

Abstract
Eukaryotic chromosomal replication is a complicated process with many origins firing at different efficiencies and times during S phase. Prereplication complexes are assembled on all origins in G1 phase, and yet only a subset of complexes is activated during S phase by DDK (for Dbf4-dependent kinase) (Cdc7-Dbf4). The yeast mcm5-bob1 (P83L) mutation bypasses DDK but results in reduced intrinsic firing efficiency at 11 endogenous origins and at origins located on minichromosomes. Origin efficiency may result from Mcm5 protein assuming an altered conformation, as predicted from the atomic structure of an archaeal MCM (for minichromosome maintenance) homologue. Similarly, an intragenic mutation in a residue predicted to interact with P83L suppresses the mcm5-bob1 bypass phenotype. We propose DDK phosphorylation of the MCM complex normally results in a single, highly active conformation of Mcm5, whereas the mcm5-bob1 mutation produces a number of conformations, only one of which is permissive for origin activation. Random adoption of these alternate states by the mcm5-bob1 protein can explain both how origin firing occurs independently of DDK and why origin efficiency is reduced. Because similar mutations in mcm2 and mcm4 cannot bypass DDK, Mcm5 protein may be a unique Mcm protein that is the final target of DDK regulation.
Eukaryotic chromosomal DNA synthesis is initiated at discrete sites called origins of replication (see references 1 and 26 for reviews). In the yeast Saccharomyces cerevisiae, specific DNA sequences that can act as origins can be identified as autonomously replicating sequences (ARS) because they confer replication to plasmids or minichromosomes. Origin activation or “firing” is regulated as a two-step process that ensures that the genome is replicated only once per cell cycle (see references 1 and 15 for reviews). In the first step, which occurs in the G1 phase of the cell cycle when the cyclin-dependent kinase (CDK) levels are low, a set of six proteins called Mcm proteins are loaded onto origins to form prereplication complexes (pre-RCs). In the second step, after the CDK levels rise, proteins of the replisome (DNA polymerases, RPA, PCNA, etc.) are recruited to the pre-RCs by Cdc45 protein, and the Mcm complex is subsequently activated by the DDK kinase to trigger bidirectional DNA replication. DDK (for Dbf4-dependent kinase) consists of two subunits, a kinase encoded by CDC7 and a regulatory subunit encoded by DBF4, both of which normally are essential for viability (see references 1, 26, and 37 for reviews). Activation by DDK occurs throughout S phase at the ∼330 yeast origins according to the temporal program that is established in the preceding M-G1 phase (32). Because DDK must be present in active form throughout S phase (21, 30, 45), it is unlikely that DDK alone can explain why different origins become active at different times within S phase. Furthermore, by limiting the amount of active DDK in the cell, it was shown that the DDK requirement at early- versus late-S activated origins is temporally rather than quantitatively different—i.e., with respect to activation by DDK, the distinction between early and late origins is when DDK acts on an origin (early or late) and not how much DDK is required (e.g., it is not the case that more DDK activity is required for the late class of origins) (9). Thus, some other regulatory event and not DDK per se regulates the temporal program.
Insight into the molecular function of DDK came from the discovery of an MCM mutation, mcm5-bob1(P83L), that bypasses the requirement for DDK in that both CDC7 and DBF4 genes can be deleted in its presence (37). The MCM genes were originally identified from mutants that showed inefficient maintenance of many ARS minichromosomes—described as the Mcm (for minichromosome maintenance) phenotype (37). They are now known to encode the members of the Mcm complex, a hexameric ring composed of six different, paralogous proteins numbered Mcm2 to Mcm7, that is essential for origin firing and is thought to act as a replicative helicase (see references 15, 24, and 38 for reviews). These observations suggested that DDK-dependent phosphorylation of one or more Mcm subunits could be a key event in origin firing. Indeed, Mcm2, Mcm4, and Mcm6 are all known to be substrates for DDK (30, 45), and it has been suggested recently that phosphorylation of any of these three proteins may fulfill the role of DDK in origin firing (30, 45).
What exactly does phosphorylation by DDK do and how does the mcm5-bob1 mutation allow bypass of DDK? One hypothesis is that in wild-type cells, DDK-dependent phosphorylation of one or more Mcm subunits causes a conformational change (a “domain-push”) in Mcm5 protein and that this altered conformation is required for Cdc45 protein loading and helicase activation (13, 40). Although Mcm5 protein itself appears not to be phosphorylated by DDK, Mcm2 protein, the preferred target of DDK, is adjacent to Mcm5 in the hexameric ring (6). This close association between Mcm2 and Mcm5 lends support to the idea of a phosphorylation-induced domain push. According to this hypothesis, the mcm5-bob1 mutation bypasses the need for DDK because it mimics the effect of DDK phosphorylation—i.e., the mcm5-bob1 mutation produces a spontaneous conformational change in Mcm5 protein, allowing for Cdc45 protein loading and helicase activation in the absence of DDK. Consistent with this hypothesis, Cdc45 protein loading (47) and initial origin unwinding/melting (18), which normally are both dependent on DDK activity, become constitutive in vivo in the presence of the mcm5-bob1 mutation (18, 40).
Evidence for the proposed conformational change comes from the atomic structure of an MCM homologue from Methanobacterium thermoautotrophicum, an archaeal species. In this species, comparison of the wild-type Mcm protein structure with that of the protein with the mcm5-bob1 mutation (P62L) shows that in the mutant, a helical bundle (“domain A”) at the very N terminus of the protein is pushed away from the body of the protein by a large amino acid side chain (13). The mutant phenotype is also reflected in an increased number of protein conformations seen in hydrodynamic and electron microscopy studies, all suggesting that the mutation results in an increased flexibility of the protein (5, 14).
In our previous report, we constructed a yeast Mcm5 protein structural model based on the archaeal Mcm atomic structure (Fig. 1) (13). The yeast Mcm5 structural model suggests that the yeast P83L mcm5-bob1 mutation, as with its archaeal counterpart, causes increased mobility of the N-terminal domain and that, as a result, domain A of the Mcm5 protein can push away from domain C of the protein and assume several different conformations independently of DDK phosphorylation, only a subset of which can recruit Cdc45 protein. Furthermore, this model posits that only mutations that cause increased flexibility of the protein should be successful in giving DDK bypass. In support of this model, we find that origin activation is inefficient at many origins in bypass mode and that mutations at the domain C “docking site” for domain A disrupt bypass of DDK. Successful bypass of DDK is limited to mutations that apparently affect Mcm5 protein flexibility; mutations at equivalent conserved positions in other Mcm proteins fail to give bypass. We also show that the effect on origin efficiency seen in the bypass mode is more drastic at later-firing origins than at early-firing ones. Thus, the presence or absence of DDK does not change the “schedule” of origin initiations but has differential effects on the “success” rate of different origins along the timeline. These observations support a model in which efficient activation of all temporal classes is dependent on events downstream of the DDK-dependent step(s) in origin activation.
FIG. 1.

Structural models of archaeal M. thermoautotrophicum MCM protein. (A) Diagram depicting the conserved secondary structure of the N terminus of M. thermoautotrophicum (archaeal) Mcm (MtMcm) and of S. cerevisiae Mcm5 (ScMcm5) (13). Boxes, α-helices; block arrows, β-sheets; gray bars, domain A; hatched bars, domain C. (B) Interaction between domains A and C in the archaeal Mcm protein. P62L is depicted in green, and F109 is depicted in light blue. Superimposed in yellow is the modeled position of the yeast P83. Notice the movement of domain A away from domain C when P62L is present (magenta lines) compared to the wild-type conformations (blue lines). (C) Yeast Mcm5 modeled on the basis of the archaeal Mcm protein. P83L is in green, and I159 is in purple. (D) Modeled P83L (green) and I159A (gray) in yeast Mcm5 protein.
MATERIALS AND METHODS
Yeast strains and media.
All S. cerevisiae strains are listed in Table 1. Yeast solid and liquid media were as described previously (3, 39, 40). Yeast strains and recombinant plasmids were manipulated as described previously (7, 8, 40). Genetic crosses using tetrad analysis were performed as described previously (39, 40).
TABLE 1.
S. cerevisiae strains and recombinant plasmids used in the present study
| Strain | Genotype | Source or reference |
|---|---|---|
| RSY299 | MATα his3 leu2 trp1 ura3 | 19, 21 |
| RSY302 | MATα leu2 trp1 his3 1 ura3 cdc7-7 | 19, 21 |
| RSY847 | MATα leu2 trp1 his3 1 ura3 cdc7-7 mcm5-P83L (mcm5-bob1-2) | 19, 21 |
| RSY864 | MATα ura3 leu2 trp1 his3 mcm4-P265L | This study |
| RSY855 | MATα leu2 trp1 his3 1 ura3 cdc7-7 mcm4-P265L | This study |
| RSY877 | MATα leu2 ura3 trp1 his7 mcm2-P264L | This study |
| RSY878 | MATα leu2 ura3 trp1 his3Δ-1 cdc7-7 mcm2-P264L | This study |
| RSY1203 | MATaura3 leu2 trp1 mcm2-P264L mcm4-P265L | This study |
| RSY1210 | MATacdc7-7 ura3 leu2 mcm2-P264L mcm4-P265 | This study |
| RSY1216 | MATaura3 leu2 trp1 mcm2-P264L mcm4-P265L mcm5-P83L | This study |
| RSY1217 | MATacdc7-7 ura3 leu2 mcm2-P264L mcm4-P265L | This study |
| RSY902 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm MCM5+TRP1) | This study |
| YRL123 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-P83L TRP1) | This study |
| YRL124 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-P83A TRP1) | This study |
| YRL125 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-I159E TRP1) | This study |
| YRL126 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-I159R TRP1) | This study |
| YRL127 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-I159A TRP1) | This study |
| YRL136 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-P83L I159A TRP1) | This study |
| YRL137 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-P83L I159E TRP1) | This study |
| YRL138 | MATα cdc7-7 leu2 trp1 his7 ura3 mcm5Δ::kanMX4 (pRS424 2μm mcm5-P83L I159R TRP1) | This study |
| YRL154 | MATaleu2 ura3 tyr1 ade1 ade2 trp1 mcm5Δ::kanMX4 (YCp50 2μm MCM5+) | This study |
| RSY311 | MATatrp1 leu2 ura3 can1 his6 bar1 | 40 |
| RSY727 | MATabar1 his6 mcm5-bob1-1 cdc7Δ::HIS3 leu2 ura3 lys2 his3-Δ1 cyh2 | 40 |
| RSY728 | MATabar1 his6 mcm5-bob1-1 trp1 leu2 ura3 lys2 his3-Δ1 | 40 |
| RSY466 | MATaleu2 ura3 trp1 his7 | 32, 40 |
| P253 | MATaura3 can1 trp1 his3 cdc7-1 mcm5-bob1-1 cyh2 | 19 |
Production of mcm2, mcm4, and mcm5 mutants.
Mutations were produced by the overlap PCR method used previously for mcm5 mutations (13). All mutations were marked by the addition or deletion of a DNA restriction site and were subsequently verified by PCR, followed by restriction digestion and DNA sequencing. To place mutations into the chromosome, plasmids were integrated at the corresponding loci by selecting for Ura+ duplications. Recombinant Ura− “pop-outs” were selected with 5-fluoroorotic acid (5-FOA) and then screened for the mutation by PCR and subsequent diagnostic restriction digests. These mutations were combined with other mutations by standard genetic crosses using PCR and a diagnostic restriction digest to verify the presence of the mutation(s).
To produce mcm2-P264L, plasmid pLPB08 (pRS306-MCM2) was made by cloning KpnI/SphI (blunted) fragment from YCp86-MCM2 into KpnI/SmaI of pRS306. YCp86-MCM2 was constructed in the laboratory of Bik Tye (Cornell University) and is described elsewhere (35). Plasmid pLPB09 (pRS306 mcm2-P264L), was then derived from plasmid pLPB08 by PCR overlap (20) using the primers MCM2-Forward (GAC GTA GAC GTG AGG AAG ATG ATT CGG), MCM2-mut2R (CGA ATA TTT TCA ACA TTT CTT CTa GAC ATT TAG CTA), MCM2-Reverse (GGA GCG TAA CCC TTT GAT AAT TTC G), and MCM2-mut2F (TAG CTA AAT GTC tAG AAG AAA TGT TGA AAA TAT TCG). (Lowercase letters in these primer sequences indicate the P264L mutation, which also introduces an XbaI site.) The full-length PCR product was then cloned into the plasmid by using BclI/MluI restriction sites within MCM2. Plasmid pLPB09 was integrated at the MCM2 locus by linearizing the plasmid with EcoNI and selecting for Ura+ transformants of strain RSY466. Recombinant Ura− “pop-outs” were selected on 5-FOA plates and screened for the mutation by PCR (MCM2-Forward and MCM2-Reverse) and diagnostic restriction digestion with XbaI.
To produce mcm4-P265L, we used plasmid pRS316-MCM4 (pLPB02), which consists of a ScaI-HindIII 4.7-kb genomic MCM4 fragment in pRS316. Plasmid pLPB03 (pRS316 mcm4-P265L) was then derived from pLPB02 by PCR overlap mutagenesis using the primers MCM4-Forward (TGT GCC TAA TCC TGA TTC TGT TCC), MCM4-Mut/Reverse (GAT AGA AAT CAC TTC cTG caG ATA ATT TAA TAG TTG), MCM4-Reverse (TTT TTA ACG TGG ACC ACA TCG AC), and MCM4-Mut/Forward (CAA CTA TTA AAT TAT Ctg CAg GAA GTG ATT TCT ATC). (Lowercase bases in these primer sequences indicate P265L mutation and the introduction of a PstI site.) The full-length PCR product was then cloned into the plasmid by using AflII/MluI restriction sites within MCM4. Plasmid pLPB04 (pRS306 mcm4-P265L) was made by cloning an NsiI-HindIII MCM4 fragment from pLPB03 into the same sites of pRS306. Plasmid pLPB04 was integrated at the MCM4 locus by linearizing the plasmid with MluI and selecting for Ura+ transformants of strain RSY466. Recombinant Ura− “pop-outs” were selected on 5-FOA and screened for the mutation by PCR (MCM4-Forward and MCM4-Reverse) and diagnostic digests with PstI.
Mutations in MCM5 were produced by the overlap PCR method used previously (13). All mutations were marked by addition of a DNA restriction site and were subsequently verified by PCR, followed by restriction digestion, and confirmed by DNA sequencing. To produce the mutations, we used the following primers in overlap PCRs: P83A (P83A-fwd [CTATCAGACGAAgctTCAGATATCA] and P83A-rev [TGATATCTGAagcTTCGTCTGATAG]) (adds an HindIII site), I159E (I159E-fwd [CTGAACACGTTTCgAAGgagGTCCGTTTAT] and I159E-rev [ATAAACGGACctcCTTcGAAACGTGTTCAG]) (adds a BstBI site), 1159A (I159A-fwd [CTGAACACGTtTCgAAGgctGTCCGTTTAT] and I159A-rev [ATAAACGGACagcCTTcGAaACGTGTTCAG]) (adds a BstBI site), and I159R (I159R-fwd [CACGTCTCCAAacgcGTCCGTTTAT] and I159R-rev [ATAAACGGACgcgtTTGGAGACGTG]) (adds a MluI site). (Lowercase bases represent the nucleotide change.) The following “outside primers” were used in the overlap reactions: pRS424-Mcm5-fwd (CACTATAGGGCGAATTGGGT) and pRS424-Mcm5-rev (TACACTGGCAGTTAACCCAG).
The full-length single mutant PCR products were cloned into pRS424-MCM5 plasmid pCH802 (19) by using XhoI/PstI restriction sites within MCM5. To make the double mutants, plasmid pPD57, which is identical to pCH802 except that it contains the mcm5-bob1-1 allele (pRS424 mcm5-bob1) (32), was digested with XhoI and BstBI, and the resulting 936-kb fragment was cloned into the XhoI/BstBI sites on pRS424-mcm5 I159E, I159A, or I159R. The resulting plasmids were checked for the I159 mutations with restriction digests by using their “unique site” restriction enzymes. The P83L mcm5-bob1-1mutation was confirmed by digesting the plasmids with Eco57I (32). For plasmid shuffle, plasmids were transformed into strain 902 (Table 1). Cells in which the TRP1 plasmid replaced the URA3 plasmid (Trp+ Ura−) were selected with 5-FOA, and Trp+ Ura− colonies were purified on Trp− and YEPD plates. Colonies were screened for mutations by PCR (pRS424-Mcm5-fwd and pRS424-Mcm5-rev), and restriction digests were done by testing for each mutation with its respective unique restriction site. Strain RSY902 transformed with pPD57 (pRS424 mcm5-bob1) was used as a positive control for cdc7ts bypass suppression. Positive transformants were checked for cdc7ts bypass by measuring growth at 37°C (13).
Plasmid loss.
Initially, minichromosome (plasmid) maintenance (Mcm phenotype) was measured by using a cdc7-1ts mcm5-bob1 strain P253 carrying the indicated ARS-CEN or 2μm plasmid (Table 2). The strains were grown in selective medium at a permissive temperature (22°C) and then replica plated onto selective or nonselective plates at either the permissive temperature or a restrictive temperature (36°C) to measure the proportion of colonies that maintained the plasmid at restrictive compared to permissive temperatures.
TABLE 2.
ARS CEN plasmids replicate poorly in bypass mode
| Plasmida | Origin | % Coloniesb at:
|
|
|---|---|---|---|
| 22°C | 36°C | ||
| pUN70 | ARS1 | 60 | 0.2 |
| YCp50 | ARS1 | 64 | 0.3 |
| pRS424 | 2μm | 64 | 29 |
| pRS314 | ARSH4 | 65 | 0.1 |
| pYES2 | 2μm | 91 | 69 |
All plasmids are in cdc7ts mcm5-bob1 strain P253. Strains were grown in selective medium at 22°C and then diluted and plated on either selective or nonselective media at 22 or 36°C and incubated for 2 to 3 days.
The numbers are the percentages of the colonies found on selective solid media compared to nonselective media.
Subsequently, plasmid loss rates were measured by molecular quantification of population-average plasmid copy number compared to genomic DNA (3). Tested for loss were three plasmids that each contained CEN5 but different ARS—p305.2 (carrying ARS305 in a 17,221-bp fragment of chromosome III [10]), pARS1 (carrying ARS1 in a 16,102-bp fragment of chromosome IV), and p12 (carrying ARS1412 in a 17,478-bp fragment of chromosome XIV [8]). The vector in each case was YIp5-5 (12) with URA3 as the selectable marker. Strains (wild type, mcm5-bob1, and cdc7Δ mcm5-bob1) were grown to mid-log phase under selection for plasmid, diluted into nonselective medium, and kept in log-phase growth over several generations in nonselective medium, with further dilutions into fresh medium as necessary. Samples were collected from the initial (selective) culture and periodically during nonselective growth. The relative amount of plasmid compared to genomic DNA within each sample was measured by Southern blotting with a 32P-labeled URA3 probe after gel electrophoresis of HaeII-digested DNA samples. Hybridization intensities (32P levels) were quantified by using an InstantImager (Packard Instruments). Plasmid loss rates were calculated from the change in the ratio of genomic to plasmid DNA levels over time in nonselective growth. In addition, plasmid retention efficiency was measured by plating cells on selective and nonselective plates after ∼7 generations of nonselective growth in liquid medium. Because the molecular and colony-based assays measure different aspects of plasmid maintenance (decrease in average copy number of plasmid in the population during nonselective growth versus the ability of cells to retain plasmid long enough to form a visible colony, respectively), we refer to the results as “plasmid loss” and “plasmid retention,” respectively.
2-D agarose gel electrophoresis.
Yeast cultures were harvested at mid-log phase growth, and DNA was extracted and tested for origin activity by two-dimensional (2-D) gel electrophoresis as described previously (16). Restriction enzymes used and the origins tested were EcoRV (ARS305 and ARS1414), FspI+SacI (ARS607 and ARS609), NcoI (ARS1, ARS1412, and ribosomal DNA ARS), PstI (ARS306 and ARS603), and XbaI (ARS501 and ARS1413). For estimation of origin firing efficiency, autoradiograms of 2-D gel blots were digitized by scanning. For each sample the intensity of the bubble-arc signal was compared to the intensity of the ascending portion of the Y-arc signal after the background signal was subtracted from similar areas of the gel. Because the bubble-to-Y ratios do not give a direct measure of origin efficiency, for each origin we normalized the observed ratio to that obtained for the wild-type sample. Note that origins show inherent differences in firing efficiency even in the wild type (e.g., the ribosomal DNA ARS is fairly inefficient).
RESULTS
Yeast recombinant minichromosomal plasmids are inefficiently replicated in the bypass mode.
Our hypothesis for Mcm5 protein function in bypass mode predicts that origin firing will likely be inefficient in bypass mode because only a subset of the conformations occupied by the N-terminal domain A of the mcm5-bob1 protein is expected to be productive for origin firing. Indeed, we had found previously that minichromosomal plasmids are maintained poorly in a cdc7Δ mcm5-bob1 strain (data not shown in reference 40), suggesting that the cdc7Δ mcm5-bob1 strain does have an MCM phenotype. This effect was verified by measuring the maintenance of recombinant plasmids in a cdc7ts mcm5-bob1 strain P253 (Table 2). Unlike normal yeast chromosomes, which have multiple origins, each of these plasmids relies on a single replication origin for its maintenance. If the origin fails to fire efficiently, then the plasmid will be lost from the cells during nonselective colony growth. By measuring colony formation at both permissive (22°C) and restrictive conditions (36°C) on both selective and nonselective media, plasmid maintenance during DDK bypass at 36°C was tested. An Mcm phenotype was seen with ARS1 and ARSH4 CEN plasmids, but not with 2μm origin plasmids, a phenotype that is similar to that found for other Mcm mutants (44).
To see whether differences in plasmid maintenance are related to the time of origin firing on the plasmids, three different ARS CEN plasmids that are all based the same plasmid backbone were used to measure the plasmid loss rates of three different replication origins during DDK bypass using the isogenic wild-type, mcm5-bob1, and cdc7Δ mcm5-bob1 strains RSY311, RSY728, and RSY727, respectively (Fig. 2). ARS305, ARS1, and ARS1412 represent early-, mid-, and late-S firing origins, respectively, and the replication times of these plasmids parallel the replication times of these origins in their native chromosomal context (17). Loss rates were measured by genomic Southern hybridization using the yeast URA3 gene: the plasmid-borne and endogenous copy could be distinguished based on fragment size differences after restriction endonuclease digestion (Fig. 2). The rates of loss were low (<2%) in the wild-type and mcm5-bob1 strains but increased more than fivefold in the cdc7Δ mcm5-bob1 strain (16 to 31%), indicating that origin usage is generally inefficient during DDK bypass, with the earliest origin ARS305 being the most stable among the three tested. Finally, the early- and late-S replicating plasmids p305.2 and p12 were retained with high efficiency (98+% retention after ∼7 generations of nonselective growth) in wild-type and mcm5-bob1 cells but both plasmids showed impaired retention in the cdc7Δ mcm5-bob1 strain (30 and 8% retention of p305.2 and p12, respectively). Retention of the late-replicating plasmid was significantly poorer than that of the early plasmid (P = 0.0003 [Student t test]).
FIG. 2.

Maintenance of minichromosomes with early-, mid-, or late-S activated origins. Wild-type (RSY311), mcm5-bob1 (RSY728), and cdc7Δ mcm5-bob1 (RSY727) yeast strains were tested for their ability to maintain early-, mid-, and late-S replicating plasmids (p305, pARS1, and p12, respectively). At the top, Southern blots show hybridization to URA3 sequences on the chromosome (Chr) and on the plasmid (Plas) after various times of growth in nonselective medium; samples were collected at approximately two-generation intervals. Bottom, plots show the ratio of plasmid to genomic DNA hybridization signal at each time, normalized to the ratio seen at the start of nonselective growth. Loss rate values (percent per generation) estimated from the plots for the different plasmids are shown in parentheses.
Endogenous yeast origins are also inefficiently replicated during DDK bypass.
Based on the plasmid results, we predicted that endogenous chromosomal replication origins should also show reduced firing efficiency in the cdc7Δ mcm5-bob1 strain, perhaps with later-firing origins showing a stronger defect. We therefore tested the efficiency of 11 different, endogenous replication origins using 2-D gel analysis of the same three strains (Fig. 3). Again, in the cdc7Δ mcm5-bob1 strain, reduced origin efficiency was seen for all origins examined, with the later-firing origins showing a greater defect than early-firing ones. Specifically, the cdc7Δ mcm5-bob1 strain shows little or no initiation at origins that are known to be Rad53 checked (i.e., those that fail to fire after a short exposure of wild-type cells to hydroxyurea [11]). This reduction in origin firing is much more pronounced than at Rad53-unchecked origins (which do fire in wild-type cells after a similar exposure to hydroxyurea) (Fig. 3). The lone exception to this trend is ARS1 (Fig. 3), which appears to be peculiarly sensitive to the loss of Mcm activity. Initiation in the mcm5-bob1 single mutant strain was slightly impaired at some origins but not at others, with no apparent pattern to the reduction with respect to time of origin initiation—some late-firing origins (e.g., ARS1412) showed robust, wild-type levels of initiation, and an early-S origin (e.g., ARS305) showed a slight reduction in origin firing in the mcm5-bob1 strain. The results of the 2-D gel analysis of ARS305 and ARS1412 are in perfect agreement with both the retention analysis and the loss rate analysis of plasmids p305.2 and p12.
FIG. 3.

2-D gel electrophoresis showing activation of endogenous early-S (A), mid-S (B), and late-S (C) activated origins. (D) Summary of the 2-D gel results. Cultures of wild-type, mcm5-bob1, and cdc7Δ mcm5-bob1 strains were harvested during log-phase growth, and the DNA was analyzed for origin firing at various origins as judged by the approximate proportions of bubble versus simple-Y replication intermediates (e.g., see bottom right panel in panel A).
Interaction between subdomains A and C is needed for bypass.
To better understand the mechanism underlying the mcm5-bob1 phenotype (bypass of Cdc7 kinase), we designed a set of targeted mutations based on the available information on the structure of the archaeal and yeast MCM complex. The “domain-push” model for activation of the Mcm hexameric ring predicts that only substitutions of large amino acid at the interface between Mcm5 subdomains A and C should allow bypass of DDK, because only large substitutions would result in subdomain A being pushed into a potentially active conformation (Fig. 1). Consistent with this prediction, changes of the MCM5 P83 residue to larger amino acids (P83L, P83K, and P83W) but not to smaller amino acids (P83G and P83A) produced bypass of DDK function (13) (Fig. 1). Our published cdc7ts mcm5 P83L and P83A mutants (13) are shown as positive and negative controls for DDK bypass, respectively (Fig. 4).
FIG. 4.
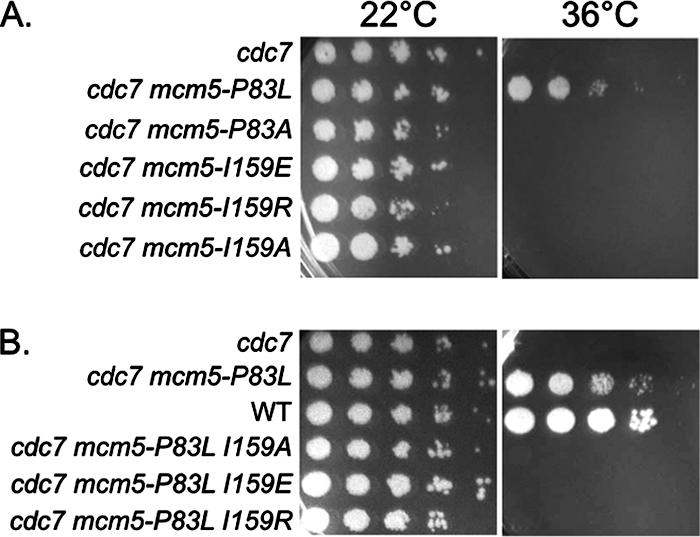
Interaction between subdomains A and C of Mcm5 protein is needed for DDK bypass. Yeast strains were diluted serially in 10-fold increments and spotted on yeast extract-peptone-dextrose plates and then incubated at 22 or 36°C for 3 days.
In the M. thermoautotrophicum MCM protein, P62 in subdomain A, analogous to the conserved P83 in S. cerevisiae Mcm5, is anchored on F109 in subdomain C (Fig. 1) (13). Mimicking the yeast mcm5-bob1 structure, P62L in this archaeal MCM protein positions the Leu side chain to push away from F109. Based on structural modeling and the hydrophobicity of phenylalanine and isoleucine, we predicted that residue I159 in the S. cerevisiae Mcm5 protein should correspond to the archaeal F109 subdomain C position (Fig. 1) and that mutations of this I159 residue should therefore show genetic interaction with the P83 position in subdomain A. In particular, we expected that smaller substitutions of I159 would fail to be “pushed” and therefore not show bypass of the requirement for DDK. Accordingly, we introduced mutations of I159 into a cdc7ts strain and tested for the bypass of DDK. Mutating this residue to a smaller amino acid (I159A) in an otherwise wild-type protein did indeed fail to give bypass of DDK (Fig. 4A); furthermore, this mutation prevented bypass of DDK by the P83L mutation (Fig. 4B). However, mutations of I159 to larger residues (I159E or I159R) also prevented bypass by the P83L mutation (Fig. 4). None of these mutations in a wild-type CDC7 background had any detectable phenotype and grow normally at 36°C (data not shown).
Why do the larger side chains at residue 159 (I159E or I159R) fail to give bypass on their own or in combination with the P83L mutation? One possibility is that the larger side chains in subdomain C are not positioned in a manner that allows subdomain A to be pushed far enough from subdomain C to produce DDK bypass. It is also possible is that these changes at I159 cause subdomain A with the P83L mutation to have reduced flexibility and thereby prevent DDK bypass. Nevertheless, these results demonstrate that the interactions between Mcm5 subdomains A and C occur and are important for DDK bypass.
Based on an alignment of Mcm protein primary sequences aided by the archaeal atomic structure, the Mcm2 and Mcm4 proteins of many species also contain the conserved proline residue (Mcm2-P264 and Mcm4-P265 in S. cerevisiae) (13). DDK bypass was specific to mutations in MCM5 in that mcm2 and mcm4 mutations at these positions (mcm2-P264L and mcm4-P265L) did not give bypass of the cdc7ts phenotype at a restrictive temperature (Fig. 5A). This failure to give bypass was not due to intrinsic temperature sensitivity of the mcm mutations, since all single mcm2, mcm4, and mcm5 mutants were temperature resistant in the presence of a wild-type CDC7 allele (Fig. 5B). Only the combination of the mcm2 and mcm4 mutations produced a temperature-sensitive phenotype and, therefore, was unable to suppress cdc7ts mutation because it is temperature sensitive on its own. The addition of the mcm5-P83L bob1 allele had no effect on the temperature sensitivity of the mcm2-P264L mcm4-P265L double mutant, suggesting that the double mutant phenotype is not a result of a failure to allow domain push in the Mcm5 protein. Similar results were also found at 30°C, which is still restrictive for the cdc7-7ts mutant (data not shown). From these results, we conclude that only mutations in the conserved proline residue in domain A of Mcm5 result in a bypass of DDK function.
FIG. 5.

Mutations in Mcm2 or Mcm4 proteins fail to give bypass. Yeast strains were diluted serially in 10-fold increments, spotted onto yeast extract-peptone-dextrose plates, and then incubated at 22 or 36°C for 3 days.
DISCUSSION
In this report we demonstrate that similar single mutations in MCM5 but not in MCM2 or MCM4 can bypass DDK function (Fig. 5). We believe MCM5 to be a unique Mcm subunit that is an important site of DDK regulation. Empirically, MCM5 is known to be unique in that neither degron mutants (23) nor N- or C-terminal green fluorescent protein fusions (28) could be made with MCM5 but were viable with all five other Mcm proteins. Although there are disagreements in the literature about which of the yeast Mcm2 to Mcm7 subunits are phosphorylated by DDK, all studies show that Mcm2 is a good substrate and that Mcm5 is probably not a substrate (21, 30, 45). Similar results were also found in Schizosaccharomyces pombe (4) and in human cells (22, 36).
The structure of an archaeal MCM protein has suggested that interaction of subdomains A and C is important for DDK bypass (Fig. 1) (13). Our mcm5 mutations are consistent with a model in which (i) DDK phosphorylation of Mcm2, which is adjacent to Mcm5 in the hexameric ring (15), is required to cause subdomain A of wild-type Mcm5 to “push away” from subdomain C and into a conformation that allows Cdc45 loading and helicase activation (40) and (ii) bypass of the DDK requirement becomes possible when mutations at residue P83 in subdomain A cause increased flexibility of that subdomain, allowing it to push away from subdomain C and assume several possible altered conformations, including the one that allows loading of Cdc45 and helicase activation (Fig. 1) (13). Because recent studies show that DDK phosphorylation of other MCM subunits, Mcm4 or Mcm6, is sufficient for DNA replication (27, 41), DDK phosphorylation of any one of three MCM subunits may favor this Mcm5 conformational change. Thus, we propose that structural changes in Mcm5 normally are the result of DDK action at the replication origins.
We propose that structural changes in Mcm5 protein due to DDK phosphorylation are more efficient than the mcm5-bob1 mutation at promoting origin initiation. In either the wild-type or the mcm5-bob1 strain, DDK still can produce the most effective conformation in Mcm5 protein by phosphorylating other MCM proteins, allowing for efficient initiation at replication origins. In the absence of DDK, mcm5-bob1 mutations could produce a series of conformations in subdomain A, of which only a subset is active. This hypothesis is analogous to one proposed from in vitro biochemical and structural studies of the archaeal MCM protein (5, 14). Thus, only a small percentage of mcm5-bob1 MCM complexes are in the proper conformation at any given moment for Cdc45 loading, which results in inefficient initiation. This explains why cdc7Δ mcm5-bob1 strains have a twofold increase in doubling time (6 h in Y-complete medium at 23°C), a slower S phase (32, 41, 45), and inefficient origin usage as shown by both 2-D gel analysis and minichromosome loss experiments (Fig. 2 and 3).
In the “domain-push” hypothesis, the yeast Mcm5 protein residues P83 (archaeal P62) and I159 (archaeal F109) would interact (Fig. 1). Our observation that mutations in the critical residue I159 in Mcm5 subdomain C can suppress DDK bypass by P83L (Fig. 4) supports the idea that these two residues interact in vivo. Thus, the data in this report represent important in vivo evidence to support our model for eukaryotic MCM helicase regulation based on the archaeal structure (5, 14). However, our results also reveal that achieving bypass of DDK may not be as simple as just having a large amino acid side chain at I159, since neither mcm5-I159R nor mcm5-I159E produced a bypass phenotype either as single mutants or in combination with mcm5-bob1 (P83L) (Fig. 4). Without more structural information, particularly on the yeast Mcm complex, we do not know whether these changes would indeed be capable of “pushing out” domain A.
Bypass of DDK by mcm5-bob1 is dependent on Cdk1-Clb5 activity (40), and mcm5-bob1 cannot bypass the role of Cdk1 in the initiation of DNA replication (29, 40). Recently, the role of CDK in promoting origin activation in yeast has been determined: yeast Cdk1 phosphorylates and loads Sld2 and Sld3 proteins, which are then used to load Dpb11, a subunit of DNA polymerase ɛ holoenzyme (43, 46). Thus, DDK and CDK perform distinct steps that converge on the same function, that is, the initiation of DNA replication by activating the MCM helicase and loading replisome components—while DDK performs the early steps in replisome assembly wherein phosphorylation of Mcm2, Mcm4, or Mcm6 protein produces a conformational change in Mcm5 protein that results in the loading of Cdc45 protein, Cdk1 completes loading of the replisome. Both events result in complete MCM helicase activation. The mcm5-bob1 mutation mimics DDK phosphorylation and allows for constitutive loading of Cdc45 protein (40) and partial origin unwinding (18), but replication cannot occur without CDK function. Thus, cell cycle regulation of DNA replication can occur either by DDK or by CDK action; when both DDK and CDK requirements are bypassed, replication will occur in the G1 phase (43, 46). In fact, G1-phase overexpression of DDK under conditions of DDK and CDK bypass results in lethality (46).
The ∼330 origins of budding yeast are more or less evenly spaced in the genome and have a large range of efficiencies (34). Why are later origins affected more in the cdc7Δ mcm5-bob1 strain (Fig. 2 and 3)? One explanation is that the Mcm5-bob1 protein bound to an early-firing origin simply has more time to sample potential conformations before an invading fork can arrive at that location—because there likely would not be forks approaching that origin early in S phase. In contrast, a late origin might have a much smaller window of opportunity to fire, since forks would already be approaching that origin. A second possibility is that there may be inherent differences in firing efficiency at different origins, as has been seen for many mcm mutants (44). In fact, similar to our findings, it is known that ARS1 is the most affected and 2μm origin is the least affected by mcm mutations (44). Because the differences we see in origin efficiency are also recapitulated on minichromosomes with single origins (Fig. 2), the differences must be inherent.
It is also possible that there is not a defect per se in activating late origins but that the observed increase in passive replication through them is due to an inherently faster rate of fork progression in the cdc7Δ mcm5-bob1 mutant. If the Mcm complex is indeed the replicative helicase, mutations in one of its subunits might confer an improvement in denaturation ahead of the fork, allowing faster fork migration rates. In this scenario, late origins are simply overrun by fast forks before their scheduled time of firing. Two observations argue against this possibility. First, the increased loss rate of single-origin plasmids indicates that reduced firing efficiency cannot be the result of just an increased fork rate. Second, we also measured fork rates on the right arm of chromosome VI and the left arm of III using density transfer methods (34) and found no evidence for increased fork rates in the cdc7Δ mcm5-bob1 strain (data not shown). In summary, the cdc7Δ mcm5-bob1 strain progresses through S phase slowly because of inefficient initiation at all origins.
Because origins fire through much of S phase (see references 1 and 26) for reviews) and DDK is needed to fire origins throughout the S phase (2, 9), DDK alone is not the regulator of the temporal program. The DDK requirement at early and late origins is temporally rather than quantitatively different, with early- and late-S origins showing equal reduction in activity under conditions where cells are producing a constant, limiting amount of Cdc7 protein throughout S phase (2, 9). Because the temporal program is established in the preceding G2/M-Start interval (33), by eliminating DDK activity from the cell using a cdc7Δ mcm5-bob1 strain, only downstream events such as Mcm2-7 activation are affected. However, we still see that late origins are more affected than early ones—presumably in this case by a reduction in Mcm2-7 efficiency (Fig. 2 and 3). This result implies that some regulatory event downstream of DDK phosphorylation of the MCM complex regulates the execution of the temporal program. We propose that the downstream event may be the “pushing out” of subdomain A of Mcm5, which is more inefficient at late origins due to other events such as changes in chromatin structure that were set up in the preceding G2/M-Start interval (33). The observation that the efficiency of mcm5-bob1 bypass is affected by changes in histone copy number (8) is consistent with this idea. The mechanism for execution of the temporal program may be quite different in S. pombe, where late-firing origins may simply be the ones that have the lowest probability of interacting with DDK (31). Another possibility is that upstream events such as the amount of Mcm2-7 complexes loaded in G1 phase dictate timing, and the effect of these events is only evident downstream of DDK action.
Finally, our results also give insight into whether origin firing contributes to the regulation of the intra-S-phase checkpoint in yeast. Origin firing is reduced in both orc2 and cdc7Δ mcm5-bob1 strains (25) (Fig. 2 and 3), but whereas the intra-S checkpoint is compromised in orc2 mutants (42), the intra-S DNA replication and damage checkpoints are intact in cdc7Δ mcm5-bob1 strains (8, 30, 32, 45). The reduction in origin firing must occur for different reasons in the two strains—due to reduced frequency of pre-RC formation in orc2 (25) as opposed to inefficient activation of pre-RCs in cdc7Δ mcm5-bob1. Indeed, because DDK is not involved with pre-RC formation (1, 15), we would expect pre-RC formation with normal efficiency and spacing in cdc7Δ mcm5-bob1. The difference in intra-S-phase checkpoint efficiency in these two strains despite the shared phenotype of reduced origin firing in both strains therefore suggests that rather than origin usage as proposed previously (42), it is the number of pre-RCs that regulates the intra-S-phase checkpoint response.
Acknowledgments
These studies were supported by Public Health Service grants GM35078 (R.A.S.), GM070403 (R.P.L.), and GM18926 (W.L.F. and B.J.B.) from the National Institute of General Medical Sciences.
We thank Bik Tye for plasmid YCp86-MCM2 and Xiaojiang Chen and Ryan Fletcher for insightful discussions.
Footnotes
Published ahead of print on 27 August 2007.
REFERENCES
- 1.Bell, S. P., and A. Dutta. 2002. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71: 333-374. [DOI] [PubMed] [Google Scholar]
- 2.Bousset, K., and J. F. X. Diffley. 1998. The Cdc7 protein kinase is required for origin firing during S phase. Genes Dev. 12: 480-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer, B. J., and W. L. Fangman. 1994. Initiation preference at a yeast origin of replication. Proc. Natl. Acad. Sci. USA 91: 3418-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, G. W., and T. J. Kelly. 1998. Purification of Hsk1, a minichromosome maintenance protein kinase from fission yeast. J. Biol. Chem. 273: 22083-22090. [DOI] [PubMed] [Google Scholar]
- 5.Chen, Y. J., X. Yu, R. Kasiviswanathan, J. H. Shin, Z. Kelman, and E. H. Egelman. 2005. Structural polymorphism of Methanothermobacter thermautotrophicus MCM. J. Mol. Biol. 346: 389-394. [DOI] [PubMed] [Google Scholar]
- 6.Davey, M. J., C. Indiani, and M. O'Donnell. 2003. Reconstitution of the mcm2-7p heterohexamer, subunit arrangement, and ATP site architecture. J. Biol. Chem. 278: 4491-4499. [DOI] [PubMed] [Google Scholar]
- 7.Dohrmann, P. R., G. Oshiro, M. Tecklenburg, and R. A. Sclafani. 1999. RAD53 regulates DBF4 independently of checkpoint function in Saccharomyces cerevisiae. Genetics 151: 965-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dohrmann, P. R., and R. A. Sclafani. 2006. Novel role for checkpoint Rad53 protein kinase in the initiation of chromosomal DNA replication in Saccharomyces cerevisiae. Genetics 174: 87-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donaldson, A. D., W. L. Fangman, and B. Brewer. 1998. Cdc7 is required throughout the yeast S phase to activate replication origins. Genes Dev. 12: 491-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donaldson, A. D., M. K. Raghuraman, K. L. Friedman, F. R. Cross, B. J. Brewer, and W. L. Fangman. 1998. CLB5-dependent activation of late replication origins in Saccharomyces cerevisiae. Mol. Cell 2: 173-182. [DOI] [PubMed] [Google Scholar]
- 11.Feng, W., D. Collingwood, M. E. Boeck, L. A. Fox, G. M. Alvino, W. L. Fangman, M. K. Raghuraman, and B. J. Brewer. 2006. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat. Cell Biol. 8: 148-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson, B. M., B. J. Brewer, A. E. Reynolds, and W. L. Fangman. 1991. A yeast replication origin is activated late in S phase. Cell 65: 507-515. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher, R. J., B. E. Bishop, R. P. Leon, R. A. Sclafani, C. M. Ogata, and X. S. Chen. 2003. The structure and function of MCM from archaeal Methanobacterium thermoautotrophicum. Nat. Struct. Biol. 10: 160-167. [DOI] [PubMed] [Google Scholar]
- 14.Fletcher, R. J., and X. S. Chen. 2006. Biochemical activities of the BOB1 mutant in Methanobacterium thermoautotrophicum MCM. Biochemistry 45: 462-467. [DOI] [PubMed] [Google Scholar]
- 15.Forsburg, S. L. 2004. Eukaryotic MCM proteins: beyond replication initiation. Microbiol. Mol. Biol. Rev. 68: 109-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman, K. L., and B. J. Brewer. 1995. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 262: 613-627. [DOI] [PubMed] [Google Scholar]
- 17.Friedman, K. L., J. D. Diller, B. M. Ferguson, S. V. Nyland, B. J. Brewer, and W. L. Fangman. 1996. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes. Dev. 10: 1595-1607. [DOI] [PubMed] [Google Scholar]
- 18.Geraghty, D. S., M. Ding, N. H. Heintz, and D. S. Pederson. 2000. Premature structural changes at replication origins in a yeast minichromosome maintenance (MCM) mutant. J. Biol. Chem. 275: 18011-18021. [DOI] [PubMed] [Google Scholar]
- 19.Hardy, C. F. J., O. Dryga, S. Seematter, P. M. B. Pahl, and R. A. Sclafani. 1997. mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc. Natl. Acad. Sci. USA 94: 3151-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51-59. [DOI] [PubMed] [Google Scholar]
- 21.Jackson, A. L., P. M. B. Pahl, K. Harrison, J. Rosamond, and R. A. Sclafani. 1993. Cell cycle regulation of the yeast Cdc7 protein kinase by association with the Dbf4 protein. Mol. Cell. Biol. 13: 2899-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang, W., D. McDonald, T. J. Hope, and T. Hunter. 1999. Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 18: 5703-5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labib, K., J. A. Tercero, and J. F. Diffley. 2000. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 288: 1643-1647. [DOI] [PubMed] [Google Scholar]
- 24.Lei, M. 2005. The MCM complex: its role in DNA replication and implications for cancer therapy. Curr. Cancer Drug Targets 5: 365-380. [DOI] [PubMed] [Google Scholar]
- 25.Liang, C., M. Weinreich, and B. Stillman. 1995. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell 81: 667-676. [DOI] [PubMed] [Google Scholar]
- 26.Machida, Y. J., J. L. Hamlin, and A. Dutta. 2005. Right place, right time, and only once: replication initiation in metazoans. Cell 123: 13-24. [DOI] [PubMed] [Google Scholar]
- 27.Masai, H., C. Taniyama, K. Ogino, E. Matsui, N. Kakusho, S. Matsumoto, J. M. Kim, A. Ishii, T. Tanaka, T. Kobayashi, K. Tamai, K. Ohtani, and K. Arai. 2006. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281: 39249-39261. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen, V. Q., C. Co, K. Irie, and J. J. Li. 2000. Clb/Cdc28 kinases promote nuclear export of the replication initiator proteins Mcm2-7. Curr. Biol. 10: 195-205. [DOI] [PubMed] [Google Scholar]
- 29.Nougarede, R., F. Della Seta, P. Zarzov, and E. Schwob. 2000. Hierarchy of S-phase-promoting factors: yeast Dbf4-cdc7 kinase requires prior S-phase cyclin-dependent kinase activation. Mol. Cell. Biol. 20: 3795-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oshiro, G., J. C. Owens, Y. Shellman, R. A. Sclafani, and J. J. Li. 1999. Cell cycle control of cdc7p kinase activity through regulation of dbf4p stability. Mol. Cell. Biol. 19: 4888-4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel, P. K., B. Arcangioli, S. P. Baker, A. Bensimon, and N. Rhind. 2006. DNA replication origins fire stochastically in fission yeast. Mol. Biol. Cell 17: 308-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pessoa-Brandao, L., and R. A. Sclafani. 2004. CDC7/DBF4 functions in the translesion synthesis branch of the RAD6 epistasis group in Saccharomyces cerevisiae. Genetics 167: 1597-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghuraman, M. K., B. J. Brewer, and W. L. Fangman. 1997. Cell cycle-dependent establishment of a late replication program. Science 276: 806-808. [DOI] [PubMed] [Google Scholar]
- 34.Raghuraman, M. K., E. A. Winzeler, D. Collingwood, S. Hunt, L. Wodicka, A. Conway, D. J. Lockhart, R. W. Davis, B. J. Brewer, and W. L. Fangman. 2001. Replication dynamics of the yeast genome. Science 294: 115-121. [DOI] [PubMed] [Google Scholar]
- 35.Ray, A., and P. Sinha. 1995. The mcm2-1 mutation of yeast causes DNA damage with a RAD9 requirement for repair. Curr. Genet. 27: 95-101. [DOI] [PubMed] [Google Scholar]
- 36.Sato, N., K.-I. Arai, and H. Masai. 1997. Human and Xenopus cDNAs encoding budding yeast Cdc7-related kinases: in vitro phosphorylation of MCM subunits by a putative human homologue of Cdc7. EMBO J. 16: 4340-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sclafani, R. 2000. Cdc7p-Dbf4p becomes famous in the cell cycle. J. Cell Sci. 113: 2111-2117. [DOI] [PubMed] [Google Scholar]
- 38.Sclafani, R. A., R. J. Fletcher, and X. S. Chen. 2004. Two heads are better than one: regulation of DNA replication by hexameric helicases. Genes Dev. 18: 2039-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sclafani, R. A., M. Patterson, J. Rosamond, and W. L. Fangman. 1988. Differential regulation of the yeast CDC7 gene during mitosis and meiosis. Mol. Cell. Biol. 8: 293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sclafani, R. A., M. Tecklenburg, and A. Pierce. 2002. The mcm5-bob1 bypass of Cdc7p/Dbf4p in DNA replication depends on both Cdk1-independent and Cdk1-dependent steps in Saccharomyces cerevisiae. Genetics 161: 47-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheu, Y. J., and B. Stillman. 2006. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24: 101-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimada, K., P. Pasero, and S. M. Gasser. 2002. ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev. 16: 3236-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka, S., T. Umemori, K. Hirai, S. Muramatsu, Y. Kamimura, and H. Araki. 2007. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445: 328-332. [DOI] [PubMed] [Google Scholar]
- 44.Tye, B.-K. 1999. MCM proteins in DNA replication. Annu. Rev. Biochem. 68: 649-686. [DOI] [PubMed] [Google Scholar]
- 45.Weinreich, M., and B. Stillman. 1999. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 18: 5334-5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zegerman, P., and J. F. Diffley. 2007. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445: 281-285. [DOI] [PubMed] [Google Scholar]
- 47.Zou, L., and B. Stillman. 2000. Assembly of a complex containing cdc45p, replication protein A, and mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol. Cell. Biol. 20: 3086-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]