

Abstract
Both transforming growth factor beta (TGF-β) and p53 have been shown to control normal cell growth. Acquired mutations either in the TGF-β signaling pathway or in the p53 protein were shown to induce malignant transformation. Recently, cross talk between wild-type p53 and the TGF-β pathway was observed. The notion that mutant p53 interferes with the wild-type p53-induced pathway and acts by a “gain-of-function” mechanism prompted us to investigate the effect of mutant p53 on the TGF-β-induced pathway. In this study, we show that cells expressing mutant p53 lost their sensitivity to TGF-β1, as observed by less cell migration and a reduction in wound healing. We found that mutant p53 attenuates TGF-β1 signaling. This was exhibited by a reduction in SMAD2/3 phosphorylation and an inhibition of both the formation of SMAD2/SMAD4 complexes and the translocation of SMAD4 to the cell nucleus. Furthermore, we found that mutant p53 attenuates the TGF-β1-induced transcription activity of SMAD2/3 proteins. In searching for the mechanism that underlies this attenuation, we found that mutant p53 reduces the expression of TGF-β receptor type II. These data provide important insights into the molecular mechanisms that underlie mutant p53 “gain of function” pertaining to the TGF-β signaling pathway.
The p53 tumor suppressor gene plays a pivotal role in the prevention of human cancer by maintaining the integrity of the genome. It is well accepted that genotoxic stress induces the stabilization of the p53 protein, leading to its activation. The biological outcomes of p53 activity include apoptosis, inhibition of cell cycle progression, senescence, differentiation, and accelerated rates of DNA repair (35, 36, 42).
It is also known that inactivation of the p53 gene is one of the critical events in tumor development. Somatic inactivation of p53 occurs in over 50% of human tumors, the majority of which arise from missense mutations in the DNA binding domain (amino acids 102 to 292). Such mutations may simultaneously lead to both the loss of wild-type p53's tumor suppressor function and the accumulation of a mutant p53 protein.
It has been suggested that mutant p53 may exert its influence by either a dominant-negative or a “gain-of-function” mechanism (5, 22, 41). The dominant-negative effect of mutant p53 is strongly supported by the observation that mutant p53 proteins oligomerize with wild-type p53 and inhibit its function (32, 46). The “gain-of-function” theory, on the other hand, suggests that p53 mutations actively promote tumorigenesis, independent of the loss of wild-type p53 function. In previous studies, it was shown that restoration of murine p53 function by means of ectopic expression of a functional p53 gene in p53-null L12 cells, followed by their injection into syngenetic mice, resulted in L12 cells with a more aggressive phenotype. This phenotype was observed by the animals’ enhanced capacity to develop tumors in vivo (47). Furthermore, it was shown that the p53R175H mutant overexpressed in a nontransformed cell line lacking p53 yields tumors in nude mice, while the parental cell line does not (11).
In recent years, a growing number of studies have provided compelling evidence that mutant p53 exerts “gain-of-function” activities in tumor cells. These activities range from enhanced cell proliferation in culture (44, 52, 53) to increased tumorigenicity in vivo, as observed in a Li-Fraumeni syndrome model generated with mice (23, 34), as well as enhanced resistance to a variety of anticancer drugs commonly used in clinical practice (1, 3, 4). These types of activities were shown to depend on the integrity of the N-terminal domain, which includes the transcriptional domain of the mutant p53 protein (31). Accordingly, it has been suggested that this “gain-of-function” effect is dependent on the ability of mutant p53 to transactivate or repress specific target genes, such as the MDR1 (multidrug resistance gp180) (39), c-Myc (16), CD95 (52), EGR1 (44), MST/MSP-1 (53), and ATF3 (4) genes.
An additional key factor known to play a critical role in preventing the initiation and progression of cancer is the tumor suppressor transforming growth factor beta (TGF-β). The TGF-β family includes three members, TGF-β1, TGF-β2, and TGF-β3, each of which is encoded by a different gene and is capable of regulating cellular processes that control the homeostasis of many tissues (30). TGF-β controls cell behavior in a variety of situations, including cell proliferation, differentiation, migration, and wound healing (26-28).
TGF-β signaling is mediated through its binding to a heteromeric complex of transmembrane serine/threonine kinases, the type Ι and type II receptors (TGF-βRI and TGF-βRII). Following ligand binding to TGF-βRII, TGF-βRI is recruited to the ligand-receptor complex and becomes activated (49). In turn, activated TGF-βRI phosphorylates the receptor-regulated proteins SMAD2 and SMAD3, which then associate with the common mediator SMAD (SMAD4), translocate to the nucleus, and induce transcriptional activation (29).
The effects of TGF-β on target cells include the negative regulation of cell proliferation in several ways, e.g., induction of G1 arrest, promotion of terminal differentiation, or activation of the cell death pathway (27). Numerous reports have described deficiencies in TGF-β responses in human tumor-derived cell lines (17). Most of these deficiencies are associated with mutations in the TGF-β signaling network which result in inactivation of the TGF-β signaling pathway (28).
A connection between wild-type p53 and the TGF-β pathway was first suggested in the early 1990s, when it was shown in thyroid epithelial cells that inactivation of wild-type p53 by means of simian virus 40 transformation induced a loss of response to TGF-β treatment (50). Furthermore, it was shown that expression of a mutant form of p53 can induce resistance to TGF-β treatment as well (18, 37, 38). Accordingly, it was speculated that expression of a mutant form of p53 blocks the tumor suppressor function of wild-type p53 and interferes with the cell's sensitivity to TGF-β.
More recently, a convergence of wild-type p53 and the SMAD signaling pathway was demonstrated. This convergence was shown to be central to the regulation of Xenopus laevis embryonic development and tissue homeostasis (6). These studies indicated that wild-type p53 associates with SMAD2 and SMAD3 in a TGF-β-dependent manner and, at the same time, contacts its own cognate DNA site on a promoter in order to facilitate TGF-β-induced transcription. Furthermore, it was recently shown that wild-type p53 can associate with SMAD2/3 via its N-terminal domain and that Ras/mitogen-activated protein kinase (MAPK)-induced phosphorylation of Ser6 and Ser9 of the protein is critical to this function (7). Accordingly, we support the possibility that TGF-β sensitivity could be reduced by means of a dominant-negative effect exerted by mutant p53. Still, the biological functions and the operating principles of mutant p53 in relation to this pathway remain poorly understood.
In the present study, we report cross talk between mutant p53 and the TGF-β1 signaling pathway. Using p53-null cells that were restored by ectopically expressed mutant p53, we noticed a reduction in the levels of TGF-β1-induced gene expression. We found that expression of mutant p53 in cells seems to attenuate their response to TGF-β1 treatment within the context of wound healing. Molecular analysis indicated that mutant p53 interferes with the SMAD-dependent TGF-β1-induced signaling pathway that is central to this process. A lowered incidence of cell migration, less metalloproteinase (MMP) production, and attenuation in TGF-β1-induced transcriptional activity were noticed in cells overexpressing the mutant p53 protein. Finally, our experiments show that mutant p53 can interact with the promoter of TGF-βRII and repress its activity, leading to a reduction in the expression of the receptor. This, in turn, most likely attenuates the TGF-β1 signaling pathway.
MATERIALS AND METHODS
Antibodies, plasmids, and reagents.
Rabbit anti-SMAD2/3, -p-SMAD2, and -p-SMAD3 antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Mouse anti-p-ERK1/2, mouse anti-SMAD4, rabbit anti-TGF-βRI and -TGF-βRII, and rabbit antihemagglutinin (anti-HA) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) antibody (MAB374) was purchased from Chemicon (Hampshire, United Kingdom), and antitubulin antibody was purchased from Sigma (St. Louis, MO). Alexa 488-goat anti-rabbit (GαR) immunoglobulin G (IgG) was purchased from Invitrogen-Molecular Probes (Leiden, The Netherlands). Cy3-goat anti-mouse (GαM) IgG and normal goat IgG were purchased from Jackson Immunoresearch Laboratories (West Grove, PA).
p3TP-Luc and p3TP-Luc(+) reporter plasmids were kindly provided by Petra Knaus (Free University of Berlin, Germany). The Myc-tagged TGF-βRII construct used for cell infection was generated by PCR amplification of the Myc-tagged TGF-βRII gene from pCDNA1 (20). The amplified product was subcloned into pBabe-puro following restriction from pGMT-Easy, using the EcoRI restriction enzyme.
Hanks’ balanced salt solution (HBSS) without bicarbonate and phenol red was obtained from Biological Industries (Beit Haemek, Israel). Bovine serum albumin (BSA; fraction V) was purchased from Roche Applied Science (Indianapolis, IN). Recombinant human TGF-β1 was obtained from R&D Systems (Minneapolis, MN) and from PeproTech (Rocky Hill, NJ) and was applied in water containing 4 mM HCl and 1 mg/ml BSA to a final concentration of 1 μg/ml. Unless otherwise indicated, all TGF-β1 treatments were performed at a concentration of 1 ng/ml.
Cell cultures.
H1299 cells obtained from the American Type Culture Collection (Manassas, VA) were stably transfected with a mutant p53R175H expression plasmid, as previously described (44), and maintained in RPMI supplemented with 10% fetal calf serum (FCS; Sigma) and antibiotics.
H1299 cells were stably transfected with pCDNA3, encoding the p53R248W mutant, together with the empty pBabe-puro plasmid used for selection. Single-cell clones were then generated following selection with puromycin (1 μg/ml). The cells were maintained in RPMI supplemented with 10% FCS and antibiotics.
Ovarian cancer SKOV3 cells stably expressing p53R175H and their vector control line were kindly provided by P. M. Chumakov (University of California at San Diego) and were grown in Dulbecco's modified Eagle's medium supplemented with 10% FCS and antibiotics. All cells were maintained in humidified incubators at 37°C and in an atmosphere of 5% CO2.
Cell invasion/migration assay.
Cells expressing either empty vector or the p53R175H mutant were serum starved overnight and then treated with TGF-β1 for 24 h. Cells were then collected and counted, and 1 × 105 cells were added, in the presence or absence of TGF-β1, to the top chambers of 8-μm-pore-sized 24-well Transwell plates (Corning Company, Corning, NY) covered with Matrigel (BD Biosciences, Franklin Lakes, NJ). The lower chambers of the Transwells were filled with serum-free medium, with or without TGF-β1. After 6 to 12 h of incubation at 37°C, nonmigrating cells which remained at the top of the wells were removed together with the Matrigel, and cells which had migrated to the bottom of the wells were fixed and stained with crystal violet. The stained cells were dissolved in 100 μl of 10% acetic acid, and the colored solution was read for optical density (OD) using an enzyme-linked immunosorbent assay reader tuned to a wavelength of 450 nm.
In vitro wound-healing assay.
p53-null and p53R175H-producing cells were seeded at confluence in a six-well plate and serum starved for 12 h. Following starvation, the cells were treated with TGF-β1 for another 24 h, and the medium of the treated and the untreated cells was collected. The confluent cells were scratched using a 200-μl tip to create an artificial wound. Cells were than washed twice with phosphate-buffered saline (PBS), and the collected medium was restored to the cells with either the addition of TGF-β1 or no treatment. Photographs were taken at hours 0 and 24, and the distance between the edges of the wound was determined using ImageJ software, version 1.34s. The percentage of wound closure was determined by subtracting the value obtained at hour 24 from that obtained at hour 0. The values obtained represent the amounts of wound closure and were calculated as percentages of that for an open wound that had healed.
siRNA transfection.
Cells were seeded to a confluence of 60 to 70% and transfected with small interfering RNA (siRNA), using DharmaFECT3 reagent (Dharmacon, Lafayette, CO) according to the manufacturer's instructions. The sequences of p53 siRNA (Ambion, Austin, TX) were 5′-GAC TCC AGT GGT AAT CTA C-3′ (sense) and 5′-GTA GAT TAC CAC TGG AGT-3′ (antisense). The sequences of LacZ siRNA (Ambion) were 5′-GTG ACC AGC GAA TAC CTG T-3′ (sense) and 5′-ACA GGT ATT CGC TGG TCA C-3′ (antisense).
Real-time reverse transcription-PCR (RT-PCR) analysis.
Total RNA was extracted using a Nucleospin RNA cell kit (Macherey Nagel, Easton, PA). Two micrograms of each RNA sample was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI) and random hexamer primers. Real-time PCR was performed on an ABI 7000 real-time PCR system (Applied Biosystems, Lincoln Center, CA), using SYBR green PCR master mix (Applied Biosystems). The following specific primers were designed for the indicated genes: for p21, 5′ GGC AGA CCA GCA TGA CAG ATT 3′ (forward) and 5′ GCG GAT TAG GGC TTC CTC T 3′ (reverse); for SM22, 5′ TTG CCC CTC CAG GAG AAC TT 3′ (forward) and 5′ CTT CTC GAT TTT GGA CTG CAC TT 3′ (reverse); for MMP2, 5′ ACT TCT TCC CTC GCA AGC C 3′ (forward) and 5′ TCC TGT ATG TGA TCT GGT TCT TGT C 3′ (reverse); for SMAD7, 5′ CTC CAG ATA CCC GAT GGA TTT TC 3′ (forward) and 5′ GCA TCT GGA CAG TCT GCA GTT G 3′ (reverse); and for PAI-1, 5′ GTC ATA GTC TCA GCC CGC ATG 3′ (forward) and 5′ GTT ATG ACT CTT TCT GAA GGA AA 3′ (reverse).
Luciferase assay.
For TGF-β1-induced reporter assay, different TGF-β-responsive elements, i.e., p3TP-Luc (48) and p3TP-Luc(+) (25), were used. The p3TP-Luc(+) construct was derived from the p3TP-Luc construct but contains a modified luciferase gene [like pSP-Luc(+) from Promega]. Both the p3TP-Luc and p3TP-Luc(+) constructs serve as specific readouts for TGF-β-mediated signaling (9, 51).
Cells were seeded into 24-well culture dishes. Each well was transfected with either p3TP-Luc (48) reporter plasmid, for H1299 cells, or p3TP-Luc(+) (25) reporter plasmid, for SKOV3 cells, together with various mutant p53 or wild-type p53 expression plasmids and a β-galactosidase (β-Gal) plasmid. Twenty-four hours following transfection, the cells were starved for 12 h and treated with TGF-β1 for another 18 h, after which luciferase activity was assayed. For p21 or TGF-βRII promoter activity, cells were transfected with a reporter plasmid expressing the firefly luciferase gene under the transcriptional control of either the p21 or TGF-βRII gene promoter, together with a wild-type p53 or various mutant p53 expression plasmids and a β-Gal plasmid. Luciferase was assayed at 48 h posttransfection. Each plasmid combination was transfected into three identical wells. Luciferase assays were performed using d-luciferin (Roche, Indianapolis, IN). Luminescence was determined with the aid of a Synergy HT luminometer (Bio-Tek, Winoosky, VT). Luciferase values were normalized to β-Gal activity.
Immunofluorescence microscopy.
Cells were plated on glass coverslips for 24 h and starved overnight without serum. After stimulation with TGF-β1 under the conditions described in the legend to Fig. 5, cells were fixed in 4% paraformaldehyde in PBS for 45 min at 22°C and permeabilized with Triton X-100 (0.2%) for 5 min. The samples were blocked with 200 μg/ml normal goat IgG in HBSS supplemented with 20 mM HEPES (pH 7.2) and 2% BSA (HBSS-HEPES-BSA) for 30 min and labeled successively in the same buffer with antibodies for SMAD4 and/or p53. All incubations with antibodies were carried out for 45 min at 22°C, with three extensive washes after each step. Cells were mounted with Prolong antifade solution (Invitrogen-Molecular Probes). Fluorescence digital images were recorded on a Zeiss Axio Imager microscope (Carl Zeiss, Göttingen, Germany) with a Plan-Apochromat 63×/1.4 oil immersion objective, using OED capture software with a CoolSNAP HQ-M charge-couple device camera (Photometrics, Tucson, AZ).
FIG. 5.
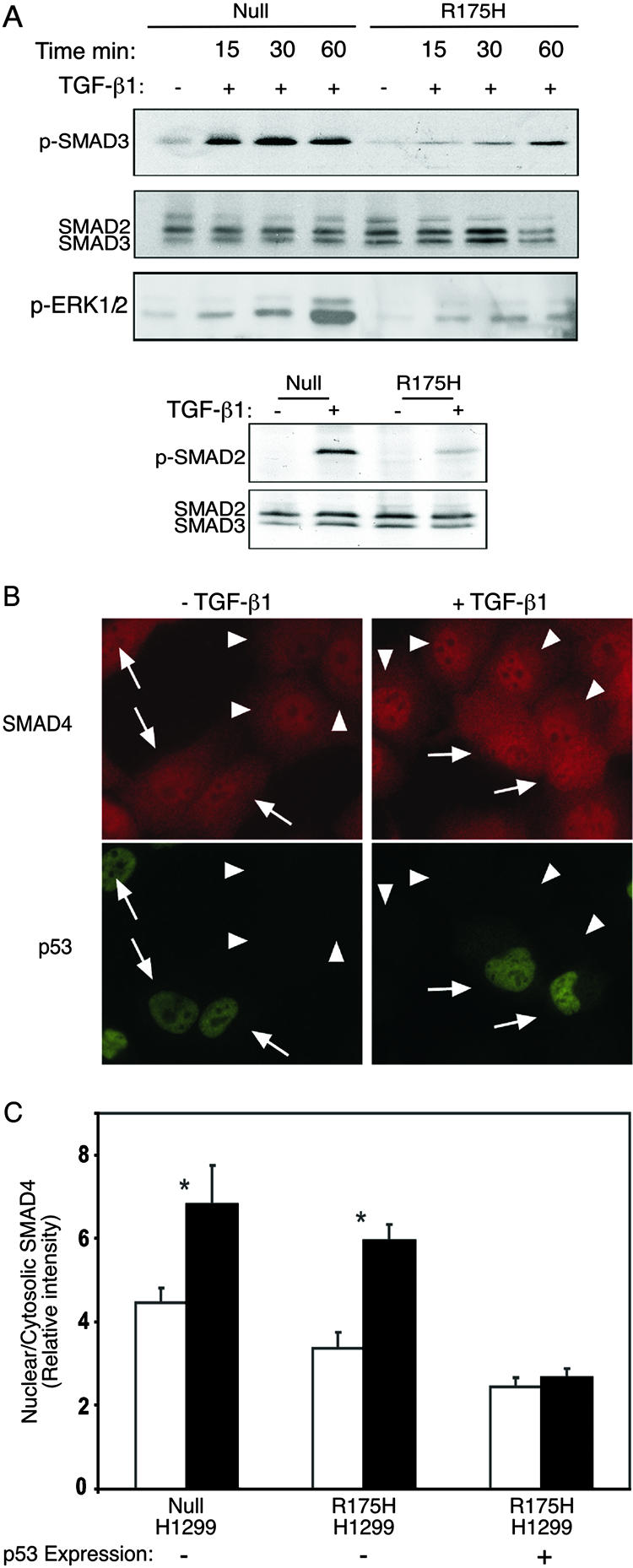
Mutant p53 attenuates TGF-β1-induced SMAD2/3 and ERK1/2 phosphorylation and abolishes the nuclear translocation of SMAD4. (A) p53-null and p53R175H-producing H1299 cells were serum starved and treated with TGF-β1 for 15, 30, and 60 min. The phosphorylated form of SMAD2 and SMAD3 was analyzed by Western blotting, using either rabbit anti-p-SMAD2 or rabbit anti-p-SMAD3 and total rabbit anti-SMAD2/3 antibody. The phosphorylation of ERK1/2 was monitored using mouse anti-p-ERK1/2. (B) p53-null or p53R175H-producing H1299 cells were serum starved and incubated with or without TGF-β1 (2 ng/ml) for 30 min at 37°C. Cells were then fixed/permeabilized and labeled for SMAD4 and p53 by successive incubations with (i) murine anti-SMAD4 (5 μg/ml) together with anti-p53 rabbit serum (1:1,000) and (ii) Cy3-GαM IgG together with Alexa 488-GαR IgG (2 μg/ml each). Arrows indicate cells expressing the p53R175H mutant, identified by fluorescence labeling for p53. Arrowheads point to cells devoid of mutant p53. (C) Quantitative measurements of the relative levels (nuclear/cytoplasmic ratio) of SMAD4 were taken using the point confocal method (see Materials and Methods). The laser beam (528.7 nm) was focused on well-defined spots within the nucleus and the cytoplasm of each cell, and the fluorescence intensity of Cy3-labeled SMAD4 was measured in both locations. The ratio of the nuclear to cytosolic intensities was recorded for each cell. Bars represent means plus standard errors of the means of 150 measurements in each case. Asterisks indicate a significant increase in the SMAD4 nuclear/cytosolic ratio when cells were stimulated with TGF-β1 (P < 0.001); the effect of TGF-β1 in p53R175H-expressing cells was not significant (P > 0.46).
Quantitative laser point confocal measurement of relative densities of SMAD4 in the nucleus and the cytoplasm.
The relative levels of fluorescence-labeled proteins (in this case, SMAD4) can be quantified by laser point confocal measurement of the fluorescence intensity, as we described earlier (12, 13). By measuring the fluorescence intensities in the nucleus and the cytoplasm of each cell and examining the ratio between the two, a direct measure of the relative distribution of the protein between the two localities could be obtained. Intensity measurements were performed using fluorescence recovery after photobleaching under nonbleaching conditions (attenuated laser beam). A 63× oil immersion objective was employed in order to focus an attenuated argon-ion laser beam (528.7 nm; 1 μW) to a Gaussian radius of 0.85 μm (12, 13) on either a nuclear or cytosolic location in each cell. The fluorescence excited in the defined focus of the laser beam passes through a pinhole located within the image plane of the microscope, which is only slightly larger than the beam itself. This makes it a true one-spot confocal measurement, collecting fluorescence exclusively from a defined plane within the nucleus or the cytoplasm and transferring it to the photomultiplier working in photon-counting mode. For each sample and treatment, 150 cells were measured and averaged.
Zymography assay.
Cells were serum starved for 12 h and incubated for another 36 h in the presence or absence of TGF-β1. Thirty-microliter aliquots of conditioned serum-free medium, normalized for the same number of cells, were subjected to 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis with a gel containing gelatin under nonreducing conditions. SDS was removed by extensive washing in 2.5% Triton X-100. The resulting gels were placed on developing buffer (50 mM Tris, pH 8, 5 mM CaCl2, 200 mM NaCl, 0.02% Brij) and incubated at 37°C overnight. Following incubation, the gels were stained with Coomassie blue reagent. MMP-dependent proteolysis of the gelatin was detected as a clear white area in a blue field.
Western blot analysis.
Cells were lysed in Triton extraction solution (15 mM Tris, pH 7.5, 120 mM NaCl, 25 mM KCl, 2 mM EGTA, 2 mM EDTA, 0.1 mM dithiothreitol, 0.5% Triton X-100, and protease inhibitor cocktail [Sigma]). Lysate aliquots were resolved by SDS-polyacrylamide gel electrophoresis on a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed sequentially with specific polyclonal antibodies against p-SMAD2, p-SMAD3, and SMAD2/3, monoclonal antibodies against p-ERK1/2, SMAD4, and p53 (DO-1), polyclonal antibodies against either TGF-βRΙ or TGF-βRII, and finally, either anti-GAPDH or antitubulin monoclonal antibody. Membranes were then incubated with secondary goat anti-mouse or goat anti-rabbit horseradish peroxidase-conjugated antibodies (1:10,000; Jackson Immunoresearch Laboratories) and developed using an ECL kit (Amersham Biosciences, Uppsala, Sweden).
Coimmunoprecipitation.
p53-null and p53R175H-producing cells were serum starved and treated with TGF-β1 for 1 h. Cells were then scraped into ice-cold PBS and lysed with lysis buffer containing 20 mM Tris-HCl, pH 8, 1 mM EDTA, 0.5% Nonidet P-40, 150 mM NaCl, 1 mM dithiothreitol, 10% glycerol, and protease inhibitors (Sigma). After being spun at top speed in a microcentrifuge to remove cellular debris, the supernatant was transferred to a clean test tube and precleared for 1 h with 50 μl of protein A. Following preclearing, rabbit anti-SMAD2/3 antibody and control rabbit anti-HA antibody, preincubated with 30 μl of protein A, were added to the lysate. Immune complexes were precipitated overnight at 4°C. The immunoprecipitated material was washed three times with 1 ml of washing buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.25% gelatin, and 0.1% NP-40. The pellet was then resuspended in SDS sample buffer and analyzed by Western blotting.
Chromatin immunoprecipitation.
Formaldehyde (Merck) was added directly to the cell culture medium at a final concentration of 1%. Fixation proceeded at room temperature for 10 min and was stopped by the addition of glycine to a final concentration of 0.125 M. Plates were rinsed with cold PBS, incubated with 10 ml of 20% trypsin-EDTA (Gibco) in PBS, and then scraped. Cells were rinsed three times in cold PBS, swelled in 5 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] (pH 8.0), 85 mM KCl, and protease inhibitor cocktail on ice for 20 min, and then disrupted with a Dounce homogenizer. Nuclei were collected by centrifugation at 4,000 rpm, resuspended in nuclear lysis buffer, and incubated on ice for 10 min. Samples were sonicated on ice to an average DNA fragment length of 1,000 bp and then microcentrifuged at 14,000 rpm. The chromatin solution was precleared by adding protein A beads for 2 h at 4°C. Precleared chromatin from 2.5 × 107 cells was diluted 1:5 in dilution buffer (0.01% SDS, 1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl, protease inhibitor cocktail), incubated with 30 μl protein A beads cross-linked via dimethyl pimelimidate to anti-p53 polyclonal antibody or to anti-HA antibody, and rotated at 4°C for 12 h. Immunoprecipitates were washed twice with dilution buffer, twice with wash buffer, and once with Tris-EDTA. After treatment with 10 μg RNase A per sample for 30 min, followed by 30 μg of proteinase K for 2 h at 50°C, cross-links were reversed by incubation at 65°C overnight. DNA samples were extracted using minicolumn PCR cleanup preps (Promega). PCR mixtures contained 2 μl of immunoprecipitated or diluted total input, 50 ng each of TGF-βRII promoter primers (forward, CAG ATT GCT CCT AGG TGC TTT AG; and reverse, CCT ACT GAA TTA GAA TCT GC), and Ready Mix PCR master mix (Promega) in a total volume of 20 μl. After 30 cycles of amplification, PCR products were run in a 2% agarose gel and analyzed by ethidium bromide staining.
Statistical analysis.
Statistical analyses were performed using functions in Microsoft Excel. Student's t test was used to assay statistical significance.
RESULTS
Mutant p53 attenuates TGF-β1-induced cell migration and wound healing.
Wild-type p53 was shown to facilitate activity in the TGF-β-induced signaling pathway (6, 7, 45) However, cells concomitantly expressing wild-type p53 and mutant p53 forms were shown to exhibit an attenuated response to TGF-β treatment (18, 38, 50). The molecular principles by which mutant p53 operates in this pathway are still unresolved.
To address this question, we first examined whether mutant p53 itself is capable of modifying the cellular response to TGF-β1 treatment in the absence of wild-type p53. To that end, we compared the effects of TGF-β1 on the migration capacity of p53-null H1299 cells and their counterpart cells that ectopically expressed mutant p53R175H protein.
Both p53-null and p53R175H-producing cells (Fig. 1A) were serum starved, treated with TGF-β1 for 24 h, and analyzed for cell migration in the presence or absence of TGF-β1. Figure 1B demonstrates that treatment of p53-null H1299 cells with TGF-β1 induced a significant increase in cell migration compared with that in untreated cells. However, treating H1299 p53R175H-producing cells with TGF-β1 did not significantly affect their cell migration rate, suggesting that mutant p53 abolishes the enhanced effect on cell migration following TGF-β1 treatment.
FIG. 1.

Mutant p53 attenuates TGF-β1-induced cell migration and wound healing. (A) Western blot analysis depicting the protein levels of p53R175H. (B) p53-null and p53R175H-producing H1299 cells were treated with TGF-β1 and subjected to a cell migration assay. Migrated cells were fixed, stained with crystal violet, dissolved in acetic acid, and analyzed for OD by using an enzyme-linked immunosorbent assay reader. The OD values represent the amounts of migrated cells (P < 0.05). (C) p53-null and p53R175H-producing H1299 cells were seeded to confluence, scratched to create an artificial wound, and treated with TGF-β1 for 24 h. The photographs indicate wound closure. (D) Wound closure, analyzed by subtracting the distance measured between the edges of the wound at hour 24 from that at hour 0 and presented as a percentage (P < 0.001).
Since migration of epithelial cells is central to the healing of injured tissue (26), we next examined the effect of TGF-β1 on cell migration by using an in vitro model of wound healing. To that end, artificial wounds were created in a monolayer of H1299 cells. The sizes of the lesions were measured immediately after their formation, at hour 0, as well as after 24 h of incubation at 37°C, with or without TGF-β1 treatment.
As demonstrated in Fig. 1C, treating H1299 cells with TGF-β1 induces cell migration, causing wounds to heal with greater rapidity. p53-null H1299 cells showed an increase of 35% in wound closure in the absence of TGF-β1 treatment. However, in the presence of TGF-β1 treatment, these cells exhibited a significant increase, of an additional 21%, in wound closure (56% total closure) (Fig. 1D). On the other hand, cells expressing the p53R175H mutant showed a 32% increase in wound closure in the absence of TGF-β1 treatment and an additional increase of only 8% in wound closure (39% total) following TGF-β1 treatment (Fig. 1D). This suggests that the p53R175H mutant confers a significant resistance to TGF-β1 treatment on H1299 cells, resulting in attenuation of cell migration and wound healing.
Mutant p53 attenuates TGF-β1-induced gene expression.
As demonstrated above, the p53R175H mutant attenuates TGF-β1-induced cell migration and wound healing in H1299 cells. Since the biological functions of TGF-β1 are tightly controlled by the expression of a well-characterized array of specific genes, it was of interest to examine whether a reduction in the cellular response to TGF-β1 treatment involves alterations in the expression of the TGF-β1-induced gene network. To that end, p53-null and p53R175H-producing H1299 (Fig. 1A) and SKOV3 (Fig. 2A) cells were treated with TGF-β1, and specific genes were measured by quantitative RT-PCR. Treating p53-null H1299 cells with TGF-β1 induced the expression of TGF-β1-specific genes, such as the p21, MMP2, SMAD7 (Fig. 2B), SM22, and PAI1 (data not shown) genes. p53-null SKOV3 cells, on the other hand, were less sensitive to TGF-β1 treatment, displaying increased expression of SM22, PAI1, and SMAD7 only (Fig. 2C), while the expression levels of p21 and MMP2 were not significantly altered in response to TGF-β1 treatment (data not shown). Both H1299 and SKOV3 cells expressing the p53R175H mutant showed a reduction in the expression of TGF-β1-specific genes compared with their p53-null counterpart controls (Fig. 2B and C, respectively). This suggests that the p53R175H mutant attenuates TGF-β1-induced gene expression.
FIG. 2.

Mutant p53 attenuates TGF-β1-induced gene expression. (A) Western blot analysis depicting protein levels of p53R175H in SKOV3 cells. H1299 (B) and SKOV3 (C) cells expressing either the p53R175H mutant or empty vector were serum starved and treated with TGF-β1 for 12 h. Total RNA was extracted and analyzed for the expression of TGF-β1-induced specific genes by RT-PCR. (D) H1299 cells expressing either p53R248W or empty vector were treated with TGF-β1 and evaluated for the expression of p21 and SMAD7 by RT-PCR analysis. The expression of the p53R248W mutant was confirmed using RT-PCR analysis. (E) H1299 cells were transiently transfected with either wild-type p53 (30 ng) or a β-Gal expression plasmid (30 ng) as a control. Twenty-four hours following transfection, the cells were starved, treated with TGF-β1, and analyzed for the expression of MMP2. The expression of wild-type p53 was confirmed using RT-PCR analysis.
All cell lines examined for TGF-β1-induced gene expression exhibited induction of a different set of genes. However, in general, treating the cells with TGF-β1 induced the expression of a TGF-β1-specific pattern of genes that were significantly attenuated by the presence of mutant p53.
Since we observed that the p53R175H mutant reduces the expression of TGF-β1-specific genes, we examined whether this reduction may also occur in the presence of an additional mutant p53 form. To that end, H1299 cells were transfected with either the p53R248W mutant or empty vector, and single-cell clones were generated. Several individual p53R248W-producing clones were then selected and analyzed for the expression of the TGF-β1-induced gene network. As depicted in Fig. 2D, the p53R248W mutant, like the p53R175H mutant, was also capable of reducing the expression of TGF-β1-specific genes, such as the SMAD7 and p21 genes. Similar results were obtained for all analyzed clones.
It was already demonstrated that wild-type p53 is able to cooperate with the TGF-β signaling components in order to facilitate TGF-β-induced gene expression (6, 7). Furthermore, it was shown by Cordenonsi et al. that when wild-type p53 was introduced into H1299 cells, TGF-β-induced p21 and p15 gene expression was augmented (7). Accordingly, we examined whether the expression of wild-type p53 protein in our hands would also increase TGF-β1-induced gene expression. To that end, H1299 cells were transiently transfected with either a wild-type p53 expression plasmid or empty vector, and the expression of TGF-β1-specific genes was measured. In agreement with published data, we found that wild-type p53 facilitated the TGF-β1 pathway, as manifested by an increase in the expression of MMP2 (Fig. 2E) and p21 (data not shown) following TGF-β1 treatment. Accordingly, we suggest that while wild-type p53 cooperates with TGF-β1 to increase the induction of specific genes, mutant p53 seems to attenuate it.
Mutant p53 attenuates the activation of a TGF-β1-induced reporter gene.
Since we observed that both p53R175H and p53R248W mutants reduced the expression of TGF-β1-specific genes, it was important to evaluate this reduction in the presence of additional forms of mutant p53 proteins. To that end, a TGF-β1-induced transcriptional assay was performed in the presence of different mutant forms of p53, using a luciferase reporter gene regulated by promoter elements that are responsive to TGF-β treatment, i.e., p3TP-Luc (48) and p3TP-Luc(+) (25).
The p3TP-Luc reporter gene was cotransfected into p53-null H1299 cells, together with various forms of mutant p53 expression plasmids, and luciferase activity was assayed following TGF-β1 treatment. In agreement with the above data (Fig. 2B, C, and D), both the p53R175H and p53R248W mutants reduced the luciferase activity of p3TP-Luc following TGF-β1 treatment compared with the corresponding control (Fig. 3A). Furthermore, reduction in the luciferase activity of p3TP-Luc was also noticed in the presence of additional forms of mutant p53, such as R179H and R273W mutants, following TGF-β1 treatment (Fig. 3A).
FIG. 3.
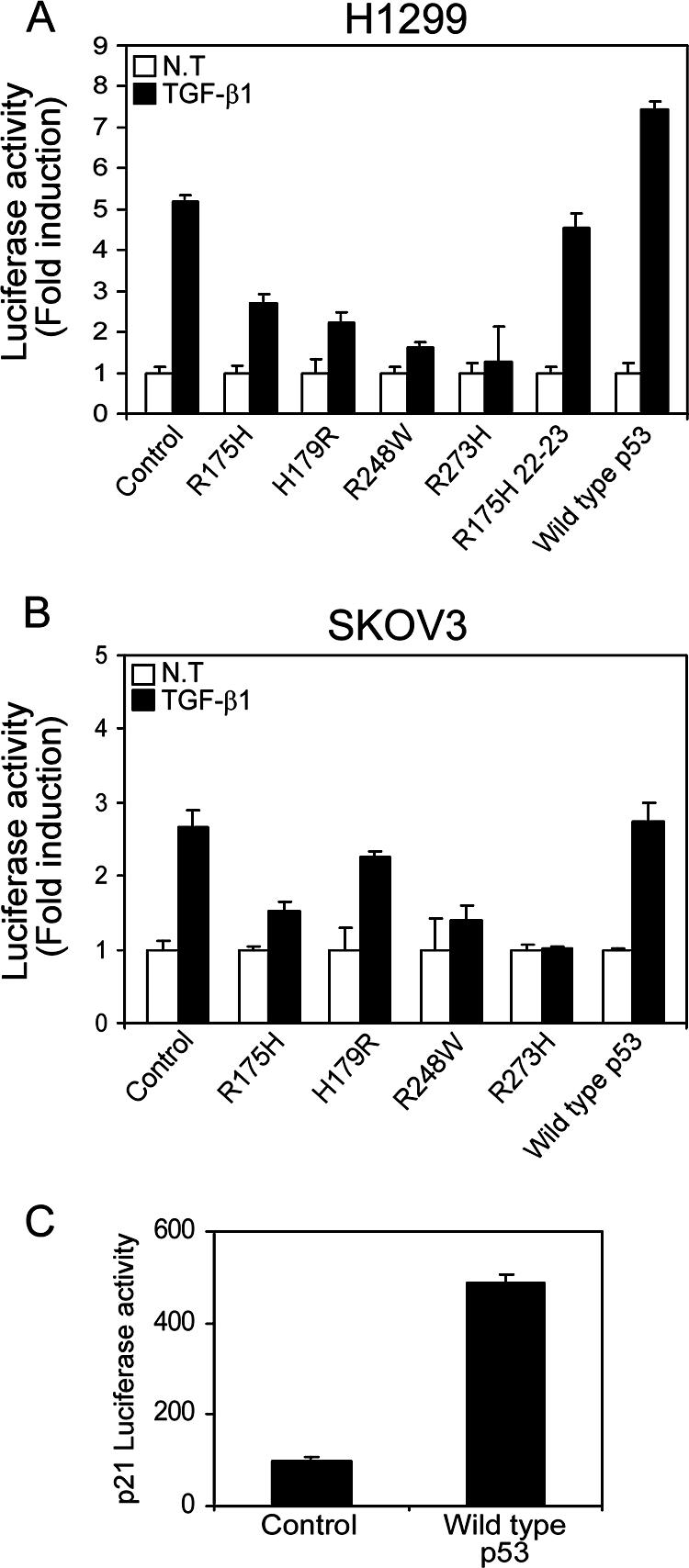
Mutant p53 attenuates the activation of TGF-β1-induced reporter gene. p53-null H1299 (A) and p53-null SKOV3 (B) cells were transfected with p3TP-Luc and p3TP-Luc(+), respectively, together with various p53 mutant or wild-type p53 expression plasmids, and luciferase activity was measured following TGF-β1 treatment. The amount of activation was calculated as the ratio of p3TP-Luc activity in TGF-β1-treated cells to p3TP-Luc activity in nontreated (NT) cells. (C) Wild-type p53-mediated induction of p21 promoter activity in H1299 cells.
A similar analysis was performed with p53-null SKOV3 cells. However, since the p3TP-Luc construct was not responsive in these cells, we used instead the derived p3TP-Luc(+) reporter gene (see Materials and Methods). Like H1299 cells, SKOV3 cells were cotransfected with the p3TP-Luc(+) reporter gene and various forms of mutant p53 expression plasmids, and luciferase activity was assayed upon TGF-β1 treatment. As demonstrated in Fig. 3B, all p53 mutants were found to reduce the TGF-β1-induced luciferase activity of p3TP-Luc(+) to a similar extent to that with the corresponding control. In contrast, expression of wild-type p53 in both H1299 and SKOV3 cells did not reduce the TGF-β1-induced luciferase activity of either p3TP-Luc or p3TP-Luc(+) (Fig. 3A and B, respectively). Furthermore, in agreement with the above data (Fig. 2E), an increase in the luciferase activity of p3TP-Luc was observed in the presence of wild-type p53 in H1299 cells (Fig. 3A). p53 function was validated by measuring the promoter activity of a well-known p53 target gene, the p21 gene (14) (Fig. 3C). These results further extend our findings that mutant p53 represses TGF-β1-induced transcriptional activation in general.
It was reported that core p53 mutants with additional mutations at residues 22 and 23 in the N-terminal transactivation domain (p53R175H, 22-23) were unable to block drug-induced apoptosis, suggesting that such p53 mutants require an intact transactivation domain for their “gain-of-function” activity (31). It was therefore of interest to examine whether such a transcription-deficient p53 mutant also lost its capacity to repress TGF-β1-induced transcriptional regulation. As shown in Fig. 3A, expression of p53R175H, 22-23 did not reduce the luciferase activity of p3TP-Luc following TGF-β1 treatment. This suggests that the integrity of the N terminus, which accounts for the transcriptional capacity of mutant p53 (31), is essential for the repression of TGF-β1-induced transcription control.
p53R175H mutant attenuates the production of MMP2 and MMP9 enzymes.
The aforementioned data indicate that mutant p53 attenuates TGF-β1-induced transcriptional regulation and gene expression. Since we found that the p53R175H mutant reduced the expression of TGF-β1-induced MMP2 (Fig. 2B) and MMP9 (data not shown), it was important to confirm this reduction by measuring the actual enzymatic activities of these proteins. To that end, p53-null and p53R175H-producing H1299 cells were serum starved and treated with TGF-β1. Gelatin substrate zymography, a method that can detect subnanogram amounts of gelatinase (40), was used to monitor MMP2 and MMP9 levels in the conditioned medium harvested from the cultures. A parallel culture of HT1080, a highly metastatic human fibrosarcoma cell line that produces both MMP2 and MMP9, was used as a control.
As shown in Fig. 4A, p53-null H1299 cells exhibited an increase in the enzymatic activities of both the 72-kDa MMP2 and 92-kDa MMP9 following treatment with TGF-β1. In contrast, p53R175H-producing cells showed little sensitivity to TGF-β1 treatment, as the enzymatic activities of the proteins were significantly attenuated following TGF-β1 treatment (Fig. 4A). In all, this shows that mutant p53R175H-producing H1299 cells exhibit reduced RNA levels as well as lower enzymatic activities of MMP2 and MMP9 upon TGF-β1 treatment.
FIG. 4.

Mutant p53R175H attenuates the production of MMP2 and MMP9 enzymes. (A) p53-null and p53R175H-producing H1299 cells were serum starved and treated with TGF-β1, and the activities of MMP2 and MMP9 were analyzed using a zymography assay. The electrophoretic positions of the 92-kDa MMP9 zymogen and the 72-kDa MMP2 zymogen are indicated. (B) siRNA for either p53 or LacZ was introduced into p53R175H-producing H1299 cells, and the expression of mutant p53 was detected by Western blot analysis using a monoclonal anti-DO1 antibody. (C) RT-PCR analysis of MMP2 in p53-null and p53R175H-producing cells that were exposed to siRNA for either p53 or LacZ and treated with TGF-β1. (D) RT-PCR analysis of SMAD7 in p53-null and p53R175H-producing cells that were exposed to siRNA for either p53 or LacZ and treated with TGF-β1. (E) Zymography analysis of MMP2 in p53R175H-producing cells expressing siRNA for either p53 or LacZ following TGF-β1 treatment. NT, not treated.
Another experimental approach that we have taken to resolve the role of mutant p53 in attenuating the TGF-β1-induced pathway is knocking down the expression of mutant p53 in H1299 cells. To that end, we transfected mutant p53R175H-producing H1299 cells with an oligonucleotide siRNA specific for either p53 or LacZ and measured the transcriptional activities of MMP2 and SMAD7 following TGF-β1 treatment. Notably, introducing siRNA for p53 into p53R175H-producing cells significantly reduced the expression of the protein compared with that in the LacZ siRNA-treated controls (Fig. 4B). In addition, p53R175H-producing cells that were exposed to siRNA for p53 and treated with TGF-β1 exhibited an increase in the transcriptional activities of both MMP2 (Fig. 4C) and SMAD7 (Fig. 4D) following the reduction in p53 expression.
Similar results were obtained by measuring the enzymatic function of MMP2 under the same conditions. As expected, knocking down the expression of mutant p53 enhanced the enzymatic function of MMP2 following TGF-β1 treatment (Fig. 4E).
To exclude the possibility that siRNA for p53 may alter the induction of MMP2 and SMAD7 in a nonspecific, p53-independent manner, we transfected p53-null H1299 cells with siRNAs for both p53 and LacZ and measured the resulting RNA expression. As demonstrated in Fig. 4C and D, introduction of siRNAs for both p53 and LacZ into p53-null H1299 cells did not significantly interfere with the transcriptional activities of both MMP2 and SMAD7 when these cells were treated with TGF-β1. These results further substantiate our conclusion that mutant p53 interferes with the TGF-β1-induced gene network. Knocking down the expression of mutant p53 caused both SMAD7 and MMP2 to regain transcriptional activity, and production of the MMP2 enzyme was also increased.
Mutant p53 attenuates the TGF-β1-induced phosphorylation of SMAD2/3 and ERK1/2.
TGF-β1-induced transcriptional regulation is mediated mainly through phosphorylation of SMAD2/3 proteins, followed by their translocation into the cell nucleus to further activate specific genes (28). Accordingly, it was speculated whether repression of TGF-β1-induced gene expression mediated by mutant p53 involves alterations in the status of SMAD2/3 following TGF-β1 treatment. To test this possibility, we treated H1299 cells expressing either the p53R175H mutant or empty vector with TGF-β1 and then performed a Western blot analysis using antibodies against the phosphorylated form of SMAD2 and SMAD3. As demonstrated in Fig. 5A, a significant induction in SMAD3 phosphorylation was already observed 15 min following treatment of p53-null cells with TGF-β1. This induction remained stable 30 and 60 min later. However, in contrast to these cells, mutant p53R175H-producing cells exhibited a significant attenuation in the phosphorylation of SMAD3 following TGF-β1 treatment. As depicted in Fig. 5A, induction of SMAD3 phosphorylation seemed to lag, and only a moderate phosphorylation was observed 60 min following TGF-β1 treatment.
By analyzing the status of the SMAD2 protein, we noticed that similar to the case for SMAD3, the p53R175H mutant reduced the phosphorylation of SMAD2 following treatment of the cells with TGF-β1 for 30 min (Fig. 5A, lower panels). Accordingly, we suggest that the p53R175H mutant attenuates the TGF-β1-induced phosphorylation of SMAD2/3 proteins.
It was already demonstrated that apart from the SMAD-dependent signaling pathway, TGF-β1 has the ability to induce MAPK or phosphatidylinositol 3-kinase signaling pathways as well (10). Accordingly, we examined whether, as with SMAD2/3, the p53R175H mutant also attenuates other TGF-β-induced signaling pathways. To that end, both p53-null and p53R175H-producing H1299 cells were treated with TGF-β1 for 15, 30, and 60 min, and ERK1/2 phosphorylation was analyzed by using specific antibodies against the phosphorylated form of the proteins. As depicted in Fig. 5A, treating p53-null H1299 cells with TGF-β1 for 15, 30, and 60 min significantly induced the phosphorylation of ERK1/2 proteins. However, mutant p53R175H-producing cells exhibited a significant attenuation in the phosphorylation of these proteins (Fig. 5A), suggesting that the p53R175H mutant also attenuates the Ras-MAPK signaling pathway.
Mutant p53 inhibits TGF-β1-mediated nuclear translocation of SMAD4 and abolishes its association with SMAD2.
Previously, it was shown that upon TGF-β1 treatment, SMAD4 associates with phosphorylated SMAD2/3 and that, together, they translocate to the cell nucleus to further activate specific genes (30). Furthermore, it was shown that TGF-β1 causes SMAD4 to promote the interaction of SMAD2/3 with the DNA and increase transcriptional efficiency (19). Given the fact that the p53R175H mutant attenuates the phosphorylation and transcriptional activation of SMAD2/3, we decided to measure the effect of the p53R175H mutant on the subcellular localization of SMAD4 following TGF-β1 treatment. To that end, we performed immunostaining of SMAD4 in H1299 cells.
It should be noted that the H1299 p53R175H-producing cell population that we have at our disposal is a pool of G418-resistant cells consisting of about 70% mutant p53-producing cells and about 30% p53-null cells. These cells therefore provide a convenient system consisting of built-in controls whereby the effects of the presence or absence of p53R175H on the nuclear translocation of SMAD4 can be compared easily.
Since SMAD4 localizes predominantly in the cytoplasmic fraction and in the nucleus both before and after TGF-β stimulation, a more quantitative approach to determine the relative distribution of SMAD4 between the nucleus and the cytoplasm was required. Therefore, the relative levels (nuclear/cytoplasmic ratio) of SMAD4 were measured by means of the laser point confocal method (12, 13). This technique involves the use of a laser beam focused on a small, well-defined spot in the nucleus or cytoplasm of each cell to quantify the fluorescence intensity, which is directly proportional to the density of the fluorescently labeled proteins in that spot.
As shown in Fig. 5B, treating the H1299 p53R175H-expressing cell population with TGF-β1 for 30 min induced the translocation of SMAD4 into the cell nucleus only in cells negatively stained for mutant p53 (arrowheads). However, cells positive for mutant p53 staining showed no alteration in the localization of SMAD4 upon TGF-β1 treatment (arrows). Furthermore, multiple values of the nuclear/cytoplasmic SMAD4 ratio, measured by the point confocal method in cells negatively stained for p53 of the p53R175H-expressing population, indicated a roughly twofold increase in the nuclear localization of SMAD4 (Fig. 5C). However, cells positive for mutant p53 staining showed no alteration in the localization of SMAD4 upon TGF-β1 treatment. As demonstrated in Fig. 5C, similar intensities of the fluorescently labeled SMAD4 protein were observed in the nuclear versus the cytoplasmic fraction of cells positively stained for mutant p53 before and after TGF-β1 treatment. This suggests that the p53R175H mutant prevents the accumulation of SMAD4 in the cell nucleus following TGF-β1 treatment.
A similar analysis performed on p53-null cells also exhibited an alteration in the subcellular localization of SMAD4 following TGF-β1 treatment. Like the negatively p53-stained cells of the p53R175H-expressing population, p53-null cells also showed a roughly twofold increase in the nuclear localization of SMAD4 (Fig. 5C).
Since TGF-β1-induced accumulation of SMAD4 in the cell nucleus involves its association with activated R-SMADs, we examined the involvement of the p53R175H mutant in the formation of a complex between SMAD2 and SMAD4 upon TGF-β1 treatment. As shown in Fig. 6A, treatment of p53-null H1299 cells with TGF-β1 induced an association between SMAD2 and SMAD4, as detected by coimmunoprecipitation followed by Western blot analysis. In contrast, treating p53R175H-producing cells with TGF-β1 yielded no such complex. However, knockdown of mutant p53R175H expression by a specific siRNA (see above) successfully restored both SMAD2/3 phosphorylation (Fig. 6B) and the association of SMAD2 with SMAD4 (Fig. 6C) compared with those in the control. These findings suggest that mutant p53, which interferes with TGF-β1-induced activation of SMAD2/3, eventually abolishes the association between SMAD2 and SMAD4 following TGF-β1 treatment.
FIG. 6.

Mutant p53 interferes with formation of the SMAD2-SMAD4 complex. (A) p53-null and p53R175H-producing H1299 cells were treated with TGF-β1 as described in Materials and Methods. Cell lysates were immunoprecipitated with rabbit anti-SMAD2 antibody, and the precipitates were analyzed with mouse anti-SMAD4 antibodies. (B) p53R175H-producing H1299 cells were transfected with siRNA for either p53 or LacZ, treated with TGF-β1 for 30 min, and tested for the presence of p-SMAD2/3 by means of Western blot analysis. (C) p53R175H-producing H1299 cells were transfected with siRNA for p53 or LacZ, immunoprecipitated with rabbit anti-SMAD2, and analyzed for the presence of SMAD4 using a mouse anti-SMAD4 antibody. IP, immunoprecipitation; IB, immunoblotting.
Mutant p53 represses the expression of TGF-βRΙΙ but not TGF-βRΙ.
Our accumulated data suggest that the p53R175H mutant can cause H1299 cells to be less responsive to TGF-β1 treatment and can attenuate the effectiveness of the TGF-β1 signaling pathway. TGF-β1 signaling activity is mediated through the binding of ligand to TGF-βRΙΙ, which then forms heterodimers with TGF-βRΙ and induces its phosphorylation (27, 28).
Next, we asked whether the p53R175H mutant affects the expression of TGF-βRΙ/ΙΙ, which in turn may attenuate the entire TGF-β1-induced signaling pathway. To that end, we employed Western blot analysis and real-time PCR to compare the total levels of TGF-βRΙ/ΙΙ in the p53R175H-producing cells and their corresponding p53-null counterpart controls. Notably, the p53R175H mutant altered the expression levels of TGF-βRII in both H1299 (Fig. 7A and C) and SKOV3 (Fig. 7B) cells. By comparing the total expression of TGF-βRI/ΙΙ in p53R175H-producing cells with that in p53-null cells, a significant reduction in the RNA level of TGF-βRII was observed in the present of mutant p53R175H protein. No alteration in the RNA level of TGF-βRI was noticed (Fig. 7A and B). A similar pattern was observed when protein levels of TGF-βRI/ΙΙ were measured (Fig. 7C).
FIG. 7.

Mutant p53 represses the expression of TGF-βRΙΙ but not TGF-βRΙ. RT-PCR analysis of the expression of TGF-βRI/II was performed with p53-null and p53R175H-producing H1299 cells (A) or p53-null and p53R175H-producing SKOV3 cells (B). (C) Western blot analysis depicting the levels of TGF-βRI and TGF-βRΙΙ in p53-null and p53R175H-producing H1299 cells. (D) H1299 cells were transiently transfected with the TGF-βRII promoter (300 ng) along with plasmids encoding the indicated p53 mutants. A β-Gal expression plasmid was also included in all transfections, and luciferase activity was normalized to β-Gal activity. (E) SKOV3 cells were transiently transfected with the promoter of TGF-βRII together with either the p53R175H mutant, p53R175H 22-23 mutant, or wild-type p53 expression plasmid, and luciferase activity was measured and normalized to β-Gal. (F) Chromatin immunoprecipitation experiment performed on H1299 cells stably expressing the p53R175H mutant. After cross-linking of proteins to DNA, DNA was fragmented, and the p53 protein was immunoprecipitated with a specific antibody against p53 or with a nonspecific antibody against HA as a control. PCR analysis was performed on the immunoprecipitated DNA samples, using either TGF-βRII- or GAPDH-specific primers.
These results led us to the conclusion that the attenuation in the TGF-β1-induced signaling pathway observed in the p53R175H-producing cells may occur due to a reduction in the expression of TGF-βRII.
It is well established that mutant p53 regulates the activities of different promoters and thus induces or represses the transcription of specific genes (4, 16, 39, 44, 52, 53). Since we observed that the p53R175H mutant reduces the RNA levels of TGF-βRII, it was important to confirm this reduction by measuring the promoter activity of this receptor in the presence of the p53R175H mutant. To that end, the promoter of TGF-βRII was cloned into a luciferase reporter plasmid, and its transcriptional regulation was analyzed by measuring luciferase activity in the presence of different mutant p53 expression plasmids or a wild-type p53 expression plasmid. As depicted in Fig. 7D and E, the p53R175H mutant repressed the promoter activity of TGF-βRII in both H1299 and SKOV3 cells, as observed by significant reductions in the luciferase activity of the promoter. This repression was also observed in the presence of other mutant p53 proteins (Fig. 7D). It should be noted that all p53 mutants were expressed at similar protein levels (Fig. 7D, lower panel). By analyzing the luciferase activity of the TGF-βRII promoter in the presence of the p53R175H mutant with an additional mutation in its N-terminal transactivation domain (R175H, 22-23), we noticed that this form of mutant p53 did not reduce the luciferase activity of the TGF-βRII promoter (Fig. 7E). Similar results were also observed in the presence of wild-type p53 protein (Fig. 7E).
Judging by evaluation of both total RNA levels and luciferase activity, it is suggested that mutant p53 reduces the expression of TGF-βRII by repressing its transcriptional regulation. Furthermore, we show that the integrity of the mutant p53 N-terminal transactivation domain is essential for this function.
Recently, it was shown that in order for mutant p53 to regulate the promoter activities of specific genes, it needs to physically interact with the promoters (43, 44, 52, 53). Therefore, we wanted to examine whether the repression in TGF-βRII promoter activity involves an interaction of mutant p53 with this promoter. To that end, we performed a chromatin immunoprecipitation analysis of p53R175H-producing H1299 cells, using antibody against either p53 or HA as a control. As shown in Fig. 7F, TGF-βRII promoter sequences were specifically immunoprecipitated with mutant p53. Thus, mutant p53 associates in vivo with the TGF-βRII promoter.
Overexpressing Myc-tagged TGF-βRII restores the TGF-β1-mediated signaling pathway.
Our findings indicate that p53R175H can associate with the promoter of TGF-βRII and thus may repress its transcriptional activity. As a result, the expression of TGF-βRII on the surfaces of cells may be reduced, thereby leading to fewer available receptors which can interact with TGF-β1. This, in turn, may attenuate the TGF-β1-induced signaling pathway.
To further examine this assumption, both mutant p53R175H-producing H1299 cells and their p53-null counterpart controls were infected with either a Myc-tagged TGF-βRII (Myc-TβRII) expression plasmid or empty vector, and stable cell pools were established. Notably, both Myc-TβRII-infected p53R175H-producing cells and their p53-null counterpart population highly expressed TGF-βRII, as observed by real-time PCR (Fig. 8A) and Western blot analysis using the 9E10 anti-Myc antibody, which specifically identifies the overexpressed receptor (Fig. 8B). Next, the phosphorylation of the SMAD2 protein was analyzed in the infected cells following treatment with TGF-β1 for 30 min. As shown in Fig. 8C, a slight increase in the phosphorylation of SMAD2 protein was observed in the Myc-TβRII-infected p53-null cells compared with their empty vector counterpart controls. However, in the case of p53R175H-producing H1299 cells, overexpressing the Myc-TβRII protein in these cells completely abolished the attenuation in the TGF-β1-induced phosphorylation of SMAD2. As shown in Fig. 8C, mutant p53R175H-producing cells exhibited a significantly smaller amount of phosphorylated SMAD2 protein following treatment of the cells with TGF-β1 for 30 min. However, expression of the Myc-TβRII protein in these cells restored the TGF-β1-induced phosphorylation of SMAD2 to a similar extent to that observed in the Myc-TβRII-infected p53-null counterpart controls. Accordingly, we assume that overexpression of ectopic Myc-TβRII protein replaced the lack of endogenous TGF-βRII, which was mediated by the negative effect of mutant p53 on its promoter.
FIG. 8.

Overexpressing Myc-tagged TGF-βRII restores TGF-β1-mediated signaling pathway and gene expression. Both p53-null and mutant p53R175H-producing H1299 cells were infected with either empty vector or Myc-TβRII expression plasmid, and the expression of TGF-βRII was measured by RT-PCR analysis (A) and Western blot analysis using the 9E10 monoclonal anti-Myc antibody (B). (C) Western blot analysis depicting the levels of phosphorylated SMAD2 protein following treatment of H1299 cells with TGF-β1 for 30 min. (D) p53-null and mutant p53R175H-producing H1299 cells were transiently transfected with p3TP-Luc, together with either empty vector or Myc-TβRII expression plasmid, and luciferase activity was measured following TGF-β1 treatment. The amount of activation was calculated as the ratio of p3TP-Luc activity in TGF-β1-treated cells to p3TP-Luc activity in nontreated (NT) cells. (E) p53-null and mutant p53R175H-producing H1299 cells were infected with either a Myc-TβRII expression plasmid or empty vector, and the expression of TGF-β1-induced SMAD7 was analyzed using RT-PCR.
Next, we examined whether recovery of the activity of SMAD2 in cells containing mutant p53 would overcome the attenuation of TGF-β1-induced transcriptional regulation. To that end, both p53-null and mutant p53-producing H1299 cells were transfected with either Myc-TβRII expression plasmid or empty vector, together with the p3TP-Luc luciferase construct, and luciferase activity was measured following TGF-β1 treatment.
Notably, expression of Myc-TβRII in p53-null cells induced a moderate increase in the TGF-β1-induced luciferase activity of p3TP-Luc compared with that in the empty vector counterpart control. Moreover, expression of Myc-TβRII in mutant p53R175H-producing cells completely restored the attenuation in the TGF-β1-induced luciferase activity of p3TP-Luc observed in these cells (Fig. 8D).
As shown in Fig. 8D, treatment of p53R175H cells lacking the Myc-TβRII protein with TGF-β1 only slightly induced the luciferase activity of p3TP-Luc compared with that in their p53-null counterpart controls. However, treatment of p53R175H-expressing cells with TGF-β1 in the presence of Myc-TβRII protein restored the luciferase activity of p3TP-Luc to a level comparable to that observed in the Myc-TβRII-expressing p53-null counterpart cells. This was further supported by the observation that similar results were obtained when the endogenous expression of SMAD7 following TGF-β1 treatment was measured in cells expressing either Myc-TβRII or empty vector (Fig. 8E).
In all, we suggest that mutant p53-mediated attenuation of the TGF-β1-induced signaling pathway and gene expression is a result of a reduction in the expression of TGF-βRII on the surfaces of cells.
DISCUSSION
Both TGF-β and wild-type p53 play key roles in regulating gene networks associated with cell growth (27, 28, 35, 42). For both, deregulation of their function causes cells to lose the ability to control normal growth, which eventually results in the development of cancer. Indeed, mutations in either the TGF-β signaling network (28) or the wild-type p53 protein (41) were shown to occur frequently in various cancerous tissues. Nevertheless, although some association between these pathways was suggested in previous studies (6, 7, 45), the precise nature of the molecular cross talk between these two gene pathways remains unresolved.
In the case of the TGF-β signaling network, proteins such as SMAD family members lose their function due to acquired mutations, such that they are no longer capable of participating in the TGF-β-induced signaling pathway (28). This loss of function, in turn, causes the cell to become unresponsive to TGF-β, thereby leading to uncontrolled proliferation, inhibition of differentiation, or inactivation of the cell death pathway.
However, in the case of p53, it is believed that acquired mutations may simultaneously lead to both the loss of wild-type p53 functions and the accumulation of active mutant p53 forms. The accumulated mutant p53 protein not only is capable of inactivating the functions of wild-type p53 (32, 46) but also can exert a “gain-of-function” activity, which in turn increases cell proliferation (23, 44, 52, 53), inhibits cell differentiation (15), and confers resistance to a variety of anticancer drugs (1, 3, 4). These “gain-of-function” activities observed in cells expressing an altered p53 gene share great similarity with the phenotype observed for the mutated TGF-β signaling pathway.
The novelty of the present study lies in its clarification of possible interactions between the TGF-β signaling pathway and the mutant p53 activity mediated by a “gain-of-function” mechanism rather than focusing on the cross talk between TGF-β and the wild-type p53 protein. In our experimental model, we tracked the activity of the TGF-β1 signaling pathway in cells that constitutively overexpress the mutant p53R175H protein. By and large, we found that the entire TGF-β1-induced signaling pathway is attenuated in cells overexpressing mutant p53 compared to that in their p53-null counterparts. We further observed that the mutant p53 protein reduces the expression of specific genes following TGF-β1 treatment. Indeed, the transcriptional activity mediated by TGF-β1 was found to be attenuated in cells expressing several types of mutant p53. In contrast, wild-type p53 was unable to repress such activity. Instead, in agreement with published data (6, 7), cooperation between wild-type p53 and the TGF-β signaling pathway was observed. Furthermore, mutation in the transactivation domain of p53R175H (p53R175H 22-23) abolished the attenuation in the TGF-β1-induced pathway.
The fact that cells expressing mutant p53 were shown to be resistant to TGF-β treatment (18, 37, 38, 50) raised the possibility that mutant p53 interferes with the functions of wild-type p53 and thereby reduces the sensitivity of cells to TGF-β. However, our findings suggest that a loss of cell response to TGF-β treatment is not due solely to the fact that mutant p53 may interfere with wild-type p53 activity but, rather, that mutant p53 interferes, actively and directly, with the TGF-β-induced signaling pathway.
Accordingly, we demonstrate herein that the mutant p53 protein is capable of reducing TGF-β1-induced transcriptional regulation and gene expression. This, in turn, may cause the cell to lose its sensitivity to TGF-β1 treatment. Indeed, by measuring the response of cells to TGF-β1 treatment in instances of wound healing, we observed that mutant p53 reduces the production and secretion of the proteases MMP2 and MMP9, which typically contribute to this process (26, 33), and attenuates TGF-β1-dependent cell migration and wound closure. The last two are the principle biological functions associated with the TGF-β1-induced signaling pathway.
Our studies show that mutant p53 reduces TGF-β1-induced MMP production and cell migration, which were shown to play a role in tumor invasion. However, it should be borne in mind that in a mouse model generated by Lang et al. and Olive et al., mutant p53 accelerates tumor metastasis (23, 34). We suggest that this apparent discrepancy may result from the possibility that metastasis in these mice might be regulated by additional factors other than TGF-β alone. It should be emphasized that our in vitro model may represent a single facet whereby mutant p53 attenuates TGF-β1-induced MMP production and cell migration. Our experiments were performed under well-defined specific conditions appropriate for measuring the response of the cells to TGF-β1 treatment only. We attribute the reduction in the capacity of mutant p53R175H-producing cells to either migrate or produce MMPs to the fact that the TGF-β1-induced signaling pathway in these cells is attenuated. Performing the same experiments under different in vitro conditions may lead to other insights regarding the role of mutant p53 in cell migration/invasion.
In searching for the mechanism underlying the reduction of TGF-β1-induced transcriptional activity, we found that mutant p53 expression caused a reduction in the phosphorylation of SMAD2 and SMAD3 following treatment of mutant p53-producing cells with TGF-β1. It was previously shown that phosphorylated SMAD2/3 oligomerizes with common SMAD4 and that, together, they translocate to the cell nucleus to further activate specific target genes (27-30). By analyzing the subcellular localization of SMAD4 following treatment of mutant p53-producing cells with TGF-β1, we noticed that mutant p53 completely abrogates the TGF-β1-induced nuclear translocation of SMAD4. Furthermore, we showed that the mutant p53 protein abolishes the formation of the complex commonly formed between SMAD2 and SMAD4 upon TGF-β1 treatment.
It was previously demonstrated for SMAD2 that the phosphorylation of serines 465 and 467 is required for SMAD2-SMAD4 complex formation and signaling (2). In addition, the formation of this complex was shown to be essential for the interaction of SMAD2/3 with the DNA, which further provides a critical trigger for transcriptional activation (19, 24). We extend these findings here, showing that by knocking down the expression of mutant p53 protein, we successfully restored the phosphorylation of SMAD2/3 and enabled the TGF-β1-induced association between SMAD2 and SMAD4, which, in turn, caused mutant p53-producing cells to regain MMP2 and SMAD7 transcriptional activity when treated with TGF-β1.
It is well established that apart from the SMAD-dependent signaling pathway, TGF-β1 is also capable of inducing SMAD-independent signaling pathways, such as the phosphatidylinositol 3-kinase or Ras-MAPK pathway (10). By analyzing the effect of mutant p53 on the TGF-β1-induced Ras-MAPK pathway, we showed that mutant p53 is capable of attenuating this pathway as well. This was observed by a significant attenuation in ERK1/2 phosphorylation upon TGF-β1 treatment. Taken together, our findings indicate that the entire TGF-β signaling pathway is significantly affected by the presence of mutant p53 and that its activity is most likely controlled by an early event which follows the specific induction of the TGF-βRs.
TGF-βRI and -ΙΙ are known to be the primary mediators of signaling to SMAD2/3 (27-30). By measuring the total expression of TGF-βRI/II, we noticed that mutant p53 represses the expression of TGF-βRII at both the RNA and protein levels, while the expression of TGF-βRI is not altered.
It is well established that the “gain-of-function” activity of mutant p53 involves alterations in the expression of specific genes (43). Genes such as the MDR1 (39), CD95 (52), EGR1 (44), MST-MP (53), and ATF3 (4) genes were shown to be either up-regulated or down-regulated by mutant p53. This kind of abnormality in gene expression mediated by mutant p53 allows cells to acquire oncogenic advantages, such as drug resistance (39, 53) and the ability to evade the fatty acid synthase legand-induced cell death pathway (52) or to induce uncontrolled cell proliferation (16).
It was shown that mutant p53 can physically associate with promoters of such genes and thus affect their regulation (43). Furthermore, mutation in the transactivation domain of the mutated protein abolishes its ability to regulate the expression of these genes, which was shown to prevent mutant p53 “gain-of-function” activity (31).
In our study, we demonstrate that mutant p53 can associate with the promoter of TGF-βRII and repress its activity. Furthermore, we show that the integrity of the N-terminal transactivation domain of the protein is essential for this function. Accordingly, we suggest that mutant p53 interacts with the promoter of TGF-βRII, represses its activity, and reduces the total expression of TGF-βRII protein. It is therefore possible that there are fewer available receptors on the surfaces of the cells to interact with TGF-β1 and to activate the signaling pathway.
By ectopic overexpression of Myc-tagged TGF-βRII in mutant p53-producing cells, we completely recovered both the TGF-β1-induced signaling pathway and gene expression. This was observed by a complete recovery of the phosphorylated SMAD2 protein and its transcriptional regulation following TGF-β1 treatment. This suggests that the attenuation in the TGF-β1-induced signaling pathway is indeed a result of a reduction in the expression of TGF-βRII on the surfaces of cells. Under these conditions, fewer receptors are activated, leading to an attenuated signaling pathway which eventually leads to a reduction in gene expression and a loss of cell response to TGF-β1 treatment.
Numerous reports have described deficiencies in the activity of TGF-βRII in human tumor-derived cell lines (28). Inactivation of the receptor leads to the loss of cell response to the growth inhibitory effect of TGF-β and therefore predisposes or causes cancer (28, 29). Recently, a high incidence of mutations in the polyadenosine tract of the TGF-βRII gene was shown to occur in colon and gastric cancers, yielding a truncated receptor which is not able to be active (28). However, not all cases of an inactive receptor involve mutations. There are some cases showing cell resistance to TGF-β due to very low expression of the TGF-βRII protein. For example, it was demonstrated that resistance of human small-cell lung carcinoma cell lines to TGF-β is due to significantly lower expression of the transcribed TGF-βRII gene. Studies in the field have revealed that the TGF-βRII gene and promoter remain intact in spite of the low expression of the receptor (8, 21). Accordingly, it is tempting to speculate that mutant p53 may greatly assist in reducing the expression of TGF-βRII in some cases of small-cell lung carcinoma and in inducing resistance to the growth inhibitory effect of TGF-β.
In conclusion, our present study provides data that support a novel molecular model by which mutant p53 protein represses the expression of TGF-βRΙΙ. This, in turn, attenuates the TGF-β1-induced signaling pathway, as demonstrated by a reduction in the transcriptional activity of SMAD proteins. We show herein for the first time that a loss of cell response to TGF-β treatment not only results from inhibiting the functions of wild-type p53, as previously suggested (18, 37, 38, 50), but that mutant p53, by means of a “gain-of-function” mechanism, contributes to this event as well.
We further demonstrate that mutant p53 is capable of conferring oncogenic properties on cells, at least some of which are mediated by reducing the expression of TGF-βRΙΙ and inducing resistance to TGF-β1 treatment.
Acknowledgments
This research was supported by a Center of Excellence grant from the Flight Attendant Medical Research Institute (FAMRI), by EC FP6 grant LSHC-CT-2004-503576, by funding from the Yad Abraham Center for Cancer Diagnosis and Therapy (to V.R.), and by a grant from the Israel Science Foundation (grant 185/05) (to Y.I.H.). V.R. is the incumbent of the Norman and Helen Asher Professorial Chair of Cancer Research at the Weizmann Institute of Science. Y.I.H. is the incumbent of the Zalman Weinberg Chair in Cell Biology at Tel Aviv University.
This publication reflects the authors’ views, which do not necessarily reflect those of the European Community (EC). The EC is not liable for any use that may be made of the information contained herein.
We thank P. Knaus for the kind gift of luciferase plasmid constructs.
Footnotes
Published ahead of print on 17 September 2007.
REFERENCES
- 1.Aas, T., A. L. Borresen, S. Geisler, B. Smith-Sorensen, H. Johnsen, J. E. Varhaug, L. A. Akslen, and P. E. Lonning. 1996. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 2:811-814. [DOI] [PubMed] [Google Scholar]
- 2.Abdollah, S., M. Macias-Silva, T. Tsukazaki, H. Hayashi, L. Attisano, and J. L. Wrana. 1997. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 272:27678-27685. [DOI] [PubMed] [Google Scholar]
- 3.Blandino, G., A. J. Levine, and M. Oren. 1999. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 18:477-485. [DOI] [PubMed] [Google Scholar]
- 4.Buganim, Y., E. Kalo, R. Brosh, H. Besserglick, I. Nachmany, Y. Rais, P. Stambolsky, X. Tang, M. Milyavsky, I. Shats, M. Kalis, N. Goldfinger, and V. Rotter. 2006. Mutant p53 protects cells from 12-O-tetradecanoylphorbol-13-acetate-induced death by attenuating activating transcription factor 3 induction. Cancer Res. 66:10750-10759. [DOI] [PubMed] [Google Scholar]
- 5.Cadwell, C., and G. P. Zambetti. 2001. The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene 277:15-30. [DOI] [PubMed] [Google Scholar]
- 6.Cordenonsi, M., S. Dupont, S. Maretto, A. Insinga, C. Imbriano, and S. Piccolo. 2003. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 113:301-314. [DOI] [PubMed] [Google Scholar]
- 7.Cordenonsi, M., M. Montagner, M. Adorno, L. Zacchigna, G. Martello, A. Mamidi, S. Soligo, S. Dupont, and S. Piccolo. 2007. Integration of TGF-{beta} and Ras/MAPK signaling through p53 phosphorylation. Science 315:840-843. [DOI] [PubMed] [Google Scholar]
- 8.de Jonge, R. R., L. Garrigue-Antar, V. F. Vellucci, and M. Reiss. 1997. Frequent inactivation of the transforming growth factor beta type II receptor in small-cell lung carcinoma cells. Oncol. Res. 9:89-98. [PubMed] [Google Scholar]
- 9.Dennler, S., S. Itoh, D. Vivien, P. ten Dijke, S. Huet, and J. M. Gauthier. 1998. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derynck, R., and Y. E. Zhang. 2003. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577-584. [DOI] [PubMed] [Google Scholar]
- 11.Dittmer, D., S. Pati, G. Zambetti, S. Chu, A. K. Teresky, M. Moore, C. Finlay, and A. J. Levine. 1993. Gain of function mutations in p53. Nat. Genet. 4:42-46. [DOI] [PubMed] [Google Scholar]
- 12.Ehrlich, M., A. Shmuely, and Y. I. Henis. 2001. A single internalization signal from the di-leucine family is critical for constitutive endocytosis of the type II TGF-beta receptor. J. Cell Sci. 114:1777-1786. [DOI] [PubMed] [Google Scholar]
- 13.Eisenberg, S., D. E. Shvartsman, M. Ehrlich, and Y. I. Henis. 2006. Clustering of raft-associated proteins in the external membrane leaflet modulates internal leaflet H-ras diffusion and signaling. Mol. Cell. Biol. 26:7190-7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.el-Deiry, W. S., J. W. Harper, P. M. O'Connor, V. E. Velculescu, C. E. Canman, J. Jackman, J. A. Pietenpol, M. Burrell, D. E. Hill, Y. Wang, et al. 1994. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 54:1169-1174. [PubMed] [Google Scholar]
- 15.Fabian, Z., M. Vecsernyes, M. Pap, and J. Szeberenyi. 2006. The effects of a mutant p53 protein on the proliferation and differentiation of PC12 rat phaeochromocytoma cells. J. Cell Biochem. 99:1431-1441. [DOI] [PubMed] [Google Scholar]
- 16.Frazier, M. W., X. He, J. Wang, Z. Gu, J. L. Cleveland, and G. P. Zambetti. 1998. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol. 18:3735-3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fynan, T. M., and M. Reiss. 1993. Resistance to inhibition of cell growth by transforming growth factor-beta and its role in oncogenesis. Crit. Rev. Oncog. 4:493-540. [PubMed] [Google Scholar]
- 18.Gerwin, B. I., E. Spillare, K. Forrester, T. A. Lehman, J. Kispert, J. A. Welsh, A. M. Pfeifer, J. F. Lechner, S. J. Baker, B. Vogelstein, and C. Curtis. 1992. Mutant p53 can induce tumorigenic conversion of human bronchial epithelial cells and reduce their responsiveness to a negative growth factor, transforming growth factor beta 1. Proc. Natl. Acad. Sci. USA 89:2759-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heldin, C. H., K. Miyazono, and P. ten Dijke. 1997. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390:465-471. [DOI] [PubMed] [Google Scholar]
- 20.Henis, Y. I., A. Moustakas, H. Y. Lin, and H. F. Lodish. 1994. The types II and III transforming growth factor-beta receptors form homo-oligomers. J. Cell Biol. 126:139-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hougaard, S., P. Norgaard, N. Abrahamsen, H. L. Moses, M. Spang-Thomsen, and H. Skovgaard Poulsen. 1999. Inactivation of the transforming growth factor beta type II receptor in human small cell lung cancer cell lines. Br. J. Cancer 79:1005-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, E., and W. Deppert. 2004. Transcriptional activities of mutant p53: when mutations are more than a loss. J. Cell Biochem. 93:878-886. [DOI] [PubMed] [Google Scholar]
- 23.Lang, G. A., T. Iwakuma, Y. A. Suh, G. Liu, V. A. Rao, J. M. Parant, Y. A. Valentin-Vega, T. Terzian, L. C. Caldwell, L. C. Strong, A. K. El-Naggar, and G. Lozano. 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119:861-872. [DOI] [PubMed] [Google Scholar]
- 24.Liu, F., C. Pouponnot, and J. Massague. 1997. Dual role of the Smad4/DPC4 tumor suppressor in TGFbeta-inducible transcriptional complexes. Genes Dev. 11:3157-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lutz, M., K. Krieglstein, S. Schmitt, P. ten Dijke, W. Sebald, A. Wizenmann, and P. Knaus. 2004. Nerve growth factor mediates activation of the Smad pathway in PC12 cells. Eur. J. Biochem. 271:920-931. [DOI] [PubMed] [Google Scholar]
- 26.Martin, P. 1997. Wound healing—aiming for perfect skin regeneration. Science 276:75-81. [DOI] [PubMed] [Google Scholar]
- 27.Massague, J. 2000. How cells read TGF-beta signals. Nat. Rev. Mol. Cell. Biol. 1:169-178. [DOI] [PubMed] [Google Scholar]
- 28.Massague, J. 1998. TGF-beta signal transduction. Annu. Rev. Biochem. 67:753-791. [DOI] [PubMed] [Google Scholar]
- 29.Massague, J., and Y. G. Chen. 2000. Controlling TGF-beta signaling. Genes Dev. 14:627-644. [PubMed] [Google Scholar]
- 30.Massague, J., and D. Wotton. 2000. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19:1745-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matas, D., A. Sigal, P. Stambolsky, M. Milyavsky, L. Weisz, D. Schwartz, N. Goldfinger, and V. Rotter. 2001. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J. 20:4163-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milner, J., E. A. Medcalf, and A. C. Cook. 1991. Tumor suppressor p53: analysis of wild-type and mutant p53 complexes. Mol. Cell. Biol. 11:12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mirastschijski, U., C. J. Haaksma, J. J. Tomasek, and M. S. Agren. 2004. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Exp. Cell Res. 299:465-475. [DOI] [PubMed] [Google Scholar]
- 34.Olive, K. P., D. A. Tuveson, Z. C. Ruhe, B. Yin, N. A. Willis, R. T. Bronson, D. Crowley, and T. Jacks. 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119:847-860. [DOI] [PubMed] [Google Scholar]
- 35.Oren, M. 2003. Decision making by p53: life, death and cancer. Cell Death Differ. 10:431-442. [DOI] [PubMed] [Google Scholar]
- 36.Oren, M. 2001. The p53 saga: the good, the bad, and the dead. Harvey Lect. 97:57-82. [PubMed] [Google Scholar]
- 37.Reiss, M., T. Munoz-Antonia, J. M. Cowan, P. C. Wilkins, Z. L. Zhou, and V. F. Vellucci. 1993. Resistance of human squamous carcinoma cells to transforming growth factor beta 1 is a recessive trait. Proc. Natl. Acad. Sci. USA 90:6280-6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reiss, M., V. F. Vellucci, and Z. L. Zhou. 1993. Mutant p53 tumor suppressor gene causes resistance to transforming growth factor beta 1 in murine keratinocytes. Cancer Res. 53:899-904. [PubMed] [Google Scholar]
- 39.Sampath, J., D. Sun, V. J. Kidd, J. Grenet, A. Gandhi, L. H. Shapiro, Q. Wang, G. P. Zambetti, and J. D. Schuetz. 2001. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 276:39359-39367. [DOI] [PubMed] [Google Scholar]
- 40.Santibanez, J. F., J. Guerrero, M. Quintanilla, A. Fabra, and J. Martinez. 2002. Transforming growth factor-beta1 modulates matrix metalloproteinase-9 production through the Ras/MAPK signaling pathway in transformed keratinocytes. Biochem. Biophys. Res. Commun. 296:267-273. [DOI] [PubMed] [Google Scholar]
- 41.Sigal, A., and V. Rotter. 2000. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 60:6788-6793. [PubMed] [Google Scholar]
- 42.Vogelstein, B., D. Lane, and A. J. Levine. 2000. Surfing the p53 network. Nature 408:307-310. [DOI] [PubMed] [Google Scholar]
- 43.Weisz, L., M. Oren, and V. Rotter. 2007. Transcription regulation by mutant p53. Oncogene 26:2202-2211. [DOI] [PubMed] [Google Scholar]
- 44.Weisz, L., A. Zalcenstein, P. Stambolsky, Y. Cohen, N. Goldfinger, M. Oren, and V. Rotter. 2004. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res. 64:8318-8327. [DOI] [PubMed] [Google Scholar]
- 45.Whitman, M., and F. McKeon. 2003. p53 and TGF-beta in development: prelude to tumor suppression? Cell 113:275-276. [DOI] [PubMed] [Google Scholar]
- 46.Willis, A., E. J. Jung, T. Wakefield, and X. Chen. 2004. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23:2330-2338. [DOI] [PubMed] [Google Scholar]
- 47.Wolf, D., N. Harris, and V. Rotter. 1984. Reconstitution of p53 expression in a nonproducer Ab-MuLV-transformed cell line by transfection of a functional p53 gene. Cell 38:119-126. [DOI] [PubMed] [Google Scholar]
- 48.Wrana, J. L., L. Attisano, J. Carcamo, A. Zentella, J. Doody, M. Laiho, X. F. Wang, and J. Massague. 1992. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71:1003-1014. [DOI] [PubMed] [Google Scholar]
- 49.Wrana, J. L., L. Attisano, R. Wieser, F. Ventura, and J. Massague. 1994. Mechanism of activation of the TGF-beta receptor. Nature 370:341-347. [DOI] [PubMed] [Google Scholar]
- 50.Wyllie, F. S., T. Dawson, J. A. Bond, P. Goretzki, S. Game, S. Prime, and D. Wynford-Thomas. 1991. Correlated abnormalities of transforming growth factor-beta 1 response and p53 expression in thyroid epithelial cell transformation. Mol. Cell. Endocrinol. 76:13-21. [DOI] [PubMed] [Google Scholar]
- 51.Yingling, J. M., M. B. Datto, C. Wong, J. P. Frederick, N. T. Liberati, and X. F. Wang. 1997. Tumor suppressor Smad4 is a transforming growth factor beta-inducible DNA binding protein. Mol. Cell. Biol. 17:7019-7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zalcenstein, A., P. Stambolsky, L. Weisz, M. Muller, D. Wallach, T. M. Goncharov, P. H. Krammer, V. Rotter, and M. Oren. 2003. Mutant p53 gain of function: repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants. Oncogene 22:5667-5676. [DOI] [PubMed] [Google Scholar]
- 53.Zalcenstein, A., L. Weisz, P. Stambolsky, J. Bar, V. Rotter, and M. Oren. 2006. Repression of the MSP/MST-1 gene contributes to the antiapoptotic gain of function of mutant p53. Oncogene 25:359-369. [DOI] [PubMed] [Google Scholar]