

Abstract
The nonenveloped polyomavirus (Py) traffics from the plasma membrane to the endoplasmic reticulum (ER), where it penetrates the ER membrane, allowing the viral genome to reach the nucleus to cause infection. The mechanism of membrane penetration for Py, and for other nonenveloped viruses, remains poorly characterized. We showed previously that the ER chaperone ERp29 alters the conformation of Py coat protein VP1, enabling the virus to interact with membranes. Here, we developed a membrane perforation assay and showed that the ERp29-activated Py perforates the physiologically relevant ER membrane, an event that likely initiates viral penetration. Biochemical analysis revealed that the internal protein VP2 is exposed in the activated viral particle. Accordingly, we demonstrate that VP2 binds to, integrates into, and perforates the ER membrane; the other internal protein, VP3, binds to and integrates into the ER membrane but is not sufficient for perforation. Our data thus link the activity of a cellular factor on a nonenveloped virus to the membrane perforation event and identify a viral component that mediates this process.
The mechanism by which nonenveloped viruses penetrate biological membranes to cause infection is a poorly characterized process. As nonenveloped viruses lack a lipid bilayer on their surface, their penetration mechanism must be fundamentally different from that of enveloped viruses. While conclusive data remain elusive, membrane penetration by nonenveloped viruses can be divided conceptually into four discrete steps (reviewed in reference 24). The virus first reaches the membrane penetration site in the host cell. Here, interaction with cellular cues (e.g., receptors, proteases, or chaperones) renders the virus hydrophobic or triggers the release of a viral membrane lytic factor. Next, binding of the hydrophobic virus or lytic factor to the limiting membrane is thought to disrupt the lipid bilayer, preparing the viral particle for transport across the membrane. In the final step, the viral (or subviral) particle is transported across the limiting membrane and released into the cytosol or the nucleus.
The abilities of cellular cues to impart structural alterations on nonenveloped viruses that stimulate membrane binding are well documented. For instance, interaction with its cell surface receptor Pvr triggers a conformational change in poliovirus that exposes the VP4 protein and the VP1 protein N terminus, allowing the virus to bind to liposomes (11). Similarly, proteolytic cleavage of the reovirus's outer protective protein, σ3, primes the virus for membrane penetration (2, 10). In the case of the murine polyomavirus (Py), where penetration of the endoplasmic reticulum (ER) membrane is essential for infection, we demonstrated recently that the ER-resident chaperone ERp29 induces a conformational change to the C terminus of the coat protein VP1 of Py, generating a hydrophobic viral particle that binds to liposomes (16, 19).
How the membrane-binding reaction leads to viral penetration is unknown. Interaction between the activated viral particle (or freed components of the activated virus) and a model membrane has been shown to disrupt the bilayer integrity. For instance, by use of the plasma membrane of erythrocytes as a model membrane, the activated reovirus was found to induce the release of hemoglobin from these cells (4), suggesting that the plasma membrane integrity was compromised. Likewise, incubating the membrane penetration protein of adenovirus, protein VI, with liposomes entrapping a fluorophore caused the release of the entrapped fluorophore (26). Similarly, the membrane-disrupting γ1 peptide of the nonenveloped Flock House virus also triggers fluorophore release from liposomes (3). While these examples indicate that a model membrane can be compromised, the ability of a physiologically activated virus to disrupt the relevant target membrane that it penetrates has not been demonstrated directly.
The possibility that Py proteins facilitate the disruption of host cell membranes to deliver Py genomes to the host nuclei during infection is suggested by several studies of simian virus 40 (SV40). The SV40 minor structural proteins VP2 and VP3 are necessary for infection and, when translated in vitro, are capable of binding to and integrating into the ER membrane (7). These proteins also demonstrate lytic properties when expressed in Escherichia coli cells, rendering the bacterial membrane permeable to the protein synthesis inhibitor hygromycin B and, in the case of VP3 only, inducing lysis of the bacterial cells (6). These findings suggest that VP2 and VP3 might play a role in ER penetration during SV40 infection. In fact, VP2 and VP3 were found exposed on the SV40 particles that reached the ER (18). Murine Py structural proteins VP2 and VP3 have been shown to associate with cellular membranes (8), but there is little additional evidence that these proteins, or the structural proteins of the human Pys JC and BK, facilitate membrane penetration.
Here, we developed a membrane perforation assay and showed that the ERp29-activated Py particle disrupts the ER membrane. Biochemical analysis further revealed that, while the activated viral particle is not disassembled, the internal protein VP2 is exposed. Accordingly, we found that VP2 binds to, integrates into, and induces perforation of the ER membrane; VP3, the other internal protein, binds to and integrates into the ER membrane but fails to perforate the ER membrane. Our data demonstrate that an activated nonenveloped virus is able to disrupt its physiologically relevant target membrane and identify a viral component mediating this process.
MATERIALS AND METHODS
Reagents.
Crude and purified murine Py and polyclonal VP1 antibody were gifts from T. Benjamin (Harvard Medical School, Boston, MA). Polyclonal VP2/VP3 antibody and the pREC-VP2 and pREC-VP3 plasmids were gifts from R. Garcea (University of Colorado, Aurora, CO). Polyclonal ERp29 antibody was a gift from S. Mkrtchian (Karolinska Institute, Stockholm, Sweden). Polyclonal protein disulfide isomerase (PDI) antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). 293T cells were obtained from D. Engel (University of Michigan, Ann Arbor, MI). Trypsin was purchased from Sigma (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), penicillin-streptomycin, Optimem, Lipofectamine 2000, and 0.05% trypsin-EDTA were purchased from Invitrogen (Carlsbad, CA). Fetalclone III (FC) was purchased from HyClone (Logan, UT). TransIT-LTI transfection reagent was purchased from Mirus Bio Corporation (Madison, WI). Digitonin was purchased from Calbiochem (San Diego, CA). MPEG-MAL-5000 was purchased from Nektar Therapeutics (Huntsville, AL).
Subcloning.
The VP2 and VP3 cDNAs were amplified using pREC-VP2 and pREC-VP3, respectively, as templates. The amplification products were subcloned into the XhoI and EcoRI sites of pcDNA3.1(−). The resulting pcDNA3.1-VP2 and pcDNA3.1-VP3 expression constructs were confirmed by sequencing.
Tissue culture and transfection.
293T cells were maintained in DMEM-5% FC with Pen-Strep in a humidified 5% CO2 incubator at 37°C. For transfection, the DMEM-5% FC-Pen-Strep medium was exchanged for DMEM-5% FC only. To transfect pcDNA3.1-VP2, complexes were prepared with Lipofectamine 2000 in Optimem and added to cells for 24 h. To transfect pcDNA3.1-VP3, complexes were prepared with TransIT-LTI transfection reagent in Optimem and added to cells for 24 h. The complexes were then removed and the media exchanged for DMEM-5% FC-Pen-Strep. At 36 h posttransfection, the proteasome inhibitor MG-132 (5 μM) was added to the medium of cells transfected with VP3 (and the corresponding vector control cells) to prevent the degradation of VP3. Cells were harvested in 0.05% trypsin-EDTA at 48 h posttransfection.
Microsome and LE preparation.
Microsomes from dog pancreas were a gift from T. Rapoport (Harvard Medical School, Boston, MA). An ER lumenal extract (LE) derived from the dog pancreatic microsomes was prepared as before (22). To prepare microsomes from mouse pancreas, tissue was harvested from mice and placed in a physiological buffer (150 mM potassium acetate [KOAc], 250 mM sucrose, 50 mM HEPES [pH 7.5], 2 mM magnesium acetate) with a protease inhibitor cocktail (Roche). The pancreas was minced with a razor blade, dounced (∼10 strokes) until homogenized, and centrifuged at 2,000 rpm in a JS 4.2 rotor (Beckman) for 10 min at 4°C. Solid and fatty materials floating at the top of the supernatant were removed, and the supernatant was centrifuged at 10,000 × g for 10 min at 4°C in an SS-34 rotor (Sorvall). The resulting supernatant was placed over a 1.25 M sucrose cushion and centrifuged at 40,000 rpm for 140 min at 4°C in a TLA 100.3 rotor (Beckman). The resulting pellet represents the microsomes and is resuspended in physiological buffer.
Preparation of an ERp29-enriched extract.
An ERp29-enriched extract was prepared by fractionating an ER LE from dog pancreatic microsomes as described before (16). The resulting fractions were analyzed for their ERp29 and PDI content by immunoblotting. Fractions in which ERp29 was enriched and in which PDI was absent were pooled.
Preparation of VP2- and VP3-containing lysates.
293T cells transfected with pcDNA3.1-VP2 or pcDNA3.1-VP3 were harvested by trypsinization and pelleted at 5,000 rpm at 4°C for 5 min. Pellets were lysed in physiological buffer with 1% Triton X-100 and protease inhibitors on ice for 30 min. Cell debris was pelleted at 13,000 rpm at 4°C for 15 min, and the VP2- and VP3-containing supernatants were incubated with BioBeads SM-2 adsorbent (Bio-Rad Laboratories Inc., Hercules, CA) overnight at 4°C with shaking at 1,000 rpm to remove the detergent. The detergent-free lysates were then removed from the beads and centrifuged at 13,000 rpm at 4°C for 15 min to remove any remaining cell debris or beads. The lysates were aliquoted and stored at −80°C.
Immunodepletion.
Immunodepletions of ERp29 and PDI from the ER LE were performed as before (16).
Trypsin digestion assay.
The trypsin digestion assay was performed as described before (16). Samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting with VP1 antibody or VP2/VP3 antibody. To analyze the sensitivity of VP2 and VP3 in the 293T lysates to trypsin digestion, the VP2 or VP3 lysates were incubated with or without urea (8 M) for 10 min at 37°C, followed by trypsin digestion (2.5, 5, 10, 12.5, or 25 μg/ml) for 30 min on ice. For the VP3 lysates, the urea was diluted prior to trypsin digestion. For the VP2-lysates, the urea was removed by dialysis prior to trypsin digestion. Trypsin was inhibited with TLCK on ice for 10 min. Samples were analyzed by reducing SDS-PAGE and immunoblotted with a VP2/VP3 antibody.
Size exclusion fractionation of Py.
Following incubation of crude Py with bovine serum albumin (BSA), LE, or 1% SDS for 30 min at 37°C, the samples were fractionated over a size exclusion column (BioSil SEC 250; Bio-Rad). Fractions (250 μl) were collected, and 50 μl of each was analyzed by SDS-PAGE and immunoblotted with a VP1 antibody. Proteins of known molecular weights were used for calibration.
Native agarose gel electrophoresis.
Following incubation of crude Py with BSA, LE, or 1% SDS for 30 min at 37°C, the samples were resolved on a 0.8% agarose gel. The agarose gel was then transferred overnight onto a nitrocellulose membrane, and the membrane was immunoblotted with VP1 antibody.
Perforation assays.
To examine perforation of ER membranes by Py, purified Py (1 μg) was incubated with the ERp29-enriched extract in the presence of dithiothreitol (DTT; 3 mM) and EGTA (10 mM) for 1 h at 37°C. One-fifth of this reaction (200 ng Py) was mixed with mouse pancreatic microsomes and the reaction mixture incubated on ice for 15 min. To examine perforation of ER membranes by VP2- or VP3-containing lysates, lysates (125 μg) were incubated with mouse pancreatic microsomes for 30 min at 37°C. For both the Py and VP2/3 perforation assays, maleimide-PEG5000 (M-PEG; 0.2 mM) was then added to the reaction mixture and incubated for 1 h at 37°C. The reactions were stopped with excess DTT (0.1 M) on ice for 5 min. For the Py perforation assay, SDS-containing sample buffer was added to the entire sample and boiled at 95°C for 10 min. For the VP2/VP3 lysate perforation assay, the microsomes were pelleted and washed three times in physiological buffer with 0.1 M DTT. The pellets were resuspended in SDS-containing sample buffer and the samples boiled at 95°C for 10 min. All samples were analyzed by SDS-PAGE followed by immunoblotting with an antibody against PDI.
VP2/VP3 binding assay and alkali or high-salt-concentration extraction.
VP2- and VP3-containing lysates were incubated with mouse pancreatic microsomes for 30 min at 37°C. Microsomes were pelleted at 9,000 rpm (Eppendorf 5415R) for 5 min at 4°C. Supernatant and pellet for each sample were separated, and SDS-containing sample buffer was added to both and then boiled at 95°C for 10 min. For the alkali and high-salt-concentration extraction of VP2 and VP3, microsomes were incubated with VP2- or VP3-containing lysates as described above to establish binding. Following the 9,000-rpm centrifugation, the microsome pellets with bound VP2 and VP3 were resuspended in 50 μl standard physiological buffer (150 mM KOAc, pH 7.5), physiological buffer at pH 11, or physiological buffer with 0.5 M KOAc and incubated on ice for 15 min. The extracted microsomes were then pelleted as described above. The supernatant and pellet fractions for each sample were separated, and SDS-containing sample buffer was added to both and then boiled at 95°C for 10 min. All samples for the binding assay and the alkali/high-salt-concentration extraction were analyzed by SDS-PAGE and immunoblotting with VP2/VP3, Ero1α, p97, or calnexin antibodies.
RESULTS
ERp29-modified Py induces perforation of the ER membrane.
Following uptake into host cells via the ganglioside receptor GD1a (12, 23), murine Py transits to the ER. In this compartment, its major coat protein, VP1, is unfolded by the PDI-like ER chaperone ERp29, an event that is necessary for viral infection (16, 19). We previously designed a trypsin digestion assay to monitor unfolding of VP1. VP1 is located at the outer surface of the virus and is subject to initial unfolding events. Information from the crystal structure of Py indicated that disulfide bonds and calcium ions stabilize the VP1 pentamer (21). Thus, reducing the disulfide bonds and removing the calcium ions should in principle lead to the partial destabilization of VP1. Hence, in this assay, Py was first incubated with the reducing agent DTT, the calcium-chelating agent EGTA, and the control protein BSA, followed by the addition of trypsin. Under this condition, a cleavage product (derived from VP1) of approximately 40 kDa called VP1a is observed (Fig. 1A, lane 2). DTT and EGTA likely mimic the activities of ER reductases (e.g., PDI) and calcium-binding proteins (e.g., calnexin) that would normally act on the virus. When Py was incubated with an extract containing ER lumenal proteins derived from pancreatic microsomes (LE) instead of BSA, a smaller cleavage product of approximately 38 kDa called VP1b was generated (Fig. 1A, lane 4). This increase in trypsin sensitivity indicates an unfolding event that reveals previously cryptic trypsin cleavage sites in the virus. We found previously that VP1b lacks the C-terminal fragment of VP1 (16), suggesting that the lumenal activity unfolds the C terminus of VP1. Formation of the VP1b peptide was shown to be ERp29 dependent, as an LE depleted of ERp29 (Fig. 1A, bottom, compare lane 8 to lane 6) but not an LE depleted of PDI generated the VP1b fragment poorly (Fig. 1A, top, compare lane 8 to lanes 6 and 7) (16).
FIG. 1.

Perforation of ER membranes by the ERp29-modified Py. (A) ERp29-dependent unfolding of Py. Py was incubated with BSA, LE, or LE depleted of either PDI or ERp29. Trypsin was added to the reaction mixture, and the samples were analyzed by SDS-PAGE and immunoblotted with antibodies against VP1 (top, lanes 1 to 8). The ERp29- and PDI-immunodepleted extracts were subjected to SDS-PAGE and immunoblotted with an antibody against ERp29 (bottom, lanes 5 to 8). (B) Generation of an ERp29-enriched extract followed the procedure described in reference 16. (C) Unfolding of Py, using the ERp29-enriched extract. Py was incubated with BSA or the ERp29-enriched extract, followed by addition of trypsin. Samples were analyzed as described for panel A. (D) Modification of PDI by M-PEG in Py-perforated microsomes. Microsomes were incubated with digitonin (lanes 1 and 2), buffer (lane 3), ERp29-enriched extract (lane 4), Py (lane 5), or Py pretreated with the ERp29-enriched extract (lane 6). M-PEG (0.2 mM) was then added to all the samples except in lane 1. The samples were analyzed by SDS-PAGE and immunoblotted with an antibody against PDI. PDI-MPEG*, -**, -***, and -**** refer to PDI modified by one, two, three, and four molecules of M-PEG, respectively.
We also found previously that the ERp29-dependent Py unfolding reaction generates a hydrophobic viral particle that binds to an artificial membrane (i.e., liposomes) (16). Because the ERp29 activity was shown to be critical for viral infection, we proposed a model in which the ERp29-dependent unfolding reaction enables Py to both bind to and penetrate the ER membrane, resulting in the delivery of the viral genome into the cytosol or possibly directly into the nucleus (16).
How might interaction of the ERp29-activated viral particle with the ER membrane allow the virus (or a subviral component) to cross the membrane? One possibility is that binding of the virus to the ER membrane disrupts its bilayer integrity, leading to membrane perforation that precedes the transport of Py across the membrane. To test this possibility, we developed an in vitro assay to detect perforation of the ER membrane induced by the ERp29-modified virus. We first generated an extract that contained a high concentration of ERp29 by fractionating the LE on an anion exchanger as described previously (16). Fractions containing ERp29 were pooled and analyzed by SDS-PAGE, followed by either Coomassie analysis (Fig. 1B, left lane) or immunoblot analysis with an antibody against ERp29 (Fig. 1B, right lane). The pooled fraction contained approximately seven visible bands (Fig. 1B, left lane), with the band corresponding to ERp29 (where indicated) verified by immunoblot analysis (Fig. 1B, right lane). This fraction is referred to as the ERp29-enriched extract. As expected, incubation of Py with the ERp29-enriched extract (but not BSA) generated the VP1b fragment potently (Fig. 1C, compare lane 2 to lane 1).
Mouse pancreatic microsomes were used as a model membrane to study the initiation of the ER-to-cytosol virus penetration process. There does not appear to be a dramatic difference in the percentages of the two major phospholipids (phosphatidylcholine and phosphatidylethanolamine) between the cytoplasmic and lumenal leaflets of the microsomal membrane (27). In addition, the orientation of the bilayer in a fraction of the microsomes is likely reversed such that the lumenal leaflet of the ER membrane becomes the outer leaflet of the microsomes. Thus, the initial steps in penetration of Py from the ER to the cytosol can be studied by monitoring the ability of the virus to interact with and disrupt these microsomes. The ER membrane was shown previously to be impermeable to the 5-kDa polar reagent M-PEG (15). This reagent modifies thiol groups in cysteines and thereby significantly increases the size of a protein such that arrival of M-PEG to the microsome lumen may be monitored by its ability to modify ER lumenal proteins, such as PDI. We asked whether the ERp29-activated Py could permeabilize the microsomes so as to allow M-PEG to reach the lumen. We note that, during preparation of the microsomes, any PDI that is not encapsulated by the microsomes is removed after washing of the membranes. Hence, any modified PDI represents PDI inside the microsomes that became accessible to M-PEG following membrane disruption.
To first test the ability of M-PEG to modify PDI, an ER lumenal protein with six cysteine residues, mouse pancreatic microsomes were incubated with a low concentration of the detergent digitonin, followed by the addition of M-PEG. Four distinct species of M-PEG-modified PDI were observed by immunoblotting (Fig. 1D, compare lane 2 to lane 1), corresponding to the different numbers of M-PEG molecules added to the free thiol groups on PDI (PDI-MPEG*, -**, -***, and -**** refer to PDI modified by one, two, three, and four molecules of M-PEG, respectively). We did not observe PDI modified by more than four molecules of M-PEG, likely because two cysteine residues are oxidized and therefore not modifiable. Thus, when the ER membrane is permeabilized artificially, M-PEG is able to cross the membrane and modify PDI efficiently.
We then asked whether ERp29-activated Py induces perforation of the microsomes. The ERp29-enriched extract itself does not disrupt the microsomes, as incubation of microsomes with the ERp29-enriched extract (which does not contain PDI) did not induce PDI modification (Fig. 1D, compare lane 4 to lane 3). Next, Py was treated with the ERp29-enriched extract to induce virus unfolding and the activated Py incubated with the microsomes, followed by the addition of M-PEG. Under this condition, we found that in contrast to native Py, ERp29-modified Py enabled microsome-encapsulated PDI to be modified by M-PEG (Fig. 1D, compare lane 6 to lane 5). Since the ERp29-enriched extract or native virus alone cannot induce microsome perforation and because the microsomes are largely impermeable to M-PEG, we conclude that the ERp29-activated Py perforated the ER membrane to allow entry of M-PEG and modification of lumenal proteins. It should be noted that this assay directly monitors the ability of a physiologically activated nonenveloped virus to perforate its target membrane.
ERp29 exposes Py VP2 without disassembling the viral particle.
We found previously that, in contrast to wild-type Py, a VP1 virus-like particle devoid of the internal proteins VP2 and VP3 did not bind to liposomes after ERp29-mediated unfolding (16). Hence, we hypothesize that VP2 and VP3 may play a role in mediating binding of Py to the ER membrane. In this scenario, these internal proteins would become exposed after ERp29-dependent unfolding of VP1. To test this hypothesis, LE-dependent exposure of VP2 and VP3 in Py particles was monitored using a trypsin digestion assay. We found that incubation of Py with LE, but not BSA or the ERp29-depleted LE, rendered VP2 sensitive to trypsin digestion while VP3 remained resistant (Fig. 2A, compare lane 2 to lanes 1 and 3). That VP2 is sensitive to trypsin digestion suggests that VP2 is exposed by an LE activity, which is ERp29 dependent. The simplest interpretation of these data is that the VP1 conformational change caused by ERp29 leads to the selective exposure of the internal protein VP2. However, the finding that VP3 does not become trypsin sensitive does not exclude its possible exposure.
FIG. 2.

ERp29 exposes VP2 of Py without disassembling the viral particle. (A) ERp29 is required to render VP2 sensitive to trypsin degradation. Py was incubated with BSA, LE, or LE immunodepleted of ERp29, followed by the addition of trypsin where indicated. Samples were analyzed by SDS-PAGE and immunoblotted with an antibody against VP2/VP3. To verify the immunodepletion of ERp29, the extracts were subjected to SDS-PAGE and immunoblotted with an antibody against ERp29 (bottom, lanes 1 to 3). The asterisk indicates an unknown protein that cross-reacts with the VP2/VP3 antibody. Lanes 4 and 5 compare trypsin-treated and untreated Py. (B) Size exclusion fractionation of Py. Py was incubated with BSA, LE, or 1% SDS and fractionated over a size exclusion column. Fractions were analyzed by SDS-PAGE and immunoblotted with an antibody against VP1. (C) Native agarose gel analysis of Py. Py was incubated with BSA, LE, or 1% SDS. The samples were resolved by native agarose gel electrophoresis, transferred to nitrocellulose, and immunoblotted with an antibody against VP1.
Exposure of VP2 may be caused by the global disassembly of Py or by a more subtle, local structural alteration. The native size of Py is predicted to be at least 20 MDa, whereas the VP1 species generated by viral disassembly are expected to range from 50 kDa (VP1 monomer) to 250 kDa (VP1 pentamer). To test whether Py is globally disassembled in the ER, the BSA- or LE-treated Py particles were subjected to size exclusion fractionation. We found the elution patterns of the BSA- and LE-treated Py to be similar (Fig. 2B, compare top and middle). Based on its fractionation pattern and the resolution of the column, the sizes of the BSA- and LE-treated viral particles are predicted to be greater than 660 kDa. In contrast, when the virus was disassembled artificially by SDS, the viral particles fractionated to a position corresponding to approximately 50 to 250 kDa (Fig. 2B, bottom), consistent with the sizes of the VP1 monomers and pentamers. This finding suggests that the LE-treated Py is not disassembled globally to generate the VP1 monomers or pentamers. The BSA-, LE-, and SDS-treated Py were also subjected to native agarose gel electrophoresis, transferred to nitrocellulose, and immunoblotted with an antibody against VP1. We found the migration pattern of the LE-treated Py to be similar to that of the BSA-treated virus (Fig. 2C, compare lane 2 to lane 1), whereas Py treated with SDS migrated faster (Fig. 2C, lane 3). These data indicate that the LE did not stimulate the global disassembly of Py, consistent with the size exclusion fractionation findings. Should the LE induce the formation of oligomers of VP1 pentamers, the resolution of neither the size exclusion nor the native gel agarose method is likely to detect these species. Nonetheless, these data suggest that the ERp29-dependent unfolding of VP1 leads to a more subtle and localized conformational change that exposes VP2. Moreover, these results raise the possibility that the exposed VP2 contributes to the ability of the ERp29-activated Py to induce ER membrane perforation.
VP2 and VP3 bind to the ER membrane.
As VP2 is exposed in the ERp29-activated virus, it may facilitate the binding, perforation, and penetration of the ER membrane by the activated virus during infection. Examination of the hydropathy plot of VP2 revealed three theoretical transmembrane domains (Fig. 3A): the first located near the N terminus of VP2 (theoretical transmembrane domain 1, residues 69 to 101), a second domain near the center of VP2 (theoretical transmembrane domain 2, residues 126 to 165), and the third domain at the C-terminal portion of VP2 (theoretical transmembrane domain 3, residues 287 to 305). Because VP3 is translated from an internal start codon in the VP2 open reading frame such that VP3 is identical to VP2 amino acids 116 to 319, VP3 is predicted to have theoretical transmembrane domains 2 and 3 only. VP2 also contains an N-terminal myristic acid, absent in VP3.
FIG. 3.

Binding and integration of VP2 and VP3 into the ER membrane. (A) Hydropathy plot of VP2 and VP3. The hydropathy plot of VP2 and the overlapping VP3 was determined by entering the VP2 amino acid sequence into the Membrane Protein Explorer3.0 program (14). Each predicted transmembrane domain is indicated by a horizontal line and numbered. The portion of the plot to the right of the dotted vertical line corresponds to VP3 (residues 116 to 319), while the portion of the plot to the left of the dotted vertical line indicates the portion unique to VP2 (residues 1 to 115). (B) Binding of VP2 to the ER membrane. A lysate from 293T cells transfected with VP2 was incubated without microsomes (lanes 1 and 2) or with microsomes (lanes 3 and 4). The samples were centrifuged, and the supernatant and pellet fractions were analyzed by SDS-PAGE and immunoblotted with an antibody against VP2/VP3. The asterisk indicates an unknown protein that cross-reacts with the VP2/VP3 antibody. (C) Binding of VP3 to the ER membrane. This was done as described for panel B, except a lysate from 293T cells transfected with VP3 was used. (D) Insertion of VP2 into the ER membrane. The pellet fraction from the binding reaction described for panel B, lane 4, was resuspended in buffers at pH 7, pH 11, or at a high salt concentration (0.5 M KOAc). The samples were centrifuged and the supernatant and pellet fractions analyzed by SDS-PAGE and immunoblotted with an antibody against VP2/VP3 (top) or calnexin (bottom). (E) Insertion of VP3 into the ER membrane. This was done as described for panel D, except the pellet fraction from the binding reaction described for panel C, lane 4, was used. (F) Alkali extraction of peripheral membrane proteins Ero1α and p97. This was done as described for panel D, except buffer was used in place of 293T lysates and antibodies to Ero1α or p97 were used. (G) Trypsin resistance of VP2 and VP3. Lysates containing VP2 or VP3 were treated with or without urea (8 M), followed by incubation with the indicated trypsin concentrations. Samples were analyzed by SDS-PAGE and immunoblotted with an antibody against VP2/VP3.
To test whether VP2 and VP3 can bind to the ER membrane, lysates from 293T cells transfected with either a VP2 or a VP3 expression construct were prepared. Since the lysates contained 1% Triton X-100, Triton X-100 was removed prior to experimentation by using SM2 beads that preferentially bind to detergents (data not shown). The detergent-free lysates were then incubated with or without microsomes. Following sedimentation of the microsomes by centrifugation, proteins bound to the microsomes should appear in the pellet fraction, whereas unbound proteins should remain in the supernatant. In the absence of microsomes, all of the VP2 in the cell lysates remained in the supernatant (Fig. 3B, compare lane 2 to lane 1). However, in the presence of microsomes, approximately 50% of the input VP2 was found in the pellet fraction (Fig. 3B, compare lane 4 to lane 3). An unidentified protein in the cell lysate that cross-reacts with the VP2/VP3 antibody remains in the soluble fraction even in the presence of microsomes, indicating that the VP2-microsome interaction was specific (Fig. 3B, asterisk row, compare lane 4 to lane 3 and lane 2 to lane 1). A smaller proportion of the input VP3 was found in the pellet fraction in a microsome-dependent manner (Fig. 3C, compare lane 4 to lane 3 and lane 2 to lane 1). These results demonstrate that both VP2 and VP3 can bind to the ER membrane, although VP2 binds with higher efficiency than VP3, consistent with the additional theoretical transmembrane domain 1 found in VP2. While it remains possible that VP2 and VP3 interacted with the microsomes indirectly via another cellular component, the fact that LE-activated Py binds to liposomes (16) suggests that VP2 and VP3 interact with the ER membrane directly.
To assess the nature of the interaction between VP2 or VP3 and the ER membrane, the pellet fractions containing microsomes with bound VP2 or VP3 were subjected to alkali extraction or high-salt-concentration treatment. Resistance to extraction by alkaline and high-salt-concentration conditions is characteristic of integral membrane proteins, while peripheral membrane proteins are extracted under these conditions. Following resuspension and incubation of the pellets in buffers at either pH 7, pH 11, or a high salt concentration, the microsomes were repelleted. Analysis of the supernatant and pellet fractions by immunoblotting revealed that all of the VP2 remained associated with the pellet when the samples were incubated at pH 7 or at a high salt concentration (Fig. 3D, top, compare lane 2 to lane 1 and lane 6 to lane 5), and a significant level of VP2 remained in the pellet when the sample was incubated at pH 11 (Fig. 3D, top, compare lane 4 to lane 3). Under all of the conditions, the ER transmembrane protein calnexin remained in the pellet, as expected (Fig. 3D, bottom, lanes 2, 4, and 6). Similar to the result obtained with VP2, essentially all of the VP3 remained associated with the pellet when the samples were incubated at pH 7 or at a high salt concentration (Fig. 3E, top, compare lane 2 to lane 1 and lane 6 to lane 5), while a significant level of VP3 remained in the pellet when the sample was incubated at pH 11 (Fig. 3E, top, compare lane 4 to lane 3). Ero1α, an ER peripheral membrane protein that binds to the lumenal side of the ER membrane, and p97, a cytosolic protein that binds to the cytosolic side of the ER membrane, are both extracted completely at pH 11 but not pH 7 (Fig. 3F, top and bottom, compare lane 3 to lane 1). These findings indicate that the resistance of VP2 and VP3 to alkali extraction is unlikely due to an interaction with a microsome-associated protein but instead reflects their integration into the membrane. It is possible that unfolded VP2 and VP3 may bind to microsomes nonspecifically and become resistant to alkali extraction. However, the VP2 and VP3 in the 293T lysates are more resistant to trypsin digestion than VP2 and VP3 pretreated with urea to mimic an unfolded state (Fig. 3G, top, compare lanes 2 through 4 to lanes 5 through 7, and bottom, compare lanes 9 through 11 to lanes 12 through 14), suggesting that VP2 and VP3 expressed in 293T cells are not grossly unfolded. Hence, we conclude that the VP2 and VP3 that remained associated with the microsomes under alkali and high-salt-concentration conditions behave as integral membrane proteins, indicating that both proteins can integrate into the ER membrane subsequent to binding.
VP2, but not VP3, perforates the ER membrane.
Binding and insertion of VP2 and VP3 into the ER membrane raises the possibility that these proteins can induce the disruption of the lipid bilayer, which is requisite for penetration by Py. We therefore employed the ER membrane perforation assay (Fig. 1D) to examine the perforation activities of VP2 and VP3. 293T cell lysates were prepared from cells transfected with an empty vector, a VP2 or a VP3 expression vector, as described above. The cell lysates were incubated with microsomes, followed by the addition of M-PEG. The microsomes were washed gently three times to remove PDI derived from the cell lysate in order to ensure that only PDI contained in the microsome lumen was evaluated for modification by M-PEG. We found that the VP2-transfected cell lysate (Fig. 4A, bottom, lane 2) modestly but reproducibly stimulated the modification of PDI to PDI-MPEG* and PDI-MPEG** compared to the vector lysate (Fig. 4A, top, compare lane 2 to lane 1). By contrast, the VP3-transfected cell lysate (Fig. 4B, bottom, lane 2) did not stimulate the perforation of microsomes over the activity of the control lysate (Fig. 4B, top, compare lane 2 to lane 1). A low level of VP3 is expressed in the VP2-transfected lysate (not shown), as the cultured cells are able to initiate translation from the internal VP3 start codon. However, the VP3 level in the VP2-transfected lysate is less than the VP3 level in the VP3-transfected lysate, which exhibits no perforation activity. Moreover, while it is possible that the low level of VP3 may enhance the VP2-mediated perforation activity, this would require physical interaction between VP2 and VP3. We therefore conclude that the perforation activity of the VP2-transfected lysate depends on VP2. It should be noted that the low level of trypsin used to harvest the VP2-expressing cells did not digest VP2 to generate a fragment of VP2 (data not shown), indicating that full-length VP2 is responsible for the perforation activity. These results demonstrate that VP2 plays a role in disrupting the ER membrane and suggest that the exposed VP2 molecules on the ERp29-activated Py are responsible for the ER membrane-perforating activity of the activated virus.
FIG. 4.
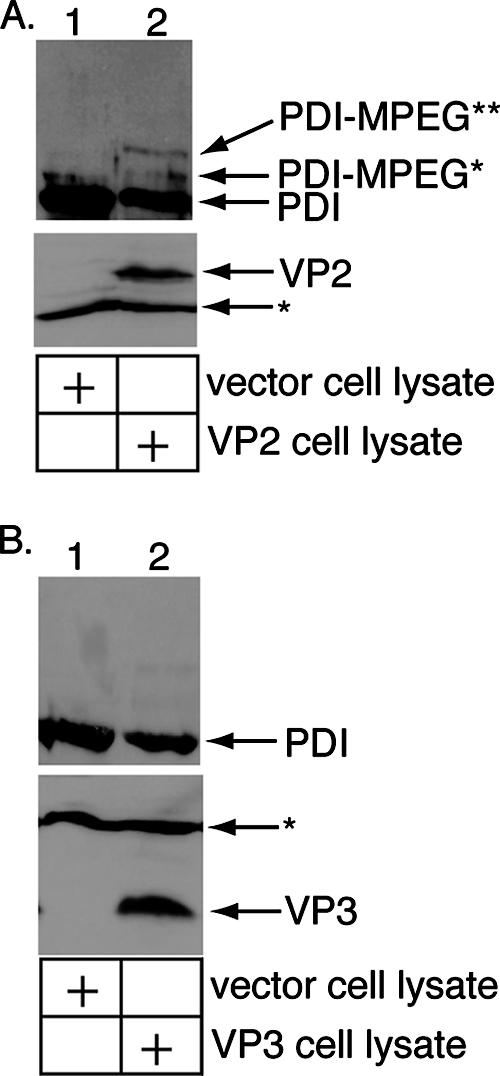
Perforation of the ER membrane by VP2 but not VP3. (A) VP2 stimulates the perforation of the ER membrane. Microsomes were incubated with a control 293T cell lysate or a lysate from 293T cells transfected with VP2. The samples were incubated with M-PEG and analyzed by SDS-PAGE and immunoblotting with an antibody against PDI (top). The lysates were also analyzed for VP2 expression by SDS-PAGE and immunoblotted with an antibody against VP2/VP3 (bottom). (B) VP3 does not stimulate ER membrane perforation. This was done as described for panel A, except a lysate from 293T cells transfected with VP3 was used.
DISCUSSION
Structural components of adenovirus (26), reovirus (1, 4), rotavirus (9, 13), poliovirus (11, 25), and SV40 (6, 7) have all been shown, following their activation, to be inserted into and/or disrupt a model membrane. However, the complete sequence of events that is necessary for penetration of the physiologically relevant membrane by a nonenveloped virus has not yet been demonstrated. We now show that Py becomes activated by a cellular factor in the ER and can subsequently perforate the ER membrane, a step that likely precedes its transport across the ER membrane. Specifically, we demonstrate that this activation alters the conformation of the VP1 capsid and reveals the underlying VP2 protein. The exposed VP2 then facilitates binding to and perforation of the ER membrane. Whether VP3 is exposed subsequent to ERp29-dependent unfolding of VP1 remains unclear. Because VP3 can bind to but not perforate the ER membrane, it may contribute only to the initial attachment of the activated viral particle to the ER membrane.
It is likely that only a subpopulation of the ER microsomes used in the assay are inverted with respect to bilayer orientation and that a larger pool are in their proper orientation (i.e., cytoplasmic leaflet exposed). We acknowledge that measuring the ability of Py to bind to and perforate these microsomes does not fully reflect the normal process of viral entry, where ER membrane penetration by Py is initiated from the lumenal leaflet of the membrane. Nonetheless, this assay provides a novel method for assessing the integrity of a biological membrane subsequent to its engagement by a nonenveloped virus.
Previous studies of Py mutants missing either VP2 or VP3 have implicated a functional role for these internal proteins in infection. For instance, Py mutants missing either VP2 (VP2−) or VP3 (VP3−) were shown to be less infectious than wild-type virus (17). Interestingly, the same study also demonstrated that the VP2− virus was significantly less infectious than the VP3− virus (17), consistent with our finding that VP2 but not VP3 plays a dominant role in the ER membrane penetration process. Structural features distinguishing VP2 and VP3 that may account for their differing significance during infection are the presence of an additional theoretical transmembrane domain in VP2 and a myristic acid at the N terminus of VP2. There is some discrepancy regarding the function of the VP2 myristic acid during the early steps of viral infection (17, 20).
Examination of the crystal structure of VP2/VP3 complexed with the VP1 pentamer shows that the C termini of VP2/VP3 are anchored to the VP1 pentamer cavity by tight hydrophobic interactions (5). According to the hydropathy plot (Fig. 3A), theoretical transmembrane domain 3 of VP2/VP3 is part of the cavity-anchored C terminus. This domain is unlikely to be exposed and contribute to membrane binding following the ERp29-stimulated conformational changes, unless global disassembly of the virus occurs. The N-terminal portions of VP2/VP3 are thought to be flexible within the viral particle (5). These flexible portions contain theoretical transmembrane domain 1, unique to VP2, and theoretical transmembrane domain 2, present in both VP2 and VP3. Thus, VP3 may bind to the ER membrane via theoretical transmembrane domain 2, while VP2 may also use theoretical transmembrane domain 2 to bind to the ER membrane and rely on theoretical transmembrane domain 1 to induce membrane perforation. The N-terminal myristic acid of VP2, absent in VP3, may also play a role in the perforation event.
Previous studies showed that VP2 and VP3 of SV40 bind to and integrate into the ER membrane (7), consistent with our finding for Py VP2 and VP3. Whether binding to and integration into the ER membrane by SV40's internal proteins caused membrane perforation was not examined. However, heterologous expression of these proteins in bacteria rendered the bacterial membrane permeable to the protein translation inhibitor hygromycin B, with VP3 inducing lysis of the bacterial cells (6). Thus, SV40's internal proteins may also possess the ability to induce ER membrane permeabilization.
The molecular mechanism by which Py penetrates the ER membrane remains elusive. Our results suggest that ERp29 unfolds VP1 to expose VP2, but the fate of VP3 in the ER is unclear as its resistance to trypsin in the digestion assay does not rule out its exposure. Because the crystal structure of Py does not indicate any obvious external features of the VP1 pentamers overlying VP2 that would distinguish them from pentamers overlying VP3, it is unclear how a cellular factor would act on VP1 pentamers overlying VP2 only. Our previous data demonstrated that the C terminus of VP1 is exposed by ERp29 (16). Since the VP1 C terminus is responsible for stabilizing interpentamer interactions, we envision a scenario in which the VP1 conformational change destabilizes the VP1 capsids, thereby exposing the flexible N termini of VP2 molecules. Extrusion of this region, which contains the N-terminal myristic acid and theoretical transmembrane domains 1 and 2, effectively generates a hydrophobic viral particle that binds to and integrates into the ER membrane. Local disruption of the bilayer integrity of the ER membrane enables the viral particle to penetrate the membrane and reach the cytosol. Alternatively, it is also possible that the activated viral particle penetrates directly into the nucleus since the ER and the nuclear envelope are contiguous. Whether the entire viral particle, a subviral particle, or the DNA genome alone is transported across the ER membrane requires further investigations.
Acknowledgments
We thank Mike Imperiale (University of Michigan) and Akira Ono (University of Michigan) for critical reviews of the manuscript.
E.K.R.-B. is supported by a Graduate Research Fellowship from the National Science Foundation. B.T. is a Biological Scholar at the University of Michigan and a Burroughs Wellcome Fund Investigator in Pathogenesis of Infectious Disease. B.T. is supported by the NIH (RO1-AI064296).
Footnotes
Published ahead of print on 19 September 2007.
REFERENCES
- 1.Agosto, M. A., T. Ivanovic, and M. L. Nibert. 2006. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc. Natl. Acad. Sci. USA 103:16496-16501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baer, G. S., and T. S. Dermody. 1997. Mutations in reovirus outer-capsid protein σ3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J. Virol. 71:4921-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bong, D. T., C. Steinem, A. Janshoff, J. E. Johnson, and M. R. Ghadiri. 1999. A highly membrane-active peptide in Flock House virus: implications for the mechanism of nodavirus infection. Chem. Biol. 6:473-481. [DOI] [PubMed] [Google Scholar]
- 4.Chandran, K., D. L. Farsetta, and M. L. Nibert. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J. Virol. 76:9920-9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, X. S., T. Stehle, and S. C. Harrison. 1998. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 17:3233-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniels, R., N. M. Rusan, A. Wilbuer, L. C. Norkin, P. Wadsworth, and D. N. Hebert. 2006. Simian virus 40 late proteins possess lytic properties that render them capable of permeabilizing cellular membranes. J. Virol. 80:6575-6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniels, R., N. M. Rusan, P. Wadsworth, and D. N. Hebert. 2006. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implication for DNA translocation out of the ER. Mol. Cell 24:955-966. [DOI] [PubMed] [Google Scholar]
- 8.Delos, S. E., L. Montross, R. B. Moreland, and R. L. Garcea. 1993. Expression of the polyomavirus VP2 and VP3 proteins in insect cells: coexpression with the major capsid protein VP1 alters VP2/VP3 subcellular localization. Virology 194:393-398. [DOI] [PubMed] [Google Scholar]
- 9.Denisova, E., W. Dowling, R. LaMonica, R. Shaw, S. Scarlatta, F. Ruggeri, and E. R. Mackow. 1999. Rotavirus capsid protein VP5* permeabilizes membranes. J. Virol. 73:3147-3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebert, D. H., J. Deussing, C. Peters, and T. S. Dermody. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609-24617. [DOI] [PubMed] [Google Scholar]
- 11.Fricks, C. E., and J. M. Hogle. 1990. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert, J., and T. Benjamin. 2004. Uptake pathway of polyomavirus via ganglioside GD1a. J. Virol. 78:12259-12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golantsova, N. E., E. E. Gorbunova, and E. R. Mackow. 2004. Discrete domains within the rotavirus VP5* direct peripheral membrane association and membrane permeability. J. Virol. 78:2037-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaysinghe, S., K. Hristova, W. Wimley, C. Snider, and S. H. White. 2006. Membrane protein explorer. http://blanco.biomol.uci.edu/mpex.
- 15.Le Gall, S., A. Neuhof, and T. Rapoport. 2004. The endoplasmic reticulum membrane is permeable to small molecules. Mol. Biol. Cell 15:447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magnuson, B., E. K. Rainey, T. Benjamin, M. Baryshev, S. Mkrtchian, and B. Tsai. 2005. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 28:289-300. [DOI] [PubMed] [Google Scholar]
- 17.Mannová, P., D. Liebl, N. Krauzewicz, A. Fetjová, J. Stokrová, Z. Palková, B. E. Griffin, and J. Forstová. 2002. Analysis of mouse polyomavirus mutants with lesions in the minor capsid proteins. J. Gen. Virol. 83:2309-2319. [DOI] [PubMed] [Google Scholar]
- 18.Norkin, L. C., H. A. Anderson, S. A. Wolfrom, and A. Oppenheim. 2002. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 76:5156-5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rainey-Barger, E. K., S. Mkrtchian, and B. Tsai. 2007. Dimerization of ERp29, a PDI-like protein, is essential for its diverse functions. Mol. Biol. Cell 18:1253-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahli, R., R. Freund, T. Dubensky, R. Garcea, R. Bronson, and T. Benjamin. 1993. Defect in entry and altered pathogenicity of a polyoma virus mutant blocked in VP2 myristylation. Virology 192:142-153. [DOI] [PubMed] [Google Scholar]
- 21.Stehle, T., Y. Yan, T. L. Benjamin, and S. C. Harrison. 1994. Structure of murine polyomavirus complexed with an oligosaccharide receptor fragment. Nature 369:160-163. [DOI] [PubMed] [Google Scholar]
- 22.Tsai, B., C. Rodighiero, W. I. Lencer, and T. A. Rapoport. 2001. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104:937-948. [DOI] [PubMed] [Google Scholar]
- 23.Tsai, B., J. M. Gilbert, T. Stehle, W. I. Lencer, T. L. Benjamin, and T. A. Rapoport. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 22:4346-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai, B. 24 April 2007, posting date. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed]
- 25.Tuthill, T. J., D. Bubeck, D. J. Rowlands, and J. M. Hogle. 2006. Characterization of early steps in the poliovirus infection process: receptor- decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J. Virol. 80:172-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiethoff, C. M., H. Wodrich, L. Gerace, and G. R. Nemerow. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 79:1992-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zachowski, A. 1993. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem. J. 294:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]