

Abstract
Background
We have recently demonstrated that mice deficient in TLR4 or its adapter molecule MyD88 have increased signs of colitis compared to wild-type (WT) mice following dextran sodium sulfate (DSS)-induced injury. We wished to test the hypothesis that Cox-2 derived PGE2 is important in TLR4-related mucosal repair.
Methods
Cox-2 expression was analyzed by real-time PCR, immunohistochemistry, western blotting, and luciferase reporter constructs. siRNA was used to inhibit expression of MyD88. TLR4−/− or WT mice were given 2.5% DSS for 7 days. Proliferation and apoptosis were assessed using BrdU staining and TUNEL assays, respectively. PGE2 was given orally to DSS-treated mice.
Results
Intestinal epithelial cell lines upregulated Cox-2 expression in a TLR4- and MyD88-dependent fashion. LPS-mediated stimulation of PGE2 production was blocked by a selective Cox-2 inhibitor or siRNA against MyD88. Following DSS injury, Cox-2 expression increased only in WT mice. TLR4−/− mice have significantly reduced proliferation and increased apoptosis following DSS injury compared to WT mice. PGE2 supplementation of TLR4−/− mice resulted in improvement in clinical signs of colitis and restoration of proliferation and apoptosis to wild-type values. The mechanism for improved epithelial repair may be through PGE2-dependent activation of the epidermal growth factor receptor.
Conclusion
We describe an important link between TLR4 signaling and Cox-2 expression in the gut. TLR4 and MyD88 signaling are required for optimal proliferation and protection against apoptosis in the injured intestine. Although TLR4 signaling is beneficial in the short-term, chronic signaling through TLR4 may lower the threshold for colitis-associated cancer.
Introduction
The intestinal mucosa coexists with a high density of luminal bacteria and pathogen-associated molecular patterns (PAMPs). Indeed, the genetic program of the epithelium is shaped by the presence of bacteria. Compared with germ-free animals, colonization with a single species of gut commensal, Bacteroides thetaiotaomicron, results in the expression of genes that enhance barrier fortification and nutrient transport1. Early and now more recent studies demonstrate that germ-free animals have reduced intestinal epithelial cell proliferation compared with colonized mice2,3. Finally, germ-free mice are more susceptible to bleeding and death following dextran sodium sulfate (DSS) induced colitis4. These data suggest a link between luminal bacteria and intestinal epithelial repair. In spite of the beneficial role of bacteria in the normal function of the intestine, bacteria have been implicated in the pathogenesis of inflammatory bowel diseases (IBD)5,6. These disorders are characterized by chronic, relapsing intestinal inflammation in the absence of a specific pathogen.
We became interested in the role of toll-like receptor (TLR) signaling in the intestine as a means to better understand the relationship between epithelial function and bacteria during inflammatory states. TLRs are pattern recognition receptors expressed by immune and non-immune cells that signal in response to PAMPs expressed by microbes7. TLR signaling provides a rapid response against pathogens. Individual or pairs of TLR recognize distinct PAMPs. For example, TLR4 is required for an immune response to LPS8,9 whereas TLR2 in combination with TLR1 recognizes lipotechoic acid10. Most TLR molecules signal through the adapter molecule MyD88 to IL-1 receptor associated kinase (IRAK) and Traf6 to transforming growth factor-beta-activated kinase 1 (TAK1) resulting in activation of mitogen activated protein (MAP) kinases and nuclear translocation of NF-κB11.
Several lines of evidence support a role for TLR signaling in intestinal homeostasis. In vitro, TLR ligands induce fortification of intestinal barrier function through redistribution of the tight junction protein ZO-112 and increase expression of beta-defensin 213. We and others have used an acute model of colitis to address the function of TLR4 in the setting of epithelial injury and inflammation. Administration of DSS to animals genetically deficient in TLR4 or MyD88 results in greater toxicity manifested by increased rectal bleeding, weight loss and mortality compared to wild-type littermates14–16. We have also found that animals deficient in TLR4 or MyD88 have decreased neutrophil recruitment to the intestine due to defective expression of chemokines and they experience bacterial translocation to mesenteric lymph nodes15. At least part of the reason for the increased bleeding and weight loss may be due to decreased intestinal epithelial cell proliferation in TLR4- or MyD88 knock-out mice3,14,15. This series of observations has led to the conclusion that recognition of luminal bacteria through the intestinal expression of TLRs is important for intestinal homeostasis.
The relationship between epithelial repair and inflammation is complex. An important mediator of both inflammation and repair in the intestine is cyclooxygenase (Cox)-2. Cox-1 and Cox-2 synthesize prostaglandins from arachidonic acid17. While intestinal epithelial cells express Cox-1 constitutively, Cox-2 is induced by inflammatory mediators. Cox-2-dependent PGE2 production is critical for epithelial repair in the intestine in a variety of contexts. In the setting of IBD, elevated Cox-2 and PGE2 have been implicated in the development of colitis-associated cancers18,19. We have recently shown that microsomal PGE synthase-1, the enzyme that catalyzes the conversion of PGH2 to PGE2 is increased in IBD mucosa18 whereas 15-hydroxyprostaglandin dehydrogenase (15-PGDH), the enzyme responsible for catabolism of PGE2, is reduced in the inflamed mucosa of IBD19. This combination results in overall increases in mucosal PGE2, and the potential for enhanced carcinogenesis in the setting of inflammation.
We wished to better understand the cellular and molecular mechanisms by which TLR4 signaling is involved in intestinal homeostasis. Studies prior to the identification of TLR4 found that systemic administration of LPS protected animals from radiation-induced injury in the gut characterized by apoptosis of intestinal stem cells20,21. The mechanism for the LPS-induced radioprotection was found to be induction of Cox-2 and prostaglandin E2 (PGE2) production21. In addition, DSS administration to Cox-2 knock-out mice results in a phenotype reminiscent of that seen in TLR4−/− mice, namely increased bleeding and increased mortality22.
In the current study, we test the hypothesis that Cox-2 derived PGE2 is important in TLR4-dependent mucosal homeostasis. Our data demonstrate that TLR4 deficient mice fail to upregulate Cox-2 expression in response to epithelial injury. Both intestinal epithelial cells and lamina propria macrophages express Cox-2 in a TLR4- and MyD88-dependent fashion. PGE2 is decreased in the mucosa of TLR4−/− mice following DSS injury. Oral supplementation with PGE2 results in increased intestinal epithelial cell proliferation and decreased apoptosis in DSS-treated TLR4−/− mice. At least part of the mechanism for TLR4-dependent mucosal healing involves activation of epidermal growth factor (EGF) receptor signaling. The results of our studies shed an important light on the previously unrecognized role of TLR signaling in regulation of Cox-2 in the gut.
Materials and Methods
Mice and interventions
TLR4−/− mice and MyD88−/− mice were purchased from Oriental Bio Service, Inc. (Kyoto, Japan). All knockout mice used were backcrossed to C57Bl/6 mice over 8 times. C57Bl/6 mice were obtained from Jackson Laboratory as controls (Jackson Laboratory, Bar Harbor, Maine). Seven to ten week old gender-matched mice were given 2.5% DSS (MW 36–50 kDa: ICN, Aurora, Ohio) in their drinking water and were sacrificed at the end of 7 days of DSS treatment. For recovery studies, DSS was administered for the first 7 days as indicated, then DSS was removed from the drinking water and mice were sacrificed 7 days after cessation of DSS treatment. PGE2 (Cayman, Ann Arbor, MI) was diluted from the ethanol stock (10 μg/μl) in PBS and 200 μg in 150μl was given twice daily by gavage feeding, starting 3hrs before first DSS administration as described previously23,24. Control mice were given PBS including same dilution of ethanol. All experiments were done according to Mount Sinai School of Medicine animal experimental ethics committee guidelines.
Assessment of colitis activity
Body weight was assessed at baseline and every day for the duration of the experiment. Weight change was calculated as percent change in weight compared to baseline. Fecal blood was tested daily using Hemoccult cards (Beckman Coulter, Inc. Fullerton, CA) and graded as follows: 0= no blood, 1= trace blood, 2=positive, and 4= gross blood. Mice were euthanized by CO2 followed by cervical dislocation. The cecum was removed and the remainder of the colon divided into proximal and distal halves. Tissue was fixed in 10% buffered formalin, paraffin-embedded, sectioned and stained with hematoxylin and eosin. Histological assessment was performed by a pathologist masked to the mouse genotype and treatment. Histologic score was a combined score of acute inflammatory cell infiltrate (0–4), chronic inflammatory cell infiltrate (0–3), and crypt damage (0–4)25–27. Specifically, the crypt damage was scored in the following manner. A score of 0 was given to an intact crypt, 1= loss of the basal 1/3 of crypt, 2= loss of basal 2/3 of crypt, 3= entire loss of crypt, and 4= loss of crypt and surface epithelium26. Since the injury from DSS is patchy, two slides from each section of the colon were assessed per mouse and at least three areas on each slide were examined.
Cell lines and reagents
Human intestinal cell lines SW480 and T84 (1 × 106 cells/well), and mouse macrophage cell line RAW264.7 cells (1.5 × 106 cells/well) (ATCC, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated FCS, 2mM L-glutamine, 5% penicillin/streptomycin and were incubated in 6-well plates overnight at 37 °C in a 5% CO2 humidified incubator. Cells were incubated with phenol-water-extracted Eschericha coli K235 LPS (Sigma, St. Louis, MO), synthetic bacterial lipoprotein (Pam3CSK4) (InvivoGen, San Siego, CA), peptidoglycan (PGN) (InvivoGen, San Siego, CA), or vehicle for the indicated periods of time. Cox inhibitors NS398, and indomethacin (Sigma, St. Louis, MO) were added at the same time as LPS. AG1478, an EGFR tyrosine kinase inhibitor, was added 30 min prior to LPS stimulation. Recombinant human EGF (R&D Systems, Minneapolis, MN) was used as a control.
Real Time PCR
Total RNA was isolated by using RNA Bee (Tel-Test, Inc., Friendwood, TX) according to the manufacturer’s instructions. A total of 1 μg RNA was used as the template for single strand cDNA synthesis utilizing the Transcriptor First Strand cDNA Synthesis Kit (Roche, Indianapolis, IN) according to the manufacturer’s instructions. Quantitative real-time PCR was performed for Cox-2, MyD88, Cox-1, and β-actin using TaqMan probes (Table 1). All TaqMan probes and primers were designed using Beacon Designer 3.0 (Premier Biosoft International, Palo Alto, CA) (Table 1). The cDNA was amplified using TaqMan universal PCR Master Mix (Roche, Indianapolis, IN) on the ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA), programmed for 95°C for 10 min, then 40 cycles of: 95 °C for 15 sec, 60 °C for 1 min. The amplification results were analyzed using SDS 2.2.1 software (Applied Biosystems, Foster City, CA) and the genes of interest were normalized to the corresponding β-actin results. Data were expressed as fold induction relative to the lowest one.
Table 1.
Sequence of the primers and probes used
| Gene | forward primer* | reverse primer* | probe* |
|---|---|---|---|
| Human | |||
| Cox-2 | GCA CGT CCA GGA ACT CCT CA | GGG GTA GGC TTT GCT GTC TG | CCT TCA GCT CCA CAG CCA GAC GCC |
| MyD88 | CTC CTC CAC ATC CTC CCT TCC | CCG CAC GTT CAA GAA CAG AGA | CGC CGC ACT CGC ATG TTG AGA GCA |
| β-actin | GAC TGA GTC TTG CTC TGT CGG | GGC ATG ATG GCT TAC GCC TAT A | AGC GAC TCC TGT GCC TCA GCC TCC |
| Mouse | |||
| Cox-1 | AAG GAG TCT CTC GCT CTG GTT T | TCT CAG GGA TGG TAC AGT TGG G | TGC TCC TGC TGC TGC CGC CGA |
| Cox-2 | ATC CTG CCA GCT CCA CCG | TGG TCA AAT CCT GTG CTC ATA CAT | ACT GCC ACC TCC GCT GCC ACC T |
| β-actin | ATG ACC CAG ATC ATG TTT GA | TAC GAC CAG AGG CAT ACA | CGT AGC CAT CCA GGC TGT GC |
Sequences are listed in 5′ to 3′ direction.
RNA interference (RNAi)
SW480 cells were plated at a density of 1.5 × 105 cells/well in 12-well plates 24hr before the first transfection. MyD88 small interfering RNA (siRNA) oligonucleotide corresponding to the sequence (GCUCAUCGAAAAGAGGUGCtt) was purchased from Ambion (Austin, TX) and 50 nM of siRNA were transfected twice every 24 hrs with X-trim gene siRNA transfection reagent (Roche, Indianapolis, IN) as per manufacturer’s instructions. Forty-eight hours after the first transfection, cells were stimulated with LPS for the indicated period of time. siRNA (50 nM), which has no significant homology to any known gene sequences from mouse, rat, or human as well as siRNA against GAPDH were used as negative controls (Ambion, Austin, TX).
Western Blot Analysis
Whole cell lysates were prepared from colonic tissue samples or SW480 cells after treatment with different stimuli using a lysis buffer containing 50mM Tris HCl, 50mM NaF, 1% Triton X100, 2mM EDTA, and 100mM NaCl, with a proteinase inhibitor cocktail (Calbiochem, San Diego, CA). Protein concentration was determined by the Bradford method using Bio-Rad Protein Assey Dye and SmartSpec™ 3000 (Bio-Rad Laboratories, Hercules, CA). 25 μg of the lysates were subjected to 8 or 10% SDS-PAGE and transferred to Immobilon-P membranes (Millipore Corporation, Bedford, MA). The membrane was blocked in 5% skim milk and was immunoblotted with the primary antibodies for 1hr, followed by HRP-conjugated secondary antibodies rabbit anti-mouse, or goat anti-rabbit IgG, (Zymed Laboratories, South San Francisco, CA). The membrane was exposed on an x-ray film using an enhanced chemiluminescent substrate SuperSignal West Pico Trial Kit (Pierce Biotechnology, Rockford, IL). Antibodies specific for murine or human Cox-2 were purchased from Cayman (Ann Arbor, MI). Anti-MyD88 antibody (HFL-296) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunofluorescent and immunohistochemical studies
Cecum, proximal, and distal colon were freshly isolated and frozen in OCT (Sakura Finetek, Torrance, CA) or fixed in 10% neutral buffered formalin and embedded in paraffin wax. Part of the samples were fixed with modified Bouin’s fixative consisting of 0.5% paraformaldehyde acetate and 15% (vol/vol) of saturated picric acid in 0.1M PBS pH 7.0. Frozen sections pre-fixed by modified Bouin’s fixation were incubated in 10% normal goat serum for 1hr and stained with anti-murine Cox-2 antibody (1: 200, Cayman, Ann Arbor, MI) overnight at 4°C, followed by TRITC-conjugated anti-rabbit IgG (1:200, Zymed Laboratories, South San Francisco, CA) for 1h at room temperature. The specificity of staining was confirmed using Cox-2 blocking peptide (Cayman, Ann Arbor, MI) according to the manufacturer’s instructions or using rabbit isotype control antibody instead of the primary antibody (Zymed Laboratories, South San Francisco, CA).
Double immunofluorescent staining of CD68 and Cox-2 was performed using pre-fixed OCT sections. Sections were incubated with 0.1% Trypsin (Sigma, St. Louis, MO) CaCl2 dissolved in 0.05M Tris-Hcl pH 7.6 for 15 min at 37 °C. Subsequently sections were blocked in a 5% skim milk for 1h and then incubated with the rat anti-CD68 antibody (1:20, MCA1957S, Serotec Ltd., Raleigh, NC) overnight at 4°C. After washing in PBS, sections were incubated with TRITC-conjugated rabbit anti-rat IgG (1:200, Sigma, St. Louis, MO) for 1h at room temperature. Then sections were re-incubated with 5% skim milk followed by Cox-2 staining as described above using FITC-conjugated goat anti-rabbit IgG (1: 200, Sigma, St. Louis, MO).
Phosphospecific EGFR staining was performed using OCT sections. After blocking with 5% milk, sections were incubated with anti-phospho EGFR antibody (1:200, Santa Cruz, CA) overnight at 4°C. Secondary antibody (FITC-conjugated anti-goat IgG, Sigma, St. Louis, MO) was used at a dilution of 1:200. As a negative control, primary antibody was omitted and tissue stained with secondary antibody alone. To quantify the expression level of phospho EGFR, staining intensity was analyzed using MetaMorph software. Epithelial cells were randomly selected and the average pixel intensities from 10 areas of gated cells per slide were analyzed to compare the expression levels of phospho EGFR.
Staining of SW480 was performed using four-chamber slides (Nalgene Nunc International, Rochester, NY), in which cells were seeded at a density of 1×105 cells/well on the day before the experiment. Cells were stained with goat anti-EGFR, or anti-phospho EGFR antibodies (dilution of 1:300) after methanol fixation and preparation with 0.5% Triton X-100. Nonspecific binding was blocked with 5% skim milk. FITC-conjugated goat anti-rabbit or sheep anti-mouse IgG (1:200, Sigma, St. Louis, MO) was used as a secondary antibody.
SW480 and double stained tissue slides were examined using a Leica TCS-SP (UV) confocal microscope. Four thin optical sections through the x-y axis were acquired. Other slides were viewed on an NIKON eclipse E600 immunofluorescence microscope and photographs were taken with a digital camera using Spot Advanced software program (Diagnostic Instruments Inc, Sterling Heights, MI).
Assessment of proliferation and apoptosis
The number of proliferating cells was detected by immunoperoxidase staining for the thymidine analog bromodeoxyuridine (BrdU). One hour and a half prior to sacrifice, mice were injected intraperitoneally with 5-bromo-2′-deoxyuridine (Sigma, St. Louis, MO) at a concentration of 100 mg/kg. Sections (4μm) of paraffin embedded colonic tissue were deparaffinized and incubated with 3% H2O2 in methanol for 15 min. Sections were incubated with 2N HCl for one hour, washed in PBS, and then incubated in 0.1% trypsin for 15 min at 37°C. Sections were stained for BrdU incorporation using BrdU staining kit (Zymed laboratories Inc, South San Francisco, CA) according to the manufacturer’s instructions. The number of BrdU-positive cells per well-oriented crypt were calculated in every 3 crypts for each colon segment at high magnification under light microscopy.
Apoptotic cells in the colonic epithelial cells were detected using the terminal deoxynucleotidyl transferase (TdT)-mediated free 3′-OH end labeling (TUNEL) assay (ApopTaq In Situ Apoptosis Detection Kit; Chemicon, Temecula, CA), following manufacturer’s instruction. Briefly, paraffin sections were prepared with proteinase K (20 μg/ml). After equilibration with TdT buffer, sections were reacted with TdT enzyme for one hour at 37 °C. Digoxigenin (DIG) labeled free 3′-OH end of DNA was detected by anti-DIG rhodamine conjugate. Sections were counterstained with DAPI. The apoptotic cells were counted as follows: three hundred epithelial cells were counted per high powered field and scored for apoptosis; a total of three fields were counted per section of mouse colon, i.e. cecum, proximal, and distal colon. Three mouse colons were counted per condition. The apoptotic index was determined by the ratio of TUNEL positive nuclei to 100 total nuclei in the epithelial cells counted. The areas of necrosis such as in an ulcer bed were identified by examining the corresponding H&E slides and were excluded from counting for apoptotic cells as previously described28.
Transient gene expression and reporter gene assays
SW480 cells were plated in 12 well plates at a density of 1.5 × 105 cells/well. Cells were transfected the following day with FuGENE 6 transfection reagent (Roche, Indianapolis, IN) as per manufacturer’s instructions. Reporter genes for pRL-TK (0.05 μg), Cox-2 promoter-luciferase constructs (phPES2 −1432/+59: 0.3 μg)29,30, which were provided by Dr. Inoue (Nara Women’s University Japan), pGL3 basic empty vector (0.3 μg) were co-transfected as indicated in figure legend. After overnight transfection, cells were stimulated with LPS (5μg/ml) for 4 hours. Cells were then lysed and firefly luciferase activity was measured with a Dual-Luciferase Reporter Assay (Promega, San Luis Obispo, CA). Luciferase measurements were normalized to the relative light units (RLU) from Renilla luciferase from pRL-KT. For experiments in which the Cox-2 promoter and siRNA against MyD88 were used, the siRNA (50 nM) was transfected on Day 1, then 24h later the promoter construct (0.3μg) and the siRNA (50nM) were transfected (Day 2). Cells were then stimulated and harvested for the indicated times. Data are reported as fold-induction over cells transfected with a control empty vector.
Measurement of PGE2
Production of PGE2 in the tissue culture supernatant was determined using a monoclonal EIA kit (Cayman, Ann Arbor, MI) according to the manufacturer’s instructions and Morteau et al.22. Briefly, colonic samples from TLR4−/− and WT mice were washed in cold PBS containing penicillin, streptomycin, and fungizone (100U/ml each). 100 mg of tissue fragments from each part of the colon were cultured for 24 hours in 12 well flat bottom plates in serum free RPMI 1640 supplemented with penicillin, streptomycin, and fungizone (100U/ml each). Culture supernatants were harvested for PGE2 measurement.
Flow cytometry
Intracellular phospho-specific flow cytometry was utilized to quantify the effect of LPS on EGFR phosphorylation as previously described31,32. Briefly, LPS-stimulated or unstimulated SW480 cells were fixed in 2% paraformaldehyde at room temperature for 10 min. After washing with wash buffer (0.5% BSA and 2mM EDTA in PBS), cells were permeabilized in 0.1% TritonX-100 for 5 min. Then cells were stained with phospho EGFR (1:500) for 30 min at room temperature. Following three washes with the wash buffer, cells were incubated with TRITC-conjugated sheep anti-mouse IgG (Zymed laboratories Inc, South San Francisco, CA) for 20 min on ice. After washing with wash buffer, cells were filtered and analyzed by FACScan. Fluorescence intensity was normalized using isotype control antibody.
Cell proliferation assay
SW480 cells (5×104 cells/well) were cultured in 96 well plates in the absence or presence of AG1478 (10 μM) in low serum condition (1% FCS), and stimulated with LPS (2 μg/ml). After 12 or 24 hours of stimulation, cell proliferation was analyzed using CellTiter 96 aqueous non-radioactive cell proliferation assay kit (Promega, WI). Cell proliferation was detected by measurement of formazan product in each well at the absorbance of 490nm following incubation with tetrazolium (MTS)/phenazine methosulfate (PMS) for 1 hour at 37 °C. The cell proliferation index was calculated as a percentage of the absorbance in relation to the untreated control cells.
Statistical analysis
Student’s t-test and standard deviation were performed using the statistics package within Microsoft Excel. Standard error was calculated with Stat View. P values were considered significant when < 0.05.
Results
Cox-2 expression and PGE2 production by intestinal epithelial cells is TLR4- and MyD88-dependent
Previous studies have demonstrated that TLR4 signaling is involved in regulating Cox-2 expression in RAW264.7 cells33. We hypothesized that TLR4 was also important for Cox-2 expression by intestinal epithelial cells. To address this question, we compared the effect of LPS on Cox-2 expression in a cell line that expresses TLR4 and activates NF-κB in response to LPS (SW480) versus one that does not express TLR4 and is LPS unresponsive (T84)13,34. Baseline expression of Cox-2 differed in these neoplastic cell lines35. Only TLR4-responsive SW480 cells increased expression of Cox-2 in response to LPS (Figure 1A). Induction of Cox-2 proceeded with rapid kinetics occurring within 30 minutes of LPS stimulation and peaking at 4h (Figure 1B); protein expression for Cox-2 remained elevated at 24h following LPS stimulation (Figure 1C). No induction of Cox-2 protein was seen following stimulation with PGN, a TLR2 ligand (Figure 1C, lower panel).
Figure 1. LPS induces Cox-2 expression in human intestinal epithelial cell lines in a TLR4- and MyD88-dependent fashion.

A. Cox-2 expression after stimulation with LPS (2μg/ml) in T84 (LPS unresponsive) and SW480 (LPS responsive) human intestinal epithelial cell lines. TaqMan real-time PCR demonstrated inducible Cox-2 expression stimulated by LPS in SW480 cells. LPS unresponsive T84 cells showed no change in Cox-2 expression in response to LPS stimulation. Data are represented as mean ± SEM of relative values of expression in 3 individual experiments of triplicate samples (NS: non significant, *P < 0.05).
B. LPS responsive SW480 cells were stimulated with LPS (2μg/ml) for indicated times. TaqMan real-time PCR demonstrates LPS-induced expression of Cox-2 mRNA with a peak at 4 hours of stimulation. Data are represented as mean ± SEM of relative values of expression in 3 individual experiments of triplicate samples (*P < 0.05, **P < 0.001).
C. Western Blot analysis of Cox-2 protein expression in SW480. Cells were stimulated with LPS for indicated periods in top panel. Lower panel demonstrates stimulation of cells with LPS (2μg/ml) or PGN (2μg/ml). Blots of whole cell lysates (25 μg/lane) were probed with Cox-2 antibody. Data are one representative experiment of three with similar results. β-actin was used as an internal control for protein loading.
D. Extent of MyD88 suppression by siRNA. SW480 cells were transiently transfected with siRNA against MyD88, or GAPDH. Negative siRNA which has no significant homology to any gene sequences was applied as a control. The knockdown efficiency of siRNA against MyD88 was assessed by real-time PCR and Western blot. The siRNA was decreased both MyD88 mRNA and protein expression. Negative siRNA as well as siRNA against GAPDH did not affect MyD88 mRNA or protein expression.
E. MyD88-dependent induction of Cox-2 in response to LPS. SW480 cells were stimulated with LPS (5μg/ml) for 4 hrs and co-transfected with either MyD88 siRNA or negative control siRNA. Untransfected control samples were not LPS treated. TaqMan real-time PCR demonstrated LPS induced expression of Cox-2 in negative control siRNA samples. This induction of Cox-2 by LPS was largely abolished in the cells in which MyD88 was blocked with siRNA, indicating a MyD88 dependent pathway. Data are represented as mean ± SEM of relative values of expression in 3 individual experiments of triplicate samples (**P < 0.001).
F. LPS regulation of Cox-2 gene promoter activity. The intestinal epithelial cell line SW480 was cotransfected with the Cox-2 (−1432/+59) luciferase reporter construct, MyD88 siRNA, or the empty pGL3 vector control, together with an internal control pRL-KT (Renilla luciferase) plasmid. Cells were stimulated with LPS (5μg/ml) for 4 hours. Reporter gene activation was significantly higher in cells stimulated with LPS than non-stimulated cells. MyD88 siRNA abrogated promoter activation in response to LPS. Data are represented as mean ± SEM of relative light units in 3 individual experiments with triplicate samples (*P < 0.05).
G. LPS induced PGE2 production in the intestinal epithelial cell line SW480. Cells were stimulated with LPS (2μg/ml) for 30 min. PGE2 concentration in supernatant was measured by monoclonal EIA. LPS stimulation resulted in PGE2 production within 30 min. MyD88 siRNA inhibited LPS induced PGE2 production as did a selective Cox-2 inhibitor (NS398 5μM) or a Cox-1/Cox-2 inhibitor (indomethacin 5μM). Data are represented as mean ± SEM of 2 individual measurements of duplicate samples taken from 3 individual experiments (*P < 0.05).
H. Effect of TLR2 ligands on production of PGE2 compared with the TLR4 ligand LPS in the intestinal epithelial cell line SW480. SW480 was stimulated with a TLR4 ligand LPS (2μg/ml), or TLR2 ligands PGN (2μg/ml) or Pam3CSK4 (500ng/ml) for indicated periods of time. Concentration of PGE2was examined by monoclonal EIA. There were significant differences in the stimulation of PGE2 between LPS versus TLR2 ligands. Data are represented as mean ± SD of triplicate samples taken from 2 individual experiments (P < 0.05, between LPS and PGN or Pam3CSK4 for each time period).
To further clarify whether LPS induction of Cox-2 requires MyD88, we transfected SW480 cells with siRNA against MyD88 or a control siRNA (Figure 1D). Only the siRNA specific for MyD88 inhibited LPS-mediated expression of Cox-2 RNA. Finally, we asked whether LPS could induce Cox-2 promoter activation and whether this also depended on MyD88. A full-length Cox-2 promoter-luciferase construct was transfected into cells in the presence or absence of siRNA against MyD88 (Figure 1E). LPS activated the Cox-2 promoter and this activation was blocked by MyD88 siRNA. We performed dose-ranging studies using the Cox-2 promoter-reporter gene and found no difference in induction of the reporter between 2 to 5μg/ml of LPS (data not shown). Taken together, these data demonstrate that LPS can directly induce Cox-2 promoter activity, mRNA expression, and protein expression in a TLR4- and MyD88-dependent fashion in colonic epithelial cells.
Cox-2 metabolizes arachidonic acid released from the plasma membrane to generate prostanoids such as PGE2. PGE2 in turn mediates many of the biologic effects of Cox-2 in the intestinal epithelium36. LPS has previously been shown to stimulate PGE2 production by intestinal epithelial cell lines37–39 although most studies have attributed the biologic effects of LPS in the gut to macrophages and other stromal cells40. We hypothesized that LPS-mediated induction of PGE2 depended on MyD88 signaling. Our results demonstrate that LPS induces PGE2 production by SW480 cells (Figure 1F). PGE2 production could be blocked by expression of MyD88 siRNA to an extent that was similar to a selective Cox-2 inhibitor or a non-selective Cox inhibitor, indomethacin. By contrast, stimulation with the TLR2 ligands PGN and PamCysk3 induced very little less PGE2 especially when compared to LPS (Figure 1G). These data suggest that LPS stimulation of TLR4 leads to Cox-2 expression and the production of PGE2 by intestinal epithelial cells in vitro.
Cox-2 and PGE2 expression are decreased in TLR4-deficient mice following DSS induced injury
Cox-2 expression is increased in human IBD and animal models of colitis including DSS22,41. Based on our in vitro findings, we hypothesized that TLR4 signaling was important for Cox-2 expression in the setting of DSS colitis. Using real-time PCR we found that Cox-2 expression is low in WT and TLR4−/− mice prior to DSS treatment (Figure 2A) whereas Cox-1 is abundantly expressed in both (data not shown). Following induction of colitis, there is a dramatic increase in expression of Cox-2 in WT mice. This upregulation is not seen in TLR4−/− mice. We confirmed protein expression of Cox-2 by Western blot analysis of tissue lysates (Figure 2B) and immunofluorescence (Figure 2C). Colonic tissue from WT mice or LPS-treated RAW cells expressed Cox-2 but little was present in TLR4−/− colon tissue. Immunofluorescent studies revealed both epithelial staining and lamina propria staining of Cox-2 in WT mice. In order to define the cell types expressing Cox-2 in the lamina propria of WT mice, we performed double staining with a macrophage marker CD68 and anti-Cox-2 Ab (Figure 2D). Most Cox-2 positive cells in the lamina propria were also CD68 positive suggesting that lamina propria macrophages express Cox-2 in the inflamed colon. These data support a role for TLR4 regulation of Cox-2 expression in the intestine.
Figure 2. Cox-2 expression is decreased in TLR4−/− mice following DSS-induced colitis.

A. TaqMan real-time PCR demonstrated up regulation of Cox-2 expression in the colon of WT mice but not in TLR4−/− mice after 7 days of DSS treatment (n=3 for each group on day 0, n=12 for the other groups). Data are represented as mean ± SEM of relative values of expression in 4 individual experiments (*P < 0.05).
B. Western Blot analysis for Cox-2 in the colon. Immunoblots of tissue lysate proteins (25 μg/lane) prepared from colonic samples of TLR4−/− and MyD88−/−, and WT control mice prior to and after 7 days of DSS treatment. Membranes were probed with Cox-2 antibody. Positive control consists of cell lysate from LPS (2μg/ml) stimulated RAW 264.7 cells (right lane). Cox-2 protein expression was greater in WT colon. Data are one representative experiment of three independent studies. β-actin was used as an internal control for protein loading.
C. Immunofluorescent staining for Cox-2 in the colon before and after 7 days of DSS treatment. Prior to DSS treatment, colonic tissue does not express detectable levels of Cox-2 in either TLR4−/− mice (C) or WT controls (A). Immunofluorescent signal (red color of rhodamine) of Cox-2 was strongly detected in the colonic epithelial cytoplasm (arrow) and lamina propria cells (arrow head) in WT mice (B) but is very low in TLR4−/− mice (D) after DSS treatment. Insets show phase contrast images identifying the orientation of colonic sections.
D. Expression of Cox-2 by lamina propria macrophages using double staining of Cox-2 (FITC green) and CD68 (TRITC red). Most Cox-2 positive lamina propria cells were double-stained with CD68 (macrophage marker). Representative data are from WT mice treated with 7 days of DSS. Arrows indicates double positive cells showing yellow cytoplasmic staining. TLR4−/− mice do not have Cox-2 positive LP macrophages (see Figure 2C).
E. PGE2 production in DSS-induced colitis. Colonic tissues from TLR4−/− and WT controls before and after 7 days of DSS were cultured in media for 24 hours and concentration of PGE2 in the supernatants were analyzed by EIA (n= 4 for each group). Data are represented as mean ± SD of duplicate samples taken from 3 individual experiments. There was a significant difference in PGE2 production in TLR4−/− mice following DSS treatment compared with WT mice (*P < 0.05).
Given that mice that are deficient in TLR4 do not express Cox-2, we hypothesized that TLR4−/− mice would have decreased production of PGE2. To test this hypothesis, we measured production of PGE2 from colonic tissue using monocolonal EIA. Our data demonstrate that PGE2 production by colonic tissues is significantly reduced in TLR4 deficient mice following DSS induced injury (Figure 2E). PGE2 production in TLR4−/− mice was decreased by 40% compared to WT mice following DSS colitis. These data suggest that at least one defect in TLR4−/− mice with respect to epithelial repair is reduced production of local PGE2.
TLR4−/− mice have decreased proliferation and increased intestinal epithelial cell apoptosis following DSS-induced colitis
Cox-2 knock-out mice demonstrate increased susceptibility to DSS colitis similar to what we have observed in TLR4−/− mice. We have previously described that TLR4−/− mice have decreased intestinal epithelial proliferation following acute DSS-induced injury15. Given our findings of decreased Cox-2 expression, we wished to examine the effect of TLR4 deficiency on baseline levels of proliferation, at the peak of injury, and 7 days after recovery (Day 14). Using BrDU labeling of proliferating intestinal epithelial cells, we demonstrate that proliferation is similar in WT and TLR4−/− mice prior to DSS treatment (Figure 3A and 3B). Following DSS-induced injury, WT mice have a large increase in proliferating cells that persists even a week after DSS is discontinued. By contrast, TLR4−/− mice have significantly fewer proliferating cells. Using computerized microscopic measurements, we found a significant decrease in crypt height in the injured TLR4−/− gut that can be appreciated in the photomicrographs (Figure 3A) (Day7: WT 291±48.2 μm, TLR4−/− 214±20.2 μm (p<0.0001); Day14: WT 257±111 μm, TLR4−/− 167± 35.1 μm (p<0.0001)). Importantly, crypt height is similar at baseline in WT and TLR4−/− mice suggesting this defect in proliferation is only apparent in the setting of injury (Day 0: WT 244±72.9 μm, TLR4−/−251±49.5 μm (p<0.321).
Figure 3. TLR4−/− mice have a persistent decrease in epithelial proliferation and increased apoptosis following 7 days of DSS treatment.
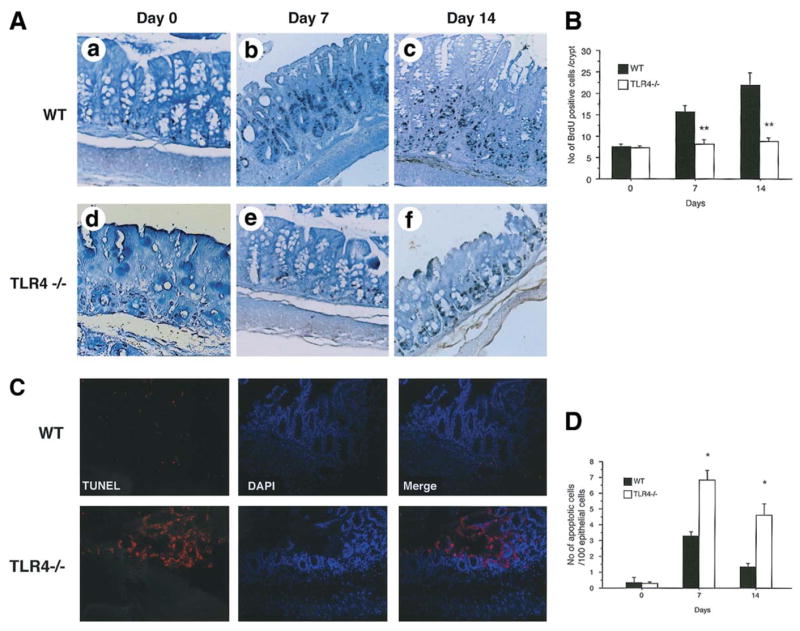
A. TLR4−/− and WT control mice were treated with 2.5% DSS for 7 days (left panels) followed by 7 days of recovery (right panels). Animals were injected with BrdU 90minutes prior to sacrifice. Colonic sections taken from day 0 (A; WT, D; TLR4−/−), day 7 (B: WT, E: TLR4−/−) and day 14 (C: WT, F: TLR4−/−) were stained with anti-BrdU. BrdU positive cells are identified by black staining of nuclei. TLR4−/− mice have fewer BrdU positive cells in the crypts on day 7 (E panel) as well as on day 14 (F panel). Tissues were counterstained with methyl green (original magnification 200x).
B. BrdU positive cells were counted in 3 crypts of each colon segment per HPF (9 crypts/mouse). Bars show mean ± SEM proliferating cells / crypts (n=3 for each group on day 0 (i.e. untreated), n=5 for the other groups). The proliferating cells in TLR4−/− mice were significantly fewer than in WT controls after 7 days of DSS treatment (day 7) as well as during recovery (day 14) from DSS-induced colitis (**P < 0.001).
C. Apoptotic cells in the colonic crypt epithelial cells of TLR4−/− and WT controls were determined by TUNEL assay. Representative sections were taken on day 7 of DSS treatment. Red staining of nuclei indicates apoptotic cells, which was observed mainly in the surface epithelium. Sections were counterstained with DAPI (blue) to identify the orientation of nuclei. TLR4−/− mice showed increased apoptotic cells compared with WT controls (original magnification 200X).
D. Numbers of apoptotic cells were counted in 300 total epithelial cells in 3 areas of each colon segment (n=3 for each group on day 0 (untreated), n=6–7 for the other groups). Bars show mean ± SEM of the number of apoptotic cells to 100 total nuclei of the epithelial cells. Significant increases of apoptotic cells in TLR4−/− mice compared with WT controls after 7 days of DSS treatment (*P < 0.05). The apoptotic cells were still increased in TLR4−/− mice even after recovery (day 14, *P < 0.05).
Colonic crypt height represents the balance of epithelial proliferation and apoptotic cell loss at the top of the crypts28,42. We reasoned therefore that TLR4−/− mice could have increased intestinal epithelial cell apoptosis in response to DSS injury. To address this question, we performed TUNEL staining on intestinal sections from DSS-treated TLR4−/− mice or their wild-type controls (Figure 3C and 3D). We found significantly more apoptotic cells per crypt in TLR4−/− intestine compared to controls. In general, the apoptotic cells were found at the tops of the colonic crypts. Similar findings were seen in MyD88−/− mice suggesting this was a MyD88-dependent phenomenon (data not shown). An increased rate of apoptosis was not seen in the intestines of mice prior to DSS treatment. We conclude from these data that TLR4 is important for intestinal epithelial repair from injury by aiding in proliferation and protecting against apoptosis.
PGE2 restores proliferation and protects against apoptosis in TLR4−/− mice
We have shown above that TLR4 is required for induction of Cox-2 and PGE2 during DSS-induced injury. We hypothesized that the decrease in PGE2 production in TLR4−/− mice was responsible for the observed phenotype, namely worse clinical signs of colitis and abnormal proliferation and apoptosis. To test this hypothesis, TLR4−/− mice were given PGE2 by oral gavage twice a day concurrently with the DSS treatment period. These mice were compared to TLR4−/− mice and WT mice given DSS alone with PBS to control for the gavage solution. Compared to TLR4−/− PBS-treated mice, TLR4−/− mice treated with PGE2 behaved like WT mice with respect to weight loss (Figure 4A) and rectal bleeding (Figure 4B). We next examined the effect of PGE2 supplementation on proliferation and apoptosis. TLR4−/− mice given PGE2 had significantly greater proliferation (Figure 4C, 4D) and reduced apoptosis (Figure 4E, 4F) than TLR4−/− mice given vehicle alone. Total histology scores were lower for PGE2-treated TLR4−/− mice compared to control TLR4−/− mice because crypt damage was less (data not shown). PGE2 treatment of wild-type DSS-treated mice showed no significant change in proliferation (WT cont 14.5 ± 1.4, WT+PGE2 12.3 ± 1.6) and a small decrease in apoptosis (WT cont 3.1 ± 2, WT+ PGE2 1.4 ± 1.3). These data suggest that PGE2 is necessary and sufficient to restore epithelial healing as measured by proliferation and apoptosis in the injured intestinal epithelium.
Figure 4. PGE2 supplementation improves signs of colitis and restores epithelial healing after DSS-induced injury in TLR4 −/− mice.

A. Weight change was examined daily during the 7 days of DSS treatment. Vehicle (PBS) treated TLR4−/− mice have significantly more weight loss than WT control mice (*P < 0.05). PGE2 treated TLR4−/− mice and have significantly less weight loss than vehicle treated TLR4−/− mice and are comparable to WT mice. The data represents the average (± SEM) of three independent experiments with a total of 19 mice (TLR4−/− Vehicle (n=6), TLR4−/− PGE2 (n=7) and WT controls (n=6)).
B. PGE2 treated TLR4−/− mice had a significant reduction in bleeding on days 2 through 6 compared with vehicle (PBS) treated TLR4−/− mice (*p<0.05). Stool blood was calculated as follows: 0=no blood, 1=trace occult blood positive, 2= strongly occult blood positive, and 4=bloody diarrhea. Standard error is shown.
C. BrdU labeling of intestinal epithelial cells was performed for vehicle treated TLR4−/−, PGE2 treated TLR4−/− and WT controls at the end of 7 days of DSS treatment. (Original magnification 200X)
D. BrdU positive cells were counted per HPF in 3 crypts of each colon segment (9 areas / mouse). Bars show mean ± SEM of proliferating cells / crypts (n=5 in each group). There is a significant increase in BrdU positive cells in PGE2 -treated TLR4−/− mice compared with vehicle treated TLR4−/− mice (**P < 0.001). PGE2-treated TLR4−/− mice are not significantly different than WT mice.
E. Apoptotic cells in the crypt epithelial cells were determined by TUNEL assay. Representative sections were taken from vehicle treated TLR4−/−, PGE2-treated TLR4−/− mice and WT controls as indicated. Red staining of nuclei indicates apoptotic cells. Sections were counterstained with DAPI (blue) to identify the nuclei of epithelial cells. PGE2 treated TLR4−/− mice showed a marked decrease of apoptotic cells compared with vehicle treated TLR−/− mice. The frequency of TUNEL positive epithelial cells in PGE2 treated TLR4−/− mice was similar to WT controls (original magnification 200X).
F. Number of apoptotic cells was counted in 300 total epithelial cells in triplicate in every 3 areas for each colon segment (n=5 in each group). Bars show mean ± SEM of the number of apoptotic cells per 100 total nuclei in the epithelial cells counted. There is a significant decrease of apoptotic cells in PGE2 treated TLR4−/− mice compared with vehicle treated TLR4−/− mice (**P < 0.001).
TLR4 regulates phosphorylation of the EGF receptor in intestinal epithelial cells
Our data point to a link between TLR4-mediated induction of Cox-2 and PGE2 production and intestinal injury. In order to understand the mechanism(s) by which PGE2 mediates an effect on the intestinal epithelium, we examined the ability of LPS to stimulate EGFR phosphorylation. EGFR phosphorylation results in a variety of biologic events including induction of proliferation and protection against apoptosis43–46. PGE2 mediates EGFR phosphorylation through induction of EGFR ligands or through intracellular mediators such as Src47,48. We hypothesized that LPS stimulation of intestinal epithelial cells would result in EGFR phosphorylation. SW480 cells were stimulated with LPS and total or phosphorylated EGFR examined by Western blot and immunofluorescent staining (Figure 5A, 5B). LPS stimulated phosphorylation of EGFR. We further quantified LPS induced EGFR phosphorylation by flow cytometric analysis (Figure 5C). LPS stimulation significantly increased EGFR phosphorylation. We then addressed whether this effect on EGFR was dependent on TLR-MyD88 signaling. EGFR phosphorylation was blocked by expression of MyD88 siRNA (Figure 5D) and partially by a specific inhibitor of Cox-2, NS398 (Figure 5E). EGF stimulation of SW480 cells resulted in EGFR phosphorylation that was not blocked by a Cox-2 inhibitor or indomethacin (Figure 5E). Since it was also possible that LPS-induced EGFR phosphorylation is upstream of Cox-2 expression, we used a specific EGFR tyrosine kinase inhibitor AG1478 and asked whether inhibition blocked LPS-mediated induction of Cox-2 protein expression. Inhibiting EGFR tyrosine kinase activity had no effect on Cox-2 induction (data not shown). Finally, we asked whether inhibition of EGFR phosphorylation affected LPS-induced cell proliferation in SW480 cells. AG1478 blocked LPS induced cell proliferation (Figure 5F). These data suggest that TLR4-mediated induction of PGE2 production may stimulate epithelial proliferation by an EGFR-dependent mechanism.
Figure 5. LPS induces EGFR phosphorylation in a MyD88-dependent fashion.

A. Western Blot analysis of EGFR and its phosphorylation in human intestinal epithelial cell line SW480. Cells were stimulated with LPS for indicated periods. Blots of whole cell lysates (25 μg/lane) were probed with either EGFR (top panel) or phosphorylated EGFR (bottom panel). LPS stimulated EGFR phosphorylation. Data are one representative experiment of three independent experiments with similar results.
B. Immunofluorescent staining for EGFR and phospho-EGFR in SW480. Cells were cultured on glass slides and treated with LPS (2μg/ml) for indicated periods. Nuclei were stained with DAPI. After 30 minutes of stimulation, confocal images show an increased intensity of staining of phospho-EGFR but not EGFR. Insets show negative control in which primary antibodies were omitted. Original magnification 400x.
C. Flow cytometric analysis of phospho EGFR (Tyr1068) after stimulation with LPS for indicated periods of time. Histograms show an increase in log fluorescence intensity. Bars represent geometric mean of fluorescence intensity of phospho EGFR positive cells ± SD based on 3 individual experiments (LPS 2μg/ml) with triplicate samples (*P < 0.05).
D. LPS-induced EGFR phosphorylation is MyD88-dependent. Western Blot analysis of EGFR and its phosphorylated form are shown. Cells were stimulated with LPS for indicated periods. Blots of whole cell lysates (25 μg/lane) were probed with EGFR or phospho-EGFR antibody. Cells transfected with MyD88 siRNA had no phosphorylation of EGFR in response to LPS. Data are one representative experiment of three independent experiments with similar results.
E. LPS induced EGFR phosphorylation is Cox-2 dependent. Cells were stimulated with LPS (2μg/ml) for 30 min with or without Cox inhibitors as indicated, and whole cell lysates (22 μg/lane) probed with EGFR or phospho EGFR antibodies. Western blot analysis demonstrates that EGFR phosphorylation is inhibited by a non-selective Cox-2 inhibitor, (NS398 5μM), or a Cox-1 and Cox-2 inhibitor (indomethacin selective (5μM). As a control, EGF (10nMol=6ng/ml) was added to cells for 30mins. EGF-mediated phosphorylation of the EGFR was not inhibited by Cox inhibitors (last three lanes). Data are one representative experiment of five independent experiments with similar results.
F. LPS induced cell proliferation via EGFR activation. SW480 cells were stimulated with LPS (2μg/ml) for indicated periods with or without EGFR specific tyrosine kinase inhibitor AG1478. Data are shown as the means ± SD of percentage of absorbance in comparison to non treated control cells from three independent experiments.
Given the results of TLR4 regulating phosphorylation of EGFR in vitro, we hypothesized that TLR4−/− mice have decreased phosphorylation of EGFR in intestinal epithelial cells in vivo. To test this hypothesis, we utilized immunohistochemistry to evaluate the expression of phosphorylated EGFR in intestinal epithelial cells following DSS-induced colitis. WT mice had significantly higher expression of phosphorylated EGFR in intestinal epithelial cells than TLR4−/− mice following DSS-induced colitis (Figure 6A). TLR4−/− mice given PGE2 treatment had restored expression of phosphorylated EGFR to nearly WT levels (Figure 6A). Omission of primary Ab did not show any staining (data not shown). Computer analysis using the MetaMorph program was used to quantify intensity of staining in the intestinal epithelium (Figure 6B). We conclude from these results that impaired epithelial repair seen in TLR4−/− mice is at least partly due to impaired activation of epithelial cell EGFR which in turn may be due to decreased mucosal production of PGE2 in response to inflammation.
Figure 6. TLR4−/− mice have decreased EGFR phosphorylation following DSS-induced colitis due to defective production of mucosal PGE2.
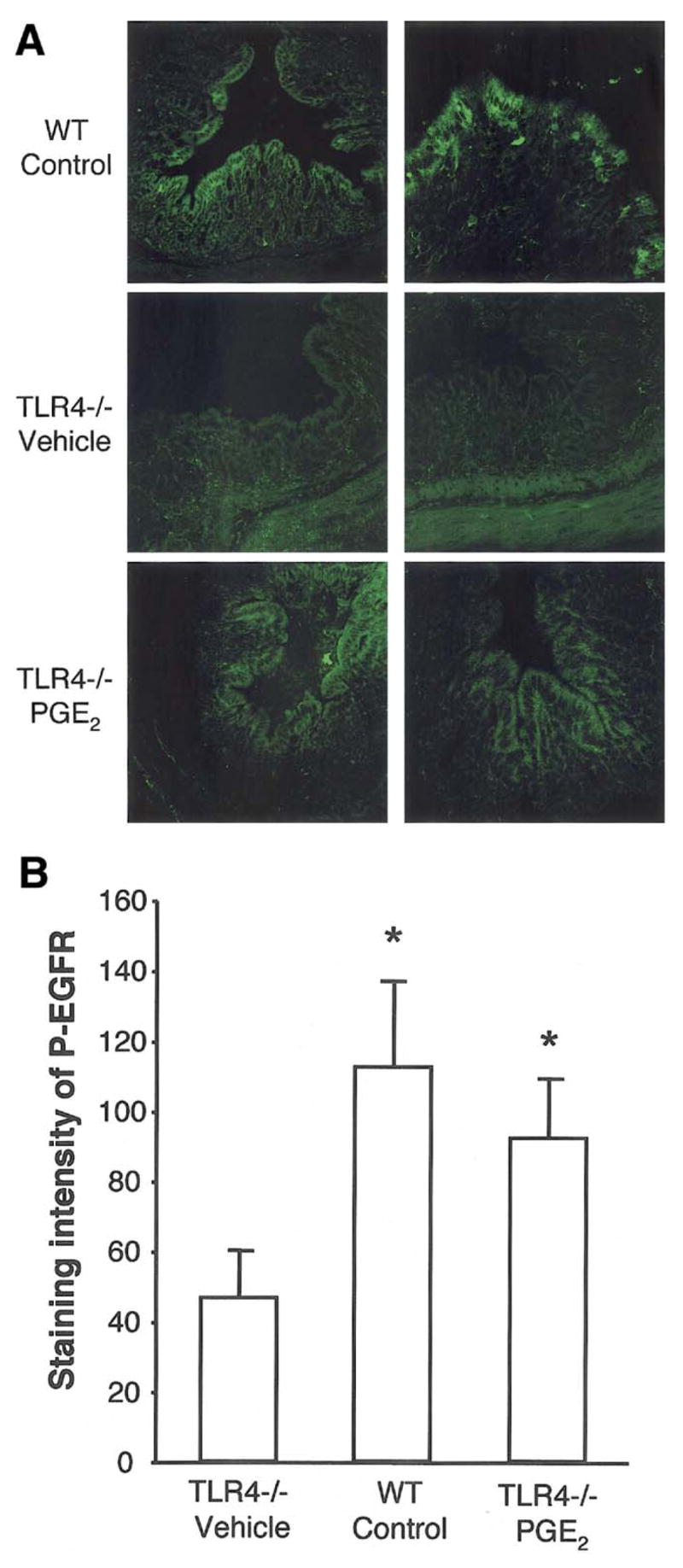
A. Immunofluorescent staining for phospho EGFR in the colon after 7 days of DSS treatment. Phosphorylated EGFR was strongly detected in surface epithelial cells in WT mice. PBS treated TLR4−/− mice had decreased epithelial cell EGFR phosphorylation following DSS colitis. PGE2 treatment partly restored the expression of phosphorylated EGFR. The pictures show two individual mouse tissue samples of each genotype with or without PGE2 treatment.
B. Quantification of the expression levels of phospho EGFR. Staining intensity of 10 randomly selected areas of epithelial cells per slide was analyzed using MetaMorph software. Staining intensity of phospho EGFR was significantly decreased in TLR4−/− mice compared with WT mice and PGE2 treated TLR4−/− mice after 7 days of DSS treatment.
Discussion
In this study, we demonstrate a requirement for TLR4 in regulating Cox-2 expression in both intestinal epithelial cells and lamina propria macrophages. In our model (Figure 7), epithelial injury permits exposure of intestinal epithelial cells and lamina propria macrophages to Gram-negative bacteria and LPS. TLR4 signaling via MyD88 activates a signaling cascade that results in enhanced transcription of Cox-2 and increased production of PGE2. The clinical manifestations of increased rectal bleeding seen in TLR4−/− mice are likely due to the epithelial defect in proliferation and apoptosis since correcting the defect with native exogenous PGE2 restored mice to wild-type levels of rectal bleeding. These data suggest that a relative deficiency in PGE2 production is likely to play an important role in the observed TLR4−/− phenotype. Our in vitro data support the idea that the effect of TLR4 on Cox-2 expression and other downstream mediators of epithelial repair can occur directly by LPS signaling in intestinal epithelial cells. In vivo, however, there is likely to be a contribution by lamina propria macrophages acting in trans on the epithelium by way of contact or secreted factors including PGE2.
Figure 7. Model of TLR4-mediated Cox-2 regulation.
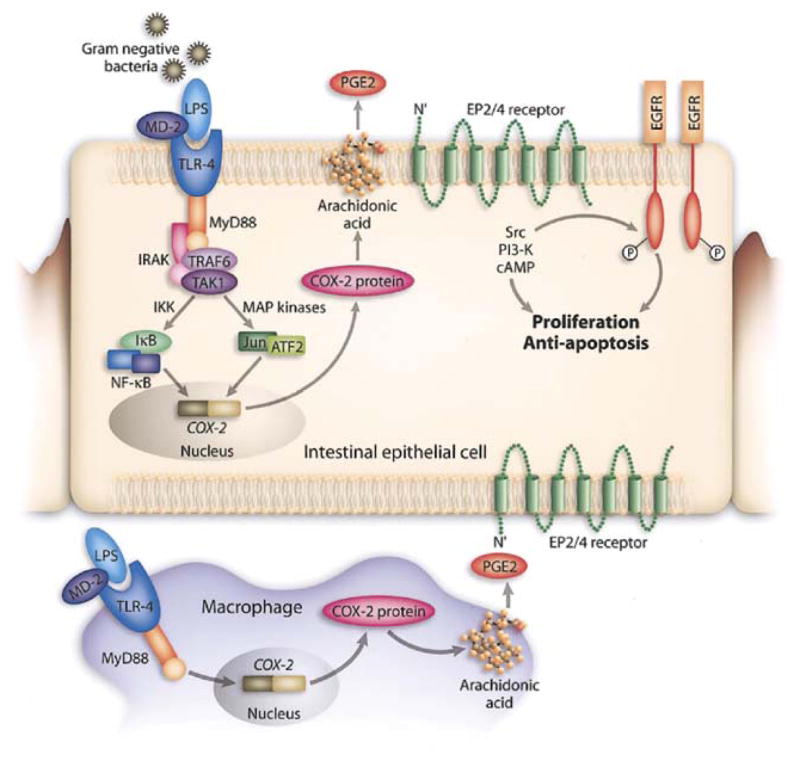
In the setting of intestinal injury, LPS exposure of intestinal epithelial cells (possibly basolaterally) and lamina propria macrophages results in TLR4 activation and signaling via MyD88. This activates a variety of signaling pathways culminating in transcription factor translocation and engagement of the Cox-2 promoter. Cox-2 is transcribed and translated; it acts on arachidonic acid to generate PGG2 which is rapidly converted to PGH2 and then microsomal PG E synthase-1 converts it to PGE2. PGE2 through its receptors EP2 or EP4 can activate downstream signaling molecules such as the tyrosine kinase Src or the lipid kinase PI’3 kinase which can lead to transactivation of the EGF receptor. EGFR signaling is associated with proliferation and protection against apoptosis in intestinal epithelial cells. PGE2 produced by macrophages may also act in trans on intestinal epithelial cells. In the absence of TLR4 signaling, Cox-2 expression is greatly decreased.
We present data that the colonic epithelium depends on bacterial-derived signals to activate the complex program involved in tissue repair. With the discovery of TLR molecules, the exact pathways by which this occurs may be better understood. The notion that TLR signaling is relevant and necessary for repair of injury is exemplified in a recent study demonstrating that mice deficient in MyD88 or TLR4/TLR2 signaling were impaired in their ability to heal after acute lung injury49.The authors describe that extracellular matrix hyaluronan, either directly or indirectly, activates TLR2 and TLR4 signaling resulting in inflammatory cell transmigration and protection against apoptosis. In the absence of TLR signaling there is decreased inflammatory infiltrate in the lungs of mice but paradoxically animals have decreased survival. The similarities of our results in a model of intestinal epithelial cell injury suggest a common theme of repair linked to an inflammatory signal.
In order to understand what role TLR4 might be playing in the setting of DSS-induced injury we examined the literature for known effects of LPS on the intestinal epithelium. Investigators have demonstrated that LPS can induce proliferation of IEC-6 cells in culture through a TNF-dependent mechanism50. Grishin et al. has recently shown that LPS stimulates Cox-2 in a rat intestinal epithelial cell line and have focused on the role this may play in necrotizing enterocolitis51,52. Although our study may not be the first to demonstrate a link between LPS and Cox-2, we believe our studies are novel in showing the degree to which TLR4 is required for intestinal expression of Cox-2 in vivo and the dependence of TLR4-mediated Cox-2 expression in repair of the damaged epithelium. Rakoff-Nahoum et al. has examined proliferation in MyD88−/− mice in the setting of radiation injury and found a decrease in proliferative response but did not investigate the underlying mechanism in detail14. Finally, Pull et al. used a bone marrow chimera model to look at the relative contribution of MyD88 expression in immune versus non-immune cells in the colonic proliferative response after DSS colitis3.They describe a prominent role for MyD88-expressing lamina propria mesenchymal cells in the proliferative response of the epithelium in DSS-treated mice. Interestingly, in supplemental material accompanying the above manuscript, Cox-2 was highly expressed in mesenchyme derived from conventionalized RAG1−/− mouse intestine compared with MyD88−/− mice suggesting that the Cox-2 from mesenchymal cells may play a role in epithelial repair. Given the broad nature of the defect in TLR, IL-1, and IL-18 signaling in MyD88−/− cells, the prior studies do not address the specific contribution of TLR4 signaling nor the mechanisms underlying the proliferative defect.
Other studies were performed before the identification of TLR4 as the receptor for LPS. Studies in C3H/HeJ mice with a mutation in TLR48 found that crypt epithelial proliferation was decreased following DSS and could be restored by PGE253. Like Wang and DuBois et al.54, we used native PGE2 administered orally rather than the longer acting dimethyl-PGE2. We used oral PGE2 because of previous evidence that this approach led to increased levels of PGE2 in the blood and intestine as well as altered colon physiology. Riehl and colleagues described that systemic LPS protected against radiation-induced epithelial apoptosis in intestinal crypts through a mechanism that depends on PGE2 production21.
The link between Cox-2 and PGE2 in protection from colitis is highlighted in a variety of studies. Cox-2−/− mice demonstrate increased susceptibility to DSS-induced colitis which correlates ith their inability to produce PGE222. Animals deficient in the PGE2 receptor, EP4, are more susceptible to DSS injury55. Several mechanisms underlie the protective effects of PGE2. PGE2 through its receptors, EP2 or EP4, may stimulate EGFR phosphorylation through intracellular kinases such as Src or by increased expression of amphiregulin, an EGFR ligand46,47,48,56. PGE2 protects against radiation-induced intestinal epithelial cell apoptosis through an EGFR and Akt-dependent effect on Bax 48. Signaling downstream of EGFR is involved in growth, repair, and barrier integrity of the gastrointestinal mucosa57–59. Increased susceptibility of EGFR deficient mice to DSS-induced colitis has also been reported60.
Studies have also shown that Cox-2 levels41 and PGE2 production61–63 are increased in inflammatory bowel disease. Cox-2 overexpression characterizes dysplasia and colon cancer64–66. Recent work demonstrates that PGE2 -dependent colon carcinogenesis involves deregulated PI-3 kinase signaling and increased expression of beta-catenin67. An increase in EGFR phosphorylation has been described in the mucosa of patients with ulcerative colitis68. This model is consistent with the known benefit of Cox-2 inhibitors and EGFR antagonists such as cetuximab in the prevention or treatment of colon cancer. In inflammatory bowel disease, it remains controversial whether Cox-2 inhibitors worsen symptoms of the disease69. Short-term studies have shown that Cox-2 inhibitors do not flare colitis70 but long-term use—as would be required for chemoprevention, might flare disease. 5-ASAs inhibit arachidonic acid metabolism to PGE2 and have a modest chemopreventive benefit in patients with ulcerative colitis71 whereas other immunomodulators such as 6-mercaptopurine do not72. These data suggest an inflammation-independent effect of 5-ASA possibly through inhibition of PGE2.
Our studies highlight the previous unknown dependence of Cox-2 expression by TLR4 in the intestine. In the acute response to injury, TLR4 signaling results in increased Cox-2 and PGE2 production that are beneficial. We may speculate that long-term, persistent TLR4 signaling may contribute to colitis-associated cancers. Strategies aimed at dampening TLR4 signaling may reduce chronic inflammation and the drive towards carcinogenesis.
Acknowledgments
Supported by NIH grants AI052266 and DK069594 (MTA), the New York Crohn’s Foundation (AJD), and a Uehara Memorial Foundation Research Fellowship (MF).
Abbreviations
- TLR
toll-like receptor
- DSS
dextran sodium sulfate
- PAMP
pathogen associated molecular pattern
- LPS
lipopolysaccharide
- Cox
cyclooxygenase
- EGFR
epidermal growth factor receptor
- PG
prostaglandin
- IEC
intestinal epithelial cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nature Immunology. 2003;4:269–73. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 2.Abrams GD, Bauer H, Sprinz H. Influence of the normal flora on mucosal morphology and cellular renewal in the ileum. A comparison of germ-free and conventional mice. Lab Invest. 1963;12:355–64. [PubMed] [Google Scholar]
- 3.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitajima S, Morimoto M, Sagara E, Shimizu C, Ikeda Y. Dextran sodium sulfate-induced colitis in germ-free IQI/Jic mice. Experimental Animals. 2001;50:387–95. doi: 10.1538/expanim.50.387. [DOI] [PubMed] [Google Scholar]
- 5.Sartor RB. Clinical Applications of Advances in the Genetics of IBD. Reviews in Gastroenterological Disorders. 2003;3:S9–S17. [PubMed] [Google Scholar]
- 6.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol. 2005;560:11–8. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- 8.Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 9.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. Journal of Immunology. 1999;162:3749–52. [PubMed] [Google Scholar]
- 10.Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, Werts C, Boneca IG. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5:1000–6. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005 doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 12.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology. 2004;127:224–38. doi: 10.1053/j.gastro.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Vora P, Youdim A, Thomas LS, Fukata M, Tesfay SY, Lukasek K, Michelsen KS, Wada A, Hirayama T, Arditi M, Abreu MT. beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. Journal of Immunology. 2004;173:5398–405. doi: 10.4049/jimmunol.173.9.5398. [DOI] [PubMed] [Google Scholar]
- 14.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–65. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 16.Araki A, Kanai T, Ishikura T, Makita S, Uraushihara K, Iiyama R, Totsuka T, Takeda K, Akira S, Watanabe M. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol. 2005;40:16–23. doi: 10.1007/s00535-004-1492-9. [DOI] [PubMed] [Google Scholar]
- 17.Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, Musiek ES, Milne GL, Katkuri S, DuBois RN. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280:3217–23. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subbaramaiah K, Yoshimatsu K, Scherl E, Das KM, Glazier KD, Golijanin D, Soslow RA, Tanabe T, Naraba H, Dannenberg AJ. Microsomal prostaglandin E synthase-1 is overexpressed in inflammatory bowel disease. Evidence for involvement of the transcription factor Egr-1. J Biol Chem. 2004;279:12647–58. doi: 10.1074/jbc.M312972200. [DOI] [PubMed] [Google Scholar]
- 19.Otani T, Yamaguchi K, Scherl E, Du B, Tai HH, Greifer M, Petrovic L, Daikoku T, Dey SK, Subbaramaiah K, Dannenberg AJ. Levels of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase are reduced in inflammatory bowel disease: evidence for involvement of TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2006;290:G361–8. doi: 10.1152/ajpgi.00348.2005. [DOI] [PubMed] [Google Scholar]
- 20.Newberry RD, McDonough JS, Stenson WF, Lorenz RG. Spontaneous and continuous cyclooxygenase-2-dependent prostaglandin E2 production by stromal cells in the murine small intestine lamina propria: directing the tone of the intestinal immune response. Journal of Immunology. 2001;166:4465–72. doi: 10.4049/jimmunol.166.7.4465. [DOI] [PubMed] [Google Scholar]
- 21.Riehl T, Cohn S, Tessner T, Schloemann S, Stenson WF. Lipopolysaccharide is radioprotective in the mouse intestine through a prostaglandin-mediated mechanism. Gastroenterology. 2000;118:1106–16. doi: 10.1016/s0016-5085(00)70363-5. [DOI] [PubMed] [Google Scholar]
- 22.Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O, Sartor RB. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. Journal of Clinical Investigation. 2000;105:469–78. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 2005;65:1822–9. doi: 10.1158/0008-5472.CAN-04-3671. [DOI] [PubMed] [Google Scholar]
- 24.Holla VR, Wang D, Brown JR, Mann JR, Katkuri S, DuBois RN. Prostaglandin E2 regulates the complement inhibitor CD55/decay-accelerating factor in colorectal cancer. J Biol Chem. 2005;280:476–83. doi: 10.1074/jbc.M407403200. [DOI] [PubMed] [Google Scholar]
- 25.Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, Strand D, Czaja J, Schlaak JF, Lehr HA, Autschbach F, Schurmann G, Nishimoto N, Yoshizaki K, Ito H, Kishimoto T, Galle PR, Rose-John S, Neurath MF. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–8. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 26.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Laboratory Investigation. 1993;69:238–49. [PubMed] [Google Scholar]
- 27.Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. 2002;122:2011–25. doi: 10.1053/gast.2002.33631. [DOI] [PubMed] [Google Scholar]
- 28.Moss SF, Holt PR. Apoptosis in the intestine. Gastroenterology. 1996;111:567–8. doi: 10.1053/gast.1996.v111.agast961110567. [DOI] [PubMed] [Google Scholar]
- 29.Inoue H, Nanayama T, Hara S, Yokoyama C, Tanabe T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994;350:51–4. doi: 10.1016/0014-5793(94)00731-4. [DOI] [PubMed] [Google Scholar]
- 30.Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J Biol Chem. 1995;270:24965–71. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- 31.Mukherji M. Phosphoproteomics in analyzing signaling pathways. Expert Rev Proteomics. 2005;2:117–28. doi: 10.1586/14789450.2.1.117. [DOI] [PubMed] [Google Scholar]
- 32.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 33.Rhee SH, Hwang D. Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase. J Biol Chem. 2000;275:34035–40. doi: 10.1074/jbc.M007386200. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular Toll-like receptor 4-MD-2 complex. Infection & Immunity. 2003;71:3503–11. doi: 10.1128/IAI.71.6.3503-3511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K, Dannenberg AJ. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res. 2001;7:3971–6. [PubMed] [Google Scholar]
- 36.Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69(Suppl 1):28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 37.Meyer TA, Noguchi Y, Ogle CK, Tiao G, Wang JJ, Fischer JE, Hasselgren PO. Endotoxin stimulates interleukin-6 production in intestinal epithelial cells. A synergistic effect with prostaglandin E2. Arch Surg. 1994;129:1290–4. doi: 10.1001/archsurg.1994.01420360080010. discussion 1294–5. [DOI] [PubMed] [Google Scholar]
- 38.Longo WE, Damore LJ, Mazuski JE, Smith GS, Panesar N, Kaminski DL. The role of cyclooxygenase-1 and cyclooxygenase-2 in lipopolysaccharide and interleukin-1 stimulated enterocyte prostanoid formation. Mediators Inflamm. 1998;7:85–91. doi: 10.1080/09629359891225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grossman EM, Longo WE, Mazuski JE, Panesar N, Kaminski DL. Role of cytoplasmic and secretory phospholipase A2 in intestinal epithelial cell prostaglandin E2 formation. Int J Surg Investig. 2000;1:467–76. [PubMed] [Google Scholar]
- 40.Newberry RD, McDonough JS, Stenson WF, Lorenz RG. Spontaneous and continuous cyclooxygenase-2-dependent prostaglandin E2 production by stromal cells in the murine small intestine lamina propria: directing the tone of the intestinal immune response. J Immunol. 2001;166:4465–72. doi: 10.4049/jimmunol.166.7.4465. [DOI] [PubMed] [Google Scholar]
- 41.Singer II, Kawka DW, Schloemann S, Tessner T, Riehl T, Stenson WF. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology. 1998;115:297–306. doi: 10.1016/s0016-5085(98)70196-9. [DOI] [PubMed] [Google Scholar]
- 42.Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, McKenna M, Rose T, Montrose MH. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902–12. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 43.Stern LE, Erwin CR, O'Brien DP, Huang F, Warner BW. Epidermal growth factor is critical for intestinal adaptation following small bowel resection. Microsc Res Tech. 2000;51:138–48. doi: 10.1002/1097-0029(20001015)51:2<138::AID-JEMT5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 44.Podolsky DK. Mechanisms of regulatory peptide action in the gastrointestinal tract: trefoil peptides. J Gastroenterol. 2000;35(Suppl 12):69–74. [PubMed] [Google Scholar]
- 45.Wu R, Abramson AL, Shikowitz MJ, Dannenberg AJ, Steinberg BM. Epidermal growth factor-induced cyclooxygenase-2 expression is mediated through phosphatidylinositol-3 kinase, not mitogen-activated protein/extracellular signal-regulated kinase kinase, in recurrent respiratory papillomas. Clin Cancer Res. 2005;11:6155–61. doi: 10.1158/1078-0432.CCR-04-2664. [DOI] [PubMed] [Google Scholar]
- 46.Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol. 2005;23:254–66. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 47.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–93. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 48.Tessner TG, Muhale F, Riehl TE, Anant S, Stenson WF. Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J Clin Invest. 2004;114:1676–85. doi: 10.1172/JCI22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 50.Ruemmele FM, Beaulieu JF, Dionne S, Levy E, Seidman EG, Cerf-Bensussan N, Lentze MJ. Lipopolysaccharide modulation of normal enterocyte turnover by toll-like receptors is mediated by endogenously produced tumour necrosis factor alpha. Gut. 2002;51:842–8. doi: 10.1136/gut.51.6.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grishin AV, Wang J, Hackam DJ, Qureshi F, Upperman JS, Zamora R, Ford HR. p38 MAP kinase mediates endotoxin-induced expression of cyclooxygenase-2 in enterocytes. Surgery. 2004;136:329–335. doi: 10.1016/j.surg.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 52.Grishin AV, Wang J, Potoka DA, Hackam DJ, Upperman JS, Boyle P, Zamora R, Ford HR. Lipopolysaccharide induces cyclooxygenase-2 in intestinal epithelium via a noncanonical p38 MAPK pathway. J Immunol. 2006;176:580–8. doi: 10.4049/jimmunol.176.1.580. [DOI] [PubMed] [Google Scholar]
- 53.Tessner TG, Cohn SM, Schloemann S, Stenson WF. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology. 1998;115:874–82. doi: 10.1016/s0016-5085(98)70259-8. [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285–95. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 55.Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, Ichikawa A, Narumiya S. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–93. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278:35451–7. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 57.Konturek PK, Brzozowski T, Konturek SJ, Dembinski A. Role of epidermal growth factor, prostaglandin, and sulfhydryls in stress-induced gastric lesions. Gastroenterology. 1990;99:1607–15. doi: 10.1016/0016-5085(90)90464-c. [DOI] [PubMed] [Google Scholar]
- 58.Romano M, Polk WH, Awad JA, Arteaga CL, Nanney LB, Wargovich MJ, Kraus ER, Boland CR, Coffey RJ. Transforming growth factor alpha protection against drug-induced injury to the rat gastric mucosa in vivo. J Clin Invest. 1992;90:2409–21. doi: 10.1172/JCI116132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Playford RJ, Wright NA. Why is epidermal growth factor present in the gut lumen? Gut. 1996;38:303–5. doi: 10.1136/gut.38.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Egger B, Buchler MW, Lakshmanan J, Moore P, Eysselein VE. Mice harboring a defective epidermal growth factor receptor (waved-2) have an increased susceptibility to acute dextran sulfate-induced colitis. Scand J Gastroenterol. 2000;35:1181–7. doi: 10.1080/003655200750056664. [DOI] [PubMed] [Google Scholar]
- 61.Carty E, De Brabander M, Feakins RM, Rampton DS. Measurement of in vivo rectal mucosal cytokine and eicosanoid production in ulcerative colitis using filter paper. Gut. 2000;46:487–92. doi: 10.1136/gut.46.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharon P, Ligumsky M, Rachmilewitz D, Zor U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology. 1978;75:638–40. [PubMed] [Google Scholar]
- 63.Wiercinska-Drapalo A, Flisiak R, Prokopowicz D. Effects of ulcerative colitis activity on plasma and mucosal prostaglandin E2 concentration. Prostaglandins Other Lipid Mediat. 1999;58:159–65. doi: 10.1016/s0090-6980(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 64.Sheehan KM, O'Connell F, O'Grady A, Conroy RM, Leader MB, Byrne MF, Murray FE, Kay EW. The relationship between cyclooxygenase-2 expression and characteristics of malignant transformation in human colorectal adenomas. Eur J Gastroenterol Hepatol. 2004;16:619–25. doi: 10.1097/00042737-200406000-00017. [DOI] [PubMed] [Google Scholar]
- 65.Konturek PC, Kania J, Burnat G, Hahn EG, Konturek SJ. Prostaglandins as mediators of COX-2 derived carcinogenesis in gastrointestinal tract. J Physiol Pharmacol. 2005;56(Suppl 5):57–73. [PubMed] [Google Scholar]
- 66.Wang D, Mann JR, DuBois RN. The role of prostaglandins and other eicosanoids in the gastrointestinal tract. Gastroenterology. 2005;128:1445–61. doi: 10.1053/j.gastro.2004.09.080. [DOI] [PubMed] [Google Scholar]
- 67.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–10. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 68.Malecka-Panas E, Kordek R, Biernat W, Tureaud J, Liberski PP, Majumdar AP. Differential activation of total and EGF receptor (EGF-R) tyrosine kinase (tyr-k) in the rectal mucosa in patients with adenomatous polyps, ulcerative colitis and colon cancer. Hepatogastroenterology. 1997;44:435–40. [PubMed] [Google Scholar]
- 69.Matuk R, Crawford J, Abreu MT, Targan SR, Vasiliauskas EA, Papadakis KA. The spectrum of gastrointestinal toxicity and effect on disease activity of selective cyclooxygenase-2 inhibitors in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:352–6. doi: 10.1097/00054725-200407000-00005. [DOI] [PubMed] [Google Scholar]
- 70.Sandborn W, Stenson W, Brynskov J, Steidle G, Robbins J. Safety of celecoxib in patients with ulcerative colitis in remission: a randomized, double-blind, placebo-controlled study. Clin Gastro and Hep. 2006 doi: 10.1016/j.cgh.2005.12.002. In Press. [DOI] [PubMed] [Google Scholar]
- 71.Velayos FS, Terdiman JP, Walsh JM. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies. Am J Gastroenterol. 2005;100:1345–53. doi: 10.1111/j.1572-0241.2005.41442.x. [DOI] [PubMed] [Google Scholar]
- 72.Matula S, Croog V, Itzkowitz S, Harpaz N, Bodian C, Hossain S, Ullman T. Chemoprevention of colorectal neoplasia in ulcerative colitis: the effect of 6-mercaptopurine. Clin Gastroenterol Hepatol. 2005;3:1015–21. doi: 10.1016/s1542-3565(05)00738-x. [DOI] [PubMed] [Google Scholar]