

Summary
By virtue of their large number, widespread distribution and important roles in cell physiology and biochemistry, G-protein-coupled receptors (GPCR) play multiple important roles in clinical medicine. Here, we focus on 3 areas that subsume much of the recent work in this aspect of GPCR biology: 1) Monogenic diseases of GPCR; 2) Genetic variants of GPCR; and 3) Clinically useful pharmacological agonists and antagonists of GPCR. Diseases involving mutations of GPCR are rare, occurring in <1/1000 people, but disorders in which antibodies are directed against GPCR are more common. Genetic variants, especially single nucleotide polymorphisms (SNP), show substantial heterogeneity in frequency among different GPCRs but have not been evaluated for some GPCR. Many therapeutic agonists and antagonists target GPCR and show inter-subject variability in terms of efficacy and toxicity. For most of those agents, it remains an open question whether genetic variation in primary sequence of the GPCR is an important contributor to such inter-subject variability, although this is an active area of investigation.
Keywords: GPCR mutations, human disease, nephrogenic diabetes insipidus, retinitis pigmentosa
Introduction
In addition to their large number, widespread expression and important mechanistic and regulatory properties, as reviewed by others in this volume, G-protein-coupled receptors (GPCR) have well-recognized roles in clinical medicine. Their expression on the plasma membrane makes GPCR readily accessible, especially by hydrophilic hormones and drugs, including both agonists and antagonists, and their non-uniformity of expression in different tissues and cell types provides selectivity (in some cases, specificity) in the targeting of these receptors for the activation or blockade of physiological events. Studies in recent years have provided a number of new insights, many of them gleaned from application of the tools of the “genetic revolution”. In this article, we will review aspects of GPCR in clinical medicine with an emphasis on recent developments and insights in 3 areas: 1) Monogenic diseases of GPCR; 2) Genetic variants of GPCR; and 3) Clinically useful pharmacological agonists and antagonists of GPCR. Each of these are large topics, have been the subject of reviews in recent years (e.g., [1–8]). We refer interested readers to such reviews for additional information that length restrictions prevent us from presenting in detail. Other sources of useful information related to these topics include a variety of web-based tools [9], including www.hapmap.org and sites accessible therefrom.
Monogenic Diseases of GPCR
Monogenic diseases and genetic variants associated with those diseases are generally quite rare, occurring in <1% of the population and often variably among subjects of different ethnicities. Since GPCR comprise ~3% of the human genome [10], it is perhaps not surprising that non-lethal mutations can occur in GPCR, especially those that are expressed in sensory and hormonal systems, where they serve as mediators of information transfer from the extracellular environment to the cell interior. One such critical action is in the visual system where rhodopsin in photoreceptor-capturing neurons, retinal rods and color (red, blue and green) opsins in retinal cones, transduce the input from photons of light into electrical impulses that then travel to the brain and are decoded. A second major class of physiologically important GPCR are those that mediate the action of hormones, especially polypeptide hormones but also including the action of such hormones, such as the calcium-sensing receptor or other chemical entities (e.g., lipids, amines, fatty acids). A third class is receptors for physiologically important neurotransmitters, such as norepinephrine (and to a lesser extent, epinephrine), acetylcholine (at muscarinic cholinergic receptors), dopamine, serotonin (at certain receptors), glutamate (at metabotropic receptors) as well as numerous peptides and lipids that function as neuromodulators. To date, mutations that lead to human disease have been identified in a relatively limited number of GPCR. We will briefly discuss 3: rhodopsin, V2 vasopressin and the calcium-sensing receptor.
A large number of monogenic mutations have been identified in rhodopsin, in particular in patients that have the disease retinitis pigmentosa; in addition, a number of hormonally responsive GPCR have been identified as pathologic entities in a variety of endocrine disorders (Table 1). The latter disorders include those with either activating mutations or mutations that block hormonal response. Studies that documented hormone resistance in patients with particular disorders were often critical in focusing attention on GPCR or their signaling pathways as the sites of lesions in such disorders, whereas in other situations excessive response in the absence of increased levels of the activating hormones provided a similar impetus to infer a role for components and events that mediate hormonal response.
Table 1.
Examples of rare mutants of GPCR that cause human diseases
| Receptor/Gene name | Mutation | Disease | Ref |
|---|---|---|---|
| Calcium-Sensing (CaS)/CaSR | Multiple (e.g. Arg185Gln) | Autosomal Dominant Hypocalcemia (ADH)
Sporadic Hypoparathyroidism Familial Hypoparathyroidism |
[15, 90] |
| CXCR4 | Multiple (e.g. Ser338X) | WHIM syndrome | [91, 92] |
| Endothelin receptor B (ETB)/EDNRB | Multiple (e.g. Trp276Cys) | Hirschsprung’s disease | [93] |
| Follicle-stimulating hormone (FSH)/FSHR | Multiple (e.g. Ala189Val) | Female infertility | [94] |
| N-formyl-peptide (FPR)/FPR1 | Phe110Ser, Cys126Trp | Juvenile periodontitis | [95] |
| Frizzled (FZD4)/FZD4 | Multiple (e.g. Arg417Gln) | Familial exudative vitreoretinopathy (FEVR) | [96, 97] |
| Goandotropin-releasing hormone (GnRH)/GNRHR | Multiple (e.g. Arg262Gln) | Hypogonadotropic hypogonadism (HH) | [98, 99] |
| GPR54/GPR54 | Multiple (e.g. Cys223Arg) | Hypogonadotropic hypogonadism (HH) | [98, 99] |
| GPR56/GPR56 | Multiple (e.g. Cys223Arg) | Bilateral frontoparietal polymicrogyria (BFPP) | [100, 101] |
| vGPCR/KSHV-GPCR | (constitutively active) | Kaposi’s sarcoma (KS) | [102, 103] |
| Relaxin family peptide receptor 2 (RXFP2)/LGR8 | Multiple (e.g. Thr222Pro) | Cryptorchidism | [104–106] |
| MASS1 (also called VLGR1, USH2C)/MASS1 | Multiple (e.g. Ser2652X)) | Usher syndrome
Febrile seizures (FS) |
[107–110] |
| Melanocortin (MC4)/MC4R | Multiple (e.g. Pro78Leu) | Dominant and recessive obesity | [111, 112] |
| Rhodopsin/RHO | Multiple (e.g. Pro23His) | Retinitis pigmentosa (RP) | [113–115] |
| Vasopressin receptor (V2)/AVPR2 | Multiple (e.g. Arg113Trp) | Nephrogenic diabetes insipidus (NDI) | [116, 117] |
The location of clinically (i.e., pathophysiologically) significant mutations are not always sites that have been suspected from mutational analyses with cloned receptors. As shown in Figure 1, which highlights the sites that are mutated in rhodopsin, a wide variety of residues have been identified in patients with retinitis pigmentosa, a leading cause of blindness and visual disability in younger people that occurs with an overall frequency of one in 4000. Retinitis pigmentosa is a disorder that leads to the progressive death of the rod photoreceptors; recent work suggests that such cell death may be a consequence of a high, constant rate of signal transduction that causes the rods to die, perhaps secondary to prolonged lowering of Ca2+ concentration [11].
Figure 1.

Schematic structure of rhodopsin. Each amino acid residue is shown as a green dot; amino acid residues that are mutated in patients with retinitis pigmentosa are shown as red dots. Seven transmembrane domains are boxed. Mutations are collected from the Human Gene Mutation Database (www.hgmd.org), the Retinal Information Network (www.sph.uth.tmc.edu/RetNet/sym-dis.htm), and the Retina International Mutation Database (www.retina-international.com/sci-news/rhomut.htm) and references [88] and [89].
Nephrogenic diabetes insipidus (NDI), which results from failure of vasopressin (antidiuretic hormone, ADH) to act on the renal collecting duct to facilitate water reabsorption, is another well-studied monogenic disorder of a GPCR, the arginine vasopressin receptor 2 (AVPR2, V2), in which mutations cause congenital nephrogenic diabetes insipidus in ~90% of patients via an X-linked recessive mode of inheritance [12]. To date, >280 families with a history of NDI have been shown to have >180 putative disease-causing mutations in AVPR2 (Figure 2 and [12]). In most cases, these mutations lead to the intracellular trapping of the V2 receptors, such that few receptors reach the plasma membrane to trigger the activation of Gs and adenylyl cyclase and thereby, the generation of cAMP. Therapeutic approaches are under investigation that involve the use of nonpeptide V2 receptor antagonists to bind intracellular receptors as what have been termed “pharmacochaperones” that will facilitate their folding, insertion and function in the plasma membrane [13].
Figure 2.
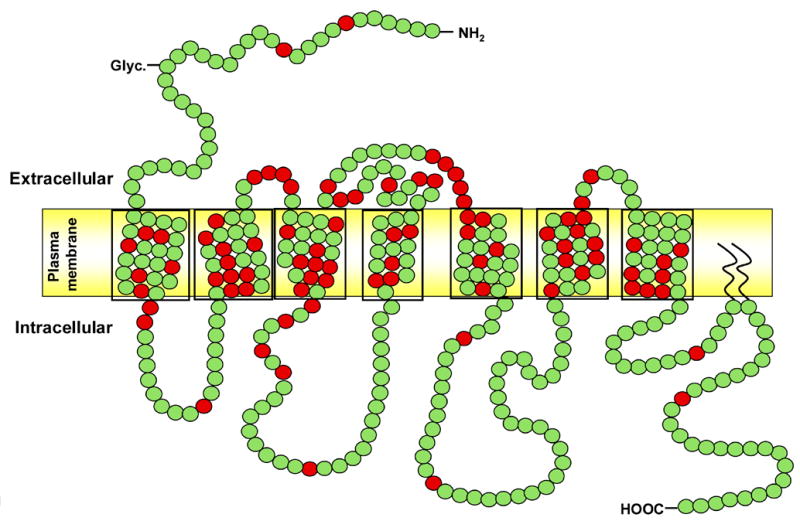
Schematic structure of the vasopressin V2 receptor (AVPR2). Each amino acid residue is shown as a green dot; amino acid residues that are mutated in patients with nephrogenic diabetes insipidus are shown as red dots. Seven transmembrane domains are boxed. Mutations found in the AVPR2 gene were collected from the Human Gene Mutation Database (www.hgmd.org) and the Diabetes Insipidus Mutation Database (www.medicine.mcgill.ca/nephros).
Rhodopsin belongs to Family A, which contains the largest group of GPCR superfamily members; receptors in this Family generally contain a relatively short extracellular N-terminus and highly conserved amino acids within each transmembrane domain. Family A members are activated by small ligands such as biogenic amines and nucleotides, although rhodopsin itself is stimulated by photons of light that activate a retinal bound in a transmembrane pocket. In contrast, the V2 receptor belongs to Family B, whose receptors recognize large peptides and are generally characterized by longer N-termini, in the case of V2 receptors one that has six conserved cysteine residues. Comparison of rhodopsin and the V2 receptor (Cf. Figure 1 and. Figure 2) reveals that disease-causing mutations occur in all portions of the two receptors with a greater number in transmembrane domains relative to mutants in non-transmembrane domains of V2 receptors in NDI than of rhodopsin in retinitis pigmentosa.
Genetic disorders of the calcium-sensing receptor (CaSR) are a third example of a monogenic disease in GPCR [14, 15]. This receptor is found on numerous tissues involved in calcium homeostasis, including the parathyroid glands, kidney, and intestine. Binding of calcium to the CaSR is the mechanism by which parathyroid cells detect changes in ionized calcium concentration and in turn, modulate parathyroid hormone secretion so as to maintain serum calcium levels within a narrow physiologic range. CaSRs in renal tubules modulate calcium reabsorption in response to alterations in extracellular calcium concentrations. Several hypocalcemic and hypercalcemic disorders have been identified that derive from rarely occurring mutations of CaSR: loss-of-function CaSR mutations in the hypercalcemic disorders of familial benign (hypocalciuric) hypercalcaemia (FHH, FBH or FBHH) and neonatal severe primary hyperparathyroidism (NSHPT) (each of which occur <1/10,000) and gain-of-function CaSR mutations in autosomal dominant hypocalcaemia with hypercalciuria (ADHH) and Bartter’s syndrome type V.
In addition to such “classical” monogenic diseases, another type of such disease can involve the generation of antibodies as monoclonal (or polyclonal) expansion of immune cells with the antibody products directed against GPCR or in some cases, their downstream targets. The most prevalent example is Graves disease, a form of hyperthyroidism with enhanced response to thyroid-stimulating hormone (TSH) that is most commonly secondary to autoimmune activation of TSH receptors [16, 17]. Another example of activating (as well as inhibitory) antibodies directed at GPCR (e.g., β-adrenergic and muscarinic cholinergic) is Chagas’ cardiomyopathy, which is triggered by infection with the protozoan Trypanosoma cruzi [18, 19].
In several other settings, antibodies directed at GPCR can blunt hormone action, preventing G-protein activation (e.g., [15, 17, 20–24]). Of note, such disorders are almost invariably ones that occur in adults rather than children whereas monogenic disorders of the receptors themselves often manifest clinical abnormalities much earlier in life.
Genetic variants of GPCR
Completion of the human genome has introduced a large amount of DNA sequence information that predicts 367 non-sensory GPCRs and an additional 380 or more chemosensory GPCRs [25]. Human genomic data also reveal that GPCR loci harbor a substantial number of genetic variants, including nucleotide insertion or deletion as well as the exchange of a single nucleotide, i.e. single nucleotide polymorphisms (SNPs). SNPs account for ~ 80% of all sequence variations and generally occur at a frequency ~ 1 in 1200 nucleotides. A polymorphism is defined as a genetic variant that occurs at a locus with an allelic frequency of greater than or equal to 1% whereas “ mutations”, such as those discussed in the prior section, designate rarer genetic variants that are germline-transmitted changes in a given individual or somatic variation identified in isolated tissues.
Genetic variants/polymorphisms identified in GPCRs can influence receptor expression, targeting, function, and receptor turnover; as well as the ability of receptors to recognize and respond to pharmacologic agents. Below we describe selected GPCRs with polymorphisms involved in human diseases, in addition to elucidating their potential for serving as future therapeutic targets. TABLE 2 lists sequence variants identified in human GPCR genes that relate to human diseases.
Table 2.
Examples of polymorphisms of GPCR associated with human diseases
| Receptor | Polymorphisms | Examples of disease associations | Ref |
|---|---|---|---|
| β1 Adrenergic receptor | Arg389Gly | Heart failure | [64, 71] |
| β2 Adrenergic receptor | Multiple | Hypertension, Asthma | [118, 119] |
| β3 Adrenergic receptor | Trp64Arg | Obesity | [120] |
| CC chemokine receptor 2 (CCR2) | Val64Ile | Delayed progression of AIDS | [121] |
| CC chemokine receptor 5 (CCR5) | Multiple | Associated with progression of AIDS | [33, 122] |
| Dopamine receptor 2 (D2) | 3’UTR52A/G | Associated with depression and anxiety | [123] |
| Dopamine receptor 3 (D3) | Ser9Gly, Promoter SNPs | Haplotype associated with schizophrenia | [124] |
| Muscarinic receptor subtype 3 (M3) | Promoter haplotype | Possible association with asthma and atopy | [125] |
| Neuropeptide S receptor (NPSR; also called GPR154, GPRA) | Haplotypes H1, H5
Asn107Ile, rs324981 |
Asthma susceptibility | [31, 32, 126] |
| P2Y12 | CA deletion at Codon 240 | Associated with bleeding diathesis | [127] |
The β2-adrenergic receptor possesses multiple polymorphisms, including several in the coding region and 5’ untranslated region, that generate 4 common haplotypes (collections of variants) in different ethnic groups (Caucasian, African-American, Hispanic-Latino and Asian) [26]. Two common (i.e., occur >20%) non-synonymous polymorphisms, Arg16Gly and the Gln27Glu, have been shown to influence regulation of receptors by agonists but not receptor binding or coupling to Gs/adenylyl cyclase. Individuals who are Arg16 homozygotes display slower/impaired bronchodilatory response upon agonist activation of the β2-adrenergic receptor than do people who are Gly16 homozygotes [27, 28]. Such results suggest that Arg16Gly may predict β2-adrenergic receptor agonist response in the therapy of asthma, although additional studies are needed [29]. Other recent data obtained with patients from the UK indicate that such variants do not contribute in a major way to asthma incidence or prevalence [30].
Another candidate GPCR associated with asthma susceptibility was recently de-orphanized and named the neuropeptide S receptor (NPSR), formerly known as orphan receptor GPRA or GPR154 [31]. One non-synonymous SNP, Asn107Ile, has a strong association with asthma but with as-yet no clear evidence to explain the genetic association. Melen et al. studied 7 polymorphisms of NPSR and inferred 7 haplotypes (H1–H7) in a case-control study of childhood allergy and asthma and found that haplotypes H1 and H5 were significantly associated with asthma [32].
The best studied example of genetic variants in a GPCR functioning as disease modifier is the CC chemokine receptor-5 (CCR5) [33]. This receptor plays a crucial role in HIV-1 pathogenesis, serving as a co-receptor for viral entry and CCR5 polymorphisms, including promoter SNP (59029A/G) and a deletion of 32 base pairs (Δ32), influence progression of HIV infection to AIDS. CCR5Δ32 causes a frame shift at amino acid 185 leading to a premature termination of the receptor between putative transmembrane domains IV and V [33]; whereas promoter SNP 59029G shows lesser activity than 59029A allele by in-vitro promoter reporter assay [34]. Thus both CCR5Δ32 and 59029G SNP seem to be protective in HIV infection due to lower expression of CCR5 receptors on the cell surface.
Significant progress has been achieved in recent years in identifying genetic polymorphisms of GPCR’s and in providing suggestive evidence of their relevance in various human diseases, in addition to yielding information regarding mechanisms of GPCR signal transduction. Given the complexity of this receptor superfamily, including structural heterogeneity, receptor multiplicity, and redundancy in signaling pathways, further efforts will be required to identify and document definitively the contributing role of polymorphisms in disease and as disease modifiers. There is as-yet incomplete information regarding the full-range of genetic variants of most GPCRs, including orphan GPCRs, and thus this is likely to remain an area of active research with potential clinical importance. In addition, two aspects of such variants that have not yet been well explored include the possible role of synonymous SNPs that may change the binding and action of exon splicing enhancer (ESE) proteins and thus affect RNA splicing even though they do not produce amino acid substitutions [3, 35] and the potential contribution of genetic variants in GPCR coding sequences that will alter the ability of receptors to oligomerize [36]. Studies of GPCR variants should be aided by the completion of the HapMap [37] and identification of tag SNPs for particular haplotypes in genes of interest [38].
Drug effects and the role of genetic variants of GPCR
As a class, GPCR have been the most successful molecular drug targets in clinical medicine. Agonists and antagonists of GPCR are used in the treatment of diseases of every major organ system including the CNS, cardiovascular, respiratory, metabolic and urogenital systems. Certain GPCRs have been particularly useful as drug targets. These include AT1 angiotensin, adrenergic, dopamine and serotonin (5-hydroxytryptamine, 5-HT) receptor subtypes as well as GPCR for hormones whose activity is increased or decreased in particular endocrine disorders. For example, antagonists of AT1 angiotensin II receptors are used to prevent diabetes mellitus-induced renal damage [39] and to treat essential hypertension and congestive heart failure. α1-Adrenergic receptor antagonists are employed for symptomatic treatment and to slow the clinical progression of benign prostatic hyperplasia [40]. β-adrenergic receptor antagonists, acting on β1- and/or β2-adrenergic receptors, increase survival in patients with congestive heart failure [41] and are used to treat essential hypertension and coronary heart disease. β2-adrenergic receptor agonists are a cornerstone in the treatment of asthma and chronic obstructive pulmonary disease and are used to delay preterm labor [42]. Dopamine receptor antagonists, primarily via their actions on D2 receptors, are standard therapeutic agents in the treatment of schizophrenia, although some representatives of this class may exert antipsychotic effects via other dopamine receptor subtypes and/or 5-HT receptors [43]. Dopamine receptor agonists (e.g., levodopa serving as a precursor for dopamine) are widely used to treat Parkinson’s disease [43]. Inhibitors of 5-HT uptake act as indirect agonists at various subtypes of 5-HT receptors and are used to treat major depressive disorders; several of these subtypes may also be involved in the effects of atypical antipsychotic drugs.
While most GPCR are expressed in a tissue-selective manner, it remains a key challenge for the safe use of drugs to identify compounds that will selectively act on a particular receptor in a target tissue and largely spare those receptors in other tissues, thereby potentially minimizing side effects. Such tissue selectivity may result from pharmacokinetic phenomena (e.g. distribution and metabolism) [44] but may also involve tissue-specific expression of GPCR splice variants and/or receptor oligomers [45, 46].
Responses to drugs targetted to GPCRs can show substantial inter-subject variability. Attempts to use clinical factors as a means to predict individual drug responses have had limited success. Therefore, much recent attention has involved assessment of genetic features that potentially influence variability in drug responses. Genetic variations of enzymes or transporters involved in the pharmacokinetics of drugs [47, 48] and polymorphisms of GPCR genes have been investigated as possible determinants of heterogeneous drug responses.
GPCR polymorphisms may have tissue-, cell type- or ligand-specific effects on drug responses. Apart from the possibility of response-specific physiological counter-regulatory mechanisms, factors intrinsic to the biochemical properties of polymorphic receptors may contribute to such heterogeneity. Tissue-selective factors may relate to the differential expression of specific biochemical properties in different cell types. For example, the human α1A-adrenergic receptor can couple to the activation of extracellular signal-regulated kinases in Chinese hamster ovary cells [49] but to its inhibition in rat-1 fibroblasts [50]. Such differences are not yet well documented for a large number of GPCR polymorphisms but may have specific consequences in different cell types and tissues. Although such differences in tissue selectivity are primarily expected from genetic variants that affect the coding region of a gene and/or its splicing, polymorphisms in the promoter region of GPCR genes may affect transcription in a tissue-selective manner [51].
GPCR polymorphisms not only may demonstrate tissue-specificity but also may act in a ligand-specific manner, termed “ligand-directed signaling”, whereby activation of a given GPCR by two chemically distinct ligands leads to differential signaling responses [52]. Indeed, some GPCR polymorphisms can affect functional responses to some but not other ligands, e.g. with β3-adrenergic receptors [53–55]. Such concepts may apply not only to acutely measured GPCR responses but also to receptor regulation, especially because certain GPCR polymorphisms alter such regulation [56]. In the following we illustrate these principles based upon examples from several drug classes acting on GPCR that are frequently used in clinical medicine (Table 3).
Table 3.
GPCR as drug targets: some examples of the impact of receptor polymorphisms
| Receptor | Drugs and some key indications | Polymorphisms | Relevance | Ref |
|---|---|---|---|---|
| AT1 angiotensin II receptor | Antagonists (e.g. losartan) in the treatment of essential hypertension or congestive heart failure | A1166C SNP in untranslated part of exon 5 | Inconclusive data for drug responses where multiple studies have been done | [57–63] |
| α1A-adrenergic receptor | Antagonists (e.g. tamsulosin) to treat micturition (bladder emptying) disorders associated with enlarged prostate glands | C1475T | Short- and long-term antagonist effects apparently not affected | [66] |
| β1-adrenergic receptor | Antagonists (e.g. propranolol, atenolol, metoprolol, carvedilol) to treat essential hypertension or congestive heart failure | Ser49Gly
Arg389Gly |
Arg389 linked to increased antagonist effect | [60, 67– 69, 73, 128] |
| β2-adrenergic receptor | Agonists (e.g. terbutaline, salbutamol, formoterol, salmeterol) for treatment of obstructive airway disease or premature labor | Arg16Gly
Gln27Glu Thr164Ile |
Possibly reduced responses with Ile164 otherwise no consistent association with drug responsiveness | [56] |
| D2 dopamine receptor | Antagonists (e.g. haloperidol and clozapine) to treat schizophrenia
Agonists (e.g. levodopa) for the treatment of Parkinson’s disease |
-141C Ins/Del
Taq1A |
Reduced antagonist response with Del or homozygous A2 allele
No consistent associations with therapeutic response or side effects of agonists |
[75–77] |
| D3 dopamine receptor | Antagonists (e.g. haloperidol) in the treatment of schizophrenia | Ser9Gly | Increased risk of tardive dyskinesia with Gly allele | [75, 76] |
| 5-HT2A receptor | Antagonists (e.g. clozapine) to treat schizophrenia
Indirect agonists (e.g. fluvoxamine) for the treatment of depression |
T102C | Reduced response to clozapine with C allele
Possibly reduced response to agonists with homozygous T allele |
[76, 82] |
| 5-HT2C receptor | Antagonists (e.g. clozapine) to treat schizophrenia | Multiple polymorphisms in promoter and coding region in linkage disequilibrium | Genotypes associate with therapeutic response and with side effects such as tardive dyskinesia and weight gain | [81, 83] |
Angiotensin II receptors
Administration of AT1 antagonists to patients with high blood pressure can lower blood pressure, increase glomerular filtration rate and decrease cardiac hypertrophy. The most widely investigated AT1 receptor polymorphism, A1166C SNP, is located in an untranslated part of exon 5; its functional relevance for the biology of AT1 receptors is unclear. This SNP has been associated with enhanced responses to antagonists of AT1 receptors or inhibition of angiotensin converting enzyme (and hence a reduction of endogenous agonist) [57–59]. However, whenever more than one study has been reported for a given response parameter, studies without significant differences [60–62] or even with a significant difference in favor of the opposite allele have been reported [63].
Thus, currently available data do not allow definitive conclusions regarding the role of the A1166C SNP of the AT1 receptor but do point to some general notions. Firstly, all of the above studies included relatively small numbers of patients (<100 in most cases) and hence probably are statistically underpowered. Second, searches for the role of a SNP for which functional correlates are not known at the molecular and cellular level are difficult to interpret, as such SNPs may only be indicative of other genetic variants, including SNPs or extended haplotypes, with which they are in linkage disequilibrium. Thirdly, efforts that emphasize a single SNP outside the coding region may not readily allow inferences that link gene structure to function.
Adrenergic receptors
α1-Adrenergic receptor antagonists are the most frequently used for the medical treatment of lower urinary tract symptoms related to benign prostatic enlargement. Various SNPs have been detected in the genes encoding the three α1-adrenergic receptor subtypes [1, 3, 64]. However, the SNPs reported thus far have only limited effects on protein function [65] and have not been found to associate with clinical responses to α1-adrenergic receptor antagonists [66].
β-Adrenergic receptor antagonists are widely used in several disorders, including the treatment of hypertension, congestive heart failure, angina pectoris and myocardial infarction. Studies on a role of β1-adrenergic receptor polymorphisms in the response to such drugs have focused on the non-synonymous SNPs Ser49Gly and Arg389Gly. Multiple studies have reported increased β1-antagonist effects associated with Arg389 [67–71], an allele exhibiting an increased function in vitro [72], although not all investigators have confirmed this [60, 73]. By contrast, no association has been observed in most studies of β1-adrenergic receptor antagonist responses with the Ser49Gly SNP, which also alters function in vitro [60, 67, 70, 73].
Selective β2-adrenergic receptor agonists are used as bronchodilators in patients with obstructive airway disease and as tocolytic agents in women with premature labor. Three β2-adrenergic receptor nonsynonymous SNPs have been studied: Arg16Gly, Gln27Glu, and Thr164Ile. Although the position 16 and 27 variants are expressed both heterozygously and homozygously, homozygous Ile164Ile subjects have not been identified, implying that this is a lethal variant. Many studies have tested the impact of these SNPs on agonist responses but have yielded inconsistent findings with the exception of a lower response of the rarer (found in < 5% subjects) Ile164 genotype [56]. However, as noted above, recent data have indicated that asthmatic patients with the Arg16 variant shows less response to short- or long-acting β2-adrenergic receptor agonists than do patients with Gly16 β2-adrenergic receptors [27–29]. Numerous inconclusive studies have reported an association of these SNPs with arterial hypertension [51] but recent data with a large population sample document association of the Arg16 variant with higher diastolic blood pressure but only in males, implying an interaction of gender with this variant in regulation of blood pressure [74]. The relevance of such a finding for the overall population may be limited since the effect appears to be small and gender-specific.
In conclusion, the emerging picture from studies of adrenergic receptor SNPs is that most have little effect on ligand recognition or receptor function in vitro and limited influence on drug responsiveness in vivo. SNPs that affect the sensitivity for receptor regulation by agonists, e.g. the Arg16Gly and Gln27Glu SNPs in the β2-adrenergic receptor, have yielded inconsistent results in terms of functional effects and disease associations in vivo. In contrast, altered drug responsiveness has been observed for certain SNPs that affect receptor function in vitro, e.g. the β1-adrenergic receptor Arg389Gly SNP and the infrequent Thr164Ile SNP in the β2-adrenergic receptor with thus far less definitive evidence for other β2-adrenergic receptor variants [29]. The findings highlight the need to carefully test the biochemical consequences of SNPs in vitro in order to enhance understanding of their potential role in clinical association studies.
Dopamine receptors
Dopamine receptor antagonists are used clinically as antipsychotic agents but frequently associated with side effects such as tardive dyskinesia and weight gain. It was initially believed that such agents act primarily via D2 receptors but some atypical antipsychotics have also been shown to act via D3 receptors or 5-HT receptors (see below). By contrast, Parkinson’s disease, which involves brain region-specific depletion of dopamine neurons, is treated with dopamine receptor agonists and compounds such as levodopa that are metabolized to dopamine receptor agonists.
Various studies have investigated a possible role of polymorphisms in dopamine receptor genes in the treatment of schizophrenia [75–77]. Such studies have found that both the Del allele of the -141C Ins/Del polymorphism and the homozygous A2 allele of the Taq1A polymorphism of the D2 receptor gene are associated with reduced therapeutic responses to both typical antipsychotics (e.g., haloperidol) and atypical antipsychotics (e.g., clozapine). Recent studies confirm such findings [78, 79]. Although side effects of typical antipsychotics, such as the motor disorder tardive dyskinesia, are classically linked to D2 receptors, studies that have assessed for these polymorphisms have not yielded consistent associations. Tardive dyskinesia has instead been linked to the Ser9Gly polymorphism of D3 receptors [75–77]. The same SNP has also been associated with therapeutic response to the atypical antipsychotic risperidone [80]. By contrast, few studies are available regarding polymorphisms of D2 and D3 receptors; therapeutic responses or side effects of agonists have not yielded consistent associations. In aggregate, studies of dopamine receptor polymorphisms have suggested that the presumed molecular target of a drug may not always be the best candidate for polymorphisms association studies, thus emphasizing anew that a drug may have molecular targets distinct from those generally assumed to mediate its effect.
5-HT receptors
Classical antipsychotics (e.g., haloperidol) are thought to be dopamine receptor antagonists while atypical antipsychotics (e.g., clozapine) have similar or even higher affinity for 5-HT receptor subtypes. 5-HT receptors are also indirectly targeted by 5-HT uptake inhibitors such as fluoxetine, i.e. drugs that increase the availability of 5-HT in the synaptic cleft. Such uptake inhibitors are a cornerstone in the treatment of major depressive disorders. Therapeutic responses and/or side effects of both drug classes have been associated with polymorphisms in the genes for 5-HT receptor subtypes, with most attention focused on 5-HT2A and 5-HT2C receptors [76, 77, 81, 82].
5-HT2A receptors may be involved in the response to atypical antipsychotics and to 5-HT uptake inhibitors. The T102C SNP in the 5-HT2A receptor gene has been associated with altered drug responses: the C allele associating with a reduced response to antagonists and the homozygous T allele with reduced response to agonists [76, 82]. Multiple polymorphisms, including a -1027(GT) repeat in the promoter and a Cys23Ser SNP in the coding region, exist in the gene for the 5-HT2C receptor and are in linkage disequilibrium [81]. Although all studies are not consistent, some of these polymorphisms, and certain haplotypes thereof, appear to be associated with differences in the therapeutic response to atypical antipsychotics and in the production of side effects such as tardive dyskinesia and weight gain [81, 83].
Conclusions and Perspective
Given their large number, widespread expression and roles in the regulation of virtually every organ system throughout the body, GPCR are physiologically important in maintaining homeostasis, in particular via their ability to mediate responses to circulating hormones and neurotransmitter input in the central, peripheral and autonomic nervous systems. The cloning and characterization of GPCR and of components involved in mediating receptor responses and in regulating receptor expression has provided a number of new insights, only some of which have been exploited in clinical medicine. For example, the physiologic agonists and functional role for a large number of orphan GPCR remain unknown, although such orphan receptors may prove to be as important—perhaps more so—than GPCR currently emphasized in physiologic and pharmacologic studies and in drug development [84–86]. Newly identified receptors (initially “orphans”) in the cloning era have already yielded pharmacologically useful drugs, for example CaSR agonists (calcimimetics) and antagonists (calcilytics) as therapies for a variety of disorders [87].
A number of monogenic diseases of GPCR have been defined, most thoroughly retinitis pigmentosa and NDI (Figs 1 and 2), and these have provided useful insights regarding the roles of particular residues and in the disease-causing potential of GPCR mutations. Other disorders that involve antibodies to GPCR are more common and have had a larger overall impact on clinical medicine.
In spite of an ever-expanding identification of genetic variants of GPCR, in particular SNPs, many studies on associations of GPCR polymorphisms with disease or drug responses are difficult to interpret because the specific consequences of a particular polymorphisms have not been unequivocally established at the molecular and cellular level and because most clinical studies are statistically underpowered to allow robust conclusions. Perhaps polymorphisms have stronger effects on therapeutic and adverse effects of their ligands than on disease because any given GPCR plays a larger role for specific drugs acting on a GPCR than for a complex disease with multiple genetic and non-genetic determinants.
GPCR polymorphisms have yielded numerous interesting mechanistic results, in particular by identifying residues not necessarily implicated as functionally important prior to their identification in sequencing studies of polymorphic receptors. To date, however, most associations are too weak to allow clinically meaningful predictions of GPCR variants and their relationship to disease onset or progression or in drug responses (the results with Arg389Gly SNP in the β1-adrenergic receptor being an exception [60–64]). Additional data are clearly needed.
GPCR will continue to be highly important in clinical medicine because of their large number, wide expression and role in physiologically important responses. We speculate that future discoveries will reveal new GPCR drugs, in part because it is relatively easy to screen for pharmacologic agents that access these receptors and stimulate or block receptor-mediated biochemical or physiological responses. In addition, further insights into GPCR biology may reveal novel, unexpected therapeutic targets that influence the GPCR life cycle or “ligand directed signaling”. The existence of a large number of orphan GPCRs provides a treasure trove of possibilities. Our bias is that the full flowering of genetic studies of GPCR variants and their clinical role has yet to be realized. Thus, we would argue, “let the future begin!”
Footnotes
Work from Dr. Insel’s laboratory is supported by grants from NIH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kirstein SL, Insel PA. Autonomic nervous system pharmacogenomics: a progress report. Pharmacol Rev. 2004;56:31–52. doi: 10.1124/pr.56.1.2. [DOI] [PubMed] [Google Scholar]
- 2.Thompson MD, Burnham WM, Cole DE. The G protein-coupled receptors: pharmacogenetics and disease. Crit Rev Clin Lab Sci. 2005;42:311–92. doi: 10.1080/10408360591001895. [DOI] [PubMed] [Google Scholar]
- 3.Tang CM, Insel PA. Genetic variation in G-protein-coupled receptors--consequences for G-protein-coupled receptors as drug targets. Expert Opin Ther Targets. 2005;9:1247–65. doi: 10.1517/14728222.9.6.1247. [DOI] [PubMed] [Google Scholar]
- 4.Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications. Pharmacol Ther. 2006 doi: 10.1016/j.pharmthera.2006.02.008. in press. [DOI] [PubMed] [Google Scholar]
- 5.Spiegel AM, Weinstein LS. Inherited diseases involving G proteins and G protein-coupled receptors. Annu Rev Med. 2004;55:27–39. doi: 10.1146/annurev.med.55.091902.103843. [DOI] [PubMed] [Google Scholar]
- 6.Schoneberg T, Schulz A, Biebermann H, Hermsdorf T, Rompler H, Sangkuhl K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104:173–206. doi: 10.1016/j.pharmthera.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 7.N.R.D.D.G.Q. Participants. The state of GPCR research in 2004. Nat Rev Drug Discov. 2004;3575:577–626. doi: 10.1038/nrd1458. [DOI] [PubMed] [Google Scholar]
- 8.Liebmann C. G protein-coupled receptors and their signaling pathways: classical therapeutical targets susceptible to novel therapeutic concepts. Curr Pharm Des. 2004;10:1937–58. doi: 10.2174/1381612043384367. [DOI] [PubMed] [Google Scholar]
- 9.Rana BK, Insel PA. Receptor databases and computational websites for ligand binding. Methods Mol Biol. 2005;306:1–15. doi: 10.1385/1-59259-927-3:001. [DOI] [PubMed] [Google Scholar]
- 10.Fredriksson R, Schioth HB. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol. 2005;67:1414–25. doi: 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- 11.Fain GL. Why photoreceptors die (and why they don’t) Bioessays. 2006;28:344–54. doi: 10.1002/bies.20382. [DOI] [PubMed] [Google Scholar]
- 12.Sands JM, Bichet DG. Nephrogenic diabetes insipidus. Ann Intern Med. 2006;144:186–94. doi: 10.7326/0003-4819-144-3-200602070-00007. [DOI] [PubMed] [Google Scholar]
- 13.Wuller S, Wiesner B, Loffler A, Furkert J, Krause G, Hermosilla R, Schaefer M, Schulein R, Rosenthal W, Oksche A. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279:47254–63. doi: 10.1074/jbc.M408154200. [DOI] [PubMed] [Google Scholar]
- 14.Tfelt-Hansen J, Brown EM. The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit Rev Clin Lab Sci. 2005;42:35–70. doi: 10.1080/10408360590886606. [DOI] [PubMed] [Google Scholar]
- 15.Thakker RV. Diseases associated with the extracellular calcium-sensing receptor. Cell Calcium. 2004;35:275–82. doi: 10.1016/j.ceca.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Schott M, Scherbaum WA, Morgenthaler NG. Thyrotropin receptor autoantibodies in Graves’ disease. Trends Endocrinol Metab. 2005;16:243–8. doi: 10.1016/j.tem.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Davies TF, Ando T, Lin RY, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J Clin Invest. 2005;115:1972–83. doi: 10.1172/JCI26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kierszenbaum F. Where do we stand on the autoimmunity hypothesis of Chagas disease? Trends Parasitol. 2005;21:513–6. doi: 10.1016/j.pt.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Wallukat G, Nissen E, Morwinski R, Muller J. Autoantibodies against the beta- and muscarinic receptors in cardiomyopathy. Herz. 2000;25:261–6. doi: 10.1007/s000590050017. [DOI] [PubMed] [Google Scholar]
- 20.Jahns R, Boivin V, Lohse MJ. beta(1)-Adrenergic receptor function, autoimmunity, and pathogenesis of dilated cardiomyopathy. Trends Cardiovasc Med. 2006;16:20–4. doi: 10.1016/j.tcm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Pearce SH, Leech NJ. Toward precise forecasting of autoimmune endocrinopathy. J Clin Endocrinol Metab. 2004;89:544–7. doi: 10.1210/jc.2003-032142. [DOI] [PubMed] [Google Scholar]
- 22.Freedman NJ, Lefkowitz RJ. Anti-beta(1)-adrenergic receptor antibodies and heart failure: causation, not just correlation. J Clin Invest. 2004;113:1379–82. doi: 10.1172/JCI21748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forges T, Monnier-Barbarino P, Faure GC, Bene MC. Autoimmunity and antigenic targets in ovarian pathology. Hum Reprod Update. 2004;10:163–75. doi: 10.1093/humupd/dmh014. [DOI] [PubMed] [Google Scholar]
- 24.Hoebeke J. Molecular mechanisms of anti-G-protein-coupled receptor autoantibodies. Autoimmunity. 2001;34:161–4. doi: 10.3109/08916930109007379. [DOI] [PubMed] [Google Scholar]
- 25.Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC, Bergmann JE, Gaitanaris GA. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A. 2003;100:4903–8. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci U S A. 2000;97:10483–8. doi: 10.1073/pnas.97.19.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Israel E, Chinchilli VM, Ford JG, Boushey HA, Cherniack R, Craig TJ, Deykin A, Fagan JK, Fahy JV, Fish J, Kraft M, Kunselman SJ, Lazarus SC, Lemanske RF, Jr, Liggett SB, Martin RJ, Mitra N, Peters SP, Silverman E, Sorkness CA, Szefler SJ, Wechsler ME, Weiss ST, Drazen JM. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet. 2004;364:1505–12. doi: 10.1016/S0140-6736(04)17273-5. [DOI] [PubMed] [Google Scholar]
- 28.Wechsler ME, Lehman E, Lazarus SC, Lemanske RF, Jr, Boushey HA, Deykin A, Fahy JV, Sorkness CA, Chinchilli VM, Craig TJ, DiMango E, Kraft M, Leone F, Martin RJ, Peters SP, Szefler SJ, Liu W, Israel E. beta-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med. 2006;173:519–26. doi: 10.1164/rccm.200509-1519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tattersfield AE, Harrison TW. beta-Adrenoceptor polymorphisms: focus moves to long-acting beta-agonists. Am J Respir Crit Care Med. 2006;173:473–4. doi: 10.1164/rccm.2512005. [DOI] [PubMed] [Google Scholar]
- 30.Hall IP, Blakey JD, Al Balushi KA, Wheatley A, Sayers I, Pembrey ME, Ring SM, McArdle WL, Strachan DP. Beta2-adrenoceptor polymorphisms and asthma from childhood to middle age in the British 1958 birth cohort: a genetic association study. Lancet. 2006;368:771–9. doi: 10.1016/S0140-6736(06)69287-8. [DOI] [PubMed] [Google Scholar]
- 31.Reinscheid RK, Xu YL, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O. Pharmacological characterization of human and murine neuropeptide s receptor variants. J Pharmacol Exp Ther. 2005;315:1338–45. doi: 10.1124/jpet.105.093427. [DOI] [PubMed] [Google Scholar]
- 32.Melen E, Bruce S, Doekes G, Kabesch M, Laitinen T, Lauener R, Lindgren CM, Riedler J, Scheynius A, van Hage-Hamsten M, Kere J, Pershagen G, Wickman M, Nyberg F. Haplotypes of G protein-coupled receptor 154 are associated with childhood allergy and asthma. Am J Respir Crit Care Med. 2005;171:1089–95. doi: 10.1164/rccm.200410-1317OC. [DOI] [PubMed] [Google Scholar]
- 33.Galvani AP, Novembre J. The evolutionary history of the CCR5-Delta32 HIV-resistance mutation. Microbes Infect. 2005;7:302–9. doi: 10.1016/j.micinf.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 34.McDermott DH, Zimmerman PA, Guignard F, Kleeberger CA, Leitman SF, Murphy PM. CCR5 promoter polymorphism and HIV-1 disease progression. Multicenter AIDS Cohort Study (MACS) Lancet. 1998;352:866–70. doi: 10.1016/s0140-6736(98)04158-0. [DOI] [PubMed] [Google Scholar]
- 35.Zheng ZM. Regulation of alternative RNA splicing by exon definition and exon sequences in viral and mammalian gene expression. J Biomed Sci. 2004;11:278–94. doi: 10.1159/000077096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park PS, Filipek S, Wells JW, Palczewski K. Oligomerization of G protein-coupled receptors: past, present, and future. Biochemistry. 2004;43:15643–56. doi: 10.1021/bi047907k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Internationl HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montpetit A, Nelis M, Laflamme P, Magi R, Ke X, Remm M, Cardon L, Hudson TJ, Metspalu A. An evaluation of the performance of tag SNPs derived from HapMap in a Caucasian population. PLoS Genet. 2006;2:e27. doi: 10.1371/journal.pgen.0020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, Mustonen J. Angiotensin-receptor-blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. New England Journal of Medicine. 2004;351:1952. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]
- 40.McConnell JD, Roehrborn CG, Bautista O, Andriole GL, Dixon CM, Kusek JW, Lepor H, McVary KT, Nyberg LM, Clarke HS, Crawford ED, Diokno AC, Foley JP, Foster HE, Jacobs SC, Kaplan SA, Kreder KJ, Lieber MM, Lucia MS, Miller GJ, Menon M, Milam DF, Ramsdell JW, Schenkman NS, Slawin KM, Smith JA. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. New England Journal of Medicine. 2003;349:2387. doi: 10.1056/NEJMoa030656. [DOI] [PubMed] [Google Scholar]
- 41.Squire IB, Barnett DB. The rational use of beta-adrenoceptor blockers in the treatment of heart failure. The changing face of an old therapy. British Journal of Clinical Pharmacology. 2000;49:1. doi: 10.1046/j.1365-2125.2000.00112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ram FSF, Sestini P. Regular inhaled short acting · 2 agonists for the management of stable chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. Thorax. 2003;58:580. doi: 10.1136/thorax.58.7.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seeman P, Niznik HB. Dopamine receptors and transporters in Parkinson’s diasease and schizophrenia. FASEB Journal. 1990;4:2737. doi: 10.1096/fasebj.4.10.2197154. [DOI] [PubMed] [Google Scholar]
- 44.Sato S, Ohtake A, Matsushima H, Saitoh C, Usuda S, Miyata K. Pharmacological effect of tamsulosin in relation to dog plasma and tissue concentrations: prostatic and urethral retention possibly contributes to uroselectivity of tamsulosin. Journal of Pharmacology and Experimental Therapeutics. 2001;296:697. [PubMed] [Google Scholar]
- 45.Hague C, Lee SE, Chen Z, Prinster SC, Hall RA, Minneman KP. Heterodimers of alpha1B - and alpha1D -adrenergic receptors form a single functional entity. Molecular Pharmacology. 2006;69:45. doi: 10.1124/mol.105.014985. [DOI] [PubMed] [Google Scholar]
- 46.Ramsay D, Carr IC, Pediani J, Lopez-Gimenez JF, Thurlow R, Fidock M, Milligan G. High-affinity interactions between human alpha1A -adrenoceptor C-terminal splice variants produce homo- and heterodimers but do not generate the ‡ 1L -adrenoceptor. Molecular Pharmacology. 2004;66:228. doi: 10.1124/mol.66.2.228. [DOI] [PubMed] [Google Scholar]
- 47.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2004;369:23. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 48.Gerloff T. Impact of genetic polymorphisms in transmembrane carrier-systems on drug and xenobiotic distribution. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2004;369:69. doi: 10.1007/s00210-003-0813-5. [DOI] [PubMed] [Google Scholar]
- 49.Keffel S, Alexandrov A, Goepel M, Michel MC. Alpha1 -Adrenoceptor subtypes differentially couple to growth promotion and inhibition in Chinese hamster ovary cells. Biochemical and Biophysical Research Communications. 2000;272:906. doi: 10.1006/bbrc.2000.2850. [DOI] [PubMed] [Google Scholar]
- 50.Alexandrov A, Keffel S, Goepel M, Michel MC. Stimulation of alpha1A -adrenoceptors in Rat-1 cells inhibits extracellular signal-regulated kinase by activating p38 mitogen-activated protein kinase. Molecular Pharmacology. 1998;54:755. doi: 10.1124/mol.54.5.755. [DOI] [PubMed] [Google Scholar]
- 51.Hahntow IN, Koopmans RP, Michel MC. The beta2 -adrenoceptor gene and hypertension: is it the promoter or the coding region or neither? Journal of Hypertension. 2006;24:1003. doi: 10.1097/01.hjh.0000226185.06063.80. [DOI] [PubMed] [Google Scholar]
- 52.Pauwels PJ, Rauly I, Wurch T. Dissimilar pharmacological responses by a new series of imidazoline derivatives at precoupled and ligand-activated alpha2A -adrenoceptor states: evidence for effector pathway-dependent differential antagonism. Journal of Pharmacology and Experimental Therapeutics. 2003;305:1015. doi: 10.1124/jpet.102.048215. [DOI] [PubMed] [Google Scholar]
- 53.Isogaya M, Nagao T, Kurose H. Enhanced cAMP response of naturally occurring mutant of human beta3 -adrenergic receptor. Japanese Journal of Pharmacology. 2002;88:314. doi: 10.1254/jjp.88.314. [DOI] [PubMed] [Google Scholar]
- 54.Hoffstedt J, Poirier O, Thîrne A, Lînnqvist F, Herrmann SM, Cambien F, Arner P. Polymorphism of the human beta3 -adrenoceptor gene forms a well-conserved haplotype that is associated with moderate obesity and altered receptor function. Diabetes. 1999;48:203. doi: 10.2337/diabetes.48.1.203. [DOI] [PubMed] [Google Scholar]
- 55.Candelore MR, Deng L, Tota LM, Kelly LJ, Cascieri MA, Strader CD. Pharmacological characterization of a recently described human beta3 -adrenergic receptor mutant. Endocrinology. 1996;137:5638. doi: 10.1210/endo.137.6.8641219. [DOI] [PubMed] [Google Scholar]
- 56.Brodde OE, Leineweber K. Beta2-adrenoceptor gene polymorphisms. Pharmacogenet Genomics. 2005;15:267–75. doi: 10.1097/01213011-200505000-00001. [DOI] [PubMed] [Google Scholar]
- 57.Kurland L, Melhus H, Karlsson J, Kahan T, Malmqvist K, ôhman P, Nystrîm F, HÑgg A, Lind L. Polymorphisms in the angiotensinogen and angiotensin II type 1 receptor gene are related to change in left ventricular mass during antihypertensive treatment: results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation versus Atenolol (SILVHIA) trial. Journal of Hypertension. 2002;20:657. doi: 10.1097/00004872-200204000-00023. [DOI] [PubMed] [Google Scholar]
- 58.Miller JA, Thai K, Scholey JW. Angiotensin II type 1 receptor gene polymorphism predicts response to losartan and angiotensin II. Kidney Int. 1999;56:2173–80. doi: 10.1046/j.1523-1755.1999.00770.x. [DOI] [PubMed] [Google Scholar]
- 59.Benetos A, Cambien F, Gautier S, Ricard S, Safar M, Laurent S, Lacolley P, Poirier O, Topouchian J, Asmar R. Influence of the angiotensin II type 1 receptor gene polymorphism on the effect of perindopril and nitrendipine on arterial stiffness in hypertensive individuals. Hypertension. 1996;28:1081. doi: 10.1161/01.hyp.28.6.1081. [DOI] [PubMed] [Google Scholar]
- 60.Liljedahl U, Kahan T, Malmqvist K, Melhus H, SyvÑnen AC, Lind L, Kurland L. Single nucleotide polymorphisms predict the change in left ventricular mass in response to antihypertensive treatment. Journal of Hypertension. 2004;22:2321. doi: 10.1097/00004872-200412000-00014. [DOI] [PubMed] [Google Scholar]
- 61.Kurland L, Melhus H, Karlsson J, Kahan T, Malmqvist K, ôhman KP, Nystrîm F, HÑgg A, Lind L. Angiotensin converting enzyme gene polymorphism predicts blood pressure response to angiotensin II receptor type 1 antagonist treatment in hypertensive patients. Journal of Hypertension. 2001;19:1783. doi: 10.1097/00004872-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 62.Hingorani AD, Jia H, Stevens PA, Hopper R, Dickerson JE, Brown MJ. Renin-angiotensin system gene polymorphisms influence blood pressure and the response to angiotensin converting enzyme inhibition. Journal of Hypertension. 1995;13:1602. [PubMed] [Google Scholar]
- 63.Coto E, Marin R, Alvarez V, Praga M, Fernandez Andrade C, Arias M, Poveda R, Valles M, Galceran JM, Luno J, Rivera F, Campistol JM. Pharmacogenetics of angiotensin system in non diabetic nephropathy. Nefrologia. 2005;25:381. [PubMed] [Google Scholar]
- 64.Small KM, McGraw DW, Liggett SB. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu Rev Pharmacol Toxicol. 2003;43:381–411. doi: 10.1146/annurev.pharmtox.43.100901.135823. [DOI] [PubMed] [Google Scholar]
- 65.Lei B, Morris DP, Smith MP, Svetkey LP, Newman MF, Rotter JI, Buchanan TA, Beckstrom-Sternberg SM, Green ED, Schwinn DA. Novel human alpha1a -adrenoceptor single nucleotide polymorphisms alter receptor pharmacology and biological function. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2005;371:229. doi: 10.1007/s00210-005-1019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mochtar CA, Laan W, van Houwelingen KP, Franke B, de la Rosette JJMCH, Schalken JA, Kiemeney LALM. Polymorphisms in the alpha1A -adrenoceptor gene do not modify the short- and long-term efficacy of alpha 1 -adrenoceptor antagonists in the treatment of benign prostatic hyperplasia. BJU International. 2006;97:852. doi: 10.1111/j.1464-410X.2006.05998.x. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz SG, Puckett BJ, Allen RC, Castillo IG, Leffler CT. Beta1 -Adrenergic receptor polymorphism and clinical efficacy of betaxolol hydrochloride in normal volunteers. Ophthalmology. 2005;112:2131. doi: 10.1016/j.ophtha.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 68.Sofowora GG, Dishy V, Muszkat M, Xie HG, Kim RB, Harris PA, Prasad HC, Byrne DW, Nair UB, Wood AJ, Stein CM. A common beta1-adrenergic receptor polymorphism (Arg389Gly) affects blood pressure response to beta-blockade. Clin Pharmacol Ther. 2003;73:366–71. doi: 10.1016/s0009-9236(02)17734-4. [DOI] [PubMed] [Google Scholar]
- 69.Liu J, Liu ZQ, Tan ZR, Chen XP, Wang LS, Zhou G, Zhou HH. Gly389Arg polymorphism of beta1 -adrenergic receptor is associated with the carviovascular response to metoprolol. Clinical Pharmacology and Therapeutics. 2003;74:372. doi: 10.1016/S0009-9236(03)00224-8. [DOI] [PubMed] [Google Scholar]
- 70.Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF. beta1 -Adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clinical Pharmacology and Therapeutics. 2003;74:44. doi: 10.1016/S0009-9236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 71.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, Weber SA, Greene SM, Hodne D, Nelson B, Morrison J, Domanski MJ, Wagoner LE, Abraham WT, Anderson JL, Carlquist JF, Krause-Steinrauf HJ, Lazzeroni LC, Port JD, Lavori PW, Bristow MR. A polymorphism within a conserved {beta}1-adrenergic receptor motif alters cardiac function and {beta}-blocker response in human heart failure. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0509937103. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leineweber K, BÅscher R, Bruck H, Brodde OE. ·-Adrenoceptor polymorphisms. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2004;369:1. doi: 10.1007/s00210-003-0824-2. [DOI] [PubMed] [Google Scholar]
- 73.Magnusson Y, Levin MC, Eggertsen R, Nystrîm E, Mobini R, Schaufelberger M, Andersson B. Ser49Gly of beta1 -adrenergic receptor is associated with effective ·-blocker dose in dilated cardiomyopathy. Clinical Pharmacology and Therapeutics. 2005;78:221. doi: 10.1016/j.clpt.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 74.Rana BK, Insel PA, Payne S, Abel K, Beutler E, Ziegler MG, Schork NJ, O’Connor DT. A large population-based sample reveals the contribution of gene-transfer interactions in blood pressure. Hypertension. 2006 doi: 10.1161/01.HYP.0000252029.35106.67. in revision. [DOI] [PubMed] [Google Scholar]
- 75.Scharfetter J. Pharmacogenetics of dopamine receptors and response to antipsychotic drugs in schizophrenia - an update. Pharmacogenomics. 2004;5:691. doi: 10.1517/14622416.5.6.691. [DOI] [PubMed] [Google Scholar]
- 76.Malhotra AK, Murphy GM, Jr, Kennedy JL. Pharmacogenetics of psychotropic drug response. AmJPsych. 2004;161:780. doi: 10.1176/appi.ajp.161.5.780. [DOI] [PubMed] [Google Scholar]
- 77.Mancama D, Arranz MJ, Kerwin RW. Genetic predictors of therapeutic response to clozapine. Current status of research. CNS Drugs. 2002;16:317. doi: 10.2165/00023210-200216050-00004. [DOI] [PubMed] [Google Scholar]
- 78.Lencz T, Robinson DG, Xu K, Ekholm J, Sevy S, Gunduz-Bruce H, Woerner MG, Kane JM, Goldman D, Malhotra AK. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. AmJPsych. 2006;163:529. doi: 10.1176/appi.ajp.163.3.529. [DOI] [PubMed] [Google Scholar]
- 79.Young R, Lawford BR, Barnes M, Burton SC, Ritchie T, Ward WK, Noble EP. Prolactin levels in antipsychotic treatment of patients with schizophrenia carrying the DRD2*A1 allele. BrJPsych. 2004;185:147. doi: 10.1192/bjp.185.2.147. [DOI] [PubMed] [Google Scholar]
- 80.Lane HY, Hsu SK, Liu YC, Chang YC, Huang Ch, Chang WH. Dopamine D 3 receptor Ser9Gly polymorphism and risperidone response. JClinPsychopharmacol. 2005;25:6. doi: 10.1097/01.jcp.0000150226.84371.76. [DOI] [PubMed] [Google Scholar]
- 81.Reynolds GP, Templeman LA, Zhang ZJ. The role of 5-HT2C receptor polymorphisms in the pharmacogenetics of antipsychotic drug treatment. ProgrNeuropsychopharmacolBiolPsychiatry. 2005;29:1021. doi: 10.1016/j.pnpbp.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 82.Serretti A, Artioli P. From molecular biology to pharmacogenetics: a review of the literature on antidepressant treatment and suggestions of possible candidate genes. Psychopharmacology. 2004;174:490. doi: 10.1007/s00213-004-1822-x. [DOI] [PubMed] [Google Scholar]
- 83.Ellingrod VL, Perry PJ, Ringold JC, Lund BC, Bever-Stille K, Fleming F, Holman TL, Miller D. Weight gain associated with the -759C/T polymorphism of the 5HT2C receptor and olanzapine. AmJMedGen. 2005;134B:76. doi: 10.1002/ajmg.b.20169. [DOI] [PubMed] [Google Scholar]
- 84.Civelli O. GPCR deorphanizations: the novel, the known and the unexpected transmitters. Trends Pharmacol Sci. 2005;26:15–9. doi: 10.1016/j.tips.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 85.Thomsen W, Leonard J, Behan DP. Orphan GPCR target validation. Curr Opin Mol Ther. 2004;6:640–56. [PubMed] [Google Scholar]
- 86.Tang CM, Insel PA. GPCR expression in the heart; “new” receptors in myocytes and fibroblasts. Trends Cardiovasc Med. 2004;14:94–9. doi: 10.1016/j.tcm.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 87.Hebert SC. Therapeutic use of calcimimetics. Annu Rev Med. 2006;57:349–64. doi: 10.1146/annurev.med.57.121304.131328. [DOI] [PubMed] [Google Scholar]
- 88.Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Teng Y, Wang W, Tian H, Wang H, Hu X, Chen Y, Bittles AH. Autosomal dominant retinitis pigmentosa in a five-generation pedigree in People’s Republic of China. Eye. 2003;17:1036–9. doi: 10.1038/sj.eye.6700499. [DOI] [PubMed] [Google Scholar]
- 90.Mittelman SD, Hendy GN, Fefferman RA, Canaff L, Mosesova I, Cole DE, Burkett L, Geffner ME. A hypocalcemic child with a novel activating mutation of the calcium-sensing receptor gene: successful treatment with recombinant human parathyroid hormone. J Clin Endocrinol Metab. 2006;91:2474–9. doi: 10.1210/jc.2005-2605. [DOI] [PubMed] [Google Scholar]
- 91.Diaz GA, Gulino AV. WHIM syndrome: a defect in CXCR4 signaling. Curr Allergy Asthma Rep. 2005;5:350–5. doi: 10.1007/s11882-005-0005-0. [DOI] [PubMed] [Google Scholar]
- 92.Diaz GA. CXCR4 mutations in WHIM syndrome: a misguided immune system? Immunol Rev. 2005;203:235–43. doi: 10.1111/j.0105-2896.2005.00226.x. [DOI] [PubMed] [Google Scholar]
- 93.Puri P, Shinkai T. Pathogenesis of Hirschsprung’s disease and its variants: recent progress. Semin Pediatr Surg. 2004;13:18–24. doi: 10.1053/j.sempedsurg.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 94.Huhtaniemi I. Mutations along the pituitary-gonadal axis affecting sexual maturation: Novel information from transgenic and knockout mice. Mol Cell Endocrinol. 2006 doi: 10.1016/j.mce.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 95.Nanamori M, He R, Sang H, Ye RD. Normal cell surface expression and selective loss of functions resulting from Phe110 to Ser and Cys126 to Trp substitutions in the formyl peptide receptor. Immunol Invest. 2004;33:193–212. doi: 10.1081/imm-120034234. [DOI] [PubMed] [Google Scholar]
- 96.Qin M, Hayashi H, Oshima K, Tahira T, Hayashi K, Kondo H. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathy with mutations in the LRP5 and/or FZD4 genes. Hum Mutat. 2005;26:104–12. doi: 10.1002/humu.20191. [DOI] [PubMed] [Google Scholar]
- 97.MacDonald ML, Goldberg YP, Macfarlane J, Samuels ME, Trese MT, Shastry BS. Genetic variants of frizzled-4 gene in familial exudative vitreoretinopathy and advanced retinopathy of prematurity. Clin Genet. 2005;67:363–6. doi: 10.1111/j.1399-0004.2005.00408.x. [DOI] [PubMed] [Google Scholar]
- 98.Dungan HM, Clifton DK, Steiner RA. Minireview: kisspeptin neurons as central processors in the regulation of gonadotropin-releasing hormone secretion. Endocrinology. 2006;147:1154–8. doi: 10.1210/en.2005-1282. [DOI] [PubMed] [Google Scholar]
- 99.Lanfranco F, Gromoll J, von Eckardstein S, Herding EM, Nieschlag E, Simoni M. Role of sequence variations of the GnRH receptor and G protein-coupled receptor 54 gene in male idiopathic hypogonadotropic hypogonadism. Eur J Endocrinol. 2005;153:845–52. doi: 10.1530/eje.1.02031. [DOI] [PubMed] [Google Scholar]
- 100.Piao X, Chang BS, Bodell A, Woods K, Benzeev B, Topcu M, Guerrini R, Goldberg-Stern H, Sztriha L, Dobyns WB, Barkovich AJ, Walsh CA. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol. 2005;58:680–7. doi: 10.1002/ana.20616. [DOI] [PubMed] [Google Scholar]
- 101.Jansen A, Andermann E. Genetics of the polymicrogyria syndromes. J Med Genet. 2005;42:369–78. doi: 10.1136/jmg.2004.023952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schulz TF. The pleiotropic effects of Kaposi’s sarcoma herpesvirus. J Pathol. 2006;208:187–98. doi: 10.1002/path.1904. [DOI] [PubMed] [Google Scholar]
- 103.Geilen CC, Husak R, Steinhoff M. Pathogenesis and clinical manifestation of Kaposi’s sarcoma. Front Radiat Ther Oncol. 2006;39:59–67. doi: 10.1159/000090804. [DOI] [PubMed] [Google Scholar]
- 104.Bathgate RA, Ivell R, Sanborn BM, Sherwood OD, Summers RJ. International Union of Pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol Rev. 2006;58:7–31. doi: 10.1124/pr.58.1.9. [DOI] [PubMed] [Google Scholar]
- 105.Kaleva M, Toppari J. Cryptorchidism: an indicator of testicular dysgenesis? Cell Tissue Res. 2005;322:167–72. doi: 10.1007/s00441-005-1143-3. [DOI] [PubMed] [Google Scholar]
- 106.Bathgate RA, Ivell R, Sanborn BM, Sherwood OD, Summers RJ. Receptors for relaxin family peptides. Ann N Y Acad Sci. 2005;1041:61–76. doi: 10.1196/annals.1282.010. [DOI] [PubMed] [Google Scholar]
- 107.Deprez L, Claes LR, Claeys KG, Audenaert D, Van Dyck T, Goossens D, Van Paesschen W, Del-Favero J, Van Broeckhoven C, De Jonghe P. Genome-wide linkage of febrile seizures and epilepsy to the FEB4 locus at 5q14.3-q23.1 and no MASS1 mutation. Hum Genet. 2006;118:618–25. doi: 10.1007/s00439-005-0077-x. [DOI] [PubMed] [Google Scholar]
- 108.Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74:357–66. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Robinson R, Gardiner M. Molecular basis of Mendelian idiopathic epilepsies. Ann Med. 2004;36:89–97. doi: 10.1080/07853890310019952. [DOI] [PubMed] [Google Scholar]
- 110.Reiners J, van Wijk E, Marker T, Zimmermann U, Jurgens K, te Brinke H, Overlack N, Roepman R, Knipper M, Kremer H, Wolfrum U. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum Mol Genet. 2005;14:3933–43. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- 111.MacKenzie RG. Obesity-associated mutations in the human melanocortin-4 receptor gene. Peptides. 2006;27:395–403. doi: 10.1016/j.peptides.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 112.Govaerts C, Srinivasan S, Shapiro A, Zhang S, Picard F, Clement K, Lubrano-Berthelier C, Vaisse C. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides. 2005;26:1909–19. doi: 10.1016/j.peptides.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 113.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–67. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–85. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 115.Kennan A, Aherne A, Humphries P. Light in retinitis pigmentosa. Trends Genet. 2005;21:103–10. doi: 10.1016/j.tig.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 116.Bichet DG. Nephrogenic diabetes insipidus. Adv Chronic Kidney Dis. 2006;13:96–104. doi: 10.1053/j.ackd.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 117.Sangkuhl K, Rompler H, Busch W, Karges B, Schoneberg T. Nephrogenic diabetes insipidus caused by mutation of Tyr205: a key residue of V2 vasopressin receptor function. Hum Mutat. 2005;25:505. doi: 10.1002/humu.9337. [DOI] [PubMed] [Google Scholar]
- 118.Litonjua AA. The significance of beta2-adrenergic receptor polymorphisms in asthma. Curr Opin Pulm Med. 2006;12:12–7. doi: 10.1097/01.mcp.0000198068.50457.95. [DOI] [PubMed] [Google Scholar]
- 119.Johnson JA, Turner ST. Hypertension pharmacogenomics: current status and future directions. Curr Opin Mol Ther. 2005;7:218–25. [PubMed] [Google Scholar]
- 120.Sawa M, Harada H. Recent developments in the design of orally bioavailable beta3-adrenergic receptor agonists. Curr Med Chem. 2006;13:25–37. [PubMed] [Google Scholar]
- 121.Puissant B, Roubinet F, Massip P, Sandres-Saune K, Apoil PA, Abbal M, Pasquier C, Izopet J, Blancher A. Analysis of CCR5, CCR2, CX3CR1, and SDF1 polymorphisms in HIV-positive treated patients: impact on response to HAART and on peripheral T lymphocyte counts. AIDS Res Hum Retroviruses. 2006;22:153–62. doi: 10.1089/aid.2006.22.153. [DOI] [PubMed] [Google Scholar]
- 122.Becker Y. The Molecular Mechanism of Human Resistance to HIV-1 Infectionin Persistently Infected Individuals-A Review, Hypothesis and Implications. Virus Genes. 2005;31:113–9. doi: 10.1007/s11262-005-2503-5. [DOI] [PubMed] [Google Scholar]
- 123.Bonci A, Hopf FW. The dopamine D2 receptor: new surprises from an old friend. Neuron. 2005;47:335–8. doi: 10.1016/j.neuron.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 124.Sokoloff P, Diaz J, Le Foll B, Guillin O, Leriche L, Bezard E, Gross C. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol Disord Drug Targets. 2006;5:25–43. doi: 10.2174/187152706784111551. [DOI] [PubMed] [Google Scholar]
- 125.Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res. 2006;7:73. doi: 10.1186/1465-9921-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feng Y, Hong X, Wang L, Jiang S, Chen C, Wang B, Yang J, Fang Z, Zang T, Xu X, Xu X. G protein-coupled receptor 154 gene polymorphism is associated with airway hyperresponsiveness to methacholine in a Chinese population. J Allergy Clin Immunol. 2006;117:612–7. doi: 10.1016/j.jaci.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 127.Murugappa S, Kunapuli SP. The role of ADP receptors in platelet function. Front Biosci. 2006;11:1977–86. doi: 10.2741/1939. [DOI] [PubMed] [Google Scholar]
- 128.Johnson JA, Lima JJ. Drug receptor/effector polymorphisms and pharmacogenetics: current status and challenges. Pharmacogenetics. 2003;13:525–34. doi: 10.1097/00008571-200309000-00001. [DOI] [PubMed] [Google Scholar]