

Abstract
The nuclear lamina is a protein meshwork lining the nucleoplasmic face of the inner nuclear membrane and represents an important determinant of interphase nuclear architecture. Its major components are the A- and B-type lamins. Whereas B-type lamins are found in all mammalian cells, A-type lamin expression is developmentally regulated. In the mouse, A-type lamins do not appear until midway through embryonic development, suggesting that these proteins may be involved in the regulation of terminal differentiation. Here we show that mice lacking A-type lamins develop to term with no overt abnormalities. However, their postnatal growth is severely retarded and is characterized by the appearance of muscular dystrophy. This phenotype is associated with ultrastructural perturbations to the nuclear envelope. These include the mislocalization of emerin, an inner nuclear membrane protein, defects in which are implicated in Emery-Dreifuss muscular dystrophy (EDMD), one of the three major X-linked dystrophies. Mice lacking the A-type lamins exhibit tissue-specific alterations to their nuclear envelope integrity and emerin distribution. In skeletal and cardiac muscles, this is manifest as a dystrophic condition related to EDMD.
Keywords: emerin, muscular dystrophy, nuclear envelope, lamins
The vertebrate nuclear lamina is a thin (10–20 nm) protein meshwork associated with the nuclear face of the inner nuclear membrane (INM). Because of its role in maintaining nuclear envelope (NE) integrity and in providing anchoring sites for chromatin domains, the lamina is considered to represent an important determinant of interphase nuclear architecture (Gerace and Burke 1988; Gant and Wilson 1997). The major components of the lamina are intermediate filament-like proteins, the nuclear lamins. Most adult mammalian somatic cells contain three major lamins, A, B1, and C, as well as several minor lamins (B2 and AΔ10). These various forms are grouped into two classes, A-type (A, AΔ10 and C) and B-type (B1 and B2). Lamins B1 and B2 are encoded by separate genes, whereas all of the A-type lamins are encoded by a single gene and arise through alternative splicing of a common transcript (Moir et al. 1995; Stuurman et al. 1998).
While B-type lamins are found in all nucleated somatic cells, the expression of A-type lamins is developmentally regulated. In the mouse, A-type lamins are absent from all preimplantation stage embryonic cells (including embryonal carcinoma [EC] cells), with their synthesis commencing at about embryonic day 9 within the visceral endoderm and trophoblast (Stewart and Burke 1987). Subsequently, A-type lamins appear asynchronously in various tissues with certain cell types not acquiring these proteins until after birth (Rober et al. 1989). A few, notably cells of the immune system, pancreatic islets and Purkinje cells, only express B-type lamins (Rober et al. 1990). These findings indicate that at the cellular level, A-type lamins are nonessential. However, the significance of these observations and how they relate to lamin function remain uncertain. While it has often been suggested that A-type lamins are involved in terminal differentiation, possibly as determinants of chromatin organization, this notion has never been tested.
Here, we report the derivation of mice in which the A-type lamins has been eliminated by gene targeting. Although these mice exhibit overtly normal embryonic development, their postnatal growth is marked by the rapid onset of muscular dystrophy. This condition is associated with aberrant localization of emerin, an INM protein linked to human Emery-Dreifuss muscular dystrophy (EDMD). Therefore, these mice provide new insights into the relationship between lamins, INM proteins, and their role in EDMD, as well as provide a unique model for the autosomal variant of this disease (Bonne et al. 1999).
Materials and Methods
Targeted Disruption of Lamin A/C Gene
An 8.6-kb fragment containing exons 2–12 was isolated from a 129/Sv mouse liver genomic library. To produce the targeting vector, an NsiI-BamHI fragment was deleted, removing exons 8 to part of 11, and replaced with the Pgkneo neomycin resistance cassette in reverse orientation to the Lmna gene. The targeting vector was linearized with ClaI and electroporated into W9.5 ES cells. Clones were picked, expanded, and screened for homologous recombinants, after digestion with EcoRI, using a probe to exon 2. Two clones were injected into C57Bl/6 blastocysts, and chimeras were derived and bred to produce germline offspring as described (Stewart 1993). Homozygotes and heterozygotes were distinguished from wild-type sibs by EcoRI digestion of tail DNA.
Transfection
A human lamin A cDNA was subcloned into the pTracer-CMV vector (Invitrogen Corp.). The linearized vector was transfected into lamin A/C −/− mouse embryonic fibroblasts (MEFs). Stable clones were selected using Zeocin, according to the manufacturer's instructions and the clones pooled. Subsequent analysis showed that the cells in the pool were heterogeneous with regard to lamin A expression.
Antibodies
The antibody to mouse emerin was provided by Dr. Glenn Morris (NE Wales Institute, UK). Dr. Erich Nigg (University of Geneva) provided the antibody against lamin B2. Dr. Larry Gerace (Scripps Institute) provided both antibodies against LAP2 and an antibody against lamin B. The antibodies SA1 (specific for Nup153) and XB10 (against the lamin A/C central rod domain) have been described previously (Horton et al. 1992; Bodoor et al. 1999). Dr. Frank McKeon provided the 1E4 antibody to the amino terminal region of lamins A and C. The rhodamine and FITC-conjugated secondary antibodies were from Tago, Inc.
Histology and Immunohistochemistry
Tissues and cells were fixed in 10% buffered formalin or 3% paraformaldehyde in PBS respectively. Tissues for immunohistochemical analysis were embedded in OCT and snap frozen. Tissues for histological analysis were dehydrated, cleared, embedded in paraffin, sectioned at 6 microns, and stained in hematoxylin/eosin. MEFs were processed for immunofluorescence microscopy as previously described (Ash et al. 1977).
Electron Microscopy
The in situ processing of cultured cells was modified from a previously described method (Gonda et al. 1976). Epon sections (50 nm), stained with uranyl acetate and lead citrate, were examined and photographed at 75 kV.
Results and Discussion
To mutate the mouse lamin A/C (Lmna) gene, we deleted a region extending from exon 8 to the middle of exon 11. This removed 114 codons as well as the 3′ untranslated sequence, including the polyadenylation signal of lamin C, whereas 152 codons were eliminated from the lamin A coding region. The deletion was introduced by homologous recombination into ES cells and six homologous recombinant clones were identified (Fig. 1, a and b). After blastocyst injection of two clones, chimeric offspring were derived with subsequent germline transmission of the mutated allele. Heterozygotes were intercrossed to derive viable homozygous offspring. At birth, these were indistinguishable from their heterozygous or wild-type siblings.
Figure 1.

Targeting of the Lmna gene. (a) Structure of the mouse Lmna gene, together with the targeting vector and homologous recombinant containing the PgkNeo cassette. (b) Southern analysis of the representative genotypes from Lmna heterozygote crosses. (c) Northern analysis from wild-type and Lmna null fibroblasts showing loss of full length forms of lamin A and C mRNAs. These are replaced by truncated transcripts at levels 10 fold below those of wild type. (d) Western analysis, with the XB10 antibody (to the central domain of lamin A/C) of nuclear extracts from MEFs of all three genotypes showing that lamin A and C proteins were undetectable in the −/− fibroblasts, whereas lamin B levels were unaffected. P19 EC cells were used as a negative control. (e) Western analysis, using the 1E4 antibody (against the lamin A/C amino-terminal region) of liver nuclei and nuclear envelopes showing, as in the MEFs' absence of the lamin A proteins.
Loss of lamin A/C expression was determined by Western blot analysis of cell extracts, nuclei, or NEs prepared from the livers of weaned offspring or from embryonic fibroblasts (MEFs) established from day 13 embryos. Whereas NEs could be readily prepared from the livers of mice heterozygous and wild-type for the Lmna gene, they could not be isolated in an intact form from the lamin null mice. Instead, the −/− NEs fragmented, leading to poor recovery. Using two independent antibodies against epitopes within either the first 250 amino acids or the central rod domain of lamin A/C (1E4 and XB10, respectively) (McKeon et al. 1986; Horton et al. 1992), proteins of the appropriate molecular masses were undetectable in any of the samples prepared from tissues homozygous for the mutated gene. Neither was there any evidence for truncated forms of the two proteins. Lamin B1 levels in all of the cell types remained unaltered (Fig. 1d and Fig. e). Northern blot analysis of poly(A)+ mRNA from −/− livers revealed two faster migrating faint bands at levels 10-fold lower than the wild-type lamin A and C transcripts (Fig. 1 c). Taken together, these results indicate that the partial deletion of the Lmna gene resulted in the absence of both full-length transcripts and stable lamin A/C proteins.
These results were confirmed by immunocytochemistry since neither of the two anti–lamin A/C antibodies labeled the NEs of the Lmna −/− MEFs (Fig. 2 a). In contrast, lamins B1 and B2 were readily detected (Fig. 2d and Fig. e). These labeling experiments also revealed dramatic changes in nuclear morphology. Whereas nuclei of wild-type MEFs are roughly circular or slightly ovoid, those of Lmna −/− MEFs are often highly elongated or irregular and exhibit loss of B-type lamins from one pole (Fig. 2d and Fig. e). Although the nuclei remain intact, the overall impression is of large-scale herniation of the nuclear membranes. This abnormality, evident in >80% of the −/− MEF nuclei, is apparent in Fig. 2 e, where Nomarski and lamin B immunofluorescence images are superimposed. Similar results were obtained with antibodies against both the inner nuclear membrane protein LAP2 (Foisner and Gerace 1993) and the nuclear pore complex (NPC) protein Nup153 (Bodoor et al. 1999) (Fig. 2b and Fig. c). The latter also revealed a slight degree of NPC clustering within some −/− nuclear envelopes. An intermediate phenotype was observed for +/− MEFs with frequent elongation of nuclei, but largely normal distribution of nuclear envelope proteins (data not shown). Ultrastructural examination of −/− MEFs and hepatocytes revealed a thinning or loss of heterochromatin at discrete regions of the nuclear face of the INM. These segments of the nuclear envelope, which also lack morphologically identifiable NPCs, likely correspond to the herniations observed in the light microscope (Fig. 3, a and b). Thus, the integrity of NEs in −/− cells is profoundly compromised and shows conclusively for the first time that A-type nuclear lamins are essential for the maintenance of normal nuclear architecture. This complements a previous study in Drosophila showing that a B-type lamin is also essential for nuclear integrity (Lenz-Bohme et al. 1997).
Figure 2.

Immunohistochemical analysis of wild-type and Lmna null MEFs. The nuclei in all experiments were labeled with DAPI; in a–d, DAPI and immunofluorescence images have been superimposed. (a) Lamins A and C were detected with the XB10 mAb. (b) Nuclear lamina–associated protein, LAP2. Note the uniform distribution of this protein in wild-type nuclear envelopes, but its loss from one pole of the irregularly shaped nuclei in the Lmna null cells. (c) Nuclear pore complex Nup153. As with LAP2, there is loss from one pole of the nucleus, indicating exclusion of NPCs from this region. (d and e) Lamin B. Exclusion of this protein from one pole of the nucleus is clearly seen in the Lmna null cells. In e, the immunofluorescence image showing lamin B distribution (Lamin B*) has been superimposed on the Nomarski image of the same cell. Note that the two panels in e show complementary images of the same Lmna null cell. The lower panel shows DAPI labeling only.
Figure 3.
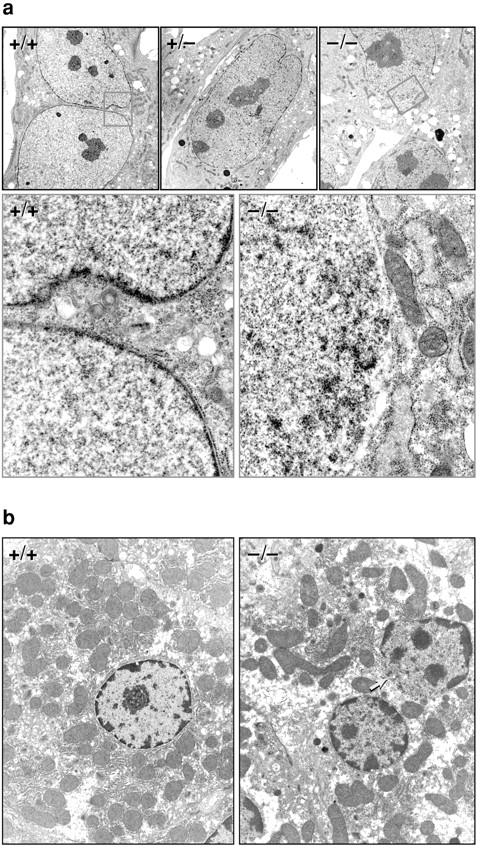
Electron microscopic analysis of MEFs (a) and hepatocytes (b). (a) In the wild-type and heterozygous MEF nuclei, a largely continuous layer of heterochromatin in contact with the inner face of the nuclear envelope is visible. In the Lmna null nuclei, there are obvious discontinuities in this layer (orange box) that are juxtaposed to distensions of the nuclear membranes. (b) Similar discontinuities can be seen in the liver nuclei (arrow).
At birth, Lmna null mice were indistinguishable from their heterozygous or wild-type sibs. However, within 2–3 wk a reduction in their growth rate was noted and by ∼4 wk, despite normal tooth development and the continued ability to eat, their growth had ceased. At this time their mean body weight was roughly 50% that of their wild-type or heterozygous littermates. At ∼3–4 wk, the homozygotes began to display an abnormal gait with a stiff walking posture, characterized by splayed hind legs and an inability to hang onto structures with their forepaws. Their overall posture became progressively more hunched, exhibiting distinct scoliosis/kyphosis. By the eighth week, all of the homozygotes had died. The heterozygotes are also apparently normal and have not exhibited any premature mortality when compared with their wild-type sibs.
Histological analysis of homozygotes revealed that the majority of their internal organs were normal, although some thymic atrophy and a reduction in spleen size was evident as was an absence of white fat, possibly as a secondary consequence of physiological stress. The wild-type and heterozygous mice exhibited no overt abnormalities. Examination of the musculature of the homozygote nulls revealed that the perivertebral muscles and those surrounding the femur (rectus femoris and semimembranous) were dystrophic. The involvement of individual fibers within each muscle was not uniform with those proximal to the bone being the most severely impaired. Many of these were atrophic with others exhibiting signs of degeneration with hyalin or flocculent cytoplasm. The dystrophic muscle fibers also exhibited variations in diameter, plus an increase in the number of nuclei with some being centrally located within the fibers (Fig. 4 b). Muscles of the head, tongue, and diaphragm were largely unaffected. In the heart, the ventricular muscle was most severely compromised although myocyte involvement was nonuniform. Some were of normal size, but had degenerated with condensed or flocculent eosinophilic or vacuolated cytoplasm (Fig. 4 d). These were often associated with patchy mineralization. Other cardiac myocytes were clearly atrophic. The Lmna null mice did not exhibit elevated serum creatine kinase levels, a feature associated with some but not all forms of muscular dystrophy (data not shown) (Bulfield et al. 1984). Overall, the Lmna −/− mice develop a cardiac and skeletal myopathy bearing a striking resemblance to human EDMD (Wehnert and Muntoni 1999).
Figure 4.
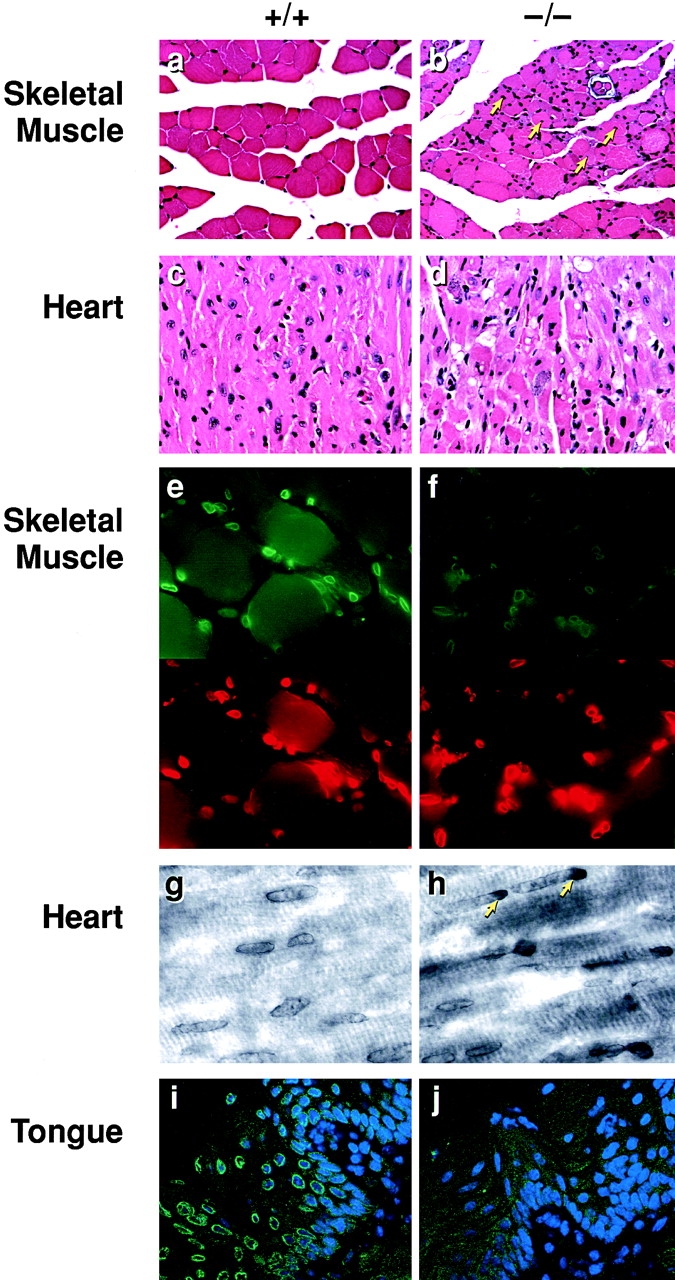
Histological and immunohistochemical analysis of tissues from Lmna null mice. (a) Wild-type perivertebral muscle showing the peripheral localization of the nuclei in the muscle fibers. (b) Perivertebral muscles from Lmna null mice showing an increase in nuclear number with many centrally located within the muscle fibers (arrows). (c) Wild-type ventricular cardiac muscle. (d) Ventricular muscle from a Lmna null mouse. (e and f) Emerin (upper panels) localization in the skeletal nuclei of wild-type (e) and lamin −/− (f) mice. In the −/− muscle, the signal intensity of emerin in the NE is weaker than in the +/+ nuclei. Anti–lamin B labeling is unchanged (lower panels rhodamine label). (g and h) Emerin staining in the cardiac nuclei of wild-type (g) and lmna −/− (h) mice. Note the polar distribution (arrows) of emerin in the −/− cardiac muscle NEs. (i and j) Emerin staining in the tongue epithelium. In wild-type epithelium, emerin localization to the NE is readily detectable (i), whereas in the lamin null mice it is lost (j). Nuclei i and j are costained with DAPI.
EDMD was originally described as an X-linked disorder that mapped to the gene for emerin, a ubiquitous INM protein (Bione et al. 1994). While the function of emerin is unknown, the finding that A-type lamin defects result in an EDMD-like disorder suggests these proteins might functionally interact. Consistent with this, a recent study by Bonne et al. 1999 revealed that in humans an autosomal variant of EDMD maps to the lamin A/C (LMNA) gene. Since the localization of integral proteins to the INM is thought to involve a process of selective retention (Powell and Burke 1990; Ellenberg et al. 1997; Yang et al. 1997), it is possible that A-type lamins serve to immobilize emerin within the INM. Therefore, we examined the distribution of emerin in wild-type and lamin A/C −/− MEFs. While the overall levels of emerin in these cells is identical (Fig. 5 a), immunofluorescence microscopy revealed dramatic differences in emerin subcellular localization. In wild-type cells, emerin is concentrated within the nuclear envelope (Manilal et al. 1996; Nagano et al. 1996) (Fig. 5 b). In the −/− cells, there is partial loss of NE-associated emerin with a more general cytoplasmic distribution identical to that observed for resident ER proteins (Fig. 5 d). Presumably, emerin is no longer retained within the INM, and is free to access the outer nuclear membrane and ER via the membrane continuities at the periphery of NPCs (Ostlund et al. 1999). MEFs from heterozygous embryos exhibited intermediate levels of cytoplasmic emerin (Fig. 5 c). These results indicate that at least in MEFs, correct emerin localization is contingent upon A-type lamin expression. In contrast, LAP2, which shares some sequence homology with emerin but which is known to interact with both chromatin and B-type lamins (Furukawa et al. 1998), still concentrates at the nuclear periphery in −/− cells (Fig. 2 c).
Figure 5.
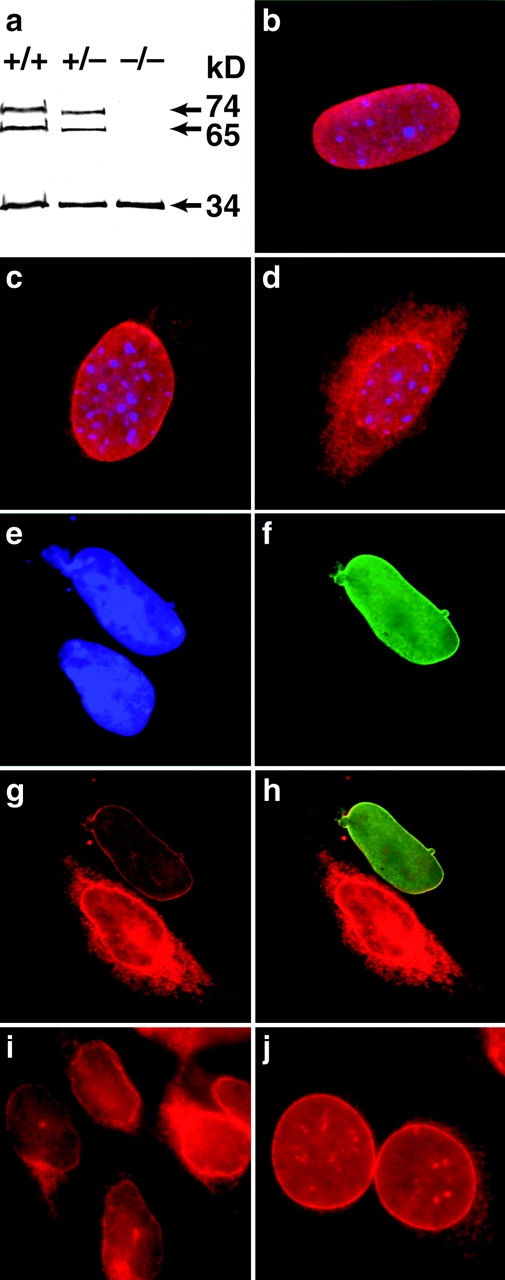
(a) Western blot analysis of emerin (34 kD) and lamins A and C (74 and 65 kD, respectively) in +/+, +/−, and −/− Lmna MEFs showing no difference in emerin levels between different genotypes. (b) In wild-type cells, emerin is localized exclusively to the nuclear envelope. (c) In the heterozygotes, some emerin localization can be detected in the cytoplasm. (d) In the lamin A/C −/− cells, emerin is largely cytoplasmic although some localization to the nuclear envelope is still apparent. Expression of human lamin A in Lmna null MEFs results in relocalization of emerin to the nuclear envelope. (e) DAPI staining revealing the nuclei of two MEFs. (f) The upper MEF expresses human lamin A localized exclusively to the nuclear envelope. (g) Both cells express emerin but in the lamin A null cell, emerin is localized predominantly in the cytoplasmic compartment, whereas in the lamin A positive MEF, emerin is localized almost entirely to the nuclear envelope. (h) Merged image showing colocalization of emerin and human lamin A in the nuclear envelope. (i and j) Emerin distribution in P19EC cells (i) and (j) their differentiated derivatives P19MES that express A-type lamins. Both cell types exhibit NE-associated emerin.
To further address the role of A-type lamins in emerin localization, we transfected a human lamin A cDNA into Lmna null MEFs and followed the distribution of emerin by double label immunofluorescence microscopy. Fig. 5e and Fig. f, shows a representative example of two −/− MEFs, only one of which is expressing the heterologous LMNA. While emerin in the nonexpressing cell is distributed between both the NE and the cytoplasm, it is restricted almost exclusively to the nuclear periphery in the cell expressing human lamin A (Fig. 5g and Fig. h). These data strongly support a role for A-type lamins in the correct localization of emerin.
Immunocytochemical examination of emerin in several tissues from the lamin A/C null mice revealed that its normal nuclear envelope localization was affected in a cell type–specific manner. In tongue epithelium, NE-associated emerin was completely lost (Fig. 4i and Fig. j), whereas in skeletal muscle NE-associated emerin was still detectable, albeit at a greatly reduced level (Fig. 4e and Fig. f). In the longitudinal fibers of the ventricular cardiac muscle, the loss of A-type lamin expression had more heterogeneous effects with ∼20% of nuclei showing a marked polar clustering of emerin staining (Fig. 4g and Fig. h) that was particularly apparent in those cells sectioned along their longitudinal axis. In heterozygous mice, <0.1% of the ventricular nuclei exhibited emerin polarization and none were detected in wild-type mice.
These observations reveal a clear role for A-type lamins in emerin localization. However, NE-associated emerin is not uniformly lost in tissues from Lmna null mice, suggesting the involvement of additional factors. This is supported by our findings that in P19 embryonal carcinoma (EC) cells, which do not express A-type lamins at any detectable level (Stewart and Burke 1987), emerin is, nevertheless, associated with the nuclear periphery (Fig. 5i and Fig. j). This implies that there is at least one other nuclear envelope component with which emerin must be able to interact, and that the level of expression of this component is likely to be cell type–specific.
EDMD arises from either loss of emerin protein or mutations resulting in its subcellular mislocalization (Ellis et al. 1998; Fairley et al. 1999), with the rarer autosomally inherited form being linked to dominant acting mutations in the LMNA gene (Bonne et al. 1999). From this study, it was not clear whether the NE was disrupted and emerin localization affected. However, this is a possibility as some lamin A-type mutants do act in a dominant form, since their injection or transfection into cells causes severe perturbations in nuclear envelope structure and organization (Spann et al. 1997).
Here, we have shown that an EDMD-like phenotype, albeit more severe and earlier acting, arises in mice after ablation of A-type lamin expression. This is associated with the mislocalization of emerin to varying degrees in different cell types. Previous observations on the fragility of purified NEs from P19 EC cells, which don't express A-type lamins, indicated a role for the lamins in NE integrity (Horton et al. 1992). In this study, we observed a similar, pronounced effect on hepatocyte NEs lacking A-type lamins with gross structural changes to the NEs and emerin distribution occurring in a variety of −/− cells and tissues. These observations indicate that in addition to the lamins, emerin itself may represent another important determinant of interphase NE organization, possibly as a link between the INM and lamina. However, the relative contributions of the A-type lamins, and emerin either alone or in combination, to NE integrity remain unclear at the present. From our studies, loss of the A-type lamins clearly affects NE integrity. Whether it does so alone or whether the phenotype is exacerbated by loss/mislocalization of emerin, possibly in conjunction with some other tissue-specific and/or developmentally regulated factors will have to await the derivation of emerin-deficient mice.
Intermediate filament proteins have long been recognized in providing structural integrity to a variety of cells and tissues. In particular, mutations in members of the cytokeratin gene family are associated with numerous pathological conditions, most notably those of epidermal and neurological origin (McLean and Lane 1995; Galou et al. 1997; Fuchs and Cleveland 1998). Here, we have shown cells lacking A-type lamins, members of the nuclear branch of the intermediate filament protein family, also develop a characteristic pathology. However, while these proteins are expressed in the majority of adult tissues, this pathology is manifest primarily in the form of muscular dystrophy. A possible explanation for this is that in the absence of functional lamins and/or emerin, muscle nuclei may be unable to withstand the mechanical stresses to which they are continually subjected. In other tissues, which do not experience the same contractile forces, while nuclear envelope integrity may still be compromised, the effects may be less deleterious. The derivation of mice with tissue-specific deficiencies in lamina-associated proteins will now allow us to test this suggestion.
Acknowledgments
We thank Glenn Morris for generous provision of the anti mouse emerin antibody, Larry Gerace, Erich Nigg (University of Geneva), and Frank McKeon (University of Geneva) for antibodies to LAP2 and lamins, Lidia Hernandez (ABL-Basic Research Program) for advice and help, Jim Resau (ABL-Basic Research Program) for help with confocal analysis, Richard Frederickson (Harvard University) for preparation of the figures and Anne Wang, Manfred Lohka, and Rhiannon Hughes for critical reading of the manuscript.
B. Burke was supported by grants from the Medical Research Council and the Alberta Heritage Foundation for Medical Research. This work was sponsored in part by the national Cancer Institute, Department of Health and Human Services, under contract with ABL and contract No. N01-C0-56000 with SAIC.
Footnotes
T. Sullivan and D. Escalante-Alcalde contributed equally to the work.
Abbreviations used in this paper: EC, embryonal carcinoma; EDMD, Emery-Dreifuss muscular dystrophy; ES, embryonic stem cell; INM, inner nuclear membrane; MEF, mouse embryonic fibroblasts; NE, nuclear envelope; NPC, nuclear pore complex.
References
- Ash J.F., Louvard D., Singer S.J. Antibody-induced linkages of plasma membrane proteins to intracellular actomyosin-containing filaments in cultured fibroblasts. Proc. Natl. Acad. Sci. USA. 1977;74:5584–5588. doi: 10.1073/pnas.74.12.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bione S., Maestrini E., Rivella S., Mancini M., Regis S., Romeo G., Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- Bodoor K., Shaihk S.A., Salina D., Raharjo W.H., Bastos R., Lohka M.J., Burke B. Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J. Cell Sci. 1999;112:2253–2264. doi: 10.1242/jcs.112.13.2253. [DOI] [PubMed] [Google Scholar]
- Bonne G., Di Barletta M.R., Varnous S., Becane H.M., Hammouda E.H., Merlini L., Muntoni F., Greenberg C.R., Gary F., Urtizberea J.A. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- Bulfield G., Siller W.G., Wight P.A., Moore K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg J., Siggia E.D., Moreira J.E., Smith C.L., Presley J.F., Worman H.J., Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cellstargeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J.A., Craxton M., Yates J.R., Kendrick-Jones J. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 1998;111:781–792. doi: 10.1242/jcs.111.6.781. [DOI] [PubMed] [Google Scholar]
- Fairley E.A., Kendrick-Jones J., Ellis J.A. The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J. Cell Sci. 1999;112:2571–2582. doi: 10.1242/jcs.112.15.2571. [DOI] [PubMed] [Google Scholar]
- Foisner R., Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 1993;73:1267–1279. doi: 10.1016/0092-8674(93)90355-t. [DOI] [PubMed] [Google Scholar]
- Fuchs E., Cleveland D.W. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Furukawa K., Fritze C.E., Gerace L. The major nuclear envelope targeting domain of LAP2 coincides with its lamin binding region but is distinct from its chromatin interaction domain. J. Biol. Chem. 1998;273:4213–4219. doi: 10.1074/jbc.273.7.4213. [DOI] [PubMed] [Google Scholar]
- Galou M., Gao J., Humbert J., Mericskay M., Li Z., Paulin D., Vicart P. The importance of intermediate filaments in the adaptation of tissues to mechanical stressevidence from gene knockout studies. Biol. Cell. 1997;89:85–97. [PubMed] [Google Scholar]
- Gant T.M., Wilson K.L. Nuclear assembly. Annu. Rev. Cell Dev. Biol. 1997;13:669–695. doi: 10.1146/annurev.cellbio.13.1.669. [DOI] [PubMed] [Google Scholar]
- Gerace L., Burke B. Functional organization of the nuclear envelope. Annu. Rev. Cell Biol. 1988;4:335–374. doi: 10.1146/annurev.cb.04.110188.002003. [DOI] [PubMed] [Google Scholar]
- Gonda M.A., Aaronson S.A., Ellmore N., Zeve V.H., Nagashima K. Ultrastructural studies of surface features of human normal and tumor cells in tissue culture by scanning and transmission electron microscopy. J. Natl. Cancer Inst. 1976;56:245–263. doi: 10.1093/jnci/56.2.245. [DOI] [PubMed] [Google Scholar]
- Horton H., McMorrow I., Burke B. Independent expression and assembly properties of heterologous lamins A and C in embryonal carcinomas. Eur. J. Cell Biol. 1992;57:172–183. [PubMed] [Google Scholar]
- Lenz-Bohme B., Wismar J., Fuchs S., Reifegerste R., Buchner E., Betz H., Schmitt B. Insertional mutation of the Drosophila nuclear lamin Dm0 gene results in defective nuclear envelopes, clustering of nuclear pore complexes, and accumulation of annulate lamellae. J. Cell Biol. 1997;137:1001–1016. doi: 10.1083/jcb.137.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manilal S., Nguyen T.M., Sewry C.A., Morris G.E. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum. Mol. Genet. 1996;5:801–808. doi: 10.1093/hmg/5.6.801. [DOI] [PubMed] [Google Scholar]
- McKeon F.D., Kirschner M.W., Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature. 1986;319:463–468. doi: 10.1038/319463a0. [DOI] [PubMed] [Google Scholar]
- McLean W.H., Lane E.B. Intermediate filaments in disease. Curr. Opin. Cell Biol. 1995;7:118–125. doi: 10.1016/0955-0674(95)80053-0. [DOI] [PubMed] [Google Scholar]
- Moir R.D., Spann T.P., Goldman R.D. The dynamic properties and possible functions of nuclear lamins. Int. Rev. Cytol. 1995;162:141–182. doi: 10.1016/s0074-7696(08)62616-9. [DOI] [PubMed] [Google Scholar]
- Nagano A., Koga R., Ogawa M., Kurano Y., Kawada J., Okada R., Hayashi Y.K., Tsukahara T., Arahata K. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat. Genet. 1996;12:254–259. doi: 10.1038/ng0396-254. [DOI] [PubMed] [Google Scholar]
- Ostlund C., Ellenberg J., Hallberg E., Lippincott-Schwartz J., Worman H.J. Intracellular trafficking of emerin, the Emery-Dreifuss muscular dystrophy protein. J. Cell Sci. 1999;112:1709–1719. doi: 10.1242/jcs.112.11.1709. [DOI] [PubMed] [Google Scholar]
- Powell L., Burke B. Internuclear exchange of an inner nuclear membrane protein (p55) in heterokaryonsin vivo evidence for the association of p55 with the nuclear lamina. J. Cell Biol. 1990;111:2225–2234. doi: 10.1083/jcb.111.6.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rober R.A., Weber K., Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animala developmental study. Development. 1989;105:365–378. doi: 10.1242/dev.105.2.365. [DOI] [PubMed] [Google Scholar]
- Rober R.A., Sauter H., Weber K., Osborn M. Cells of the cellular immune and hemopoietic system of the mouse lack lamins A/Cdistinction versus other somatic cells. J. Cell Sci. 1990;95:587–598. doi: 10.1242/jcs.95.4.587. [DOI] [PubMed] [Google Scholar]
- Spann T.P., Moir R.D., Goldman A.E., Stick R., Goldman R.D. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J. Cell Biol. 1997;136:1201–1212. doi: 10.1083/jcb.136.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C.L. Production of chimeras between embryonic stem cells and embryos. Methods Enzymol. 1993;225:823–855. doi: 10.1016/0076-6879(93)25053-5. [DOI] [PubMed] [Google Scholar]
- Stewart C., Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell. 1987;51:383–392. doi: 10.1016/0092-8674(87)90634-9. [DOI] [PubMed] [Google Scholar]
- Stuurman N., Heins S., Aebi U. Nuclear laminstheir structure, assembly, and interactions. J. Struct. Biol. 1998;122:42–66. doi: 10.1006/jsbi.1998.3987. [DOI] [PubMed] [Google Scholar]
- Wehnert M., Muntoni F. 60th ENMC international workshopnon X-linked Emery-Dreifuss muscular dystrophy 1998. Neuromuscul. Disord. 1999;9:115–121. doi: 10.1016/s0960-8966(98)00095-9. [DOI] [PubMed] [Google Scholar]
- Yang L., Guan T., Gerace L. Integral membrane proteins of the nuclear envelope are dispersed throughout the endoplasmic reticulum during mitosis. J. Cell Biol. 1997;137:1199–1210. doi: 10.1083/jcb.137.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]