

Synopsis
Beta2 adrenergic receptors were identified in keratinocytes more than 30 years ago, but their function in the epidermis continues to be elucidated. Abnormalities in their expression, signaling pathway, or in the generation of endogenous catecholamine agonists by keratinocytes have been implicated in the pathogenesis of cutaneous diseases such as atopic dermatitis, vitiligo and psoriasis. New studies also indicate that the beta2AR also modulates keratinocyte migration, and thus can function to regulate wound re-epithelialization. This review focuses on the function of these receptors in keratinocytes and their contribution to cutaneous physiology and disease.
Keywords: beta-adrenergic, keratinocyte, atopic dermatitis, psoriasis, vitiligo, wound
Physiology of the beta2 adrenergic receptor in keratinocytes
Of the several identified classes of adrenergic receptors (alpha and beta, and their subtypes), it is of particular interest to note that human keratinocytes, the cells the make up the majority of the epidermis, express only beta2 adrenergic receptors (beta2AR)[1-4]. The beta adrenergic receptor is a classic seven transmembrane G protein coupled receptor. These receptors serve as the endogenous receptor to the catecholamine hormones norepinephrine and epinephrine, and can be further subdivided into beta1, beta2, and beta3 subtypes, based on their differential pharmacological response to catecholamines and specific antagonists, as well as differences in their protein sequences[5-7].
The first studies to suggest the presence of betaAR in the epidermis were functional ones that demonstrated an increase in levels of cAMP in keratinocytes after stimulation with non-specific betaAR agonists [8, 9]. Subsequently, work using agonists and antagonists with specificity for the different adrenergic receptor subtypes demonstrated that the increase in keratinocyte intracelluar cAMP levels was observed only with agents specific for the beta2AR, suggesting that keratinocytes mainly expressed this receptor subtype[2]. This finding was confirmed by later studies that investigated binding affinities of selective agonists and antagonists[10-12], radioligand-binding assays of epidermal membrane fragments[3], and autoradiographic mapping of the beta adrenergic receptor subtypes on epidermal keratinocytes[4]. Since only the beta2AR is expressed in keratinocytes, this review will focus on this receptor subtype and its physiological role in the epidermis.
The beta2AR transduces signals via a G-protein coupled signaling cascade that classically includes adenylate cyclase-mediated increases in cAMP, although cAMP independent signaling cascades have recently been described and their regulatory roles are becoming clearer (reviewed in [13]). Stimulation of the beta2AR leads to a transient increase in the keratinocyte intracellular calcium concentration[14], and this likely occurs through several signaling cascades (Figure 1). Activation of the cAMP-dependent pathway by direct activation of adenylate cyclase has been shown to reproduce the increase in intracellular calcium that is seen with ligand stimulation of the beta2AR[15, 16]. Although beta2AR activation in other cell types, notably cardiac myocytes, results in modulation of activity of voltage-gated calcium channels[17, 18], only recently has the presence of a voltage gated calcium channel been observed in keratinocytes[19]. Thus, the question of whether or not the beta2AR-induced calcium transient in keratinocytes is mediated by this channel has not yet been addressed. The rise in intracellular calcium induced by beta2AR activation in keratinocytes may also be the result of release of intracellular stores, as suggested by Koizumi and colleagues[15, 20], and this intracellular release may in turn trigger calcium-activated calcium influx through as yet unidentified store-operated calcium channel within the keratinocyte membrane (reviewed in [21]). Regardless of the specific channels that mediate the calcium influx secondary to beta2AR stimulation, it is clear that an increase in intracellular calcium results from the activation of the beta2AR, and that this increase can alter keratinocyte differentiation. Increases in the intracellular calcium concentration inhibit keratinocyte proliferation while increasing phenotypic differentiation [22-24]. And indeed, Mammone and colleagues have demonstrated that activation of the beta2AR on a human keratinocyte cell line results not only in increases in cAMP levels but also in increased expression of keratins 1 and 10, involucrin and transglutaminase, all markers of terminal differentiation in keratinocytes[25]. Taken together, the evidence suggests that beta2AR activation can upregulate keratinocyte differentiation by the cAMP-mediated signaling pathway, but does not rule out contributions of the less classical non-cAMP mediated signaling pathway. Future work will be needed to clarify the role of each signaling arm in the differentiation process.
Figure 1. Keratinocyte Beta2 Adrenergic Receptor Signaling Pathways.
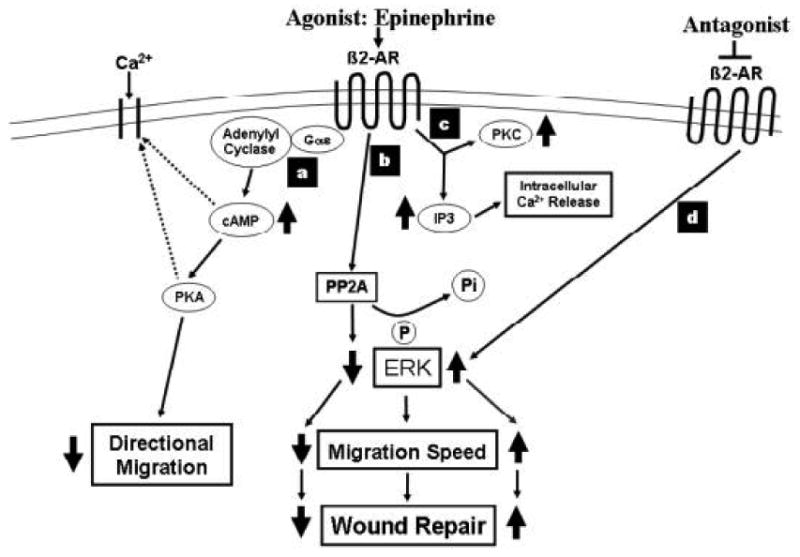
(a) cAMP dependent signaling, (b) PP2A dependent pathway, (c) IP3 and PKC dependent pathway, and (d) pathway for beta2 adrenergic receptor blockade. The signaling depicted in the diagram is unique for keratinocytes. In other cell types, agonist activation of the receptor results in different downstream effector cascades. Dotted lines indicate signaling pathways where causal associations have been demonstrated but definitive mechanisms have not yet been defined (described in text).
In the epidermis, calcium concentrations are lower in the basal layer and increase toward the stratum corneum[26, 27], correlating with the level of keratinocyte differentiation. Interestingly, it has been shown that keratinocytes are capable of producing epinephrine[28, 29], an endogenous adrenergic agonist with selectivity for the beta2AR, and basal layer keratinocytes produce higher levels of epinephrine than suprabasal keratinocytes[29]. Thus, it has been proposed that epinephrine activates keratinocyte beta2AR to modulate calcium influx and begin the differentiation cascade crucial to the native architecture of the epidermis[29]. The beta2AR desensitizes upon repeated activation through several mechanisms, including downregulation of the number of beta2AR receptors (reviewed in [30, 31]). Indeed, beta2AR expression is more highly expressed at the basal layers of the epidermis and decreases in expression toward the stratum corneum[12], suggesting that epinephrine may be activating the receptor to increase intracellular calcium levels and induce differentiation. However, further studies will be needed to determine how essential the autocrine epinephrine/beta2AR signaling cascade is for keratinocyte differentiation and formation of the epidermal architecture.
Several cAMP-independent signaling pathways have also been described for the beta2AR in many cell types, and this is true in keratinocytes as well. Epinephrine stimulation of the beta2AR has been shown to increase the activity of protein kinase C and formation of inositol-1,4,5-trisphosphate[20] which can stimulate calcium release from internal reserves. In keratinocytes, beta2AR activation results in a decrease in the activity of extracellular signal-related kinase (ERK), mediated not by the cAMP-dependent pathway, but rather by its dephosphorylation which is accomplished through the action of beta2AR-induced activation of the serine/threonine phosphatase PP2A[32, 33]. This is in contradistinction to beta2AR activation in multiple other cell types, which typically results in ERK phosphorylation and activation[34-37]. Classical, cAMP-dependent signaling with beta2AR activation has also been described in keratinocytes[38, 39]. Thus, both cAMP –dependent and independent signal transduction govern keratinocyte physiology, and continuing investigations will clarify which pathway controls which cellular functions.
Atopic Dermatitis
Numerous disparate theories have been proposed for the pathogenesis of atopic dermatitis in the past decades. Although the definitive mechanism of atopic dermatitis remains uncertain, the possible role of beta adrenoceptor has been implicated for many years. As early as 1968, Szentivanyi hypothesized that the exaggerated vascular activity and increased sensitivity of pilomotor smooth muscle seen in the skin of patients with atopic dermatitis could be attributed to a decreased response to beta adrenergic receptor blockade, and he suggested that this was like the result of a deficiency of adenylate cyclase [40]. An alternative explanation for the reduced cAMP responsiveness to beta adrenergic agonists (such as isoproterenol) seen in leukocytes from atopic individuals has been proposed by Hanifin and colleagues, who showed an increase in cAMP-phosphodiesterase activity in these patients' leukocytes[41, 42]. In vitro studies by Reed et al. showed that beta adrenergic agonists inhibited DNA synthesis in normal epidermis but had no inhibitory effect on DNA synthesis of either lichenified or normal-appearing skin in atopic dermatitis patients[43]. Failure to evoke normal inhibition of cell division of basal cells thus seems plausible in the pathogenesis of epidermal hyperproliferation seen in atopic dermatitis. While some investigators proposed that there might be lower densities of beta2 adrenergic receptors in undifferentiated keratinocytes [44], there is limited evidence of this, and in fact, other investigators have found normal adrenoceptor densities but lower receptor binding affinities, albeit on lymphocytes[45]. Nonetheless, these early studies have provided provocative evidence suggesting the involvement of beta adrenoceptors as contributing to the pathogenesis of atopic dermatitis.
Focusing on the keratinocyte defects, Schallreuter and colleagues have demonstrated that cell extracts from epidermal suction blister roofs of the uninvolved skin of atopic dermatitis patients have a significantly increased concentrations of norepinephrine and the cofactor (6R)-L-erythro-5,6,7,8,-tetrahydrobiopterin (6BH4), required for the conversion of L-phenylalanine to L-tyrosine, and for the subsequent conversion of L-tyrosine to L-dopa—two of the early steps in the catecholamine synthetic pathway (Figure 2), compared to aged-matched controls[44]. In addition, in this study there was a parallel threefold induction of the catecholamine degrading enzyme monoamine oxidase-A (MAO-A) activity with half the normal activity of the enzyme phenylethanolamine-N-methyl transferase (PNMT) required for conversion of norepinephrine into epinephrine (figure 2) in patients with atopic dermatitis. A prior study demonstrated an increase in the catecholamine degrading enzyme catechol-o-methyl transferase (COMT) levels in atopic epidermis[46]. Although circulating levels of norepinephrine have been reported to be between 1.5 to 2.5 fold increased in patients with atopic dermatitis as compared to controls[44, 47], given the increase in the catecholamine degrading enzymes COMT and MAO-A in atopic epidermis, the source of the circulating catecholamines is likely not epidermal. Taken together these results support earlier observations of a perturbed catecholamine/ beta adrenoceptor signaling in atopic dermatitis. Moreover, since gene polymorphisms of the beta2 adrenergic receptor gene have been found to influence the structure and function of the receptor in other tissues [48], it is possible that these may also contribute to the altered responses in the beta2AR pathway seen in atopic epidermis. In a small preliminary study of 4 patients, this was found to be the case, with a single nucleotide polymorphism of the beta2AR noted in keratinocytes isolated from these patients, and the investigators propose that this can alter binding and perhaps function in affected keratinocytes[49]. Recently, Maestroni investigated the interplay between beta adrenergic system and immune system[50]. He found that beta2 AR activation in skin and dendritic cells results in downregulation of inflammatory cytokine production and receptor expression, leading to reduced TH1 priming. He thus postulated that the beta adrenergic system plays a key role in modulating cytokine production and antigen presentation and can contribute to pathologic disorders of the skin such as atopic dermatitis. Overall, the evidence is suggestive of a defect in the beta2 AR system as contributing to the pathogenesis of atopic dermatitis, but the exact biochemical pathways remain to be clarified. Further investigation of beta adrenoceptors of the skin and their interaction with the immune system may offer new therapeutic approaches to atopic dermatitis.
Figure 2. Catecholamine synthesis pathway in atopic dermatitis and vitiligo.
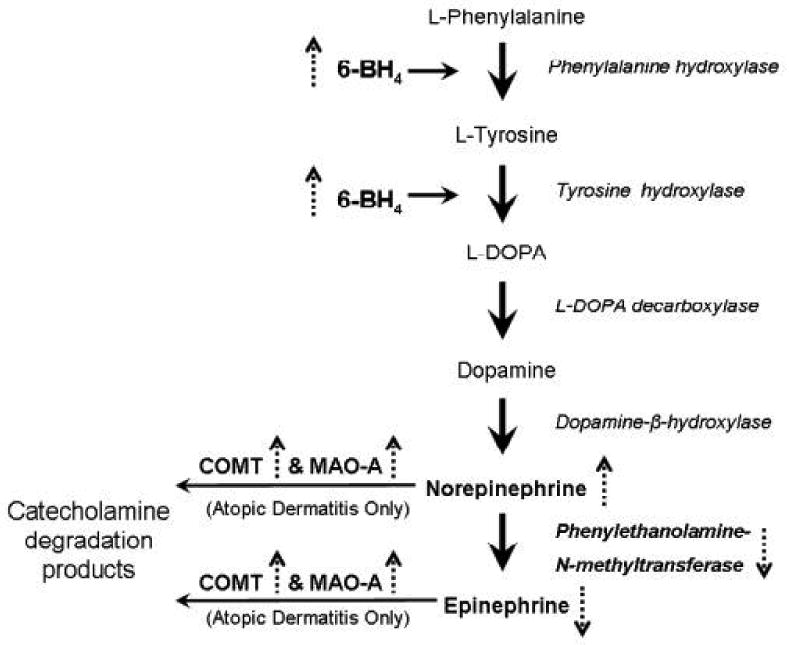
The solid arrows represent the normal pathway in the synthesis of norepinephrine and epinephrine from L-phenylalanine. The dashed arrows represent how this pathway is modulated in atopic dermatitis and vitiligo except that epidermal increases in the level COMT and MAO-A have only been shown for atopic dermatitis.
Vitiligo and Beta2AR
Many theories have been proposed for the pathogenesis of vitiligo, including genetic, autoimmune, and oxidative damage etiologies (reviewed in [13, 51, 52]). One pathogenic mechanism advanced by the work of Schallreuter and colleagues implicates the beta2AR signaling pathway and catecholamine synthetic network within the epidermis (recently reviewed in [13]). Both keratinocytes and melanocytes express beta2AR [4, 10, 11, 53], both cell types have the enzymatic machinery for catecholamine synthesis[53, 54], and indeed, both have been demonstrated to generate norepinephrine (keratinocytes, but not melanocytes can generate epinephrine) [53-55]. Stimulation of the beta2AR on melanocytes results in an increase in intracellular levels of cAMP, and subsequently, increased melanogenesis [53]. Since the normal epidermal keratinocytes can endogenously generate epinephrine, they may be providing local stimulation of this beta2AR-mediated pathway for melanogenesis in the normal melanocyte. In vitiligo, there is accumulating evidence generated by the Schallreuter group that implicates both the catecholamine synthetic pathway and subsequent beta2AR signaling in the pathogenesis of the disease. First, it is interesting to note that systemic beta blockers can lead to exacerbation of vitiligo in some patients[56]. Since beta2AR-stimulation activates melanogenesis in vitro, it is tempting to propose that blockade of beta2AR on melanocytes in vivo may contribute to the decrease in melanin synthesis seen in vitiliginous skin. Second, patients with vitiligo exhibit increased plasma and urine concentrations of norepinephrine and its degradation products[57-59]. One possible source may be from the keratinocytes within the vitiliginous areas, that have increased levels of 6BH4[58, 60](Figure 2). This defect, along with a concomitantly noted reduction in the activity of PNMT, in keratinocytes in affected vitiliginous epidermis[58] (Figure 2) may explain, in part, the accumulation of norepinephrine. Third, epidermal keratinocytes derived from the affected skin of patients with vitiligo (and not the clinically normal appearing skin in these patients), demonstrate increased expression of the beta2AR in culture[12]. The expression of these receptors on keratinocytes are regulated by extracellular levels of calcium[12] as well as by 6-biopterin, the redox product of 6BHR, which is increased in the epidermis in patients with vitiligo (reviewed in [13, 61]). Since beta2AR activation by catecholamines also induces calcium influx into keratinocytes[14], one might have anticipated that intracellular calcium levels are increased in keratinocytes in vitiliginous epidermis. However, a defect in calcium influx has been demonstrated in keratinocytes derived from involved vitiliginous epidermis[62], although this data may be represented by a limited number of patient samples. How, or if, this possible defect in keratinocyte calcium flux plays a role in the pathogenesis of vitiligo, is not clear at this time. Nevertheless, the cumulative evidence points to an associated defect in the catecholamine synthetic pathway and to abnormal beta2AR expression in the vitiliginous epidermis, and it is likely that continued investigation will uncover the mechanisms by which these converge to dysregulate melanogenesis in vitiligo. Pharmacological manipulation of the beta2AR may then offer another tool for the treatment of vitiligo.
Psoriasis
Psoriasis is yet another disease in which the beta2AR has been implicated in its pathogenesis. Studies by Voorhees and colleagues and Flaxman and Harper over 30 years ago demonstrated the regulation of keratinocyte proliferation by intracellular levels of cAMP, and that elevation of intracellular cAMP by catecholamine stimulation resulted in a decrease in proliferation[8, 9, 63-65]. Work by these laboratories also suggested that a decrease in the ability of psoriatic keratinocytes to respond to beta adrenergic agonists with an increase in cAMP could be, in part, responsible for the increase in cell proliferation characteristic of the disease[66-69]. These studies relied on keratome sections of psoriatic epidermis, but a later study by Iizuka and colleagues demonstrated similar findings in epidermis microdissected from involved psoriatic skin free of stratum corneum, dermis and appendages. These preparations also generated decreased levels of cAMP after stimulation with epinephrine, when compared to epidermal preparation microdisseccted from uninvolved skin[70]. Generation of cAMP in response to other activators of adenylate cyclase, such as histamine, was intact, suggesting that the defect in the psoriatic epidermis resided in the beta adrenergic receptor pathway. Similar findings were obtained by other investigators measuring the generation of cAMP in response to beta adrenergic agonist stimulation of cell membrane fractions derived from psoriatic and normal epidermis[71]. This reduced response is likely a function of decreased expression of receptors on keratinocytes from involved psoriatic epidermis, as has been demonstrated by direct binding studies[72]. Consistent with these finding is the more recent report of decrease expression of beta 2 adrenergic receptor mRNA in involved psoriatic epidermis shown by RT-PCR[73]. Together, these findings suggest that the downregulation of the number of beta adrenergic receptors, rather than an inherent defect in the receptor itself, is the mechanism that is responsible for the reduced beta-adrenergic responsiveness seen in psoriatic epidermis. This decreased response to endogenous agonists then results in a decrease in intracellular cAMP and thus an increase in keratinocyte proliferation.
Adding support to the hypothesis that beta adrenergic receptor dysregulation contributes to the pathogenesis of psoriasis, are the multiple reports that beta adrenergic antagonists can exacerbate the disease. Systemic use of beta blockers, a common approach for hypertension, or even their topical use in eyedrops for glaucoma, has been reported to either exacerbate existing psoriasis, or to initiate the disease (reviewed in [74, 75]). The drug-induced psoriasis often remits when the drug is withdrawn. A guinea pig model for psoriasis has been proposed, using animals treated with systemic beta blockers[76, 77], although not every histologic finding of the disease is replicated in this model[78]. It has been speculated that in addition to the direct effect of the beta blockers on keratinocyte proliferation, these drugs may be inducing an immunologic component that exacerbates the disease[79] and in fact, in patients who have had psoriatic exacerbations in response to treatment with the beta blocker propanol, a lymphocytic hypersensitivity to the drug has been demonstrated[80, 81].
Recently, data suggesting that beta2AR polymorphisms may contribute to the pathogenesis of some psoriatics by altering beta2AR has been reported[82]. Polymorphisms that involve amino acid substitutions at position 16 and 27 have previously been shown to alter functional properties of beta2AR in humans causing beta2AR desensitization. The polymorphic Gly16 beta2AR demonstrates prolonged downregulation following long-term agonist exposure as compared to the wild type Arg16 receptor[83]. Interestingly, in a recent study of 50 subjects, it was found that the Arg16 allele frequency was significantly higher in the sporadic psoriasis group as compared to controls[82]. Since increased circulating levels of catecholamines have been observed in psoriatic patients[84], and a 10-fold increase in the expression of the epinephrine sythetic enzyme PNMT is also found in basal keratinocytes in involved psoriatic epidermis[85], it is tempting to propose that long-term exposure to increased levels of catecholamines, in the circulation or locally derived by the keratinocytes themselves, in combination with increased desensitization of beta 2AR in individuals with the Arg16 allele, may predispose to psoriasis.
In view of the noted abnormalities in the beta2AR response in psoriatic epidermis, it is somewhat surprising that pharmacologic approaches addressing this target have not been vigorously pursued. One U.S. patent for the use of isoproterenol for the treatment of psoriasis was approved in 1977[86], but no commercial product appears to have resulted as a consequence. However, it may be that currently utilized therapies also work by modifying this signaling pathway. For example, vitamin D, currently used as a topical treatment of psoriasis, has been shown to increase the generation of cAMP in response to betaAR agonists[87]. Glucocorticoids, the mainstay of topical therapy for psoriasis, increase both the expression of beta2AR in keratinocytes, and the generation of cAMP in response to agonists[88]. UVB irradiation, another mainstay in the treatment of psoriasis, has been shown to increase beta2AR-mediated cAMP accumulation[89]. Thus it is possible that the various effective treatments for psoriasis in some way improve the disease by modulating the beta2 AR response in keratinocytes. Since the safety profiles of beta agonists are well-documented, it may be worthwhile re-visiting this pharmaceutical class of drugs for efficacy in the treatment of psoriasis, perhaps as adjunctive therapies.
Wound Healing
The identification of beta AR on keratinocytes in the 1970's was followed by studies that focused on correlating their activation and subsequent generation of cAMP with cellular consequences. Thus, early studies correlated beta AR agonist stimulation and intracellular increases of cAMP in keratinocytes with a decrease in cell proliferation [9, 63]. Studies by Dunlap and Donaldson, demonstrated that incubation of newt keratinocytes with beta agonists inhibited their migration [90, 91], but other studies failed to confirm the presumed relationship between the increase in intracellular cAMP and inhibition of cell migration in either human[92] or newt keratinocytes[90, 93, 94]. Recent work in our laboratory has focused on the role of the beta2 AR in keratinocyte migration, and particularly with respect to cutaneous wound repair. We found that activation of the beta2 AR by isoproterenol both increased intracellular cAMP concentrations and inhibited keratinocyte migration in a concentration-dependent manner[32]. However, we demonstrated that the beta2AR-mediated decrease in migration is not regulated by the induced increase in cAMP, but rather by a cAMP-independent signaling pathway (Figure 1). Evidence for this conclusion included the findings that adenylate cyclase inhibitors that decreased intracellular cAMP failed to prevent the inhibition of keratinocyte migration induced by isoproterenol, and agents that directly activate adenylate cyclase, such as forskolin, induced an increase in intracellular concentrations of cAMP yet had no inhibitory effect on migration. In keratinocytes, activation of the beta2 AR results in dephosphorylation and inactivation of phospho-ERK MAP kinase, through the activation of the protein phosphatase PP2A[33]. ERK is activated by wounding[95] and is required for wound repair both in confluent keratinocyte cultures[96] and in human epidermis[97, 98]. Thus, inactivation of ERK by its dephosphorylation as a direct consequence of activation of the beta2 AR results in a reduced capacity of the keratinocytes to migrate. This inability to migrate in the presence of beta agonists then translates to in an inability to re-epithelialize a cutaneous wound, either in ex vivo cultured human skin, or in vivo in a murine tail wound model[55]. Taken together, these studies demonstrate that beta2 AR agonists impair cutaneous wound epithelialization.
The finding that beta2 AR agonists impair wound healing prompted the logical question of whether beta2 AR antagonists (beta blockers) can, on the other hand, improve it. This is indeed the case. Cultured human keratinocytes treated with beta2 AR antagonists demonstrate an increase in migratory speed, and this translates to better ability to re-epithelialize cutaneous wounds in either ex vivo organ cultured human skin[28], or in vivo in murine wounded skin[99, 100]. As might be anticipated, in keratinocytes, beta blockers increase the activation of the pro-motogenic ERK and its phosphorylation[28]. If beta blockers can improve wound epithelialization, the question of what endogenous ligand they are blocking naturally ensues. Two sources of ligand can be proposed. Circulating levels of epinephrine are increased post- trauma[101, 102] and thus exogenously added beta blockers could be preventing their action on beta2AR in keratinocytes, thereby enhancing migratory speed and wound epithelialization. Alternatively, since keratinocytes have the enzymatic machinery to generate catecholamines[1, 28], they can, and do synthesize endogenous epinephrine[28, 103], which could be locally secreted into the wound and function in an autocrine manner. In either scenario, the pharmacologic use of beta blockers would result in the observed increase in wound epithelialization.
Other studies have also provided additional evidence that beta blockers may be beneficial to cutaneous wound repair. Recently, Denda and colleagues reported that beta2 AR antagonists could repair the epidermal permeability barrier, a function required for the restoration of epidermal integrity after wounding[104]. Additionally, beta blockers are also widely used in the post-burn wound recovery period, and a retrospective outcome analysis by Arbabi et al. has demonstrated a shorter time for burn wound healing in a cohort of patients that received beta blockers during their hospital stay[105]. Thus, there are suggestions in the literature that both the endogenous beta2 AR signaling network and exogenously supplied beta2 AR antagonists may modulate wound repair in the clinical setting. Interestingly, preliminary epidemiologic studies have suggested a decrease in the incidence of one type of chronic wound, venous stasis ulcers, in patients in whom beta blockers are prescribed[106]. Clearly, this provocative finding will need further investigation to determine if systemic or topically administered beta blockers can be useful therapeutic approaches to healing chronic wounds. A more systematic epidemiological analysis of wound healing in cohorts of patients treated with beta2AR antagonists is warranted, and a clinical trial of these agents in patients with chronic wounds could help answer the question of clinical usefulness of this class of drugs. These studies may be on the horizon.
Perspective
Beta2 adrenergic receptors were identified in keratinocytes more than 30 years ago, but their function in the epidermis continues to be elucidated. Abnormalities in their expression, signaling pathway, or in the generation of endogenous catecholamine agonists by keratinocyes have been implicated in the pathogenesis of cutaneous diseases such as atopic dermatitis, vitiligo and psoriasis. Though the keratinocyte defects appear to be similar in some of these diseases, it may be that in patients with an associated defect in melanocyte beta2 AR signaling, vitiligo ensues, while in patients with associated abnormalities in lymphocytic immunologic function, atopic dermatitis is the outcome. New studies indicate that the beta2 AR also modulates keratinocyte migration, and thus can function to regulate wound re-epithelialization. Cumulatively, these studies suggest a role for beta 2AR active drugs in the management of cutaneous disease and wound healing. It is interesting to note that there appear to be only three U.S. patents registered for the use of beta adrenergic agents for skin disease: one for beta agonists for the treatment of psoriasis[86], one for beta agonists to decrease skin edema in acute surgical wounds[107], and a third for the topical use of beta agonists in cosmetics or topical medications to decrease substance P-mediated pain or irritation [108]. Thus, ample opportunity exists for development of beta2AR active drugs as therapeutic agents for a number of dermatological indications.
Table 1. Proposed modulation of the keratinocyte beta2 adrenergic receptor and the catecholamine synthesis pathway in selected dermatological diseases.
| Disorder | Beta2AR expression/activation | 6BH4 | PNMT activity | Norepinephrine | Epinephrine |
|---|---|---|---|---|---|
| Atopic Dermatitis | NA | ↑ | ↓ | ↑ | ↓ |
| Vitiligo | ↑ | ↑ | ↓ | ↑ | ↓ |
| Psoriasis | ↓ | NA | ↑ | NA* | NA* |
NA= not yet definitively addressed.
Elevated circulating levels of norepinephrine and epinephrine have been reported in psoriasis but epidermal or keratinocyte levels have not yet been studied.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schallreuter KU. Epidermal adrenergic signal transduction as part of the neuronal network in the human epidermis. J Investig Dermatol Symp Proc. 1997 Aug;2(1):37–40. doi: 10.1038/jidsymp.1997.9. [DOI] [PubMed] [Google Scholar]
- 2.Orenberg EK, Pfendt EA, Wilkinson DI. Characterization of alpha- and beta-adrenergic agonist stimulation of adenylate cyclase activity in human epidermal keratinocytes in vitro. J Invest Dermatol. 1983 Jun;80(6):503–507. doi: 10.1111/1523-1747.ep12535068. [DOI] [PubMed] [Google Scholar]
- 3.Steinkraus V, Steinfath M, Korner C, Mensing H. Binding of beta-adrenergic receptors in human skin. J Invest Dermatol. 1992 Apr;98(4):475–480. doi: 10.1111/1523-1747.ep12499860. [DOI] [PubMed] [Google Scholar]
- 4.Steinkraus V, Mak JC, Pichlmeier U, Mensing H, Ring J, Barnes PJ. Autoradiographic mapping of beta-adrenoceptors in human skin. Arch Dermatol Res. 1996 Aug;288(9):549–553. doi: 10.1007/BF02505253. [DOI] [PubMed] [Google Scholar]
- 5.Frielle T, Daniel KW, Caron MG, Lefkowitz RJ. Structural basis of beta-adrenergic receptor subtype specificity studied with chimeric beta 1/beta 2-adrenergic receptors. Proc Natl Acad Sci U S A. 1988 Dec;85(24):9494–9498. doi: 10.1073/pnas.85.24.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lands AM, Arnold A, McAuliff JP, Luduena FP, Brown TG., Jr Differentiation of receptor systems activated by sympathomimetic amines. Nature. 1967 May 6;214(88):597–598. doi: 10.1038/214597a0. [DOI] [PubMed] [Google Scholar]
- 7.Zheng M, Zhu W, Han Q, Xiao RP. Emerging concepts and therapeutic implications of [beta]-adrenergic receptor subtype signaling. Pharmacology & Therapeutics. 2005;108(3):257. doi: 10.1016/j.pharmthera.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Powell JA, Duell EA, Voorhees JJ. Beta adrenergic stimulation of endogenous epidermal cyclic AMP formation. Arch Dermatol. 1971 Oct;104(4):359–365. [PubMed] [Google Scholar]
- 9.Flaxman BA, Harper RA. In vitro analysis of the control of keratinocyte proliferation in human epidermis by physiologic and pharmacologic agents. J Invest Dermatol. 1975 Jul;65(1):52–59. doi: 10.1111/1523-1747.ep12598043. [DOI] [PubMed] [Google Scholar]
- 10.Gazith J, Cavey MT, Cavey D, Shroot B, Reichert U. Characterization of the beta-adrenergic receptors of cultured human epidermal keratinocytes. Biochemical Pharmacology. 1983;32(22):3397. doi: 10.1016/0006-2952(83)90368-4. [DOI] [PubMed] [Google Scholar]
- 11.Steinkraus V, Korner C, Steinfath M, Mensing H. High density of beta 2-adrenoceptors in a human keratinocyte cell line with complete epidermal differentiation capacity (HaCaT) Arch Dermatol Res. 1991;283(5):328–332. doi: 10.1007/BF00376622. [DOI] [PubMed] [Google Scholar]
- 12.Schallreuter KU, Wood JM, Pittelkow MR, Swanson NN, Steinkraus V. Increased in vitro expression of beta 2-adrenoceptors in differentiating lesional keratinocytes of vitiligo patients. Arch Dermatol Res. 1993;285(4):216–220. doi: 10.1007/BF00372012. [DOI] [PubMed] [Google Scholar]
- 13.Grando SA, Pittelkow MR, Schallreuter KU. Adrenergic and cholinergic control in the biology of epidermis: physiological and clinical significance. J Invest Dermatol. 2006 Sep;126(9):1948–1965. doi: 10.1038/sj.jid.5700151. [DOI] [PubMed] [Google Scholar]
- 14.Koizumi H, Yasui C, Fukaya T, Ohkawara A, Ueda T. Beta-adrenergic stimulation induces intracellular Ca++ increase in human epidermal keratinocytes. J Invest Dermatol. 1991 Feb;96(2):234–237. doi: 10.1111/1523-1747.ep12462120. [DOI] [PubMed] [Google Scholar]
- 15.Yasui C, Koizumi H, Fukaya T, Kumakiri M, Ohkawara A, Ueda T. Adenylate cyclase induces intracellular calcium increase in single human epidermal keratinocytes measured by fluorescence microscopy using Fura 2-AM. Br J Dermatol. 1992 Dec;127(6):589–594. doi: 10.1111/j.1365-2133.1992.tb14871.x. [DOI] [PubMed] [Google Scholar]
- 16.Osawa Y, Koizumi H, Fukaya T, Yasui C, Ohkawara A, Ueda T. Adenylate cyclase induces intracellular Ca2+ increase in single human epidermal keratinocytes of the epidermal sheet as measured by digital imaging microscopy using Fura 2-AM. Arch Dermatol Res. 1991;283(2):91–95. doi: 10.1007/BF00371615. [DOI] [PubMed] [Google Scholar]
- 17.Findlay I. Physiological modulation of inactivation in L-type Ca2+ channels: one switch. J Physiol. 2004 Jan 15;554(Pt 2):275–283. doi: 10.1113/jphysiol.2003.047902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou YY, Cheng H, Bogdanov KY, et al. Localized cAMP-dependent signaling mediates beta 2-adrenergic modulation of cardiac excitation-contraction coupling. Am J Physiol. 1997 Sep;273(3 Pt 2):H1611–1618. doi: 10.1152/ajpheart.1997.273.3.H1611. [DOI] [PubMed] [Google Scholar]
- 19.Denda M, Fujiwara S, Hibino T. Expression of voltage-gated calcium channel subunit alpha1C in epidermal keratinocytes and effects of agonist and antagonists of the channel on skin barrier homeostasis. Exp Dermatol. 2006 Jun;15(6):455–460. doi: 10.1111/j.0906-6705.2006.00430.x. [DOI] [PubMed] [Google Scholar]
- 20.Koizumi H, Tanaka H, Ohkawara A. beta-Adrenergic stimulation induces activation of protein kinase C and inositol 1,4,5-trisphosphate increase in epidermis. Exp Dermatol. 1997 Jun;6(3):128–132. doi: 10.1111/j.1600-0625.1997.tb00159.x. [DOI] [PubMed] [Google Scholar]
- 21.Mauro T. ‘The discovery channel’: CRAC'king the code of calcium influx. J Invest Dermatol. 2003 Jul;121(1):IX–X. doi: 10.1046/j.1523-1747.2003.12336.x. [DOI] [PubMed] [Google Scholar]
- 22.Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell. 1980 Jan;19(1):245–254. doi: 10.1016/0092-8674(80)90406-7. [DOI] [PubMed] [Google Scholar]
- 23.Boyce ST, Ham RG. Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J Invest Dermatol. 1983 Jul;81 1:33s–40s. doi: 10.1111/1523-1747.ep12540422. [DOI] [PubMed] [Google Scholar]
- 24.Tsao MC, Walthall BJ, Ham RG. Clonal growth of normal human epidermal keratinocytes in a defined medium. J Cell Physiol. 1982 Feb;110(2):219–229. doi: 10.1002/jcp.1041100217. [DOI] [PubMed] [Google Scholar]
- 25.Mammone T, Marenus K, Maes D, Lockshin RA. The induction of terminal differentiation markers by the cAMP pathway in human HaCaT keratinocytes. Skin Pharmacol Appl Skin Physiol. 1998 May-Jun;11(3):152–160. doi: 10.1159/000029821. [DOI] [PubMed] [Google Scholar]
- 26.Menon GK, Elias PM. Ultrastructural localization of calcium in psoriatic and normal human epidermis. Arch Dermatol. 1991 Jan;127(1):57–63. [PubMed] [Google Scholar]
- 27.Menon GK, Grayson S, Elias PM. Ionic calcium reservoirs in mammalian epidermis: ultrastructural localization by ion-capture cytochemistry. J Invest Dermatol. 1985 Jun;84(6):508–512. doi: 10.1111/1523-1747.ep12273485. [DOI] [PubMed] [Google Scholar]
- 28.Pullar CE, Rizzo A, Isseroff RR. beta-Adrenergic receptor antagonists accelerate skin wound healing: evidence for a catecholamine synthesis network in the epidermis. J Biol Chem. 2006 Jul 28;281(30):21225–21235. doi: 10.1074/jbc.M601007200. [DOI] [PubMed] [Google Scholar]
- 29.Schallreuter KU, Lemke KR, Pittelkow MR, Wood JM, Korner C, Malik R. Catecholamines in human keratinocyte differentiation. J Invest Dermatol. 1995 Jun;104(6):953–957. doi: 10.1111/1523-1747.ep12606218. [DOI] [PubMed] [Google Scholar]
- 30.Broadley KJ. Review of mechanisms involved in the apparent differential desensitization of beta1- and beta2-adrenoceptor-mediated functional responses. J Auton Pharmacol. 1999 Dec;19(6):335–345. doi: 10.1111/j.1365-2680.1999.tb00006.x. [DOI] [PubMed] [Google Scholar]
- 31.Johnson M. Beta2-adrenoceptors: mechanisms of action of beta2-agonists. Paediatric Respiratory Reviews. 2001;2(1):57. doi: 10.1053/prrv.2000.0102. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Hoffman BB, Isseroff RR. Beta-adrenergic receptor activation inhibits keratinocyte migration via a cyclic adenosine monophosphate-independent mechanism. J Invest Dermatol. 2002 Dec;119(6):1261–1268. doi: 10.1046/j.1523-1747.2002.19611.x. [DOI] [PubMed] [Google Scholar]
- 33.Pullar CE, Chen J, Isseroff RR. PP2A activation by beta2-adrenergic receptor agonists: novel regulatory mechanism of keratinocyte migration. J Biol Chem. 2003 Jun 20;278(25):22555–22562. doi: 10.1074/jbc.M300205200. [DOI] [PubMed] [Google Scholar]
- 34.Kim N, Kim H, Youm JB, et al. Site specific differential activation of ras/raf/ERK signaling in rabbit isoproterenol-induced left ventricular hypertrophy. Biochim Biophys Acta. 2006 Oct;1763(10):1067–1075. doi: 10.1016/j.bbamcr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Zheng M, Hou R, Han Q, Xiao RP. Different regulation of ERK1/2 activation by beta-adrenergic receptor subtypes in adult mouse cardiomyocytes. Heart Lung Circ. 2004 Jun;13(2):179–183. doi: 10.1016/j.hlc.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Shenoy SK, Drake MT, Nelson CD, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006 Jan 13;281(2):1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 37.Yeh CK, Ghosh PM, Dang H, et al. beta-Adrenergic-responsive activation of extracellular signal-regulated protein kinases in salivary cells: role of epidermal growth factor receptor and cAMP. Am J Physiol Cell Physiol. 2005 Jun;288(6):C1357–1366. doi: 10.1152/ajpcell.00370.2004. [DOI] [PubMed] [Google Scholar]
- 38.Pullar CE, Isseroff RR. Cyclic AMP mediates keratinocyte directional migration in an electric field. J Cell Sci. 2005 May 1;118(Pt 9):2023–2034. doi: 10.1242/jcs.02330. [DOI] [PubMed] [Google Scholar]
- 39.Pullar CE, Isseroff RR, Nuccitelli R. Cyclic AMP-dependent protein kinase A plays a role in the directed migration of human keratinocytes in a DC electric field. Cell Motil Cytoskeleton. 2001 Dec;50(4):207–217. doi: 10.1002/cm.10009. [DOI] [PubMed] [Google Scholar]
- 40.Szentivanyi A. The beta adrenergic theory of the atopic abnormality in bronchial asthma. J Allergy. 1968;42:203–232. [Google Scholar]
- 41.Grewe SR, Chan SC, Hanifin JM. Elevated leukocyte cyclic AMP-phosphodiesterase in atopic disease: a possible mechanism for cyclic AMP-agonist hyporesponsiveness. J Allergy Clin Immunol. 1982 Dec;70(6):452–457. doi: 10.1016/0091-6749(82)90008-2. [DOI] [PubMed] [Google Scholar]
- 42.Hanifin JM. Pharmacophysiology of atopic dermatitis. Clin Rev Allergy. 1986 Feb;4(1):43–65. doi: 10.1007/BF02991187. [DOI] [PubMed] [Google Scholar]
- 43.Reed CE, Busse WW, Lee TP. Adrenergic mechanisms and the adenyl cyclase system in atopic dermatitis. J Invest Dermatol. 1976 Sep;67(3):333–338. doi: 10.1111/1523-1747.ep12514494. [DOI] [PubMed] [Google Scholar]
- 44.Schallreuter KU, Pittelkow MR, Swanson NN, et al. Altered catecholamine synthesis and degradation in the epidermis of patients with atopic eczema. Arch Dermatol Res. 1997 Nov;289(12):663–666. doi: 10.1007/s004030050258. [DOI] [PubMed] [Google Scholar]
- 45.Pochet R, Delaspesse G, DeManbuege J. Characterization of beta adrenoceptors on intact circulating lymphocytes from patients with atopic dermatitis. Acta Derm Venereol Suppl (Stockh) 1980;92:26–29. [Google Scholar]
- 46.Bamshad J. Catechol-O-methyl transferase in skin of patients with atopic dermatitis. J Invest Dermatol. 1969 Jan;52(1):100–102. doi: 10.1038/jid.1969.14. [DOI] [PubMed] [Google Scholar]
- 47.Buske-Kirschbaum A, Geiben A, Hollig H, Morschhauser E, Hellhammer D. Altered responsiveness of the hypothalamus-pituitary-adrenal axis and the sympathetic adrenomedullary system to stress in patients with atopic dermatitis. J Clin Endocrinol Metab. 2002 Sep;87(9):4245–4251. doi: 10.1210/jc.2001-010872. [DOI] [PubMed] [Google Scholar]
- 48.Small KM, McGraw DW, Liggett SB. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu Rev Pharmacol Toxicol. 2003;43:381–411. doi: 10.1146/annurev.pharmtox.43.100901.135823. [DOI] [PubMed] [Google Scholar]
- 49.Schallreuter KU, Wei Y, Pittelkow MR, et al. Atopic ezcema can be associated with a mutation in the beta2 adrenergic gene. J Invest Dermatol. 1996;106:902. [Google Scholar]
- 50.Maestroni GJ. Sympathetic nervous system influence on the innate immune response. Ann N Y Acad Sci. 2006 Jun;1069:195–207. doi: 10.1196/annals.1351.017. [DOI] [PubMed] [Google Scholar]
- 51.Le Poole IC, Wankowicz-Kalinska A, van den Wijngaard RM, Nickoloff BJ, Das PK. Autoimmune aspects of depigmentation in vitiligo. J Investig Dermatol Symp Proc. 2004 Jan;9(1):68–72. doi: 10.1111/j.1087-0024.2004.00825.x. [DOI] [PubMed] [Google Scholar]
- 52.Njoo MD, Westerhof W. Vitiligo. Pathogenesis and treatment. Am J Clin Dermatol. 2001;2(3):167–181. doi: 10.2165/00128071-200102030-00006. [DOI] [PubMed] [Google Scholar]
- 53.Gillbro JM, Marles LK, Hibberts NA, Schallreuter KU. Autocrine catecholamine biosynthesis and the beta-adrenoceptor signal promote pigmentation in human epidermal melanocytes. J Invest Dermatol. 2004 Aug;123(2):346–353. doi: 10.1111/j.0022-202X.2004.23210.x. [DOI] [PubMed] [Google Scholar]
- 54.Schallreuter KU, Wood JM, Lemke R, et al. Production of catecholamines in the human epidermis. Biochem Biophys Res Commun. 1992 Nov 30;189(1):72–78. doi: 10.1016/0006-291x(92)91527-w. [DOI] [PubMed] [Google Scholar]
- 55.Pullar CE, Grahn JC, Liu W, Isseroff RR. Beta2-adrenergic receptor activation delays wound healing. Faseb J. 2006 Jan;20(1):76–86. doi: 10.1096/fj.05-4188com. [DOI] [PubMed] [Google Scholar]
- 56.Schallreuter KU. Beta-adrenergic blocking drugs may exacerbate vitiligo. Br J Dermatol. 1995 Jan;132(1):168–169. doi: 10.1111/j.1365-2133.1995.tb08660.x. [DOI] [PubMed] [Google Scholar]
- 57.Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190(2):109–115. doi: 10.1159/000246657. [DOI] [PubMed] [Google Scholar]
- 58.Schallreuter KU, Wood JM, Ziegler I, et al. Defective tetrahydrobiopterin and catecholamine biosynthesis in the depigmentation disorder vitiligo. Biochim Biophys Acta. 1994 May 25;1226(2):181–192. doi: 10.1016/0925-4439(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 59.Cucchi ML, Frattini P, Santagostino G, Preda S, Orecchia G. Catecholamines increase in the urine of non-segmental vitiligo especially during its active phase. Pigment Cell Res. 2003 Apr;16(2):111–116. doi: 10.1034/j.1600-0749.2003.00015.x. [DOI] [PubMed] [Google Scholar]
- 60.Schallreuter KU, Wood JM. The Importance of -Phenylalanine Transport and Its Autocrine Turnover to -Tyrosine for Melanogenesis in Human Epidermal Melanocytes. Biochemical and Biophysical Research Communications. 1999;262(2):423. doi: 10.1006/bbrc.1999.1241. [DOI] [PubMed] [Google Scholar]
- 61.Hasse S, Gibbons NC, Rokos H, Marles LK, Schallreuter KU. Perturbed 6-tetrahydrobiopterin recycling via decreased dihydropteridine reductase in vitiligo: more evidence for H2O2 stress. J Invest Dermatol. 2004 Feb;122(2):307–313. doi: 10.1046/j.0022-202X.2004.22230.x. [DOI] [PubMed] [Google Scholar]
- 62.Schallreuter KU, Pittelkow MP. Defective calcium uptake in keratinocyte cell cultures from vitiliginous skin. Arch Dermatol Res. 1988;280(3):137–139. doi: 10.1007/BF00456842. [DOI] [PubMed] [Google Scholar]
- 63.Harper RA, Flaxman BA. Effect of pharmacological agents on human keratinocyte mitosis in vitro. II. Inhibition by catecholamines. J Cell Physiol. 1975 Oct;86(2 Pt 1):293–299. doi: 10.1002/jcp.1040860213. [DOI] [PubMed] [Google Scholar]
- 64.Voorhees JJ, Duell EA, Bass LJ, Harrell ER. Role of cyclic AMP in the control of epidermal cell growth and differentiation. Natl Cancer Inst Monogr. 1973 Jul;38:47–59. [PubMed] [Google Scholar]
- 65.Voorhees JJ, Duell EA, Kelsey WH. Dibutyryl cyclic AMP inhibition of epidermal cell division. Arch Dermatol. 1972 Mar;105(3):384–386. [PubMed] [Google Scholar]
- 66.Voorhees JJ, Duell EA. Psoriasis as a possible defect of the adenyl cyclase-cyclic AMP cascade. A defective chalone mechanism? Arch Dermatol. 1971 Oct;104(4):352–358. [PubMed] [Google Scholar]
- 67.Voorhees JJ, Duell EA. Imbalanced cyclic AMP-cyclic GMP levels in psoriasis. Adv Cyclic Nucleotide Res. 1975;5:735–758. [PubMed] [Google Scholar]
- 68.Voorhees JJ, Duell EA, Bass LJ, Powell JA, Harrell ER. Decreased cyclic AMP in the epidermis of lesions of psoriasis. Arch Dermatol. 1972 May;105(5):695–701. [PubMed] [Google Scholar]
- 69.Voorhees JJ, Duell EA, Bass LJ, Powell JA, Harrell ER. The cyclic AMP system in normal and psoriatic epidermis. J Invest Dermatol. 1972 Jul;59(1):114–120. doi: 10.1111/1523-1747.ep12625885. [DOI] [PubMed] [Google Scholar]
- 70.Iizuka H, Umeda K, Koizumi H, Aoyagi T, Miura Y. Epinephrine-induced cyclic AMP accumulation in the psoriatic epidermis. Acta Derm Venereol. 1981;61(5):391–395. [PubMed] [Google Scholar]
- 71.Mahrle G, Orfanos CE. Lack of beta-adrenergic stimulation of membrane bound adenyl cyclase in psoriasis as compared to normal epidermis (author's transl) Arch Dermatol Res. 1975 Sep 12;253(2):195–202. doi: 10.1007/BF00582071. [DOI] [PubMed] [Google Scholar]
- 72.Steinkraus V, Steinfath M, Stove L, Korner C, Abeck D, Mensing H. Beta-adrenergic receptors in psoriasis: evidence for down-regulation in lesional skin. Arch Dermatol Res. 1993;285(5):300–304. doi: 10.1007/BF00371601. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi H, Kinouchi M, Tamura T, Iizuka H. Decreased beta 2-adrenergic receptor-mRNA and loricrin-mRNA, and increased involucrin-mRNA transcripts in psoriatic epidermis: analysis by reverse transcription-polymerase chain reaction. Br J Dermatol. 1996 Jun;134(6):1065–1069. [PubMed] [Google Scholar]
- 74.O'Brien M, Koo J. The mechanism of lithium and beta-blocking agents in inducing and exacerbating psoriasis. J Drugs Dermatol. 2006 May;5(5):426–432. [PubMed] [Google Scholar]
- 75.Tsankov N, Kazandjieva J, Drenovska K. Drugs in exacerbation and provocation of psoriasis. Clin Dermatol. 1998 May-Jun;16(3):333–351. doi: 10.1016/s0738-081x(98)00005-4. [DOI] [PubMed] [Google Scholar]
- 76.Gaylarde PM, Brock AP, Sarkany I. Psoriasiform changes in guinea-pig skin from propranolol. Clin Exp Dermatol. 1978 Jun;3(2):157–160. doi: 10.1111/j.1365-2230.1978.tb01479.x. [DOI] [PubMed] [Google Scholar]
- 77.Van de Kerkhof P. Psoriasiform changes in guinea-pig skin from propanalol. Clin Exp Dermatol. 1979 Jun;4(2):267–268. doi: 10.1111/j.1365-2230.1979.tb01631.x. [DOI] [PubMed] [Google Scholar]
- 78.Wolf R, Shechter H, Brenner S. Induction of psoriasiform changes in guinea pig skin by propranolol. Int J Dermatol. 1994 Nov;33(11):811–814. doi: 10.1111/j.1365-4362.1994.tb01007.x. [DOI] [PubMed] [Google Scholar]
- 79.Wolf R. A new concept in the pathogenesis of drug-induced psoriasis. Med Hypotheses. 1986 Nov;21(3):277–279. doi: 10.1016/0306-9877(86)90021-6. [DOI] [PubMed] [Google Scholar]
- 80.Halevy S, Livni E. Psoriasis and psoriasiform eruptions associated with propranolol--the role of an immunological mechanism. Arch Dermatol Res. 1991;283(7):472–473. doi: 10.1007/BF00371785. [DOI] [PubMed] [Google Scholar]
- 81.Halevy S, Livni E. Beta-adrenergic blocking drugs and psoriasis: the role of an immunologic mechanism. J Am Acad Dermatol. 1993 Sep;29(3):504–505. doi: 10.1016/s0190-9622(08)82012-9. [DOI] [PubMed] [Google Scholar]
- 82.Ozkur M, Erbagci Z, Nacak M, Tuncel A, Gorucu S, Aynacioglu AS. Association of the Arg16Gly polymorphism of the beta-2-adrenergic receptor with psoriasis. J Dermatol Sci. 2004 Aug;35(2):162–164. doi: 10.1016/j.jdermsci.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 83.Liggett SB. beta(2)-adrenergic receptor pharmacogenetics. Am J Respir Crit Care Med. 2000 Mar;161(3 Pt 2):S197–201. doi: 10.1164/ajrccm.161.supplement_2.a1q4-10. [DOI] [PubMed] [Google Scholar]
- 84.Schmid-Ott G, Jacobs R, Jager B, et al. Stress-induced endocrine and immunological changes in psoriasis patients and healthy controls. A preliminary study. Psychother Psychosom. 1998;67(1):37–42. doi: 10.1159/000012257. [DOI] [PubMed] [Google Scholar]
- 85.Johansson O, Olsson A, Enhamre A, Hammar H, Goldstein M. Phenylethanolamine N-methyltransferase-like immunoreactivity in psoriasis. An immunohistochemical study on catecholamine synthesizing enzymes and neuropeptides of the skin. Acta Derm Venereol. 1987;67(1):1–7. [PubMed] [Google Scholar]
- 86.Nelson EL Nelson Research & Development Company. Method for treatment of psoriasis. US patent 4038417. 1977 assignee.
- 87.Takahashi H, Tamura T, Iizuka H. 1,25-Dihydroxyvitamin D3 increased beta-adrenergic adenylate cyclase response of fetal rat keratinizing epidermal cells (FRSK cells) J Dermatol Sci. 1996 Feb;11(2):121–128. doi: 10.1016/0923-1811(95)00428-9. [DOI] [PubMed] [Google Scholar]
- 88.Takahashi H, Iizuka H. Regulation of beta 2-adrenergic receptors in keratinocytes: glucocorticoids increase steady-state levels of receptor mRNA in foetal rat keratinizing epidermal cells (FRSK cells) Br J Dermatol. 1991 Apr;124(4):341–347. doi: 10.1111/j.1365-2133.1991.tb00594.x. [DOI] [PubMed] [Google Scholar]
- 89.Iizuka H, Kajita S, Ohkawara A. Ultraviolet radiation augments epidermal beta-adrenergic adenylate cyclase response. J Invest Dermatol. 1985 May;84(5):401–403. doi: 10.1111/1523-1747.ep12265501. [DOI] [PubMed] [Google Scholar]
- 90.Dunlap MK. Cyclic AMP levels in migrating and non-migrating newt epidermal cells. J Cell Physiol. 1980 Sep;104(3):367–373. doi: 10.1002/jcp.1041040310. [DOI] [PubMed] [Google Scholar]
- 91.Donaldson DJ, Mahan JT. Influence of catecholamines on epidermal cell migration during wound closure in adult newts. Comp Biochem Physiol C. 1984;78(2):267–270. doi: 10.1016/0742-8413(84)90081-1. [DOI] [PubMed] [Google Scholar]
- 92.Iwasaki T, Chen JD, Kim JP, Wynn KC, Woodley DT. Dibutyryl cyclic AMP modulates keratinocyte migration without alteration of integrin expression. J Invest Dermatol. 1994 Jun;102(6):891–897. doi: 10.1111/1523-1747.ep12383031. [DOI] [PubMed] [Google Scholar]
- 93.Dunlap MK, Donaldson DJ. Effect of cAMP and related compounds on newt epidermal cell migration both in vivo and in vitro. J Exp Zool. 1980 Apr;212(1):13–19. doi: 10.1002/jez.1402120103. [DOI] [PubMed] [Google Scholar]
- 94.Donaldson DJ, Dunlap MK, Mahan JT. Effects of concanavalin A and cholera toxin on epidermal cAMP and migration rate during wound closure in adult newts. Comp Biochem Physiol C. 1984;79(2):243–248. doi: 10.1016/0742-8413(84)90193-2. [DOI] [PubMed] [Google Scholar]
- 95.Matsubayashi Y, Ebisuya M, Honjoh S, Nishida E. ERK activation propagates in epithelial cell sheets and regulates their migration during wound healing. Curr Biol. 2004 Apr 20;14(8):731–735. doi: 10.1016/j.cub.2004.03.060. [DOI] [PubMed] [Google Scholar]
- 96.Davidoff MS, Ungefroren H, Middendorff R, et al. Catecholamine-synthesizing enzymes in the adult and prenatal human testis. Histochem Cell Biol. 2005 Sep;124(34):313–323. doi: 10.1007/s00418-005-0024-x. [DOI] [PubMed] [Google Scholar]
- 97.McCaig CD, Rajnicek AM, Song B, Zhao M. Controlling cell behavior electrically: current views and future potential. Physiol Rev. 2005 Jul;85(3):943–978. doi: 10.1152/physrev.00020.2004. [DOI] [PubMed] [Google Scholar]
- 98.Providence KM, Higgins PJ. PAI-1 expression is required for epithelial cell migration in two distinct phases of in vitro wound repair. J Cell Physiol. 2004 Aug;200(2):297–308. doi: 10.1002/jcp.20016. [DOI] [PubMed] [Google Scholar]
- 99.Pullar CE, Rizzo AE, Isseroff RR. Beta-adrenergic receptor antagonists accelerate skin wound healing: evidence for a catecholamine synthesis network in the epidermis. J Invest Dermatol. 2006;126:64. doi: 10.1074/jbc.M601007200. abstract. [DOI] [PubMed] [Google Scholar]
- 100.Sivamani RK, Pullar CE, Griffiths B, Baier BS, Greenhalgh DG, Isseroff RR. Beta2-adrenergic receptor blockade accelerates burn wound healing. J Invest Dermatol. 2006;126:59. abstract. [Google Scholar]
- 101.Sedowofia K, Barclay C, Quaba A, et al. The systemic stress response to thermal injury in children. Clin Endocrinol (Oxf) 1998 Sep;49(3):335–341. doi: 10.1046/j.1365-2265.1998.00553.x. [DOI] [PubMed] [Google Scholar]
- 102.Wilmore DW, Long JM, Mason AD, Jr, Skreen RW, Pruitt BA., Jr Catecholamines: mediator of the hypermetabolic response to thermal injury. Ann Surg. 1974 Oct;180(4):653–669. doi: 10.1097/00000658-197410000-00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schallreuter KU, Wood JM, Lemke R, et al. Production of catecholamines in the human epidermis. Biochemical and Biophysical Research Communications. 1992;189(1):72. doi: 10.1016/0006-291x(92)91527-w. [DOI] [PubMed] [Google Scholar]
- 104.Denda M, Fuziwara S, Inoue K. Beta2-adrenergic receptor antagonist accelerates skin barrier recovery and reduces epidermal hyperplasia induced by barrier disruption. J Invest Dermatol. 2003 Jul;121(1):142–148. doi: 10.1046/j.1523-1747.2003.12310.x. [DOI] [PubMed] [Google Scholar]
- 105.Arbabi S, Ahrns KS, Wahl WL, et al. Beta-blocker use is associated with improved outcomes in adult burn patients. J Trauma. 2004 Feb;56(2):265–269. doi: 10.1097/01.TA.0000109859.91202.C8. discussion 269-271. [DOI] [PubMed] [Google Scholar]
- 106.Margolis D, Isseroff RR, Hofstad O. Beta adrenergic drug use is associated with decreased incidence of venous leg ulcers. J Invest Dermatol. 2006;126:49. abstract. [Google Scholar]
- 107.McDonald DM The Regents of the University of California. Use of formoterol for treatment of tissue injury. US patent 5135954. 1992 assignee.
- 108.Leclaire J, de Lacharriere O, Breton L Societe L'Oreal S.A. Cosmetic/pharmaceutical compositions comprising β-adrenergic agonists/substance P antagonists. US patent 5958432. 1999 assignee.