

SUMMARY
The facultative pathogen Vibrio cholerae can exist in both the human small bowel and in aquatic environments. While investigation of the infection has revealed many factors important for pathogenesis, little is known regarding transmission of this nor of other water-borne pathogens. Here we focus on the late stage of infection using a temporally controlled reporter of transcription, and identify a unique class of V. cholerae genes that are specific to this stage. Mutational analysis revealed few roles for these genes in infection. However, using a novel host-to-environment transition assay we detected roles for six of ten genes examined in persistence within cholera stool and/or within aquatic environments. Our results further indicate that passage through the intestinal tract is necessary to observe this phenotype. Thus, V. cholerae has evolved mechanisms that are advantageous for life in aquatic environments, which are expressed prior to exiting the host intestinal tract.
INTRODUCTION
Cholera is caused by the Gram-negative bacterium Vibrio cholerae (Koch, 1884). This facultative pathogen resides in two dissimilar habitats: aquatic ecosystems and the human gastrointestinal (GI) tract. The ability to survive harsh conditions in the GI tract is facilitated by the in situ induction of genes, as well as by the induction of genes in previous cholera victims that increase the infectivity of V. cholerae during transmission (Butler et al., 2006; Merrell et al., 2002). V. cholerae uses motility to make contact with the small bowel epithelium (Freter et al., 1981) whereupon lipopolysaccharide (LPS) and the carbohydrate binding protein GbpA aid in attachment to the lumenal surface (Benitez et al., 1997; Kirn et al., 2005; Schild et al., 2005). Later in infection cholera toxin (CT) is expressed causing a massive secretory diarrhea that can lead to hypotensive shock and death within 12h of the onset of symptoms (Bennish, 1994). The diarrhea aids in expulsion of bacteria from the infected host thereby facilitating transmission to new hosts.
A variety of V. cholerae colonization factors have been identified and characterized using the infant mouse and rabbit ligated ileal loop models of infection. These include the toxin-coregulated pilus (TCP), accessory colonization factors, outer membrane porins and LPS (Reidl and Klose, 2002). Induction of TCP, CT and other virulence factors during infection is dependent on two transmembrane regulators ToxR and TcpP and on cytoplasmic regulators AphA/ B and ToxT (Reidl and Klose, 2002).
It was recently shown that virulence gene expression is also modulated by the intracellular second messenger cyclic diguanylate (c-di-GMP), whereby low levels of c-di-GMP lead to increased toxT expression and thus virulence gene activation (Tischler and Camilli, 2005). In addition, quorum sensing regulates virulence gene expression. At high cell density in the late stage of infection, virulence genes are repressed while the HapA protease involved in detachment from the epithelium is induced. This inverse regulation is mediated by the regulator HapR (Zhu et al., 2002) whose expression, in turn, is controlled by the Lux quorum sensing system. However, since virulent V. cholerae strains with null mutations in hapR have been isolated (Joelsson et al., 2006), the function of HapR is not required for pathogenesis.
Upon being shed by humans into aquatic ecosystems, V. cholerae faces nutrient limitation and shifts in temperature and osmolarity. In the environment it is believed that V. cholerae is associated primarily with phytoplankton, zooplankton, crustaceans and insect egg masses (Reidl and Klose, 2002). The mannose sensitive hemagglutinin (MSHA), another type IV pilus, has been shown to promote adherence to the chitinous surfaces of aquatic organisms (Chiavelli et al., 2001), and other adherence factors like GbpA and PilA have also been implicated in this process (Kirn et al., 2005; Meibom et al., 2004). Recently an important regulon in chitin utilization was identified, which includes secreted chitinases, a chitoporin, and uptake systems for breakdown products of chitin that serve as carbon and nitrogen sources (Berg et al., 2007; Li and Roseman, 2004; Meibom et al., 2004). Additionally, persistence of V. cholerae in the environment is likely aided by the formation of surface-attached communities called biofilms (Watnick and Kolter, 1999; Yildiz and Schoolnik, 1999). Biofilm formation is favored by high levels of c-di-GMP and is therefore inversely regulated with virulence genes (Tischler and Camilli, 2004). The concentration of c-di-GMP is controlled by the opposing activities of diguanylate cyclases (GGDEF proteins) and phosphodiesterases (Romling and Amikam, 2006).
While much has been revealed regarding the environmental persistance of V. cholerae and its transition from the environment into the GI tract, little is known about the equally important reverse transition. Recent studies indicate that V. cholerae leaves the host in a hyperinfectious state, and that the transcriptome of human shed V. cholerae differs substantially from in vitro grown V. cholerae (Alam et al., 2005; Butler et al., 2006; Merrell et al., 2002). Using the rabbit ileal loop model, in which V. cholerae grows in an anatomically closed compartment, a distinct phenotype was recently described whereby V. cholerae undergo a stationary growth phase controlled detachment from the mucosal surface into the lumen (Nielsen et al., 2006a).
We wished to explore in greater detail the physiology and virulence of V. cholerae during late stages of infection prior to its transition into aquatic environments using a more natural, anatomically open intestinal system. Specifically, we asked whether V. cholerae expresses genes late in the infection of infant mice that either facilitate exit from the host or enhance subsequent survival in aquatic environments. There exist several methods to identify genes expressed during infection (Chiang and Mekalanos, 1998; Hang et al., 2003; Merrell et al., 2002; Nielsen et al., 2006a; Xu et al., 2003a). We used a variant of one of these, called recombination-based in vivo expression technology (RIVET), which functions as a genetic switch reporter of gene induction (Camilli et al., 1994). The temporal expression patterns of several of the identified in vivo induced genes of V. cholerae have been studied in detail and shown to have an early induction within the first 5h of infection of mice (Lee et al., 1998; Lee et al., 1999). In this report, we adapt RIVET to specifically identify V. cholerae genes induced at later times of infection. Although some of these late genes are shown to play roles in fitness within the GI tract, the majority are dispensable for infection. Instead, we observed that many late genes enhance the fitness of V. cholerae in aquatic environments, indicating that this pathogen initiates a program of gene expression late in the infection that augments survival upon entry into aquatic environments.
RESULTS
Identification of late genes
The RIVET is used to identify genes expressed during infection based on the TnpR resolvase-mediated excision of a gene reporter cassette flanked by res sequences (Fig. 1A and (Camilli and Mekalanos, 1995)). To identify V. cholerae genes that are transcriptionally induced late in the infection, we used a previously constructed plasmid library containing random genomic fragments inserted upstream of a promoterless tnpR (Osorio et al., 2005). Mobilization of this library into a res -cassette containing V. cholerae strain results in homologous recombination into the genome and thus the generation of random transcriptional fusions of chromosomal genes to tnpR. Induction of a tnpR fusion leads to excision and loss of the res-cassette containing two reporter genes: neo and sacB which confer kanamycin resistance (KnR) and sucrose sensitivity (SucS), respectively. Therefore, the loss of the res-cassette can be monitored by a phenotypic change to KnS and SucR. Because we were interested in the later stages of infection when cell density may be high, we used the HapR+ V. cholerae strain E7946, hereafter referred to as AC53 (Miller et al., 1989; Vance et al., 2003).
Figure 1. RIVET screen for late genes.
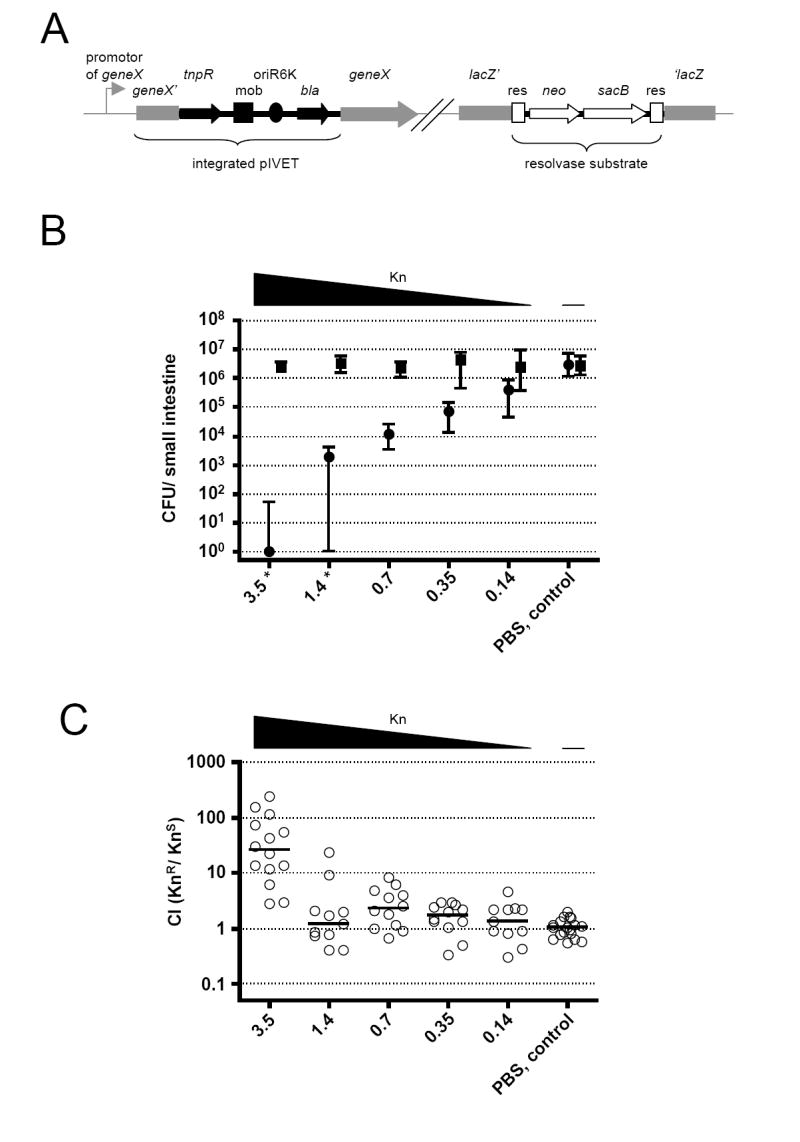
(A) Illustration of the genetic components of RIVET (Osorio et al., 2005). Chromosomal sequences are in gray, pIVET parts are in black, and the res-cassette parts are in open shapes. pIVET is integrated into V. cholerae hypothetical geneX via homologous recombination resulting in a merodiploid in which geneX and tnpR (resolvase) are transcriptionally fused and controlled by the chromosomal promotor of geneX. The mobilization (mob), origin of replication (oriR6K) and ApR (bla) regions of pIVET, as well as the gene for KnR (neo), SucS (sacB) and the target sites of resolvase (res) of the res-cassette are indicated.
(B) Shown are the medians of recovered KnR (boxes) and KnS (circles) bacteria at 24h post-infection. The different amounts of Kn given at 7h post-infection are indicated on the x-axis. ≥10 mice from three independent experiments comprise each data set. Error bars indicate interquartile ranges. There was a significant decrease in colonization for the KnS strain compared to the PBS control for Kn concentrations ≥0.35mg/kg bw (p<0.01, using a Kruskal-Wallis test and a post-hoc Dunn’s Multiple comparisons), whereas the KnR bacteria were unaffected (p>0.05, using a Kruskal-Wallis test). * The level of KnS bacteria was below detection limit of 1 CFU for some animals, but in order to plot the data the CFU/ml were set to 1.
(C) Competition indices (CI) at 24h post-infection from the small bowel of infant mice. 4h before infection mice were given orally the same concentrations of Kn used in Panel B. Each circle represents the CI from a single animal. Horizontal bars indicate the median of each data set. Only the 3.5mg/kg bw data set is significantly different from the PBS control (p<0.01, using tests in panel B legend).
Induction of tnpR fusions early during infection leads to KnS bacteria appearing early, whereas strains with uninduced tnpR fusions remain KnR. Therefore, to remove all strains harboring early gene-tnpR fusions, mice were treated with Kn at 7h post-infection. This time point is after attachment and other early colonization events have occurred, including the induction of known virulence genes such as ctxAB and tcpA (Angelichio et al., 2004), which encode CT and the TCP pilin subunit, respectively. The early induction of these genes was confirmed for the strain used in this study (Table 1). The removal of early gene-tnpR fusion strains demands a certain Kn concentration that can reduce the number of KnS, but not KnR bacteria, without remaining active in the small bowel over a long period. To identify such a concentration mice were infected with a 1:1 mixture of KnR:KnS V. cholerae followed by treatment at 7h with Kn at various concentrations. The number of V. cholerae colony-forming units (CFU) recovered from the small bowel at 24h are shown in Fig. 1B. As expected in the PBS control group the KnR and KnS strains colonized and proliferated equally well. With increasing amounts of Kn the KnS CFU decreased, eventually falling over a million-fold to <1. This occurred for 3/11 mice in the 1.4mg/kg body weight (bw) and for 8/10 mice in the 3.5mg/kg bw groups. In contrast the KnR CFU recovered did not change at any of the Kn concentrations used, implying that colonization of KnR bacteria was unaffected.
Table 1. V. cholerae genes induced during infection.
Shown are medians of the percent resolution for individual gene-tnpR fusion strains in each condition and/or time point of at least three independent resolution assays. Also shown are ratios of in vivo/in vitro resolution medians. The criteria used to define early versus late genes are listed. Each group is sorted by the gene locus.
| gene locusb | operonc | annotation/ gene symbolb | resolution in % a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| median (in vitrod) | in vivo | median (21h)/median (in vitro)e | median (21h)/median (7h)e | median (21h)/median (5h) | |||||
| median (5h) | median (7h) | median (21h) | |||||||
| early infection-induced control genes | |||||||||
| VC0828 | toxin co-regulated pilin (tcpA) | 0.9 | 99.0 | 98.5 | n.d.f | n.d. | n.d. | n.d. | |
| VC1457 | VC1456-7 | cholera enterotoxin, A subunit (ctxA) | 8.5 | 91.5 | 98.9 | n.d. | n.d. | n.d. | n.d. |
| late infection-induced genes - (median 21h/median 7h) >2 | |||||||||
| VC0044 | VC0041-6 | sun protein (sun) | 26.1 | n.d. | 8.6 | 61.1 | 2.3 | 7.1 | n.d. |
| VC0130 | GGDEF family protein | 0 | n.d. | 2.3 | 63.9 | 63.9 | 27.7 | n.d. | |
| VC0203 | VC0200-3 | iron(III) ABC transporter, permease protein | 18.9 | n.d. | 13.3 | 79.9 | 4.2 | 6 | n.d. |
| VC0280 | VC0280-1 | cadaverine/lysine antiporter CadB, putative | 0 | n.d. | 0 | 28.3 | 28.3 | 28.3 | n.d. |
| VC0353* | conserved hypothetical protein, 40bp to VC0351 on minus strand | 1.1 | n.d. | 1.2 | 50 | 47.2 | 42.4 | n.d. | |
| VC0376 | conserved hypothetical protein | 2 | n.d. | 2.9 | 19 | 9.5 | 6.6 | n.d. | |
| VC0384 | VC0383-6 | sulfite reductase (NADPH) flavoprotein alpha-component (cysJ) | 14 | n.d. | 14.8 | 50.9 | 3.6 | 3.4 | n.d. |
| VC0404 | VC0398-414 | MSHA biogenesis protein MshN | 42.4 | n.d. | 39.9 | 93.6 | 2.2 | 2.3 | n.d. |
| VC0426 | hypothetical protein | 14.5 | n.d. | 5.7 | 54.3 | 3.7 | 9.5 | n.d. | |
| VC0554 | VC0553-4 | protease, insulinase family-protease, insulinase family | 1.5 | n.d. | 5.3 | 75.3 | 51.2 | 14.3 | n.d. |
| VC1093 | VC1091-5 | oligopeptide ABC transp., permease (oppC) | 1.4 | n.d. | 0 | 75.4 | 55.8 | 75.4 | n.d. |
| VC1169 | VC1169-75 | tryptophan synthase, alpha subunit (trpA) | 0.9 | n.d. | 20.7 | 84.7 | 92.1 | 4.1 | n.d. |
| VC1212* | DNA polymerase II (polB), overlap to VC1211 | 0 | n.d. | 5.8 | 30.9 | 30.9 | 5.3 | n.d. | |
| VC1317 | conserved hypothetical protein | 6.6 | n.d. | 4.3 | 53.9 | 8.2 | 12.6 | n.d. | |
| VC1822 | PTS system, fructose-specific IIABC component (frwABC) | 20.4 | n.d. | 19.9 | 85.3 | 4.2 | 4.3 | n.d. | |
| VC1906 | hypothetical protein | 5.8 | n.d. | 3.3 | 30.3 | 5.2 | 9.1 | n.d. | |
| VC1926 | VC1924-6 | C4-dicarboxylate transport transcriptional regulatory protein (dctD -1) | 7.1 | n.d. | 5.3 | 78 | 10.9 | 14.8 | n.d. |
| VC1975 | VC1971-6 | 2-oxoglutarate decarboxylase /2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase (menD) | 1.4 | n.d. | 47.3 | 99.5 | 70.1 | 2.1 | n.d. |
| VC2072* | peptidase, insulinase family, overlap to VC2073 on minus strand | 5.6 | n.d. | 0 | 51.6 | 9.2 | 51.6 | n.d. | |
| VC2118 | adenine-specific methylase, putative | 2.7 | n.d. | 0 | 12.9 | 4.9 | 12.9 | n.d. | |
| VC2161 | methyl-accepting chemotaxis protein | 13.7 | n.d. | 3.9 | 35.9 | 2.6 | 9.2 | n.d. | |
| VC2167 | VC2164-7 | hypothetical protein | 0 | n.d. | 30.2 | 67.1 | 67.1 | 2.2 | n.d. |
| VC2209 | nonribosomal peptide synthetase VibF, authentic frameshift (vibF) | 7.6 | n.d. | 1.7 | 15.4 | 2 | 9.1 | n.d. | |
| VC2294 | VC2288-95 | NADH:ubiquinone oxidoreductase | 20 | n.d. | 25.9 | 78.3 | 3.9 | 3 | n.d. |
| VC2300 | AmpG protein, putative | 39.3 | n.d. | 10.5 | 88.9 | 2.3 | 8.4 | n.d. | |
| VC2369 | VC2369-71 | sensor histidine kinase FexB (fexB) | 7.3 | n.d. | 31.8 | 66.2 | 9.1 | 2.1 | n.d. |
| VC2376 | VC2376-7 | glutamate synthase, large subunit (gltB -2) | 49.43 | n.d. | 49.01 | 98.93 | 2.0 | 2.0 | n.d. |
| VC2538 | VC2537-9 | thiamine transport system permease protein | 37.3 | n.d. | 25 | 85.3 | 2.3 | 3.4 | n.d. |
| VC2697 | GGDEF family protein | 13.9 | n.d. | 2.3 | 57.9 | 4.2 | 25.3 | n.d. | |
| VC2739 | conserved hypothetical protein | 3.9 | n.d. | 0.6 | 43.1 | 11.2 | 74.4 | n.d. | |
| VCA0033 | hypothetical protein | 1.6 | n.d. | 4.2 | 21.8 | 13.9 | 5.2 | n.d. | |
| VCA0043 | conserved hypothetical protein | 6.8 | n.d. | 2.1 | 60.5 | 8.9 | 29.5 | n.d. | |
| VCA0083 | multidrug resistance protein D (emrD -1) | 37.4 | n.d. | 32.3 | 94.3 | 2.5 | 2.9 | n.d. | |
| VCA0182* | sigma-54 dependent transcriptional regulator, VCA0181 on minus | 1.4 | n.d. | 8.6 | 55.7 | 40.4 | 6.5 | n.d. | |
| VCA0183 | ferrisiderophore reductase (hmpA) | 6.3 | n.d. | 3.7 | 25 | 4 | 6.7 | n.d. | |
| VCA0630 | VCA0630-1 | D-isomerspecific 2-hydroxyacid dehydrogenase family protein | 37.3 | n.d. | 12.7 | 92.5 | 2.5 | 7.3 | n.d. |
| VCA0686 | VCA0685-7 | iron(III) ABC transporter, permease protein | 1.6 | n.d. | 12 | 46.3 | 29.8 | 3.9 | n.d. |
| VCA0744 | glycerol kinase (gplK) | 7.5 | n.d. | 8.6 | 26.7 | 3.6 | 3.1 | n.d. | |
| VCA0766 | VCA0764-6 | cytochrome c554 | 2.2 | n.d. | 6 | 17.7 | 8.2 | 3 | n.d. |
| VCA0907 | VCA0907-9 | heme-binding protein HutZ (hutZ) | 3.6 | n.d. | 0 | 32 | 9 | 32 | n.d. |
| VCA0920 | conserved hypothetical protein | 6.3 | n.d. | 6 | 47.9 | 7.6 | 8 | n.d. | |
| VCA0955 | transcriptional regulator, MarR family | 7.1 | n.d. | 9.2 | 69.2 | 9.8 | 7.5 | n.d. | |
| VCA0956* | GGDEF family protein, 100bp to VCA0957 on minus strand | 9 | n.d. | 1 | 26.6 | 2.9 | 28 | n.d. | |
| VCA0980 | hypothetical protein | 2.4 | n.d. | 8.8 | 67.3 | 28.1 | 7.6 | n.d. | |
| VCA1046 | VCA1045-7 | mannitol-1-phosphate 5-dehydrogenase (mtlD) | 21.4 | n.d. | 7.7 | 68.8 | 3.2 | 8.9 | n.d. |
| VCA1071 | VCA1071-2 | sodium-proline symporter (putP) | 18.6 | n.d. | 12.4 | 68.2 | 3.7 | 5.5 | n.d. |
| VCA1110 | VCA1105-11 | long-chain-fatty-acid--CoA ligase, putative | 3.2 | n.d. | 3.6 | 12.3 | 3.9 | 3.5 | n.d. |
| VCA1115* | ParA family protein, 100bp to VCA1113 on minus strand | 0 | n.d. | 8.6 | 72.8 | 72.8 | 8.5 | n.d. | |
| late infection-induced genes - (median 21h/median 5h) >2 | |||||||||
| VC0612 | VC0611-20 | cellulose degradation product phosphorylase | 28.0 | 6.5 | 43.8 | 75.0 | 2.7 | 1.7 | 11.5 |
| VC1593 | VC1592-3 | GGDEF family protein | 8.8 | 7.5 | 76.5 | 94.1 | 10.7 | 1.2 | 12.5 |
| VC2191 | VC2190-200 | flagellar hook-associated protein FlgM | 13.4 | 5.4 | 47.4 | 88.4 | 6.6 | 1.9 | 16.5 |
| VC2330 | conserved hypothetical protein | 9.8 | 3.1 | 16.7 | 21.4 | 2.2 | 1.3 | 7.0 | |
| VC2371 | VC2369-71 | conserved hypothetical protein | 12.7 | 22.3 | 86.8 | 98.8 | 7.8 | 1.1 | 4.4 |
| VC2621 | hypothetical protein | 0.0 | 3.4 | 51.7 | 90.1 | 90.1 | 1.7 | 26.4 | |
| VCA0601 | VCA0601-2 | ABC transporter, permease protein | 6.2 | 1.3 | 53.8 | 88.2 | 14.3 | 1.6 | 66.0 |
| VCA0756 | transcriptional regulator, LysR family | 3.0 | 7.6 | 10.1 | 17.3 | 5.8 | 1.7 | 2.3 | |
| VCA0985 | VCA0984-5 | oxidoreductase-iron-sulfur cluster-binding protein | 13.0 | 22.3 | 43.3 | 88.0 | 6.7 | 2.0 | 3.9 |
| early infection-induced genes - (median 21h/median 5h) <2 | |||||||||
| VC0066 | VC0061-6 | thiH protein (thiH) | 7.3 | 48.2 | 60.0 | 78.6 | 10.8 | 1.3 | 1.6 |
| VC1667 | VC1664-7 | conserved hypothetical protein | 48.1 | 64.5 | 73.8 | 97.9 | 2.0 | 1.3 | 1.5 |
| VC1687 | VC1686-7 | conserved hypothetical protein | 27.9 | 93.2 | 92.3 | 94.3 | 3.4 | 1.0 | 1.0 |
| VC1692 | VC1692-94 | trimethylamine-N-oxide reductase (torA) | 6.6 | 93.7 | 93.8 | 92.0 | 14.0 | 1.0 | 1.0 |
| VC2712 | xanthine-uracil permease family protein | 18.2 | 89.7 | 96.5 | 100.0 | 5.5 | 1.0 | 1.1 | |
percentage of resolution is given by the amount of SmR/KnS CFU (given by SmR/ApR CFU minus SmR/KnR CFU) divided by the amount of SmR/ApR CFU
gene locus and annotation/gene symbol according to The Institute of Genomic Research TIGR (http://cmr.tigr.org/tigr-scripts/CMR/GenomePage.cgi?database=gvc)
operon prediction according to http://www.microbesonline.org/operons/gnc666.named (Alm et al., 2005)
percentage of resolution after 8h growth in LB with aeration
in cases where the median of the divisor would be 0 it was set to 1 to allow calculating ratios of the medians.
not determined (n.d.)
the orientation of tnpR was the opposite of the transcription of indicated genes, however genes in the same orientation as tnpR overlap or are within 100bp of the insertion site
To address how long the Kn remains active in vivo, we treated mice with Kn or PBS 4h prior to infection with a 1:1 mix of KnR:KnS strains. The results expressed as a competitive index (CI) (KnR/KnS CFU) are shown in Fig. 1C. The median CI for the PBS control group and the groups treated with Kn up to 1.4mg/kg bw was ~1, indicating no significant effect on colonization of either strain. However, 3.5mg/kg bw caused a 25-fold decrease in colonization of the KnS strain indicating that Kn was still active in the small bowel at the time of infection.
Taken together these experiments revealed that 1.4mg/kg bw is an optimal Kn concentration, allowing a 1000-fold reduction of the KnS bacteria without affecting the colonization of KnR bacteria, and not remaining active longer than 4h after application. Therefore this concentration was chosen for the RIVET screen for late genes. A total of 12,000 tnpR fusion strains were screened in mice. After sequencing the genomic insertion site of tnpR in ~200 resolved strains we identified 116 fusions to different genes. For 107 of these we were able to recover the integrated plasmid containing the tnpR-fusion, which we then used to reconstruct the original unresolved strain. To eliminate false positives we assayed resolution of the reconstructed strains after 8h growth in Luria broth (LB). 19 of the reconstructed strains gave an in vitro resolution of ≥50% and were not tested in vivo (herein in vivo refers to mouse infection). 79 of the remaining 88 gene fusions were plus stranded and were tested for resolution in vivo. 62 of the fusions gave an in vivo resolution ≥2-fold higher than that in vitro and were considered to be infection-induced (Table 1).
We determined the time interval of late gene induction in vivo . To compare the resolution data to previously known early genes we included tnpR fusions to ctxA and tcpA, which resolve to >90% within the first 5h (Table 1 and (Lee et al., 1999)). In contrast to ctxA and tcpA, most of the fusions identified in this screen showed a low or moderate in vivo resolution at 5 or 7h compared to at 24h. Late genes were defined by a ≥2-fold increase in resolution in vivo from 7 to 24h, or from 5 to 24h. 57 fusions fulfilled these requirements whereas 5 fusions already showed high expression levels at 5h and were designated early genes (Table 1). 14 of the late genes are hypothetical or conserved hypothetical, 14 are predicted to be involved in metabolic pathways, 10 are associated with transport systems, 3 are putative transcriptional regulators, 2 are implicated in motility and 4 are involved (or are within operons involved) in c-di-GMP metabolism.
Quantitation of induction of late genes
To confirm and quantify the in vivo induction of late genes identified in our screen we measured steady-state transcript levels by quantitative real-time RT-PCR (qRT-PCR). We measured the expression of VCA0980 as a representative of late genes that play roles in colonization, as well as six late genes that play roles in fitness in stool and/or pond water (see below). All but one of the genes tested show a significant increase in transcript level in vivo compared to in vitro (Fig. 2A). The 2-fold increase observed for VC0612 was not significant. However, since this is the penultimate gene of a large operon (Alm et al., 2005; Meibom et al., 2004), we also measured the transcript level for the predicted first gene in the operon VC0620, which revealed a 7-fold in vivo induction over in vitro. The in vivo induction of the other genes ranged from 3-fold for VCA0686 to 300-fold for VCA0601. In order to eliminate the possibility that the observed gene regulation is strain specific, we also obtained qRT-PCR data on three late genes in another V. cholerae strain AC51, which was isolated from a cholera patient in a different continent (Fig. 2B). The results show that these late genes are induced in vivo at a magnitude similar to that observed for strain AC53.
Figure 2. Validation of late genes by qRT-PCR.
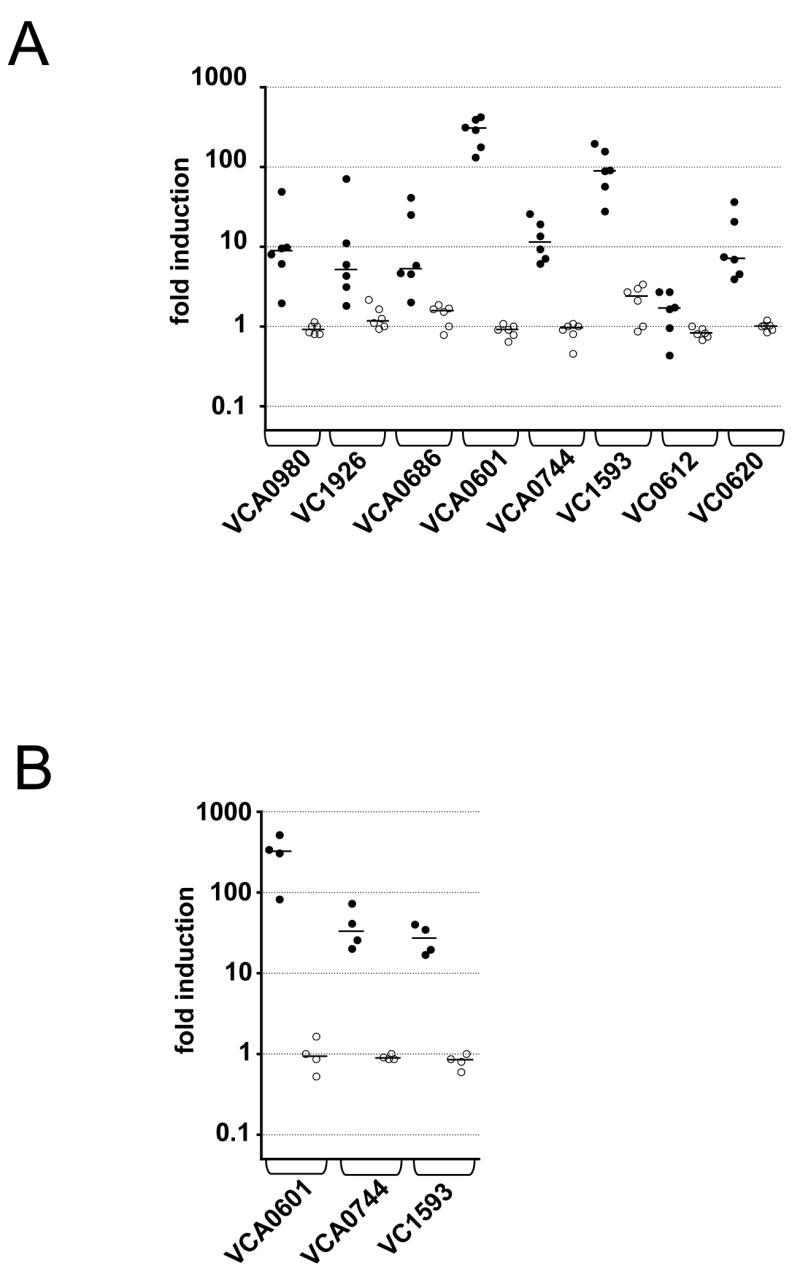
Shown is the ratio of transcripts of the late genes indicated on the x-axis to rpoB (control) in vivo at 24h post-infection (filled circles) and for log-phase in vitro LB cultures (open circles). Each data set was normalized to one randomly selected in vitro reference sample. Expression was analyzed in two different V. cholerae strains; (A) AC53 and (B) AC51. Each circle represents the qRT-PCR result from an independent culture or mouse output. The horizontal bar indicates the median of each data set. Except for VC0612 all in vivo data sets are significant different compared to the respective in vitro data set (p<0.05, using a Mann-Whitney U-test).
Roles of late genes in infection
To investigate if some of the identified late genes are involved in colonization of the infant mouse small bowel, we constructed in-frame deletions of 22 genes and tested each mutant against the virulent, marked strain AC53res1 in in vivo and in vitro competitions. Interestingly, only three mutants had an in vivo defect (Fig. 3A, supplementary data Fig. S1A and data not shown). Two of these, ΔVCA0920 and ΔVCA0980, exhibited a 2-3-fold colonization defect at 24h, but not at 7h nor in vitro. Expression of the respective genes in trans rescued colonization for both mutants. The third, VC0201, is the ATPase component of a ferrichrome transport system (Rogers et al., 2000). Deletion of this gene resulted a 40-fold colonization defect at 24h, but only a mild 3-fold defect at 7h. Expression of VC0201 in trans gave a partial rescue of the defect. Although we did not directly hit VC0201 in our screen, it is in an operon with the identified VC0203, which encodes the permease component of the transport system. This same operon was recently found to be induced during human infection as well (Lombardo et al.). Interestingly, the colonization defect of all three late gene mutants was most pronounced late in the infection. This stands in contrast to the early gene tcpA mutant where there is already a massive 400-fold attenuation by 7h (Fig. 3B) and the defect increases only another 25-fold between 7 and 24h.
Figure 3. Some late gene mutants are attenuated late in infection.
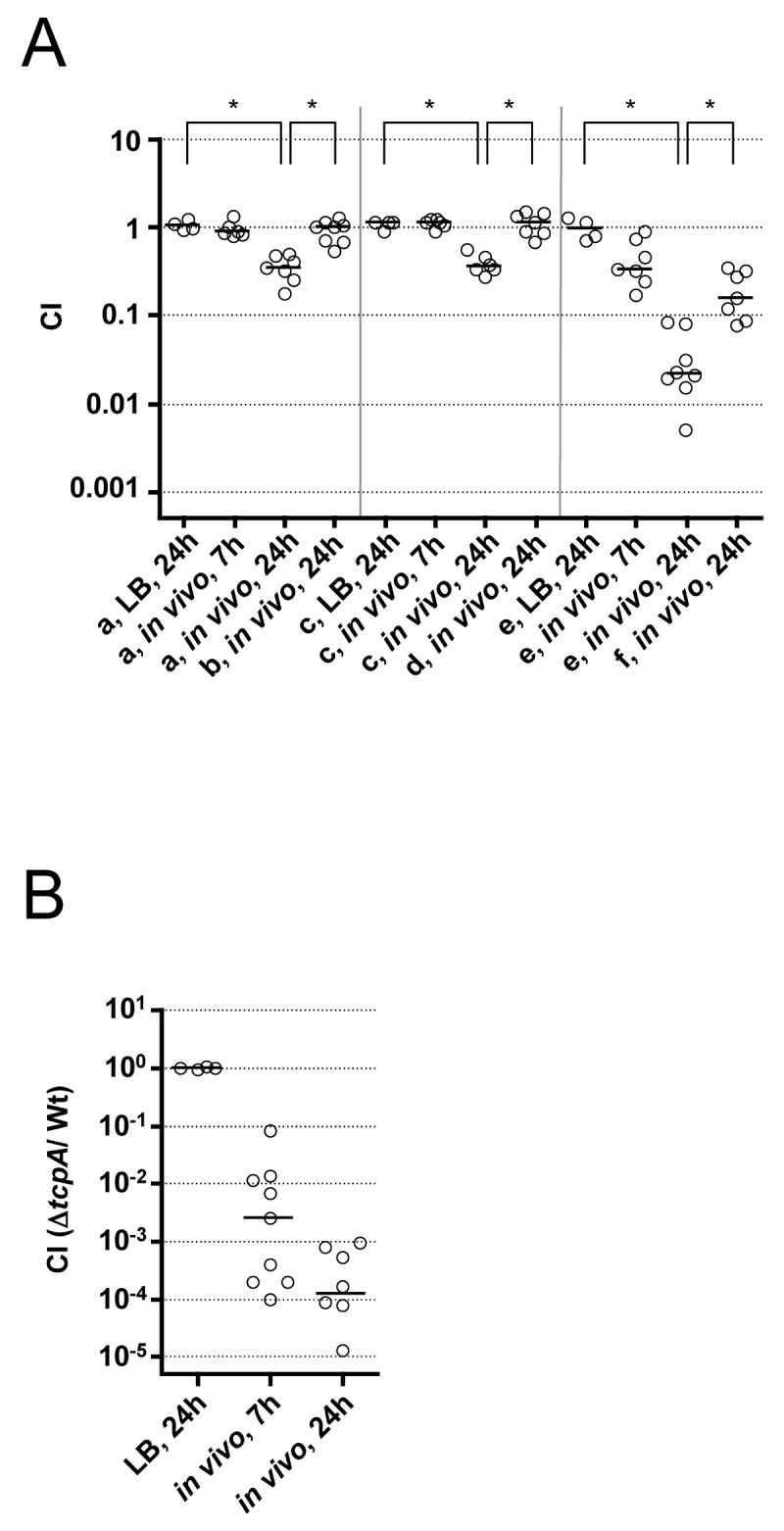
Results are shown as the CI at given time points for competitions in LB and in vivo using the infant mouse model. Each circle represents the CI from a single assay. The horizontal bars indicate the median of each data set. The asterisks indicate significantly different medians of the compared data sets indicated by the line below each asterisk (p<0.01, using a Mann-Whitney U-test).
(A) Abbreviations stand for the following competitions: a) ΔVCA0920/AC53res1; b) ΔVCA0920 (pVCA0920)/AC53res1 (pMMB67EH); c) ΔVCA0980/AC53res1; d) ΔVCA0980 (pVCA0980)/AC53res1 (pMMB67EH); e) ΔVC0201/AC53res1; f) ΔVC0201 (pVC0201)/AC53res1 (pMMB67EH). (B) CI for the early infection-induced gene mutant Δ tcpA competed against AC53res1 in LB and in vivo .
Roles of late genes in metabolism
The predicted functions for three of the late genes suggested phenotypes that could be tested in vitro. These include VC1926, annotated as a C4-dicarboxylate transport transcriptional regulator, and VCA0744, which shows 79.5% amino acid identity to a glycerol kinase of Escherichia coli. VC1926 is predicted to form a two component system with VC1925. By analogy to an orthologous system in Rhodobacter capsulatus, VC1926 should act as an activator of the cis dctPQM operon (VC1927-9) encoding a putative transporter of malate, succinate and fumarate (Forward et al., 1997; Hamblin et al., 1993). To determine the functions of VC1926 and VCA0744 in carbon acquisition we competed the respective mutants against AC53res1 in M9 minimal medium (M9) with various sole carbon sources (Fig. 4A and B). ΔVC1926 grew normally in M9 plus glycerol (Gly) but was defective for growth in M9 plus the C4-dicarboxylate succinate. In contrast, ΔVCA0744 failed to grow in M9 plus Gly but grew in M9 plus glucose (Glc). The observed growth defects could be complemented by expression of VC1926 or VCA0744 in trans. Furthermore, VC0612 was identified to be a late gene (Table 1), which encodes an orthologue of a Vibrio furnissii cytoplasmic N, N -diacetylchitobiose phosphorylase (Park et al., 2000). However, a ΔVC0612 strain shows only a mild growth defect in M9 plus chitin (Fig. 4C). Since the upstream gene VC0613 encodes an orthologue of a V. furnissii periplasmic β-N-acetylglucosaminidase (Keyhani and Roseman, 1996), we tested the ΔVC0612-3 double mutant and observed a 40-fold attenuation in M9 plus chitin (Fig. 4C). Expression of VC0612 in trans in the double mutant partially rescued the phenotype. As expected, the double mutant grows normally if Gly or N-acetylglucosamine (GlcNAc) is given as a carbon source. The growth defect of ΔVC0612-3 is consistent with induction of this operon in V. cholerae in the presence of chitin or GlcNAc-oligomers and with their predicted role in utilization of the chitin-derived substrates chitobiose (GlcNAc2) and GlcNAc-oligomers (Meibom et al., 2005).
Figure 4. In vitro growth defects of late gene mutants using specific carbon sources.
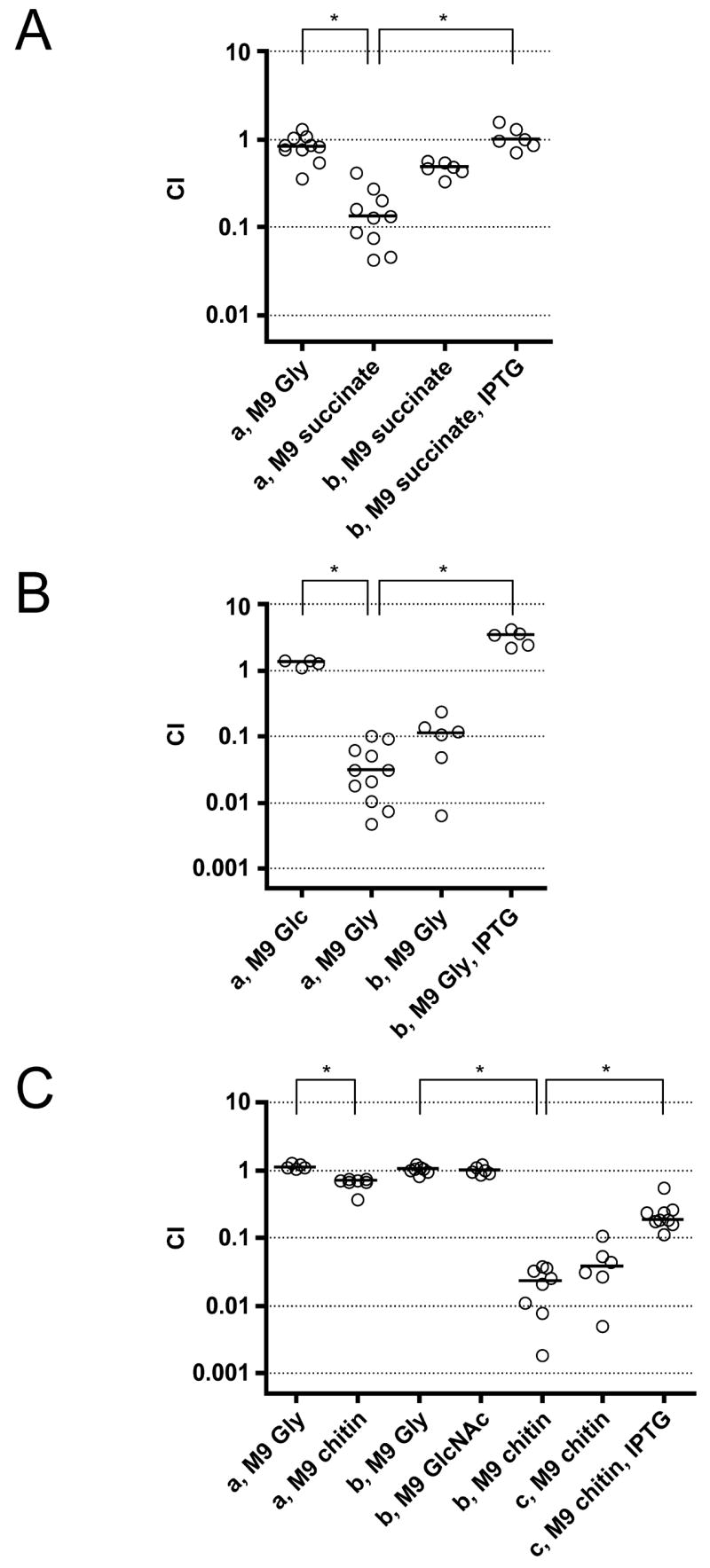
Competition assays performed in M9 minimal media with the carbon sources indicated. Each circle represents the value from a single competition experiment. The horizontal bar shows the median of each data set. The asterisks indicate significantly different medians between the data sets indicated (p<0.01, using a Mann-Whitney U-test).
- (A) a) ΔVC1926/AC53res1, b) ΔVC1926 (pVC1926)/AC53res1 (pMMB67EH)
- (B) a) ΔVCA0744/AC53res1, b) ΔVCA0744 (pVCA0744)/AC53res1 (pMMB67EH)
- (C) a) ΔVC0612/AC53res1, b) ΔVC0612-3/AC53res1, c) ΔVC0612-3 (pVC0612)/AC53res1 (pMMB67EH)
Roles of late genes in the transition to aquatic environments
Since most of the late genes did not play a detectable role in colonization or maintenance of the infection we hypothesized that some of these genes may play a role in transitioning to aquatic environments, and furthermore, that pre-induction of those genes late in infection may provide a fitness advantage when the bacterium encounters the aquatic environment. To test this hypothesis, we passaged AC53res1 and late gene deletion mutants in single strain infections. At 24h bacteria were harvested from the small bowels and mutant and AC53res1 were mixed 1:1. This mix was used as the inoculum in a variety of survival and growth assays. We focused on two different conditions V. cholerae is likely to face after detaching from the small bowel epithelium: a short incubation in rice-water stool and a longer persistence in pond water. We detected a 2-3-log drop in CFU after the 2h incubation in stool indicating this is a hostile environment. In contrast, the recovered CFU after 24h incubation in pond water were similar to the input CFU, and in LB or M9 plus Gly the bacteria multiplied ~10,000-fold.
Fig. 5 displays the results obtained from competitions of six late gene mutants, for which we obtained the clearest phenotypes out of ten tested. These include ΔVC1926, ΔVCA0744 and the ΔVC0612-3 double mutants (Fig. 5A-C, respectively), described above. In addition we tested mutants of VCA0686 and VCA0601 (Fig. 5D and E), both annotated as permeases in putative FeSO4 uptake systems, and a triple mutant of three late genes encoding GGDEF proteins (Fig. 5F). Although VC2370 was not a direct hit in our screen, the flanking genes VC2369 and VC2371, within the same operon, were identified to be late in vivo induced. The observed phenotypes for the GGDEF mutants increased from a single to the triple mutant, and therefore only the data for the triple mutant is shown. The ΔVCA0601 and triple GGDEF mutants had a respective 2- and 3-fold survival defect after 2h incubation in stool compared to LB (Fig. 5E and F). In addition, all six mutants exhibited a significant fitness defect in pond water after 24h. The attenuation ranged from 4-fold for ΔVC1926 (Fig. 5A) to 10-fold for ΔVCA0601 (Fig. 5E). None of the phenotypes in pond water were observed in LB or M9 plus Gly, except for the ΔVCA0744 glycerol kinase mutant, which was attenuated in the latter medium as expected (Fig. 5B). In addition, we competed the late gene mutants with AC53res1 after in vivo passage in a re-infection assay. None of the six late gene mutants demonstrated any significant defect in this second round of infection (Fig. S1B). In all cases, the observed defects of these mutants in stool and pond water could be rescued by expression of the respective gene in trans (Fig. 5A-F).
Figure 5. Some late gene mutants are attenuated in stool and/or pond water survival after in vivo passage.

Competition assays in various conditions using in vivo or in vitro passaged strains as indicated. Each circle represents the value from a single competition assay. The horizontal bar indicates the median of each data set. A significant decrease of the CI compared to that in LB is indicated above the data sets (* p<0.01 or ** p<0.05, using a Mann-Whitney U-test). A plus sign indicates a significant difference of the CIs between mutants expressing the respective gene in trans and AC53res1 with vector alone compared to the mutant/AC53res1 (p<0.05, using a Mann-Whitney U-test).
- (A) a) ΔVC1926/AC53res1, b) ΔVC1926 (pVC1926)/AC53res1 (pMMB67EH)
- (B) a) ΔVCA0744/AC53res1, b) ΔVCA0744 (pVCA0744)/AC53res1 (pMMB67EH)
- (C) a) ΔVC0612-3/AC53res1, b) ΔVC0612-3 (pVC612)/AC53res1 (pMMB67EH)
- (D) a) ΔVCA0686/AC53res1, b) ΔVCA0686 (pVCA0686)/AC53res1 (pMMB67EH)
- (E) a) ΔVCA0601/AC53res1, b) ΔVCA0601 (pVCA0601)/AC53res1 (pMMB67EH)
- (F) a) ΔVC1593-ΔVC2697-ΔVC2370/AC53res1, b) ΔVC1593-ΔVC2697-ΔVC2370 (pGPVC1593-VC2370-VC2697)/AC53lacZ
All pond water samples used were hypoosmolar, having conductivities ~50-fold lower than LB or M9. To test whether the observed transition phenotypes correlate with changes in osmolarity we added 100 mM betaine or M9 minus a carbon source to an equal volume of pond water. In general all mutants performed better under these conditions (Fig. 5A-F). However, since most mutants still retained a significant fitness defect, changes in osmolarity are not the sole cause of the fitness defects observed in pond water. Only the defects of ΔVCA0601 and ΔVCA0686 can be fully rescued by adding M9, but not by adding betaine (Fig. 5D and E). We also competed each mutant against AC53res1 in 1/50 M9, however, no significant defect in fitness for any of the mutants was detected (Fig. 5A-F).
We hypothesized that pre-induction of V. cholerae late genes within the intestinal tract is important for their subsequent roles in stool and pond water survival. To test this, in addition to competing late gene mutants with AC53res1 after an in vivo passage, we also performed the assays with in vitro passaged cultures of the same strains where no pre-induction had occurred. In contrast to the observed fitness defects in stool or pond water after in vivo passage, none of the six in vitro passaged late gene mutants exhibited any significant attenuation in these assays (Fig. 5A-F) or in LB (data not shown).
DISCUSSION
The major goals of this study were to identify V. cholerae genes induced in late stages of intestinal infection and characterize their function in the life cycle of this facultative pathogen. We chose RIVET in combination with the infant mouse model to address these questions for several reasons. DNA microarrays on bacteria recovered from the infant mouse small bowel have not been feasible thus far due to low yield of bacterial RNA and massive contamination with host RNA. DNA microarray data is available for the rabbit ileal loop model of cholera (Nielsen et al., 2006a; Xu et al., 2003a), a model based on growth of V. cholerae in a surgically closed section of the ileum. Although the ileal loop model may simulate some aspects of the human infection, it cannot reflect the dynamic progression of the natural disease in the open GI tract. One striking point is that, according to both published DNA microarray data sets, the genes encoding CT are not induced in the rabbit ileal loop (Nielsen et al., 2006a; Xu et al., 2003a).
We validated our results by qRT-PCR in two different V. cholerae strains and by comparing the identified late genes to previously published data sets of infection-induced genes. Since our reporter system is based on detection of infection-induced promotors, we searched by operons containing late genes (Table 1) rather than only by the genes directly identified by a tnpR fusion. Among the 57 identified late gene operons, 30 have been demonstrated before to be induced in cholera patient stool samples (Bina et al., 2003; Merrell et al., 2002), and/or identified in a RIVET screen in human volunteers (Lombardo et al.). Furthermore 29 of the 57 late gene operons have previously been shown to be induced in animal models of cholera (Bina et al., 2003; Nielsen et al., 2006b; Xu et al., 2003b). These findings suggest that the induction of these late genes is a general phenomenon in V. cholerae and is likely to be observed in a human infection. In the rabbit ileal loop model it has recently been demonstrated that in the late phase of infection the stationary phase sigma factor RpoS, which is under the control of HapR, controls a mucosal escape process (Nielsen et al., 2006a). According to that report, ten of the late gene operons identified in this study are under the control of RpoS. However, seven of those are repressed by RpoS, suggesting that we identified a distinct set of genes, maybe due to the different dynamics in the infant mouse compared to the rabbit ileal loop.
Unexpectedly, seven late gene operons were in common with those reported to be repressed at 12h in the infant mouse model using a FACS-based screen (Hsiao et al., 2006), including the chitin degradation operon (VC0611-20) and the MSHA operon (VC0398-414). The authors did not confirm the in vivo repression of most of these genes. However, they did provide convincing data for repression of the MSHA operon and did demonstrate that constitutive expression of this operon is deleterious to colonization due to a failed evasion of host immunity. Repression of the MSHA operon at 12h post-infection is not directly in conflict with our data as we detected an increase in expression only at the 24h time point. Combining these results we propose that V. cholerae represses this operon in the early- to mid-stages of infection to evade host defenses, but induces it late in infection to allow synthesis of MSHA, which then facilitates adherence to chitin after release into the environment (Chiavelli et al., 2001).
Previous studies showed that ~25% of infection-induced gene deletion mutants are attenuated in the infant mouse model (Camilli and Mekalanos, 1995; Osorio et al., 2005). In contrast only three out of 22 late gene deletion mutants revealed a significant in vivo defect. One explanation for this difference could be that a competition assay in infant mice mostly queries for colonization early in the infection. Consequently the early gene tcpA mutant lacking TCP shows a massive attenuation early on in the infection. In contrast, we demonstrated that the attenuation for late gene mutants, if it occurred, was apparent late in the infection consistent with their late induction.
The majority of the late genes that we identified did not appear to contribute to infection. Rather they assist in preparing the bacterium for the transition to aquatic environments. Using a novel host-to-pond transition assay we confirmed this hypothesis for six late genes. In contrast there was no fitness defect when the mutant strains were grown in vitro and competed in stool and/or pond water. Thus, we conclude that the induction of these genes late in infection serves to prepare V. cholerae for survival in rice-water stool and in aquatic environments.
In trying to identify specific roles for these late genes, we found that the defects of the ΔVCA0686 and ΔVCA0601 mutants could be fully restored by adding M9 salts to pond water. The genes are part of two operons annotated as FeSO4 uptake systems. However these annotations are not supported by recent data demonstrating that the major regulator of iron homeostasis in Gram-negative bacteria, Fur, does not control these operons and expression of them in a Shigella flexneri strain deficient in iron uptake fails to rescue growth (Wyckoff et al., 2006). Our data suggest that these transport systems rather facilitate the uptake of a component of M9 salts. The other four late gene mutants show only a partial rescue in pond water when M9 salts or betaine is added, and none of the mutants was attenuated in diluted, hypoosmotic M9. Therefore we conclude that hypoosmolarity is only one of several stress factors in the transition to pond water.
Carbon sources and their concentrations differ dramatically between the intestinal tract and aquatic environments. We demonstrated that three late genes mutants involved in carbon source utilization exhibited a defect in the transition to pond water. The observed growth defects of ΔVC1926 on succinate, ΔVCA0744 on Gly and ΔVC0612-3 on chitin are consistent with their respective roles as an activator of C4-dicarboxylate transport, a glycerol kinase and degradation enzymes in the chitin utilization cascade, respectively. Although, the concentrations of Gly and the specific C4-dicarboxylate (or precursors) that would feed into these pathways are unknown, it is known that chitin is a major carbon source in aquatic environments (Keyhani and Roseman, 1999). Given the absence of chitin in mammals, the identification of a chitin catabolic operon together with the MSHA chitin-binding pilus operon (Chiavelli et al., 2001) as late genes clearly indicates that V. cholerae prepares its metabolic program prior to its encounter with this carbon and nitrogen source.
We identified three late genes encoding GGDEF proteins, whose expression is predicted to lead to increased c-di-GMP concentration. It is believed that the concentration of this signaling molecule in V. cholerae is high in aquatic environments to facilitate biofilm formation (Tischler and Camilli, 2004). On the other hand, it is likely that V. cholerae must reduce the concentration of c-di-GMP upon entry into the human host since high levels of c-di-GMP inhibit virulence gene expression (Tischler and Camilli, 2005). The observed fitness defect for the triple GGDEF mutant in stool and pond water suggests an important role for c-di-GMP in the transition from host to environment. Our data suggest that V. cholerae already increases the concentration of c-di-GMP inside the host, and not only after release into the environment.
Taken together our results extend the model of the life cycle of pathogenic V. cholerae as we propose in Fig. 6. Upon entry into humans, V. cholerae induces early genes critical for colonization of the small bowel, such as the biosynthetic genes for TCP. After colonization and proliferation, late genes are induced, which increase fitness in the late stage of infection and/or in the transition to aquatic environments. When shed into fresh water, V. cholerae will experience a severe drop in osmolarity and carbon source availability. Large-scale changes to its proteome and metabolic state in response to this harsh environment will be energetically costly. Thus, the preinduction of V. cholerae genes in the GI tract that are advantageous for environmental survival provides an overall increase in fitness. Additionally, the expression of late genes might allow V. cholerae to hoard factors in the host that are limited in aquatic environments. Evolution is likely to have selected for expression of those genes important for transition while V. cholerae is still in the host. After release in stool, V. cholerae can either persist for long periods, for example by attaching to surfaces and using chitin as a carbon and nitrogen source, or it can persist short-term awaiting ingestion by a new human host. Interestingly, the late genes identified in this study that are important for environmental fitness were found not to be necessary for a second round of infection and are therefore not involved in host-passaged hyperinfectivity. However, we cannot exclude that other late genes play roles in hyperinfectivity, and this possibility will be investigated in future studies.
Figure 6. Model of the pathogenic V. cholerae life cycle.
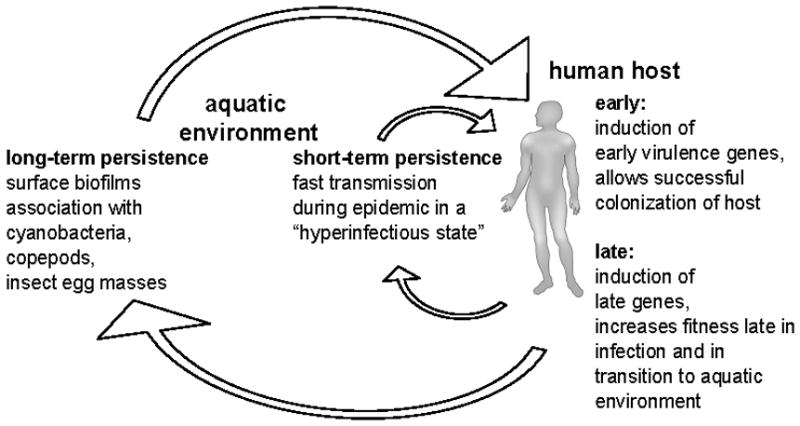
Human infection occurs by ingestion of V. cholerae. Induction of early genes is crucial for survival and successful colonization of the small bowel. Induction of late genes allows maintenance of the infection and increases fitness for the transition step into aquatic environments. After the release into the environment V. cholerae can infect a new human host in a short time period (short-term persistance) or form associations with chitinous material allowing long-term persistence in the environment.
As demonstrated in this study, the ability to prepare for a change in environment enables V. cholerae to better survive the transition from host to aquatic environment. To our knowledge this is the first identification of such transition factors for a facultative pathogen.
EXPERIMENTAL PROCEDURES
Reagents, DNA manipulations and construction of mutant and complemented strains
Bacterial strains and plasmids are listed in Table S1, PCR primers used to construct plasmids in Table S2. Information about general growth conditions, reagents, standard DNA manipulations and strain or library construction methods are stated in supplementary data.
Screening for late genes
An aliquot of each pool of the library was spread in triplicate on LB-Ap/Sm/Kn plates. After O/N incubation ~5000 colonies were collected from each plate, diluted in LB to ~106 CFU/ml and 50μl used to intragastrically inoculate 5-day-old CD-1 mice (anesthetized by isoflurane) as previously described (Camilli et al., 1994). 7h post-infection, mice were anesthetized and a Kn dose of 1.4mg/kg bw in 50μl PBS was given intragastrically. 24h post-infection, mice were euthanized and their small bowels were removed and homogenized in 1ml of LB plus 20% Gly. Serial dilutions of the homogenate were plated on LB agar lacking NaCl and supplemented with 10% Suc and Sm to select for resolved strains. After incubation O/N at 30°C eight SucR KnS colonies were picked from each mouse output, grown in LB and stored at -80°C in 96-well plates (Costar) in LB plus 20% Gly. Gene fusions to tnpR were PCR amplified and sequenced as described (Lombardo et al.). Sequences were compared to the V. cholerae N16961 genome database (Heidelberg et al., 2000) with blastN (http://tigrblast.tigr.org/cmr-blast/). We considered transcriptional fusions of tnpR to any annotated ORF within which it had inserted in the same orientation, as well as any annotated ORF in the same orientation lying ≤100bp from the RBS of tnpR as long as no factor-independent transcriptional terminators were present. Fusion strain reconstructionans quntifacation of resolution is described in supplementary data.
Competition assays
Competition assays in infant mice and in LB broth were performed with late gene in-frame deletion mutants (lacZ+) competed for 7 or 24h against the isogenic AC53res1 strain (lacZ-) as described (Camilli and Mekalanos, 1995). Results are shown by the competition index (CI), which is the ratio of mutant CFU to AC53res1 CFU normalized for the input ratio. Competitions in M9 with various carbon sources were done by inoculation of 2mL of medium with ~105 CFU of V. cholerae prepared in saline (0.85% NaCl, pH 7.5). The M9 cultures were incubated 24h at 25°C.
To show complementation in trans in all assays in this study, deletion mutants (lacZ+) with the respective pMMB67EH expression derivative were competed against AC53res1 (lacZ-) containing the empty pMMB67EH, with the exception of strain ΔVC1593- ΔVC2697- ΔVC2370 (pGPVC1593-VC2697-VC2370), which was competed against AC53lacZ.
RNA Purification and qRT-PCR
RNA was isolated at 24h post-infection from small bowels of six mice infected with V. cholerae and mock-inoculated mice that received PBS. Small bowels were homogenized in Trizol (1ml). Following chloroform extraction, ethanol was added to the aqueous phase to a final concentration of 35% and purified using the RNeasy® Mini Kit (Qiagen). RNA was also isolated from six independent mid-exponential phase cultures of bacteria grown in LB. DNA was removed using a DNA-free kit (Ambion). cDNA was synthesized from 1μg RNA using SuperScript First Strand Synthesis System for qPCR (Invitrogen). Controls lacking reverse transcriptase were included.
qRT-PCR experiments were done using Brilliant SYBR Green qPCR Master Mix (Stratagene). Each reaction contained 300nM primers, 100ng template and ROX reference dye. The six independent in vivo and in vitro samples were tested in triplicate. The sequences of the primers used in these studies are in Table S1, labeled as x-qF and x-qR, in which x stands for the respective gene. All primer pairs amplified the target gene with efficiencies of 97% or greater (data not shown).
For each sample, the mean cycle threshold of the test transcript was normalized to that of rpoB and to one randomly selected in vitro reference sample. Values >1 indicate that the transcript is present in higher numbers in the test sample than in the in vitro reference.
Transition assays
The fitness of late gene deletion mutants (lacZ+) in transitioning from mouse intestinal tract to rice-water stool or pond water was determined in competition assays against the isogenic AC53res1 strain (lacZ-). Each strain was grown on LB-Sm plates O/N, diluted in LB to OD600=0.002 and used to intragastrically inoculate three infant mice as above. 24h post-infection mice were euthanized, their small bowels were removed and homogenized in 1ml of saline. The three homogenates of each strain were combined and filtered through a 100μm cell strainer. 150μl of the filtrate of each competing strain was mixed together with 700μl saline and used as inoculum (~5×106 CFU/ml) in various fitness assays performed in 96-well polysterene plates (Costar). For survival in filtered rice-water stool, 10μl of the inoculum was mixed with 100μl of stool and incubated for 2h at 25°C. In all other assays, 10μl of inoculum was diluted in 100μl saline and then diluted a further 1/10 and 1/100 in test solution and incubated for 24h at 25°C. For reinfection 50μl of the inoculum was used to intragastrically inoculate infant mice as described above.
In all cases incubation in LB was used as a control. After incubation, serial dilutions were plated on LB-Sm/X-Gal plates. The results are shown as the CI as described above.
We also determined the fitness in transition assays without prior in vivo passage of the competing strains. In this case, strains were grown on LB-Sm plates O/N, diluted in saline to OD600=0.02, mixed 1:1 and diluted to OD600=0.02 (~5×106 CFU/ml), and used as the inoculum as described above.
Collection and preparation of stool and pond water samples
Rice-water stool samples were collected from patients (>15yrs of age) in Bangladesh with acute watery diarrhea and no prior treatment with antibiotics during a 2006 spring outbreak. The two samples included in this study were selected on the basis of positive culture for V. cholerae O1, and negative tests for lytic vibriophage and enterotoxigenic E. coli, each assayed by standard methodology. Upon collection stool supernatants were prepared by centrifugation at 15,000rpm for 15min at 4°C, filtration of the supernatant through a 0.22μM polystyrene filter (Millipore), and stored at -80°C. The protocol for the collection of these samples was approved by the Research Review Committee and the Ethical Review Committee at the International Centre for Diarrhoeal Disease Research in Bangladesh and by the Human Research Committee at the Massachusetts General Hospital. Pond water from Dhaka, Bangladesh and Boston, Massachusetts was filtered through a 0.45μM polystyrene filter (Millipore) and frozen at -80°C. All pond water samples used tested negative for lytic vibriophage. Both pond water samples had similar chemical compositions (Table S3).
Supplementary Material
Acknowledgments
Author contributions: S. Schild and A. Camilli designed research and wrote the paper; S. Schild performed research except for qRT-PCR done by R. Tamayo; E. Nelson and F. Qadri collected stool and/or pond water samples from Bangladesh; S. B. Calderwood wrote and directs the human subjects protocol under which the stool samples were collected in Bangladesh. We are grateful to M. J. Lombardo for the strain VC0201, L. Bourassa for providing Boston pond water samples, A. Bishop and D. W. Lazinski for critically reading this manuscript as well as the Camilli lab for helpful suggestions and discussion. This research was supported by NIH Grant AI55058 to A. C. and the Center for Gastroenterology Research on Absorptive and Secretory Processes, NEMC (P30 DK34928). A. Camilli is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam A, Larocque RC, Harris JB, Vanderspurt C, Ryan ET, Qadri F, Calderwood SB. Hyperinfectivity of human-passaged Vibrio cholerae can be modeled by growth in the infant mouse. Infect Immun. 2005;73:6674–6679. doi: 10.1128/IAI.73.10.6674-6679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm EJ, Huang KH, Price MN, Koche RP, Keller K, Dubchak IL, Arkin AP. The MicrobesOnline Web site for comparative genomics. Genome Res. 2005;15:1015–1022. doi: 10.1101/gr.3844805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelichio MJ, Merrell DS, Camilli A. Spatiotemporal analysis of acid adaptation-mediated Vibrio cholerae hyperinfectivity. Infect Immun. 2004;72:2405–2407. doi: 10.1128/IAI.72.4.2405-2407.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez JA, Spelbrink RG, Silva A, Phillips TE, Stanley CM, Boesman-Finkelstein M, Finkelstein RA. Adherence of Vibrio cholerae to cultured differentiated human intestinal cells: an in vitro colonization model. Infect Immun. 1997;65:3474–3477. doi: 10.1128/iai.65.8.3474-3477.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennish ML. Cholera: pathophysiology, clinical features, and treatment. In: Wachsmuth KI, Blake PA, Olsik O, editors. Vibrio cholerae and Cholera: molecular to global persepectives. Washington, D.C.: ASM Press; 1994. pp. 229–255. [Google Scholar]
- Berg T, Schild S, Reidl J. Regulation of the chitobiose-phosphotransferase system in Vibrio cholerae. Arch Microbiol. 2007 doi: 10.1007/s00203-006-0207-4. [DOI] [PubMed] [Google Scholar]
- Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc Natl Acad Sci U S A. 2003;100:2801–2806. doi: 10.1073/pnas.2628026100. Supplemental data: Table S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SM, Nelson EJ, Chowdhury N, Faruque SM, Calderwood SB, Camilli A. Cholera stool bacteria repress chemotaxis to increase infectivity. Mol Microbiol. 2006;60:417–426. doi: 10.1111/j.1365-2958.2006.05096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Beattie DT, Mekalanos JJ. Use of genetic recombination as a reporter of gene expression. Proc Natl Acad Sci U S A. 1994;91:2634–2638. doi: 10.1073/pnas.91.7.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Mekalanos JJ. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol Microbiol. 1995;18:671–683. doi: 10.1111/j.1365-2958.1995.mmi_18040671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SL, Mekalanos JJ. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol. 1998;27:797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- Chiavelli DA, Marsh JW, Taylor RK. The mannose-sensitive hemagglutinin of Vibrio cholerae promotes adherence to zooplankton. Appl Environ Microbiol. 2001;67:3220–3225. doi: 10.1128/AEM.67.7.3220-3225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forward JA, Behrendt MC, Wyborn NR, Cross R, Kelly DJ. TRAP transporters: a new family of periplasmic solute transport systems encoded by the dctPQM genes of Rhodobacter capsulatus and by homologs in diverse gram-negative bacteria. J Bacteriol. 1997;179:5482–5493. doi: 10.1128/jb.179.17.5482-5493.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freter R, O’Brian PCM, MS M. Role of chemotaxis in the association of motile bacteria with intestinal mucosa: in vivo studies. Infect Immun. 1981;34:234–240. doi: 10.1128/iai.34.1.234-240.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MJ, Shaw JG, Kelly DJ. Sequence analysis and interposon mutagenesis of a sensor-kinase (DctS) and response-regulator (DctR) controlling synthesis of the high-affinity C4-dicarboxylate transport system in Rhodobacter capsulatus. Mol Gen Genet. 1993;237:215–224. doi: 10.1007/BF00282803. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Hang L, John M, Asaduzzaman M, Bridges EA, Vanderspurt C, Kirn TJ, Taylor RK, Hillman JD, Progulske-Fox A, Handfield M, et al. Use of in vivo-induced antigen technology (IVIAT) to identify genes uniquely expressed during human infection with Vibrio cholerae. Proc Natl Acad Sci U S A. 2003;100:8508–8513. doi: 10.1073/pnas.1431769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, L U, et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao A, Liu Z, Joelsson A, Zhu J. Vibrio cholerae virulence regulator-coordinated evasion of host immunity. Proc Natl Acad Sci U S A. 2006;103:14542–14547. doi: 10.1073/pnas.0604650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joelsson A, Liu Z, Zhu J. Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect Immun. 2006;74:1141–1147. doi: 10.1128/IAI.74.2.1141-1147.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyhani NO, Roseman S. The chitin catabolic cascade in the marine bacterium Vibrio furnissii. Molecular cloning, isolation, and characterization of a periplasmic beta-N-acetylglucosaminidase. J Biol Chem. 1996;271:33425–33432. doi: 10.1074/jbc.271.52.33425. [DOI] [PubMed] [Google Scholar]
- Keyhani NO, Roseman S. Physiological aspects of chitin catabolism in marine bacteria. Biochim Biophys Acta. 1999;1473:108–122. doi: 10.1016/s0304-4165(99)00172-5. [DOI] [PubMed] [Google Scholar]
- Kirn TJ, Jude BA, Taylor RK. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature. 2005;438:863–866. doi: 10.1038/nature04249. [DOI] [PubMed] [Google Scholar]
- Koch R. An address on cholera and its bacillus. Br Med J. 1884;2:403–407. doi: 10.1136/bmj.2.1235.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter R, Inuzuka M, Helinski DR. Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell. 1978;15:1199–1208. doi: 10.1016/0092-8674(78)90046-6. [DOI] [PubMed] [Google Scholar]
- Lee SH, Angelichio MJ, Mekalanos JJ, Camilli A. Nucleotide sequence and spatiotemporal expression of the Vibrio cholerae vieSAB genes during infection. J Bacteriol. 1998;180:2298–2305. doi: 10.1128/jb.180.9.2298-2305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Hava DL, Waldor MK, Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625–634. doi: 10.1016/s0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- Li X, Roseman S. The chitinolytic cascade in Vibrios is regulated by chitin oligosaccharides and a two-component chitin catabolic sensor/kinase. Proc Natl Acad Sci U S A. 2004;101:627–631. doi: 10.1073/pnas.0307645100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo M, Michalski J, Martinez-Wilson H, Maroncle N, Osorio C, Nataro J, Tacket C, Camilli A, Kaper J. A Vibrio cholerae IVET (In Vivo Expression Technology) Screen in Human Volunteers. doi: 10.1073/pnas.0705636104. in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meibom KL, Blokesch M, Dolganov NA, Wu CY, Schoolnik GK. Chitin induces natural competence in Vibrio cholerae. Science. 2005;310:1824–1827. doi: 10.1126/science.1120096. [DOI] [PubMed] [Google Scholar]
- Meibom KL, Li XB, Nielsen AT, Wu CY, Roseman S, Schoolnik GK. The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci U S A. 2004;101:2524–2529. doi: 10.1073/pnas.0308707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. Host-induced epidemic spread of the cholera bacterium. Nature. 2002;417:642–645. doi: 10.1038/nature00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VL, DiRita VJ, Mekalanos JJ. Identification of toxS, a regulatory gene whose product enhances toxR-mediated activation of the cholera toxin promoter. J Bacteriol. 1989;171:1288–1293. doi: 10.1128/jb.171.3.1288-1293.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales VM, Backman A, Bagdasarian M. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene. 1991;97:39–47. doi: 10.1016/0378-1119(91)90007-x. [DOI] [PubMed] [Google Scholar]
- Nielsen AT, Dolganov NA, Otto G, Miller MC, Wu CY, Schoolnik GK. RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2006a;2:e109. doi: 10.1371/journal.ppat.0020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AT, Dolganov NA, Otto G, Miller MC, Wu CY, Schoolnik GK. RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2006b;2:e109. doi: 10.1371/journal.ppat.0020109. Supplemental data: Table S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio CG, Crawford JA, Michalski J, Martinez-Wilson H, Kaper JB, Camilli A. Second-generation recombination-based in vivo expression technology for large-scale screening for Vibrio cholerae genes induced during infection of the mouse small intestine. Infect Immun. 2005;73:972–980. doi: 10.1128/IAI.73.2.972-980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JK, Keyhani NO, Roseman S. Chitin catabolism in the marine bacterium Vibrio furnissii. Identification, molecular cloning, and characterization of A N, N’-diacetylchitobiose phosphorylase. J Biol Chem. 2000;275:33077–33083. doi: 10.1074/jbc.M001042200. [DOI] [PubMed] [Google Scholar]
- Reidl J, Klose KE. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol Rev. 2002;26:125–139. doi: 10.1111/j.1574-6976.2002.tb00605.x. [DOI] [PubMed] [Google Scholar]
- Rogers MB, Sexton JA, DeCastro GJ, Calderwood SB. Identification of an operon required for ferrichrome iron utilization in Vibrio cholerae. J Bacteriol. 2000;182:2350–2353. doi: 10.1128/jb.182.8.2350-2353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romling U, Amikam D. Cyclic di-GMP as a second messenger. Curr Opin Microbiol. 2006;9:218–228. doi: 10.1016/j.mib.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Schild S, Lamprecht AK, Fourestier C, Lauriano CM, Klose KE, Reidl J. Characterizing lipopolysaccharide and core lipid A mutant O1 and O139 Vibrio cholerae strains for adherence properties on mucus-producing cell line HT29-Rev MTX and virulence in mice. Int J Med Microbiol. 2005;295:243–251. doi: 10.1016/j.ijmm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Thelin KH, Taylor RK. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect Immun. 1996;64:2853–2856. doi: 10.1128/iai.64.7.2853-2856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler AD, Camilli A. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol Microbiol. 2004;53:857–869. doi: 10.1111/j.1365-2958.2004.04155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler AD, Camilli A. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun. 2005;73:5873–5882. doi: 10.1128/IAI.73.9.5873-5882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance RE, Zhu J, Mekalanos JJ. A constitutively active variant of the quorum-sensing regulator LuxO affects protease production and biofilm formation in Vibrio cholerae. Infect Immun. 2003;71:2571–2576. doi: 10.1128/IAI.71.5.2571-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsmuth IK, Evins GM, Fields PI, Olsvik O, Popvic T, Bopp CA, Wells JG, Carrillo C, Blake PA. The molecular epidemiology of cholera in Latin America. J Infect Dis. 1993;167:621–626. doi: 10.1093/infdis/167.3.621. [DOI] [PubMed] [Google Scholar]
- Watnick P, Kolter R. Steps in the develompent of a Vibrio cholerae El Tor biofilm. Mol Microbiol. 1999;34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff EE, Mey AR, Leimbach A, Fisher CF, Payne SM. Characterization of ferric and ferrous iron transport systems in Vibrio cholerae. J Bacteriol. 2006;188:6515–6523. doi: 10.1128/JB.00626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Dziejman M, Mekalanos JJ. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc Natl Acad Sci U S A. 2003a;100:1286–1291. doi: 10.1073/pnas.0337479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Dziejman M, Mekalanos JJ. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc Natl Acad Sci U S A. 2003b;100:1286–1291. doi: 10.1073/pnas.0337479100. Supplemental data: data set 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz FH, Schoolnik GK. Vibrio cholerae O1 El Tor: Identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc Natl Acad Sci USA. 1999;96:4028–4033. doi: 10.1073/pnas.96.7.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A. 2002;99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.