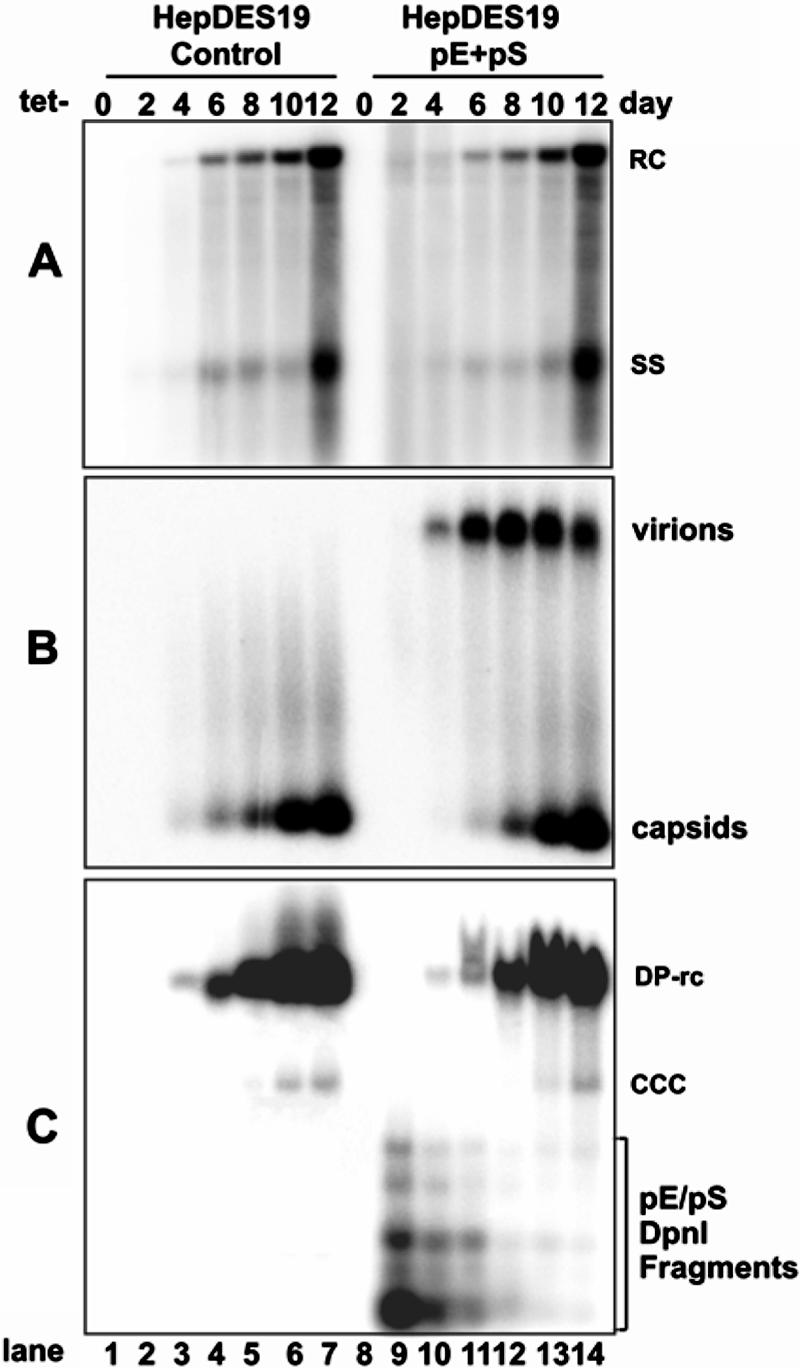
HBV envelope proteins regulate viral cccDNA accumulation in cell culture. HepDES19 cells were seeded in 35-mm dishes at a density of 1 × 106. Six hours later, cells were transfected with a total of 4 μg of plasmid pE (2 μg) and pS (2 μg) or 4 μg of control vector plasmid pUC19. After transfection, cells were cultured in the absence of tetracycline (Tet−). Cells and culture medium were harvested at the indicated time points. Intracellular HBV core DNA (A), viral particle-associated DNA (B), and protein-free viral DNA (C) were subjected to analysis as described in the legend of Fig. 2. Protein-free DNA samples were treated with DpnI prior to loading to digest input plasmid DNA. Fragments from the DpnI-digested plasmids are indicated. RC, rcDNA; CCC, cccDNA; SS, single-strand DNA.
Viral DNA analysis.
Intracellular viral core DNA was extracted as described previously (10). Briefly, cells from one 35-mm diameter dish were lysed with 0.5 ml of lysis buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% NP-40, and 2% sucrose at 37°C for 10 min. Cell debris and nuclei were removed by centrifugation, and the supernatant was mixed with 130 μl of 35% polyethylene glycol (PEG) 8000 containing 1.5 M NaCl. After a 1-h incubation in ice, viral nucleocapsids were pelleted by centrifugation at 10,000 × g for 5 min at 4°C, followed by a 1-h digestion at 37°C in 400 μl of digestion buffer containing 0.5 mg/ml pronase (Calbiochem), 0.5% sodium dodecyl sulfate (SDS), 150 mM NaCl, 25 mM Tris-HCl (pH 8.0), and 10 mM EDTA. The digestion mixture was extracted twice with phenol, and DNA was precipitated with ethanol and dissolved in TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) buffer. One-third of the DNA sample from each plate was resolved by electrophoresis into a 1.5% agarose gel. The gel was then subjected to denaturation in a solution containing 0.5 M NaOH and 1.5 M NaCl, followed by neutralization in a buffer containing 1 M Tris-HCl (pH 7.4) and 1.5 M NaCl. DNA was then blotted onto Hybond-XL membrane (GE Healthcare) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer.
Extraction of protein-free viral DNA (cccDNA and protein-free rcDNA) was carried out by using a modified Hirt extraction procedure (14, 58). Briefly, cells from one 35-mm diameter dish were lysed in 3 ml of 10 mM Tris-HCl (pH 7.5), 10 mM EDTA, and 0.7% SDS. After a 30-min incubation at room temperature, the lysate was transferred into a 15-ml tube, and this step was followed by the addition of 0.8 ml of 5 M NaCl and incubation at 4°C overnight. The lysate was then clarified by centrifugation at 12,000 × g for 30 min at 4°C and extracted twice with phenol and once with phenol:chloroform. DNA was precipitated with two volumes of ethanol overnight at room temperature and dissolved in TE buffer. One-third of the protein-free DNA sample was then resolved in a 1.2% agarose gel and transferred onto Hybond-XL membrane.
For the detection of HBV DNA, membranes were probed with a [α-32P]UTP (800 Ci/mmol; Perkin Elmer)-labeled minus- or plus-strand-specific full-length HBV riboprobe. Hybridization was carried out in 5 ml of EKONO hybridization buffer (Genotech) with a 1-h prehybridization at 65°C and overnight hybridization at 65°C, followed by a 1-h wash with 0.1× SSC and 0.1% SDS at 65°C. The membrane was exposed to a phosphorimager screen, and hybridization signals were quantified with QuantityOne software (Bio-Rad).
Viral particle assay.
The secreted viral particles in culture medium were assayed as previously described with modifications (22). Briefly, the viral particles were precipitated from 1 ml of culture medium by adding PEG 8000 to a final concentration of 10% and incubated at 4°C for 1 h. The precipitates were collected by centrifugation at 925 × g for 20 min and dissolved in 40 μl of DMEM-F12 medium. The viral particles were fractionated by electrophoresis through nondenaturing 1% agarose gels and transferred to a nitrocellulose filter by blotting with TNE buffer (10 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 1 mM EDTA). To detect HBV surface and core antigens, membrane was soaked in phosphate-buffered saline buffer containing 2.5% formaldehyde at room temperature for 10 min. After being briefly rinsed with water, the membrane was then fixed with 50% methanol at room temperature for 30 min and washed three times with water. Membranes were blocked with phosphate-buffered saline containing 0.1% Tween 20 and 5% nonfat dry milk and probed with a monoclonal antibody against HBV surface antigen ([HBsAg] catalog no. M3506; Dako) or with polyclonal antibody against HBV core protein (Zymed catalog no. 18-0002). Bound antibodies were revealed by horseradish peroxidase (HRP)-labeled secondary antibodies and visualized with an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech) according to the protocol of the manufacturer. For the detection of HBV DNA, the DNA-containing particles on the membrane were denatured with a solution containing 0.5 N NaOH and 1.5 M NaCl, and this step was followed by neutralization with a solution containing 1 M Tris-HCl (pH 7.6) and 1.5 M NaCl. HBV DNA was detected by hybridization with a [α-32P]UTP (800 Ci/mmol; Perkin Elmer)-labeled minus-strand-specific full-length HBV riboprobe.
Alkaline gel analysis of HBV DNA.
Total and protein-free viral DNAs were extracted from dstet5 or HepDES19 cells as described above and mock treated or digested with EcoRI or BspEI. Viral DNAs were then resolved in 1.5% agarose gel containing 50 mM NaOH and 1 mM EDTA and blotted onto nylon membrane. Viral DNAs were detected by hybridization.
Cell fractionation. (i) DHBV-infected liver tissue.
Approximately 70 mg of frozen duck liver tissue was homogenized with a dounce homogenizer (Bellco) in 1 ml of solution containing 10 mM Tris-HCl-1 mM EDTA. Nuclei and cytoplasmic contents were separated by low-speed centrifugation (1,000 × g for 5 min at 4°C). Cell nuclei were suspended in TE buffer. Total and protein-free viral DNAs were extracted from whole-cell lysate or nuclear and cytoplasmic fractions with or without prior DNase I digestion. Viral DNA was analyzed by Southern blot hybridization.
(ii) HepDES19 cells.
The cytoplasmic and nuclear fractions of HepDES19 cells that were cultured in the absence of tetracycline for 10 days were separated with a Qproteome Cell Compartment Kit (item 37502; QIAGEN) by following the manufacturer's directions. Purity of the cytoplasmic and nuclear fractions was confirmed by measuring cytoplasm- and nuclear-specific protein markers, annexin I and lamin A/C, respectively, by Western blot assay. Total and protein-free viral DNAs were extracted from both nuclear and cytoplasmic fractions with or without prior DNase I digestion. For the immunoprecipitation assay, 1 ml of cell fractions prepared from approximately 1 × 106 cells was mixed with 50 μl of Sepharose 4B-protein A beads that were preabsorbed with 160 μg of rabbit polyclonal antibody against HBV core antigen (HBcAg) (catalog no. B0586; Dako) and incubated at 4°C overnight. Beads were washed three times with TNE buffer, and protein-free DNA was extracted with or without prior digestion of DNase I. Viral DNA was analyzed by Southern blot hybridization.
Sedimentation equilibrium gradient analysis.
Gradients were formed by overlaying 3 ml of 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, and 60% (wt/vol) sucrose solutions in TNE buffer. Three milliliters of the cytoplasmic fraction of cell lysates obtained from approximately 2 × 107 HepDES19 cells cultured in the absence of tetracycline for 10 days was overlaid on the gradients. Samples were centrifuged at 24,000 rpm for 16 h at 4°C by using Beckman SW28 rotor (Palo Alto, CA). Seventeen fractions of approximately 2.25 ml were collected from the bottom of the tube.
For detection of HBcAg, 2 μl of each fraction was spotted onto nitrocellulose membrane and probed with rabbit anti-HBc (Dako), and bound antibody was detected with HRP-labeled goat anti-rabbit immunoglobulin G and chemiluminescence. For detection of HBV DNA, 50 μl of each fraction was spotted onto nylon membrane and denatured with a solution containing 0.5 N NaOH and 1.5 M NaCl, followed by neutralization with a solution containing 1 M Tris-HCl (pH 7.6) and 1.5 M NaCl. HBV DNA was revealed by hybridization. A total of 100 μl of each fraction was subjected to total viral DNA extraction with pronase digestion. Five hundred microliters of each fraction was used to extract protein-free DNA without or with prior DNase I digestion. For immunoprecipitation analysis, 500 μl of each fraction was mixed with an equal volume of TNE buffer and then combined with 50 μl of Sepharose 4B-protein A beads that had preabsorbed 160 μg of rabbit anti-HBcAg. The mixture was incubated at 4°C overnight. Beads were washed three times with TNE buffer, and protein-free DNA was extracted without or with prior DNase I digestion. Viral DNA was resolved on 1.2% agarose gel and detected by Southern blot hybridization.
Primer extension assay.
A primer extension reaction was carried out essentially as described by Loeb and Tian (25). Briefly, approximately 1 ng of DHBV virion DNA extracted from DHBV-positive duck serum or cytoplasmic DP-rcDNA extracted from DHBV-infected duck liver tissue was mixed with 20 ng of 32P end-labeled oligonucleotide primer that annealed to the sequence spanning nt 2375 to 2396 of DHBV minus-strand DNA (57) plus 0.5 μl of Advantage-HF 2 DNA polymerase and the accompanying polymerase buffer and deoxynucleoside triphosphates (Clontech). Water was supplied to bring the reaction mixture volume to 25 μl. The reaction mixture was denatured at 95°C for 5 min and then transferred to a thermocycler (Biometra) for 30 cycles of 95°C for 30 s, 60°C for 30 s, and 70°C for 30 s, with an additional 2-min extension at 70°C after the last cycle. DNA sequence ladders were generated with plasmid pCMVDHBV (1) as a template by using a SequiTherm EXCEL II DNA Sequencing Kit (Epicenter biotechnologies) under the same thermocycling conditions. The reaction products were resolved on a DNA sequencing gel and visualized by a PhosphorImager (Bio-Rad).
RESULTS
Establishment and characterization of cell lines that support inducible HBV replication.
The nuclei of hepatocytes naturally infected with hepadnaviruses contain approximately 10 to 50 copies of cccDNA per cell, with the pool size of cccDNA being regulated by viral and cellular factors (47, 48, 55). For example, previous studies indicate that the DHBV L protein regulates cccDNA formation. Replication of L protein-deficient DHBV genomes in both cultured primary duck hepatocytes and LMH cells in vitro and of L protein-deficient virus-infected duck livers in vivo leads to high levels of cccDNA accumulation, which is detrimental to host cells (21, 22, 44). However, due to the relatively low efficiency of cccDNA formation in HBV DNA-transfected cells and the technical difficulty of accurately measuring HBV cccDNA, the regulatory effects of the envelope proteins on HBV cccDNA formation have not been unambiguously determined (24).
To investigate the effects of HBV envelope proteins on cccDNA formation, we established HepG2-derived stable cell lines supporting the tetracycline-inducible replication of HBV genomes that are deficient in producing HBV e antigen (HBeAg) from the transgene (HepDE19) or both HBeAg and three envelope proteins (HepDES19). HepAD38, a HepG2-derived cell line supporting the tetracycline-inducible wild-type HBV genome replication (18), was used as a control. The cell lines were cultured in the presence of tetracycline until cells reached confluence, followed by tetracycline removal from culture medium to induce viral pgRNA transcription and DNA replication. Cells and culture medium were harvested at the indicated time points before and after tetracycline removal (Fig. 2). Intracellular HBV core DNA and Hirt DNA were extracted and analyzed by Southern blot hybridization. Viral particles secreted into cultured medium were determined by an agarose gel-based particle assay. Our results, shown in Fig. 2, led to several observations.
FIG. 2.

Kinetics of HBV DNA synthesis, cccDNA formation, and virion secretion in HepAD38, HepDE19, and HepDES19 cell lines upon induction. Cells were seeded in 35-mm dishes and cultured in the presence of tetracycline (1 μg/ml) until cells became confluent; cells continued to be cultured in tetracycline-free medium for 11 days. Cells and culture fluids were harvested at the indicated time points before and after tetracycline removal. Cytoplasmic core DNA (A), secreted viral particle-associated DNA (B), and Hirt DNA (C) were analyzed by Southern blot hybridization and particle gel assay with [α-32P]UTP-labeled minus-strand-specific HBV riboprobe. The Hirt DNA samples were denatured at 85°C for 5 min before loading to improve the detection sensitivity of cccDNA. As a consequence, the protein-free rcDNA (labeled DP-rcDNA) migrates at the position of the single-strand DNA. (D) Culture medium was harvested from HepAD38 cells 4, 6, and 8 days after removal of tetracycline, and viral particles were pelleted with PEG 8000 and separated in a 1% agarose gel and transferred onto nitrocellulose membrane as described in Materials and Methods. The membrane was probed sequentially with anti-HBs (left panel) and anti-HBc (middle panel) antibodies and hybridized with minus-strand-specific full-length HBV riboprobe (right panel). (E) Hirt DNA preparations made from HepDES19 cells cultured in the absence of tetracycline for 8 days were separated in a 1.5% agarose gel without treatment (lane 3), after denaturalization at 85°C for 5 min (lane 4), or digestion with EcoRI after denaturalization at 85°C for 5 min (lane 5). Core DNA prepared from the same culture (lane 2) served as a control. Unit-length linear HBV DNA (50 pg; lane marker) served as a hybridization standard for estimating the copy number of viral DNA. RC, rcDNA; ccc, cccDNA; SS, single-strand DNA; DSL, double-strand linear DNA.
First, upon the removal of tetracycline from culture medium, HBV DNA replicative intermediates (core DNA) became detectable at days 2 to 4 and continued increasing to approximately 2,000, 3,000, and 4,000 copies per cell in HepAD38, HepDE19, and HepDES19 cells, respectively, at day 11. Interestingly, HepDES19 cells accumulate higher levels of capsid DNA, possibly due to the deficiency of envelope proteins. This is most obviously observed after 7 days of tetracycline removal (Fig. 2A, compare lanes 5 to 7 and 17 to 19).
Second, particle gel assays reveal that virion secretion is detectable at 4 days after tetracycline removal from both HepAD38 and HepDE19 cells. As expected, HepDES19 cells do not secrete virions (Fig. 2B). To further validate the identity of virions, viral particles prepared from HepAD38 cells were separated by agarose gel and transferred onto a membrane. The membrane was subjected to sequential detection of HBsAg, HBcAg, and HBV DNA as described in Materials and Methods. As shown in Fig. 2D, virions can be detected by both anti-HBs and anti-HBc antibodies and HBV DNA hybridization. The lower band can be detected only by anti-HBc antibody and HBV DNA hybridization. Thus, it should be naked capsid. Although the reason is currently unknown, naked capsids are found in the media of all three cell lines (Fig. 2B), as has been reported previously (58).
Third, cccDNA becomes detectable between days 7 to 9 after the removal of tetracycline and slowly increases to approximately 10 copies per cell in HepAD38 and HepDE19 cells and 60 copies per cell in HepDES19 (Fig. 2C). The identity of the observed cccDNA was confirmed by heat denaturation and further EcoRI digestion, which converted cccDNA into a unit-length linear DNA as shown in Fig. 2E, compare lane 3 to 5) (58).
Finally, in Hirt DNA preparations, in addition to cccDNA, there is a DNA species that migrates at the same position with viral capsid (core) mature rcDNA (Fig. 2E, lane 3). After heat denaturation at 85°C for 5 min (a condition that does not denature cccDNA) (58), this DNA species migrates to the position of single-strand DNA (Fig. 2E, lane 4). This rcDNA species had been observed previously in Hirt DNA preparations from cells and livers infected with HBV or animal hepadnaviruses (10, 28, 36, 37, 41, 48, 51, 54). Extraction of viral DNA by the Hirt procedure involves lysis of cells with SDS and high concentrations of salts to disassociate physically associated proteins, such as capsid protein, from DNA, and this procedure is followed by direct phenol extraction of DNA from cell lysates without protease digestion. Therefore, DNA covalently linked to protein is partitioned into phenol or interphase, and only the DNA molecules that are not covalently attached to protein are selectively extracted. Hence, the HBV DNA species in the Hirt preparations should be free of covalently genome-bound viral DNA polymerase (48, 51). This notion is supported by the fact that not all viral DNA replication intermediates known to be covalently attached to polymerase are extracted by this method (Fig. 2C and E) (34, 48, 51). Moreover, DHBV DNA in secreted virion particles is known to covalently attach to polymerase (29) and cannot be extracted with this method (Fig. 3, compare lanes 2 and 3). Thus, we designated this protein-free rcDNA species DP-rcDNA.
FIG. 3.
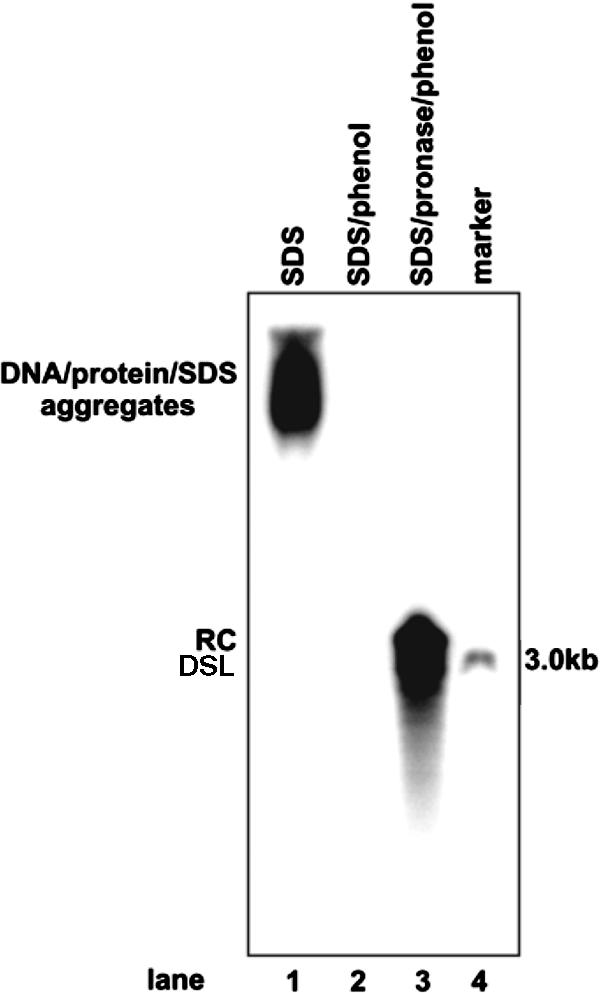
Hirt DNA extraction procedure extracts only protein-free viral DNA. DHBV viral particles were purified from 1 ml of DHBV-positive duck serum by centrifugation through a 10 to 20% sucrose cushion and lysed in a 60-μl solution containing 0.1% SDS, 10 mM Tris-HCl (pH 7.5), and 10 mM EDTA at 37°C for 1 h. Viral DNA was extracted from the lysates by direct phenol extraction (Hirt procedure; lane 2) or by phenol extraction after digestion with 1 mg/ml pronase at 37°C for 1 h (core DNA procedure; lane 3). The lysate of viral particles (lane 1) and purified viral DNA were resolved on a 1.2% agarose gel. Viral DNA was detected by Southern blot hybridization with [α-32P]UTP-labeled full-length minus-strand-specific DHBV riboprobe. Fifty picograms of unit-length DHBV DNA served as a hybridization standard (lane 4). DSL, double-strand linear DNA.
As shown in Fig. 2A and C, DP-rcDNA appears at the same times as the mature capsid rcDNA, between 2 to 4 days after tetracycline removal, and constitutes approximately 15% of the total pool of intracellular mature rcDNA. Moreover, DP-rcDNA accumulates at much higher levels in the HBV envelope-defective HepDES19 cells than it does in the two other envelope-competent cell lines.
HBV envelope proteins regulate both DP-rcDNA and cccDNA formation.
To further determine the effects of HBV envelope proteins on the accumulation of DP-rcDNA and cccDNA, HepDES19 cells were either cotransfected with two plasmids that express HBV L protein and M/S proteins or transfected with a control vector plasmid. After transfection, cells were cultured in tetracycline-free medium to induce viral DNA replication. Cells and culture medium were harvested at the indicated time points before and after tetracycline removal (Fig. 4). Intracellular HBV core DNA, secreted viral particles, and Hirt DNA were analyzed as described above. As expected, expression of viral envelope proteins in trans rescues viral particle secretion from HepDES19 cells (Fig. 4B). Moreover, the accumulation kinetics of both DP-rcDNA and cccDNA in cells exogenously expressing the envelope proteins is slightly slower than that in mock-transfected cells (Fig. 4C, compare lanes 4 to 6 with lanes 11 to 13). Presumably, due to the degradation of transfected plasmids that occurs after several days, virion particle secretion is decreased in the transfected cells (Fig. 4B, lane 14), and the differences in cccDNA and DP-rcDNA levels between envelope genes and vector plasmid-transfected cultures are diminished (Fig. 4C, compare lanes 7 to 14). Considering that less than 40% of cells were transfected and expressed the intended proteins, as judged by transfection with a green fluorescent protein expression plasmid (data not shown), the observed 30% reduction of DP-rcDNA and cccDNA accumulation in the cultures that were transfected with envelope protein-expressing plasmids should represent a significant regulatory effect of the viral proteins on the formation of both DP-rcDNA and cccDNA.
The results presented above suggest that, as observed with DHBV, HBV envelope proteins regulate cccDNA formation in HepG2 cells but with distinctive properties. In marked contrast to the dramatically increased accumulation of cccDNA in envelope protein-deficient DHBV genome-replicating cells (21, 22, 44), lack of HBV envelope proteins leads to a drastic accumulation of DP-rcDNA, with a less profound increase of cccDNA formation (Fig. 2C). A possible explanation for this observation is that HepG2 cells are inefficient in converting DP-rcDNA into cccDNA.
Consistent with the above interpretation, we noticed that DP-rcDNA is present in excess over cccDNA in all three HepG2-derived cell lines. The ratios of DP-rcDNAs to cccDNAs in Hirt DNA preparations are 13, 20, and 25 in HepAD38, HepDE19, and HepDES19 cells, respectively, at day 11 after tetracycline removal (Fig. 2C). These values are in marked contrast to the ratios observed both in the livers of human beings and animals infected with HBV, woodchuck hepatitis virus, ground squirrel hepatitis virus, and DHBV and in cultured cells infected with DHBV or transfected with DHBV genomes. Under these conditions, the ratios are approximately 1 or, in the most cases, less than 1 (10, 28, 35-37, 48, 51, 54).
Nevertheless, the overwhelming accumulation of DP-rcDNA in HepDES19 cells provided an opportunity to biochemically characterize this DNA species and determine the molecular mechanism by which the viral genome-bound DNA polymerase is removed.
DP-rcDNA is not derived from nicked cccDNA, and its plus-strand DNA is completely synthesized.
To characterize the biochemical property of the observed DP-rcDNA species, an important question concerns where DP-rcDNA originates. In principle, it can be derived from either the deproteinization of viral capsid rcDNA or from nicks in cccDNA. These two possibilities can be distinguished by mapping the terminal locations of the rcDNA. If the DNA species is derived from the deproteinization of capsid rcDNA, the ends of both its plus strand and minus strand should be at fixed positions. Otherwise, the ends resulting from nicked cccDNA ought to be randomly distributed.
Therefore, the prediction is that digestion of viral rcDNA with a single-cut restriction enzyme will produce two distinct DNA species in alkaline gel electrophoresis, while the same treatment of nicked cccDNA should yield DNA fragments that are heterogeneous in length (34). To distinguish these two possibilities, Hirt DNA prepared from HepDES19 cells was digested with EcoRI (Fig. 5A), and the restriction fragments were resolved by alkaline agarose gel electrophoresis. Viral DNA was detected by Southern blot hybridization with minus- and plus-strand-specific full-length riboprobes. The results show that when probed with either a minus-strand- or plus-strand-specific riboprobe, the undigested viral protein-free DNA including rc and cccDNA migrates as a single species at a 3.2-kb position. EcoRI digestion yields three minus-strand DNA species of 3.2, 1.8, and 1.4 kb in length (Fig. 5B) and two plus-strand DNA species of 3.2 and 1.6 kb in length (Fig. 5C). While the 3.2-kb plus- and minus-strand DNAs should be the products of EcoRI digestion of cccDNA, the 1.8- and 1.4-kb minus-strand and 1.6-kb plus-strand DNA species are in agreement with the predicted products of rcDNA digested by the enzyme (Fig. 5A). Hence, the results indicate that the observed DP-rcDNA is derived from the deproteinization reaction of nucleocapsid rcDNA rather than nicking of cccDNA. Furthermore, Southern blot hybridization with plus-strand-specific probe does not reveal detectable DNA species that are shorter than full-length (Fig. 5C, lane 2), suggesting that the plus strand of protein-free rcDNA is completely, or at least almost completely, synthesized. To further confirm this observation, Hirt DNA prepared from HepDES19 cells was digested with BspEI that cuts HBV DNA at nt 2331 (Fig. 5A). The restriction fragments were detected by alkaline agarose gel electrophoresis and Southern blot hybridization with a plus-strand-specific full-length riboprobe. While the undigested Hirt DNA and purified cccDNA migrate as a single full-length species with an apparent molecular mass of 3.2 kb (Fig. 5D, lanes 2 and 3), BspEI digestion yields three plus-strand HBV DNA species of 3.2, 2.5, and 0.7 kb in length (Fig. 5D, lane 4). This result is in agreement with the predicted size of full-length plus-strand DNA and cccDNA digestion products with the enzyme.
FIG. 5.

Protein-free HBV and DHBV rcDNA species are not derived from nicked cccDNA. (A and E) Schematic representation of the HBV and DHBV rcDNA structures, respectively. EcoRI and BspEI restriction sites are indicated. (B, C, and D) Hirt DNA preparations from HepDES19 cells cultured in the absence of tetracycline for 10 days were left untreated (B and C, lane 2; D, lane 3) or digested with EcoRI (B and C, lane 3) or BspEI (D, lane 4) and then resolved in a 1.5% alkaline agarose gel. Purified HBV cccDNA (D, lane 2) and unit-length linear HBV DNA (lane 1) served as controls. DNAs were transferred onto nylon membrane and hybridized with [α-32P]UTP-labeled full-length minus-strand-specific (B) or plus-strand-specific (C and D) HBV riboprobe. (F) Hirt DNA preparations from dstet5 cells cultured in the absence of tetracycline for 7 days were left untreated (lane 1) or digested with EcoRI (lane 2) and then resolved in a 1.5% alkaline agarose gel. Unit-length linear DHBV DNA served as a hybridization standard (lane 3). DNAs were transferred onto nylon membrane and hybridized with [α-32P]UTP-labeled full-length minus-strand-specific DHBV riboprobe.
To test if this observation is true in DHBV, Hirt DNA was prepared from dstet5 cells, an LMH-derived stable cell line supporting inducible DHBV replication (11), cultured in the absence of tetracycline for 7 days, and digested with EcoRI (Fig. 5D). The restriction products were analyzed as described above, and DHBV DNA was detected with a minus-strand-specific full-length riboprobe. As predicted, undigested protein-free DHBV DNA migrates as a single DNA species of unit length, and EcoRI digestion yields three DNA species of 3.0, 2.5, and 0.5 kb in length, matching perfectly with the predicted products of cccDNA and rcDNA digested by the enzyme (Fig. 5E).
Taken together, the results presented above suggest the following: (i) the DP-rcDNA is derived from the deproteinization reaction of rcDNA; (ii) neither gap in the plus and minus strands of DP-rcDNA has been covalently ligated; (iii) DP-rcDNA contains complete plus-strand DNA, suggesting that the removal of covalently genome-bound polymerase may require the completion of plus-strand DNA synthesis.
Transfection of DHBV DP-rcDNA into LMH cells initiates viral DNA replication.
To determine if the observed DP-rcDNA species is a functional precursor of cccDNA formation, Hirt DNA was prepared from dstet5 cells cultured in the absence of tetracycline for 6 days and resolved on 1% agarose gel. DP-rcDNA and cccDNA bands were visualized by ethidium bromide staining (Fig. 6A) and recovered with a QIAGEN DNA gel purification kit. Purified DP-rcDNA and cccDNA were confirmed and quantified by Southern blot hybridization assay (Fig. 6B). Approximately 5 ng of DP-rcDNA and cccDNA was mixed with 2 μg of pUC19 plasmid and transfected into LMH cells cultured in a 35-mm plate. Seven days after transfection, cells were harvested, and capsid-associated DNAs were extracted and detected by Southern blot hybridization. The results show that all DNA replication intermediates are detected in both DP-rcDNA- and cccDNA-transfected cells (Fig. 6C), suggesting that, like cccDNA, DP-rcDNA is able to initiate viral DNA replication and is, thus, a functional precursor of cccDNA formation.
FIG. 6.

Transfection of DHBV DP-rcDNA into LMH cells initiates viral DNA replication. (A) Hirt DNA preparations from dstet5 cells cultured in the absence of tetracycline for 7 days were resolved in a 1.5% agarose gel, and DHBV cccDNA and DP-rcDNA were visualized by ethidium bromide staining. (B) Recovered HBV cccDNA and DP-rcDNA were resolved on agarose gel and detected by Southern blot hybridization. (C) LMH cells were transfected with approximately 5 ng of gel-purified DHBV cccDNA and DP-rcDNA mixed with 2 μg of plasmid pUC19 or with 2 μg of pUC19 alone (mock) by using Lipofectamine 2000. Cells were harvested at day 7 posttransfection, and capsid-associated viral DNA was extracted and determined by Southern blot hybridization with [α-32P]UTP-labeled full-length minus-strand-specific DHBV riboprobe. Fifty picograms of unit-length DHBV DNA served as a hybridization standard. RC, rcDNA; CCC, cccDNA; SS, single-strand DNA; DP-rc, DP-rcDNA; DSL, double-strand linear DNA.
DP-rcDNA is present in both the cytoplasm and nucleus.
To determine whether the deproteinization reaction of viral rcDNA occurs in the cytoplasm or nucleus, cell fractionation studies were performed. HepDES19 cells were cultured in the absence of tetracycline for 10 days. Cytoplasmic and nuclear lysates were prepared with a Qproteome Cell Compartment kit. Western blot analysis of cytoplasmic protein (annexin I) and nuclear protein (lamin A/C) markers in the cell fractions demonstrated that there is no cross-contamination in either the cytoplasmic or the nuclear fraction (Fig. 7A, lower panel). Intracellular capsid DNA and Hirt DNA extracted from whole-cell, cytoplasmic, and nuclear lysates without or with prior DNase I digestion were analyzed by Southern blot hybridization. As expected, cccDNA was detected only in the nuclear fraction and was sensitive to DNase I digestion (Fig. 7A, upper panel). In marked contrast, however, DP-rcDNA could be found in similar amounts in both the cytoplasmic and nuclear fractions. Curiously, while all nuclear DP-rcDNA is completely DNase I sensitive, a minority (approximately 14%) of cytoplasmic DP-rcDNA is resistant to the enzyme treatment. Furthermore, the cytoplasmic but not the nuclear DP-rcDNA can be precipitated with an antibody against HBcAg and not with the antibody against DHBcAg (Fig. 7A and data not shown), suggesting that this DNA species remains associated with core protein and that the immunoprecipitation was specific. Moreover, approximately 15% of immunoprecipitated cytoplasmic DP-rcDNA is DNase I resistant. Consistent with previous reports, partially double-stranded genomic DNA in serum-derived HBV virions covalently attached to viral polymerase and thus was not present in the Hirt DNA preparation (Fig. 7B, compare lanes 2 and 3) (9).
FIG. 7.
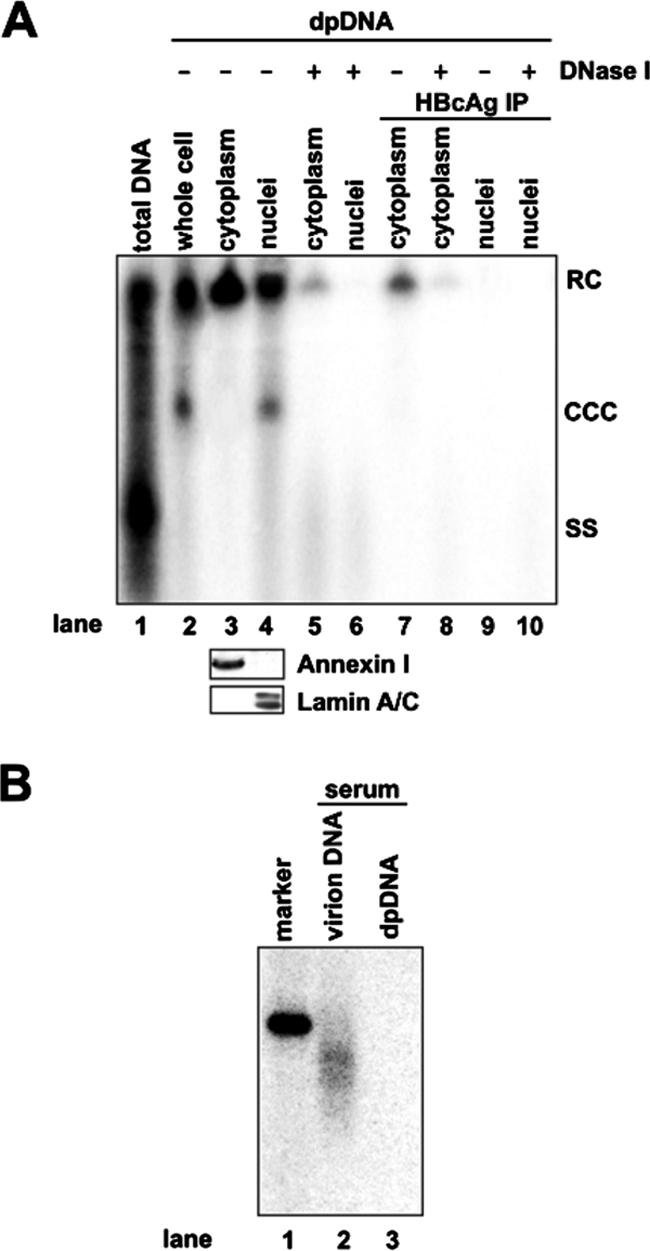
Subcellular distribution of HBV DP-rcDNA in HepDES19 cells. (A) HepDES19 cells were cultured in the absence of tetracycline for 10 days. Intracellular HBV capsid DNA (lane 1) and Hirt DNA (lanes 2 to 10) extracted from the lysates of whole cell, cytoplasm, and nuclei of HepDES19 cells with or without prior DNase I digestions were analyzed by Southern blot hybridization. Lane 1 was loaded with one-quarter of the capsid DNA extracted from a 35-mm dish. Lane 2 was loaded with one-half of total Hirt DNA from a 35-mm dish. Lanes 3 to 10 were each loaded with Hirt DNA prepared from the cytoplasmic or nuclear fraction from one 35-mm dish with the treatments described below. Hirt DNAs were extracted from both fractions without (lanes 3 and 4) or with (lanes 5 and 6) prior DNase I digestion or extracted after immunoprecipitation (IP) with a polyclonal antibody against HBV capsid (lanes 7 and 9) and digestion with DNase I (lanes 8 and 10). Total proteins of each fraction were resolved by SDS-polyacrylamide gel electrophoresis, transferred on polyvinylidene difluoride membrane, and probed with antibodies against annexin I and lamin A/C, respectively. Bound antibodies were revealed by HRP-labeled secondary antibodies and visualized with an enhanced chemiluminescence detection system (lower panel). (B) HBV DNAs extracted from 250 μl of HBV-positive human serum with (lane 2) or without (lane 3) prior protease digestion were resolved in a 1.5% agarose gel, transferred onto membrane, and hybridized with an HBV minus-strand DNA-specific riboprobe. Fifty picograms of unit-length HBV DNA served as a hybridization standard (lane 1). RC, rcDNA; CCC, cccDNA; SS, single-strand DNA.
To confirm this observation in hepadnavirus-infected livers in vivo, Hirt DNA was extracted from cytoplasmic and nuclear fractions prepared from DHBV-infected duck liver tissue and analyzed by Southern blot hybridization. In agreement with the observations presented above, cccDNA exists only in the nuclear fraction, but DP-rcDNA is present in both the cytoplasmic and nuclear fractions. Surprisingly, both the cytoplasmic and nuclear DHBV DP-rcDNA is DNase I sensitive (Fig. 8). Serving as a control, DP-rcDNA is not present in serum-derived DHBV particles (Fig. 8, lanes 7 and 8). Failure to detect cytoplasmic DNase I-resistant DHBV DP-rcDNA could be due to distinct properties of HBV and DHBV; alternatively, this DNA species may exist in such a small amount in duck livers that it is beneath the detection limit.
FIG. 8.
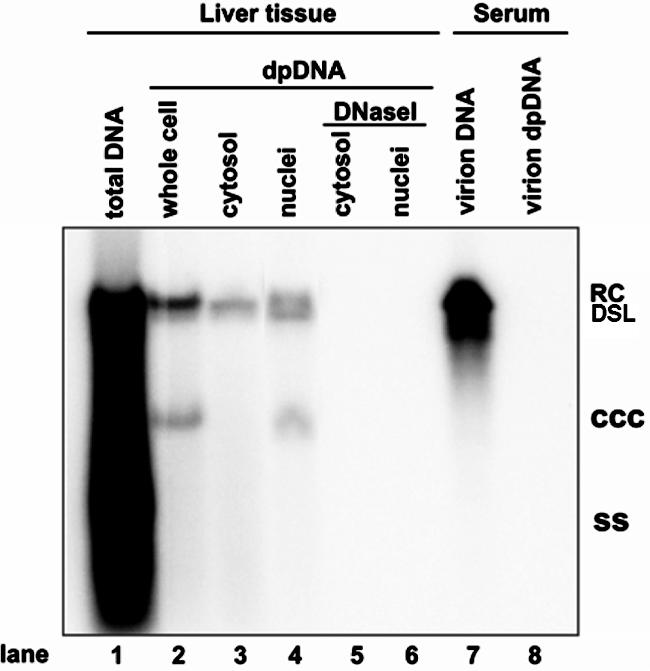
Subcellular distribution of DHBV DP-rcDNA in the liver of virally infected duck. Total intracellular capsid DNA (lane 1), Hirt DNA extracted from total lysate (lane 2), or cytoplasmic and nuclear fractions of a DHBV-positive duck liver without (lanes 3 and 4) or with (lanes 5 and 6) prior DNase I digestion and DHBV DNA extracted from 250 μl of DHBV-positive duck serum with (lane 7) or without (lane 8) prior pronase digestion were resolved in a 1.5% agarose gel, transferred onto membrane, and hybridized with a DHBV minus-strand DNA-specific riboprobe. Total DNA loaded in lane 1 was prepared from 7 mg of duck liver tissue, and each Hirt DNA sample was prepared from lysate prepared from 15 mg of liver tissue. RC, rcDNA; CCC, cccDNA; SS, single-strand DNA; DSL, double-strand linear DNA.
Cytoplasmic DP-rcDNA cosediments with nucleocapsids.
To further examine the relationship between cytoplasmic HBV DP-rcDNA and the viral capsid, cytoplasmic lysates prepared from HepDES19 cells were separated by centrifugation through a 10 to 60% continuous sucrose gradient. Seventeen fractions were collected from the bottom of the tube, and the presence of HBcAg, core DNA, and DP-rcDNA in each fraction was determined by enzyme immunoassay and DNA hybridization assays.
The results reveal that HBcAg appears in two peaks. The first peak, spanning fractions 5 to 8, cosediments with core DNA and presumably represents intact capsids. The second peak, spanning fractions 14 to 16, presumably represents free capsid protein dimers (Fig. 9A, B, and C). Moreover, both DNase I-sensitive and -resistant DP-rcDNAs appear in the same fractions as do core DNA and intact capsids (Fig. 9D and E). Consistent with the results presented in Fig. 7A, at least a fraction of DP-rcDNA can be immunoprecipitated with HBV core antibody, and a small portion of the immunoprecipitated DP-rcDNA species is DNase I resistant (Fig. 9F and G).
FIG. 9.

HBV cytoplasmic DP DNA cosediments with nucleocapsids. Three milliliters of cytoplasmic fraction prepared from 2 × 107 HepDES19 cells cultured in the absence of tetracycline for 10 days was overlaid on 33 ml of a 10 to 60% (wt/vol) sucrose gradient and centrifuged at 24,000 rpm for 16 h at 4°C using a Beckman SW28 rotor. Seventeen 2.25-ml fractions were collected from the bottom. HBV core antigen (A), total HBV DNA (B), and capsid DNA (C) in each fraction were assayed as described in Material and Methods. DP-rcDNA was extracted from each fraction without (D) or with (E) prior DNase I digestion. For the immunoprecipitation (IP) assay, each faction was mixed with Sepharose 4B-protein A beads preabsorbed with rabbit anti-HBc and incubated at 4°C overnight. Beads were washed three times with TNE buffer, and protein-free DNA was extracted without (F) or with (G) prior DNase I digestion. The Hirt DNAs were resolved in a 1.2% agarose gel and detected by Southern blot hybridization. RC, rcDNA; SS, single-strand DNA; EIA, enzyme immunoassay.
A similar experiment was performed with a cytoplasmic lysate prepared from dstet5 cells and shows that, similar to the observations made with HBV, the cytoplasmic DHBV DP-rcDNA cosediments with DHBV capsid and can be immunoprecipitated by an antibody against DHBV core protein (data not shown).
In summary, the results presented in the above two sections reveal that DP-rcDNA is present in both the cytoplasm and nucleus and that there are two distinct populations of cytoplasmic DP-rcDNA in HepDES19 cells. While the majority of cytoplasmic DP-rcDNA cosediments with capsids, can be immunoprecipitated with core antibody, and is sensitive to DNase I digestion, a minor population of cytoplasmic DP-rcDNAs cosediment with capsids and can be precipitated by core antibody but is resistant to DNase I treatment. These results suggest that although the majority of cytoplasmic DP-rcDNA may exist in partially disassembled capsids or in particles (or complexes) that contain viral capsid proteins, leaving the DNA vulnerable to DNase I digestion, a minor population of cytoplasmic DP-rcDNAs may still be within intact capsids.
Mapping the 5′ end of the minus strand of cytoplasmic DHBV DP-rcDNA.
The results presented above suggest that removal of viral DNA polymerase from the 5′ end of the minus strand of nucleocapsid rcDNA occurs in the cytoplasm. However, the mechanism whereby the polymerase is removed from rcDNA remains an open question. As mentioned previously, this reaction could occur via either nucleolytic cleavage of viral DNA close to the 5′ end by an endonuclease or hydrolysis of the phosphodiester bond between the hydroxyl on the tyrosine side chain of the polymerase and the 5′ phosphoryl group of minus-strand DNA. While hydrolysis of the phosphodiester bond between tyrosine and minus-strand DNA would be expected to yield a 5′ end of minus-strand DNA at its authentic position, an endonuclease reaction will result in a trimmed 5′ end of minus-strand DNA.
To distinguish between these two possibilities, we performed a primer extension assay to map the end of the minus strand of cytoplasmic DHBV DP-rcDNA prepared from a DHBV-infected duck liver. The result shows that primer extension with DHBV DNA prepared from serum-derived virions yields two bands that map the 5′ ends of minus-strand DNA to nt 2537G and 2538T. Interestingly, the longest primer extension product of DP-rcDNA corresponds to nucleotide 2537G (Fig. 10). While the previous work has mapped the 5′ end of DHBV minus strand DNA to position 2537G (12, 46, 49), heterogeneity of the 5′ ends of minus strand has been observed with DHBV and HBV DNA by primer extension and DNA cloning and sequence assays (13, 38, 52). It is possible that the observed heterogeneity either is due to the initiation of minus-strand DNA synthesis at an alternative site or is an artifact of the assays. Nevertheless, the result indicates that the removal of DNA polymerase leaves an authentic 5′ end of minus-strand DNA and thus supports a model that DNA polymerase is removed via a reaction that cleaves the phosphodiester bond between tyrosine of polymerase and the 5′ phosphoryl group of minus-strand DNA.
FIG. 10.

Mapping the 5′ end of the minus strand of DHBV DP-rcDNA. Virion DNA prepared from DHBV-positive duck serum (lane 1) and Hirt DNA prepared from the cytoplasmic fraction of DHBV-infected (lane 2) and normal (lane 3) duck liver tissues were annealed with a 5′ end-labeled oligonucleotide coordinating with nt 2375 to 2396 in the DHBV genome. The primer extension reaction was carried out as described in Materials and Methods. Products were resolved with a 6% acrylamide-8 M urea gel alongside a sequencing ladder (lanes C, T, A, and G) and visualized by a PhosphorImager (Bio-Rad).
DISCUSSION
Maintenance of cccDNA in the nuclei of HBV-infected hepatocytes is the molecular basis of persistent infections as well as the source of viral replication rebound following the discontinuation of antiviral therapy (16). Although the molecular mechanisms of HBV replication, especially, reverse transcription, have been studied in great detail, little is known about the molecular mechanism by which rcDNA is converted into cccDNA, the first replication intermediate made upon the entry of the virus into hepatocytes (3, 40, 45). The work reported herein is intended to determine the molecular pathway of cccDNA formation and its regulation by viral and host cellular factors.
Using our recently developed cell lines that support the inducible replication of wild-type and envelope protein-deficient HBV genomes, we obtained evidence suggesting that, as with DHBV L protein, the HBV envelope proteins negatively regulate cccDNA formation in HepG2 cells. However, to our surprise, the deficiency of HBV envelope proteins only modestly increases the level of cccDNA but leads to a dramatic accumulation of DP-rcDNA. Consistent with the results reported recently by Gao and Hu (8), we independently confirmed that the DP-rcDNA is derived from rcDNA upon removal of its covalently attached polymerase protein. Furthermore, transfection of DHBV DP-rcDNA into LMH cells initiates viral DNA replication, strongly suggesting that the DP-rcDNA is a functional precursor of cccDNA.
The results presented in this report (Fig. 2 and 4) thus reveal several important properties of HBV cccDNA formation and its regulation in HepG2 cells. First, HBV envelope proteins may regulate a step before rcDNA deproteinization, most possibly the intracellular traffic of mature nucleocapsids. This notion is consistent with the finding that the regulatory effect of DHBV L protein on cccDNA formation could not be genetically separated from its role in virion secretion (22). Second, the conversion of DP-rcDNA into cccDNA is a rate-limiting step during cccDNA formation in HepG2 cells. One explanation for this observation is that DNA repair pathway(s) required for this process may be impaired in the hepatoma cells, which is commonly observed in many types of tumor cells (5, 7, 15). Third, the nonpermissiveness of HepG2 and perhaps other tumor-derived cell lines to HBV infection could be, at least in part, due to the inefficiency of cccDNA formation.
In deciphering the molecular pathway of cccDNA formation, we initially focused on determining where and how the covalently HBV genome-bound polymerase protein is removed. Cell fractionation studies suggest that while cccDNA is present only in the nucleus, which is consistent with previous reports, the DP-rcDNA exists in both cytoplasm and nucleus, implying that the removal of DNA polymerase from the minus strand of rcDNA occurs in the cytoplasm. Moreover, our results suggest that the majority of cytoplasmic DP-rcDNA is present in partially disassembled capsids or particles (or complexes) that contain viral capsid proteins and that a minor population of cytoplasmic DP-rcDNA is present within intact capsids. These results support a hypothesis that the removal of genome-bound DNA polymerase protein occurs in intact nucleocapsids and is followed by capsid disassembly, which might actually be triggered by the deproteinization reaction.
Interestingly, a protein-free rcDNA species had been detected in both the cytoplasmic and nuclear fractions of the liver tissues from HNF1α-knockout HBV transgenic mice that are able to synthesize low levels of cccDNA. But, surprisingly, the protein-free rcDNA species is undetectable under the same conditions in the livers of HBV transgenic wild-type mice (34). While consistent with our results that the nuclear protein-free rcDNA is nuclease sensitive, the cytoplasmic protein-free rcDNA in the transgenic mouse livers is exclusively nuclease resistant (34). Moreover, as we demonstrate in this report, the cytoplasmic DP-rcDNA in the livers of DHBV-infected ducks is completely sensitive to DNase I treatment (Fig. 8). While the reasons for these discrepancies are not clear, the observations do suggest that both viral and host cellular factors regulate the processes of rcDNA deproteinization and capsid disassembly.
The possibility that the deproteinization reaction occurs within nucleocapsids raises questions regarding how the reaction is properly timed and triggered, which is essential to prevent the premature removal of polymerase. Since DP-rcDNA contains only full-length plus-strand DNA (Fig. 5) (8), the completion of plus-strand DNA synthesis may be required for this reaction to occur. However, as mentioned above, DP-rcDNA constitutes only approximately 15% of total mature rcDNA, and, thus, the completion of plus-strand DNA synthesis alone is apparently not sufficient to trigger the deproteinization reaction and/or capsid disassembly.
Moreover, our hypothesis predicts that deproteinization of rcDNA may be catalyzed by polymerase itself and/or cellular proteins that are packaged in nucleocapsids. The primer extension result suggests that removal of polymerase protein from the viral rcDNA genome leaves an authentic 5′ end of minus-strand DNA. It thus suggests that the protein is most probably removed by cleaving the phosphodiester bond between the tyrosine in the TP domain of polymerase and the 5′ phosphoryl group of minus-strand DNA. This reaction is chemically similar to the reaction that removes topoisomerase II from topoisomerase II inhibitor-induced DNA strand break (4). Interestingly, a recent report suggests that a cellular enzyme, TDP1 (tyrosyl-DNA phosphodiesterase 1), is able to cleave the tyrosine and 5′ phosphotyrosyl linkage between topoisomerase II and DNA (2, 32). Hence, it would be interesting to determine whether TDP1 plays a role in the rcDNA deproteinization reaction.
In summary, based on our observations and the interpretations presented above, a working model for the molecular pathway of cccDNA formation from nucleocapsid rcDNA can be proposed (Fig. 11). Upon the completion of plus-strand DNA synthesis, deproteinization of rcDNA occurs inside nucleocapsids, which may serve as a trigger for the partial disassembly of nucleocapsids. Alternatively, as observed with DHBV, deproteinization of rcDNA may either be tightly linked to partial capsid disassembly or occur during and/or after partial capsid disassembly. The partial disassembly of capsid may lead to the exposure of a nuclear localization signal that mediates the import of DP-rcDNA into the nucleus through nuclear pore complex (33, 60), and subsequently DP-rcDNA is converted into cccDNA by cellular DNA repair machinery (17). This model provides a framework for further experiments to determine the molecular pathway of cccDNA formation and its regulation by viral and host cellular factors.
FIG. 11.

Proposed model for the molecular pathway of hepadnavirus cccDNA formation. See text for a detailed explanation. pol, polymerase; NLS, nuclear localization signal.
Acknowledgments
We thank Pamela Norton, Jinhong Chang, and Anand Mehta for critical reading of the manuscript and helpful suggestions.
This work was supported by grants from the Commonwealth of Pennsylvania through the Hepatitis B Foundation and the NIH.
Footnotes
Published ahead of print on 5 September 2007.
REFERENCES
- 1.Barrasa, M. I., J. T. Guo, J. Saputelli, W. S. Mason, and C. Seeger. 2001. Does a cdc2 kinase-like recognition motif on the core protein of hepadnaviruses regulate assembly and disintegration of capsids? J. Virol. 75:2024-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barthelmes, H. U., M. Habermeyer, M. O. Christensen, C. Mielke, H. Interthal, J. J. Pouliot, F. Boege, and D. Marko. 2004. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J. Biol. Chem. 279:55618-55625. [DOI] [PubMed] [Google Scholar]
- 3.Beck, J., and M. Nassal. 2007. Hepatitis B virus replication. World J. Gastroenterol. 13:48-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connelly, J. C., and D. R. Leach. 2004. Repair of DNA covalently linked to protein. Mol. Cell 13:307-316. [DOI] [PubMed] [Google Scholar]
- 5.Dixon, K., and E. Kopras. 2004. Genetic alterations and DNA repair in human carcinogenesis. Semin. Cancer Biol. 14:441-448. [DOI] [PubMed] [Google Scholar]
- 6.Fallows, D. A., and S. P. Goff. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fritz, G., K. Tano, S. Mitra, and B. Kaina. 1991. Inducibility of the DNA repair gene encoding O6-methylguanine-DNA methyltransferase in mammalian cells by DNA-damaging treatments. Mol. Cell. Biol. 11:4660-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao, W., and J. Hu. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J. Virol. 81:6164-6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerlich, W. H., and W. S. Robinson. 1980. Hepatitis B virus contains protein attached to the 5′ terminus of its complete DNA strand. Cell 21:801-809. [DOI] [PubMed] [Google Scholar]
- 10.Guo, H., W. S. Mason, C. E. Aldrich, J. R. Saputelli, D. S. Miller, A. R. Jilbert, and J. E. Newbold. 2005. Identification and characterization of avihepadnaviruses isolated from exotic anseriformes maintained in captivity. J. Virol. 79:2729-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo, J. T., M. Pryce, X. Wang, M. I. Barrasa, J. Hu, and C. Seeger. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 77:1885-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habig, J. W., and D. D. Loeb. 2003. The conformation of the 3′ end of the minus-strand DNA makes multiple contributions to template switches during plus-strand DNA synthesis of duck hepatitis B virus. J. Virol. 77:12401-12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Havert, M. B., and D. D. Loeb. 1997. cis-acting sequences in addition to donor and acceptor sites are required for template switching during synthesis of plus-strand DNA for duck hepatitis B virus. J. Virol. 71:5336-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 15.Hoglund, P. 2006. DNA damage and tumor surveillance: one trigger for two pathways. Sci. STKE 2006:pe2. [DOI] [PubMed] [Google Scholar]
- 16.Hu, J., and D. Nguyen. 2004. Therapy for chronic hepatitis B: the earlier, the better? Trends Microbiol. 12:431-433. [DOI] [PubMed] [Google Scholar]
- 17.Kann, M., A. Schmitz, and B. Rabe. 2007. Intracellular transport of hepatitis B virus. World J. Gastroenterol. 13:39-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ladner, S. K., M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo, C. Seeger, and R. W. King. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai, C. L., M. Rosmawati, J. Lao, H. Van Vlierberghe, F. H. Anderson, N. Thomas, and D. Dehertogh. 2002. Entecavir is superior to lamivudine in reducing hepatitis B virus DNA in patients with chronic hepatitis B infection. Gastroenterology 123:1831-1838. [DOI] [PubMed] [Google Scholar]
- 20.Lee, W. M. 1997. Hepatitis B virus infection. N. Engl. J. Med. 337:1733-1745. [DOI] [PubMed] [Google Scholar]
- 21.Lenhoff, R. J., and J. Summers. 1994. Construction of avian hepadnavirus variants with enhanced replication and cytopathicity in primary hepatocytes. J. Virology. 68:5706-5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenhoff, R. J., and J. Summers. 1994. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J. Virol. 68:4565-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liaw, Y. F., N. W. Leung, T. T. Chang, R. Guan, D. I. Tai, K. Y. Ng, R. N. Chien, J. Dent, L. Roman, S. Edmundson, C. L. Lai, et al. 2000. Effects of extended lamivudine therapy in Asian patients with chronic hepatitis B. Gastroenterology 119:172-180. [DOI] [PubMed] [Google Scholar]
- 24.Ling, R., and T. J. Harrison. 1997. Production of hepatitis B virus covalently closed circular DNA in transfected cells is independent of surface antigen synthesis. J. Gen. Virol. 78:1463-1467. [DOI] [PubMed] [Google Scholar]
- 25.Loeb, D. D., and R. Tian. 2001. Mutations that increase in situ priming also decrease circularization for duck hepatitis B virus. J. Virol. 75:6492-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcellin, P., T. T. Chang, S. G. Lim, M. J. Tong, W. Sievert, M. L. Shiffman, L. Jeffers, Z. Goodman, M. S. Wulfsohn, S. Xiong, J. Fry, and C. L. Brosgart. 2003. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N. Engl. J. Med. 348:808-816. [DOI] [PubMed] [Google Scholar]
- 27.McMahon, B. J. 2005. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 25(Suppl. 1):3-8. [DOI] [PubMed] [Google Scholar]
- 28.Miller, R. H., and W. S. Robinson. 1984. Hepatitis B virus DNA in nuclear and cytoplasmic fractions of infected human liver. Virology 137:390-399. [DOI] [PubMed] [Google Scholar]
- 29.Molnar-Kimber, K. L., J. Summers, J. M. Taylor, and W. S. Mason. 1983. Protein covalently bound to minus-strand DNA intermediates of duck hepatitis B virus. J. Virol. 45:165-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moraleda, G., J. Saputelli, C. E. Aldrich, D. Averett, L. Condreay, and W. S. Mason. 1997. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 71:9392-9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newbold, J. E., H. Xin, M. Tencza, G. Sherman, J. Dean, S. Bowden, and S. Locarnini. 1995. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 69:3350-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nitiss, K. C., M. Malik, X. He, S. W. White, and J. L. Nitiss. 2006. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc. Natl. Acad. Sci. USA 103:8953-8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabe, B., A. Vlachou, N. Pante, A. Helenius, and M. Kann. 2003. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 100:9849-9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raney, A. K., C. M. Eggers, E. F. Kline, L. G. Guidotti, M. Pontoglio, M. Yaniv, and A. McLachlan. 2001. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1α-null hepatitis B virus transgenic mice. J. Virol. 75:2900-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren, S., and M. Nassal. 2001. Hepatitis B virus (HBV) virion and covalently closed circular DNA formation in primary tupaia hepatocytes and human hepatoma cell lines upon HBV genome transduction with replication-defective adenovirus vectors. J. Virol. 75:1104-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Opazo, N., P. R. Chakraborty, and D. A. Shafritz. 1982. Characterization of viral genomes in the liver and serum of chimpanzee long-term hepatitis B virus carriers: a possible role for supercoiled HBV-DNA in persistent HBV infection. J. Cell Biochem. 19:281-292. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz-Opazo, N., P. R. Chakraborty, and D. A. Shafritz. 1982. Evidence for supercoiled hepatitis B virus DNA in chimpanzee liver and serum Dane particles: possible implications in persistent HBV infection. Cell 29:129-136. [DOI] [PubMed] [Google Scholar]
- 38.Saldanha, J. A., H. Qiu, H. C. Thomas, and J. Monjardino. 1992. Mapping of 5′ ends of virion-derived HBV DNA. Virology 188:358-361. [DOI] [PubMed] [Google Scholar]
- 39.Schormann, W., A. Kraft, D. Ponsel, and V. Bruss. 2006. Hepatitis B virus particle formation in the absence of pregenomic RNA and reverse transcriptase. J. Virol. 80:4187-4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seeger, C., and W. S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sells, M. A., A. Z. Zelent, M. Shvartsman, and G. Acs. 1988. Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J. Virol. 62:2836-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 43.Summers, J., A. O'Connell, and I. Millman. 1975. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc. Natl. Acad. Sci. USA 72:4597-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Summers, J., P. M. Smith, and A. L. Horwich. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 64:2819-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tagawa, M., M. Omata, O. Yokosuka, K. Uchiumi, F. Imazeki, and K. Okuda. 1985. Early events in duck hepatitis B virus infection. Sequential appearance of viral deoxyribonucleic acid in the liver, pancreas, kidney, and spleen. Gastroenterology 89:1224-1229. [PubMed] [Google Scholar]
- 46.Tavis, J. E., S. Perri, and D. Ganem. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turin, F., C. Borel, M. Benchaib, A. Kay, C. Jamard, C. Guguen-Guillouzo, C. Trepo, and O. Hantz. 1996. n-Butyrate, a cell cycle blocker, inhibits early amplification of duck hepatitis B virus covalently closed circular DNA after in vitro infection of duck hepatocytes. J. Virol. 70:2691-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tuttleman, J. S., C. Pourcel, and J. Summers. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451-460. [DOI] [PubMed] [Google Scholar]
- 49.Wang, G. H., and C. Seeger. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang, G. H., and C. Seeger. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663-670. [DOI] [PubMed] [Google Scholar]
- 51.Weiser, B., D. Ganem, C. Seeger, and H. E. Varmus. 1983. Closed circular viral DNA and asymmetrical heterogeneous forms in livers from animals infected with ground squirrel hepatitis virus. J. Virol. 48:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Will, H., W. Reiser, T. Weimer, E. Pfaff, M. Buscher, R. Sprengel, R. Cattaneo, and H. Schaller. 1987. Replication strategy of human hepatitis B virus. J. Virol. 61:904-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu, T. T., L. Coates, C. E. Aldrich, J. Summers, and W. S. Mason. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255-261. [DOI] [PubMed] [Google Scholar]
- 54.Yang, W., W. S. Mason, and J. Summers. 1996. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J. Virol. 70:4567-4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeh, C. T., H. T. Chiu, C. M. Chu, and Y. F. Liaw. 1998. G1 phase dependent nuclear localization of relaxed-circular hepatitis B virus DNA and aphidicolin-induced accumulation of covalently closed circular DNA. J. Med. Virol. 55:42-50. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, Y. Y., B. H. Zhang, D. Theele, S. Litwin, E. Toll, and J. Summers. 2003. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 100:12372-12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng, Y., J. Li, and J. H. Ou. 2004. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J. Virol. 78:6908-6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou, T., H. Guo, J. T. Guo, A. Cuconati, A. Mehta, and T. M. Block. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res. 72:116-124. [DOI] [PubMed] [Google Scholar]
- 59.Zhu, Y., T. Yamamoto, J. Cullen, J. Saputelli, C. E. Aldrich, D. S. Miller, S. Litwin, P. A. Furman, A. R. Jilbert, and W. S. Mason. 2001. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J. Virol. 75:311-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zlotnick, A., N. Cheng, S. J. Stahl, J. F. Conway, A. C. Steven, and P. T. Wingfield. 1997. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: implications for morphogenesis and organization of encapsidated RNA. Proc. Natl. Acad. Sci. USA 94:9556-9561. [DOI] [PMC free article] [PubMed] [Google Scholar]