

Abstract
About 50% of spinal motoneurons undergo programmed cell death (PCD) after target contact, but little is known about how this process is initiated. Embryonic motoneurons coexpress the death receptor Fas and its ligand FasL at the stage at which PCD is about to begin. In the absence of trophic factors, many motoneurons die in culture within 2 d. Most (75%) of these were saved by Fas-Fc receptor body, which blocks interactions between Fas and FasL, or by the caspase-8 inhibitor tetrapeptide IETD. Therefore, activation of Fas by endogenous FasL underlies cell death induced by trophic deprivation. In the presence of neurotrophic factors, exogenous Fas activators such as soluble FasL or anti-Fas antibodies triggered PCD of 40–50% of purified motoneurons over the following 3–5 d; this treatment led to activation of caspase-3, and was blocked by IETD. Sensitivity to Fas activation is regulated: motoneurons cultured for 3 d with neurotrophic factors became completely resistant. Levels of Fas expressed by motoneurons varied little, but FasL was upregulated in the absence of neurotrophic factors. Motoneurons resistant to Fas activation expressed high levels of FLICE-inhibitory protein (FLIP), an endogenous inhibitor of caspase-8 activation. Our results suggest that Fas can act as a driving force for motoneuron PCD, and raise the possibility that active triggering of PCD may contribute to motoneuron loss during normal development and/or in pathological situations.
Keywords: motoneuron, Fas ligand, APO-1/CD95, neuron killing, caspases
The existence of a phase of programmed cell death (PCD) in the normal development of spinal motoneurons has been known for many decades, since the pioneering work of Hamburger and Levi-Montalcini (for review see Hamburger 1992). A considerable body of work has since been devoted to the study of this phenomenon, making the motoneuron a locus classicus of cell death research in the nervous system (for reviews see Oppenheim 1991; Pettmann and Henderson 1998). During the 4–6 d after target muscle contact, an average of 50% of the motoneurons initially generated undergo PCD at most levels of the spinal cord. This death has many features that classify it as apoptotic: (a) characteristic morphological changes such as shrinkage and condensation have been reported (Chu-Wang and Oppenheim 1978a,Chu-Wang and Oppenheim 1978b); (b) administration of caspase inhibitors to chicken embryos protects many motoneurons against PCD in ovo (Milligan et al. 1995; Li et al. 1998); and (c) motoneuron death is almost completely abrogated in mouse embryos deficient for Bax, a proapoptotic member of the Bcl-2 family (Deckwerth et al. 1996; White et al. 1998).
The fraction of motoneurons that survive the phase of PCD is tightly linked to the presence of the target tissue: early removal of a limb bud leads to death of nearly all motoneurons that would normally have innervated it (Hamburger 1934). Thus, limb tissues normally produce neurotrophic factors and when these are absent, motoneurons activate a cell death process. What drives the death of motoneurons (or indeed other neurons) deprived of trophic support is not clear. It is possible that in the absence of signaling through survival-related cascades, caspase activation occurs spontaneously. However, specific molecular events may also serve as a driving force for death induced by trophic deprivation.
The fact that the 50% of motoneurons that normally survive require trophic support led to the hypothesis that the other 50% die through lack of sufficient support. Indeed, there is support for this idea, since grafting a supernumerary limb bud can save some of the motoneurons that normally die (Hollyday and Hamburger 1976). Similar effects are observed when neurotrophic factors or tissue extracts are exogenously administered to chicken embryos in ovo (Oppenheim et al. 1991, Oppenheim et al. 1995). Nevertheless, it is striking that it has not been possible to save all motoneurons in this manner, and that to date, transgenic overexpression of neurotrophic factors in muscle has not been reported to save motoneurons (Nguyen et al. 1998). Furthermore, many agents capable of reducing naturally occurring motoneuron death in chicken embryos are not effective in saving motoneurons that have been deprived of trophic support by limb ablation (Calderò et al. 1998). Thus, it is possible that at least a proportion of naturally occurring motoneuron PCD may be triggered by mechanisms other than trophic deprivation.
Different mechanisms for controlled elimination of neurons during development are indeed known to exist. In nematodes, genes such as ces-2 seem to act cell-autonomously to trigger PCD (Ellis and Horvitz 1991; Metzstein et al. 1996). Another possibility in vertebrates is that exogenous influences may trigger the death of specific populations. Although this type of mechanism has been widely analyzed in lesioned adult brain (e.g., excitotoxicity), it had not been intensely studied during development until recently. However, a series of recent results on PCD triggered by NGF signaling through the low-affinity neurotrophin receptor p75NTR have radically changed our vision of this question (for review see Carter and Lewin 1997). In the developing retina for instance, the early role of NGF seems not to be as a trophic factor, but rather as a trigger of PCD: in the absence of NGF or p75 function, cell death among the population of immature retinal neurons that express p75 is markedly reduced (Frade and Barde 1999). The source of NGF appears to be macrophages that invade the developing retina at early stages (Frade and Barde 1998). Therefore, the PCD of at least some neuronal populations seems to be regulated by molecules that actively trigger cell death.
p75NTR is a member of the tumor necrosis factor (TNF) receptor family of transmembrane receptors, many of which carry in their cytoplasmic domain a group of about 80 amino acids referred to as the death domain, which allows them to interact with adaptor molecules and thereby indirectly mediate caspase activation in the cell (Ashkenazi and Dixit 1998). Another well-studied member of this family is the Fas/Apo1/CD95 receptor (referred to here as Fas), which with its ligand FasL has well-studied roles in instructive apoptosis (for review see Nagata 1997). In the immune system, Fas and FasL are involved in deletion of mature T cells to end an immune response, in triggering death of inflammatory cells, and in elimination of infected cells or tumors by cytotoxic T lymphocytes (Nagata and Golstein 1995). FasL, which can act either in membrane-bound or soluble form, triggers cell death by clustering the Fas receptor at the cell surface. In these conditions, the cytoplasmic domain of Fas can bind the adaptor protein Fas-associated death domain (FADD)/Mort1 (Chinnaiyan et al. 1995). FADD in turn binds procaspase-8, which can thereby self-activate (Muzio et al. 1996). Cleavage of downstream substrates by caspase-8 rapidly triggers the cell death cascade in the cell that expressed Fas (Scaffidi et al. 1998). FasL can activate Fas both in trans and in cis: T lymphocytes induced to die by T cell receptor ligation upregulate FasL and Fas at their surface and thus trigger cell death in an autocrine manner (Brunner et al. 1995; Dhein et al. 1995). Partial loss-of-function mutations for Fas (lpr) and FasL (gld) in mouse lead to accumulation of peripheral lymphoid cells and to an autoimmune disorder (Watanabe-Fukunaga et al. 1992; Takahashi et al. 1994).
The potential role of Fas in the nervous system has been the object of relatively little study. It is known that FasL is quite widely expressed in the nervous system (Becher et al. 1998a), but its presence has been related to a role in conferring immune privilege on the central nervous system through its ability to trigger death of invading lymphocytes (French et al. 1996; French and Tschopp 1996; Saas et al. 1997). However, there have been reports of expression of Fas itself in the nervous system. After spinal cord ischemia in adult rabbits, increases in Fas immunoreactivity of spinal motoneurons precede cell death (Sakurai et al. 1998). Furthermore, specific neuronal populations in postnatal mouse brain were shown to express Fas mRNA and protein even in nonlesioned animals (Park et al. 1998). Very recently, the Fas–FasL system has been implicated in cell death induced by ischemia (Martin-Villalba et al. 1999), as well as in death of PC12 cells and cerebellar granule neurons (Le-Niculescu et al. 1999) and cortical neurons (Cheema et al. 1999) in vitro.
We wished to investigate the possibility that some motoneuron cell death may be actively triggered. We show here that both Fas and FasL are expressed by motoneurons at the time at which PCD is about to begin in the spinal cord. In vitro, interaction of endogenous FasL with Fas drives death induced by trophic deprivation, and exogenous activation of Fas triggers PCD of motoneurons even in the presence of neurotrophic factors. The extent and timing of the Fas-induced cell death are close to those observed in vivo, and neurotrophic factors regulate synthesis of both death-inducing and death-inhibitory factors. Our results are consistent with the possibility that motoneuron cell death in vivo may be actively triggered via Fas.
Materials and Methods
Reagents and Animals
Recombinant human APO-1/Fas–Fc IgG (Fas-Fc), soluble Fas-ligand (sFasL), and enhancer antibodies used for aggregating tagged sFasL were from Alexis Corp. Rabbit polyclonal antibodies against Fas (M-20) and against FasL (N-20) and the related control peptides were from Santa Cruz Biotechnology, Inc. Polyclonal rabbit antibodies to mouse FLICE-inhibitory protein (FLIP) were from R & D Systems, Inc., and mouse mAbs to neurofilament 68-kD subunit (NF-68) were from Sigma Chemical Co. mAb against FasL (H11) was purchased from Alexis Corp. Rat anti–mouse Fas mAbs (RMF6) were from Chemicon International, Inc. Control rat IgG 2a were obtained from PharMingen International and control rabbit Ig from Jackson ImmunoResearch Laboratories, Inc. DEVD-fmk (Asp-Glu-Val-Asp-fluoro methyl ketone) was purchased from Enzyme Systems Products and IETD-fmk (Ile-Glu-Thr-Asp-fluoro methyl ketone) from Clontech Laboratories, Inc. All mutant mice and control strains were purchased from Jackson Laboratories, Inc.
RNA Detection by Reverse Transcription PCR
Total RNA was isolated from different tissues and cells using the guanidine isothiocyanate-phenol chloroform method (Chomczynski and Sacchi 1987). Reverse transcription (RT) steps were performed using Expand™ reverse transcriptase (Roche Diagnostics) and random hexamer primers. Normalization of cDNA amounts was performed by PCR on a Perkin-Elmer Thermal Cycler, using primers for β-actin (sense primer TT GTA ACC AAC TGG GAC GAT ATG G and antisense primer GAT CTT GAT CTT CAT GGT GCT AGG). PCR reactions were performed for a total of 24 cycles.
Fas and FasL cDNA were amplified using primers for rat Fas (nucleotides 147–168, CTG TCC TGC CTC TGG TGC TTG, and nucleotides 682–702, CAT CTG AGA CAT TCA TTG GC; sequence data available from EMBL/GenBank/DDBJ under accession no. D26112) and rat FasL (nucleotides 30–51, CCA CAA GAC TGA GAG GAG GAA A, and nucleotides 814–836, TAA ATG GTC AGC AAC GGT AAG A; sequence data available from EMBL/GenBank/DDBJ under accession no. U03470). The reaction mixture contained 20 mM Tris-HCl, pH 8.4, 50 mM KCl, 0.2 mM each dNTP, 1.5 mM MgCl2, 1.5 U of Taq platinum polymerase (Roche Diagnostics), 20 pmol of each primer and a normalized amount of cDNA template. The PCR conditions for both sets of primers were 30 s at 94°C, 30 s at 60°C, 1 min at 72°C for a total of 35 cycles.
Samples were then run on a 1.2% agarose gel, treated with 0.4 N NaOH and transferred to Hybond N+ nylon membranes (Amersham Pharmacia Biotech). Blots were hybridized with 32P-labeled probes corresponding to nucleotides 174–196, CCG ACA ACA ACT GCT CAG AAG G for rat Fas, and nucleotides 347–368, ACA TTC CTA ACC CCA TTC CAA C for FasL, at 42°C for 2 h. Membranes were then washed in decreasing concentrations of saline sodium citrate buffer (SSC), and exposed for autoradiography.
For RT-PCR from cultures, a total of 50,000 motoneurons were seeded in five 35-mm dishes for each culture condition and rinsed with diethyl pyrocarbonate (DEPC)-treated PBS. Total RNA was isolated using TRIzol (Life Technologies, Inc.) reagent, and cDNA normalization was performed as above. Primers used were as follows: mouse Fas, nucleotides 273–292, CAC CAA CCT GTG CCC CAT GC, and nucleotides 999–1019, GTC CTT CAT TTT CAT TTC CAG (sequence data available from EMBL/GenBank/DDBJ under accession no. M83649); mouse FLIP, nucleotides 226–247, TGA TGA AGA CGA GAA GGA GAT, and nucleotides 795–815, AAT CTT GGC TCT TTA CTT CGC (sequence data available from EMBL/GenBank/DDBJ under accession no. U97076). The PCR conditions were 30 s at 94°C, 30 s at 58°C for Fas or 54°C for FLIP, and 45 s at 72°C for a total of 35 cycles, using the same reaction conditions as described above. PCR products were separated on a 1.2% agarose gel and visualized by ethidium bromide staining. The relative intensity of ethidium bromide–stained bands was determined using an AGFA densitometer, and data were analyzed using the NIH Image 1.62 software.
Immunoblot Purification of Antibodies to Fas
Rabbit polyclonal antibodies against Fas antigen (M-20) were immunoblot-purified using a procedure described previously (La Bella et al. 1998). In brief, 5 mg of mouse thymus protein extract was submitted to preparative PAGE electrophoresis and Western blotting. The horizontal strip corresponding to Fas (45 kD) was cut out of the blot and incubated with 1 μg/ml M-20, then washed. Subsequently, bound antibody was eluted using a glycine buffer, pH 2.8, and rapidly buffered to pH 7.5. This step greatly decreased nonspecific staining observed using the initial preparation (data not shown). An identical immunopurification was performed using blots of E12.5 mouse ventral spinal cord extracts (see text).
Western Blotting
Tissues were collected in cold PBS, centrifuged to remove excess PBS and placed in lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 100 mM DTT, 1 mM PMSF, 2% SDS, 500 mM NaCl and Complete™ EDTA-free, a protease inhibitor mix purchased from Roche Diagnostics). After mechanical dissociation followed by sonication, lysates were centrifuged (12,000 rpm for 5 min) and the supernatant was heated at 95°C for 2 min in loading buffer (10% [vol/vol] glycerol, 2% [wt/vol] 2-mercaptoethanol, 250 mM Tris, pH 6.8, 0.005% bromophenol blue). Loading was 20 μg of proteins for adult tissue extracts and 40 μg for all embryonic tissues and cells, as determined using a modified Bradford reaction (Bio-Rad Laboratories). Proteins were separated by SDS-PAGE (12% acrylamide gel) and transferred to Immobilon membranes (Millipore). Membranes were blocked with 10% nonfat dry milk in PBS 0.1% Tween 20 (PBT) for 2 h at room temperature, then incubated with primary antibodies diluted in PBT containing 4% milk (100 ng/ml for FasL N-20 and Fas M-20 antibodies, 450 ng/ml for caspase-8 antibodies, 1 μg/ml for c-FLIP antibodies, and 50 ng/ml for NF-68 antibodies) overnight at 4°C. After washing with PBT, membranes were incubated with HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) diluted in PBT containing 4% milk, for 1 h, washed with PBT and then developed using the ECL system (Amersham Pharmacia Biotech). Scanning was performed as for RT-PCR.
Motoneuron Purification and Culture
E14 rat and E12.5 mouse spinal motoneurons were purified as described previously (Henderson et al. 1995; Arce et al. 1999). In brief, cells were dissociated from ventral spinal cord after trypsin treatment. The largest cells were isolated by centrifugation on a 6.5% metrizamide density gradient. The immunoaffinity purification step performed previously by immunopanning (Henderson et al. 1995) was replaced by a cell-sorting step using magnetic microbeads (Arce et al. 1999). For rat, cells in the metrizamide fraction were incubated with a mouse mAb against rat p75NTR (Ig192); for mouse, with a rat mAb against mouse p75NTR (Chemicon). Subsequently, motoneurons were incubated with magnetic microbeads conjugated to either mouse anti–rat or rat anti–mouse secondary antibodies, thus allowing the purification of motoneurons on separating columns (Miltenyi Biotec, Inc.). After a final centrifugation through a BSA cushion, motoneurons were resuspended in either L15 or neurobasal medium and plated in 4-well dishes (Nunc) previously treated with polyornithine/laminin as described (Henderson et al. 1995).
Neurobasal medium (Life Technologies, Inc.) was supplemented with 2% (vol/vol) horse serum, 25 μM l-glutamate, 25 μM β-mercaptoethanol, 0.5 mM l-glutamine, and 2% (vol/vol) B-27 supplement (Life Technologies, Inc.). L15 (Leibovitz) medium (Life Technologies, Inc.) was supplemented with 2% (vol/vol) horse serum, 3.6 mg/ml glucose, 100 U/ml penicillin-streptomycin, 20 nM progesterone, 5 μg/ml insulin, 0.1 mM putrescine, 0.1 mg/ml conalbumin, 30 nM sodium selenite.
For rat motoneurons, the different neurotrophic factors were used at the optimal concentrations as follows: human brain-derived neurotrophic factor (BDNF) (R & D Systems, Inc.) at 1 ng/ml, rat glial cell line–derived neurotrophic factor (GDNF) (Sigma Chemical Co.) at 100 pg/ml, mouse cardiotrophin-1 (CT-1) (Genentech, Inc.) at 10 ng/ml. Mouse motoneurons were cultured in the presence of a cocktail of trophic factors (NTFs) consisting of ciliary neurotrophic factor (CNTF; 10 ng/ml). All factors were added at the time of cell seeding.
Assay of Motoneuron Survival
Rat or mouse motoneurons were seeded in 4-well plates and cultured in the indicated medium at the density of 1,000–2,000 cells per 16-mm well. Motoneurons were grown for 24 h at 37°C before being incubated with Fas activators at the indicated concentrations diluted in neurobasal medium. Treatments with sFasL were always performed in the presence of 1 μg/ml enhancer antibody. The activity of the different death-inducing agents was determined after different periods of treatment by direct counting of living cells using a phase-contrast microscope. Fas-Fc or IETD-fmk were added at the time of seeding, and the surviving motoneurons were counted after 24 h at 37°C in the indicated culture medium. Counts were performed using a phase-contrast microscope: ∼100 motoneurons in 2 diameters of each well were evaluated as described previously (Henderson et al. 1994; Pennica et al. 1996). All assays were performed in duplicate or triplicate wells in each of two to four independent experiments.
Immunohistochemistry
Approximately 3,000 purified motoneurons were seeded per polyornithine/laminin–coated 12-mm diameter glass coverslip, and grown for 1 or 3 d at 37°C in neurobasal medium with trophic factors. Cultures were fixed on ice using freshly made paraformaldehyde, first for 10 min with 2% paraformaldehyde in PBS-neurobasal medium (1:1), then for 10 min with 4% paraformaldehyde in PBS. Cultures were washed three times with PBS, permeabilized with 50 mM l-Lysine, 0.1% Triton X-100 in PBS for 10 min, and incubated overnight at 4°C in PBS containing 4% BSA, 2% goat or donkey serum (species depending on the secondary antibodies). The primary antibodies were diluted in PBS containing 4% BSA, 2% donkey and goat serum at the following concentrations: 0.2 μg/ml for rabbit polyclonal antibodies against FasL (N-20) and Fas (M-20), 20 μg/ml for anti-Fas (RMF-6), 0.9 μg/ml for caspase-8 rabbit polyclonal antibodies, and matching concentrations for each control antibody. Cells were incubated with primary antibodies overnight at 4°C, then washed with PBS containing 0.1% Triton and incubated for 30–45 min at room temperature with fluorophore-conjugated anti–rabbit, anti–mouse, or anti–rat secondary antibodies (Jackson ImmunoResearch, Inc.) diluted in PBS 10% horse serum. After washing with PBS containing 10% horse serum and PBS, cultures were rinsed with double-distilled water before being mounted on glass slides using DABCO mounting solution and examined by fluorescence microscopy. Immunostaining using the polyclonal antibody CM1 to detect the activated form of caspase-3 was performed as described (Srinivasan, 1998b). To label all nuclei, immunostained preparations were incubated with a solution of 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) at 200 ng/ml in PBS for 15 min in the dark before washing and mounting.
Results
Fas and FasL Are Expressed by Embryonic Motoneurons as PCD Begins
We first asked whether Fas and FasL are expressed at the stage at which motoneuron PCD is about to begin. By RT-PCR followed by Southern blotting using specific oligonucleotide probes, we showed that, as reported (Suda et al. 1993; French et al. 1996), Fas and FasL were both expressed in a variety of adult tissues (Fig. 1A and Fig. B; data not shown), and that FasL was expressed by ventral spinal cord of E14 rat embryos (Fig. 1 A). In addition, we showed that Fas was expressed at significant levels by freshly isolated E14 ventral spinal cord (Fig. 1 B).
Figure 1.

Embryonic motoneurons express Fas and FasL both in situ and after purification. Total RNA was prepared from the indicated rat tissues, and RT-PCR analysis followed by Southern blotting was performed using specific primers and internal probes for FasL (A) and Fas (B). RT-PCR for β-actin was used as an internal control to ensure that similar concentrations of mRNA were present in all samples. For each mRNA, the first panel corresponds to positive and negative control samples prepared from indicated tissues of adult rat; the results are in agreement with several reports in the literature. The second panel shows results from E14 rat embryos, using either freshly dissected ventral spinal cord or motoneurons purified by a metrizamide-immunoaffinity method in conditions that limit de novo synthesis of mRNA. Fas and FasL are present at significant levels in motoneurons. Alternate unlabeled lanes in all panels are control in which reverse transcriptase was omitted from the incubation.
Because levels of mRNA were not high enough to be reproducibly detected by in situ hybridization (data not shown), we used an alternative approach. To determine whether motoneurons themselves expressed Fas and FasL, we purified E14 rat motoneurons to near-homogeneity using the metrizamide-immunoaffinity method (see Materials and Methods). All purification steps were performed below 20°C to reduce de novo synthesis. mRNAs for both Fas and FasL were clearly present in freshly isolated motoneurons (Fig. 1A and Fig. B), demonstrating that motoneurons about to undergo PCD in vivo potentially express the elements of the Fas–FasL system.
Embryonic Motoneurons Coexpress Fas and FasL Polypeptides
Next, we determined whether motoneurons express significant levels of Fas and FasL protein. By Western blotting using a polyclonal antibody to FasL, we detected a band of the appropriate molecular mass (38 kD) in adult thymus but not in heart, as expected from previous reports (Suda et al. 1993) and our RNA data (Fig. 1 A). The same antibody clearly detected FasL in ventral spinal cord from both E14 rat and E12.5 mouse embryos and in purified rat motoneurons of the same age (Fig. 2 A).
Figure 2.
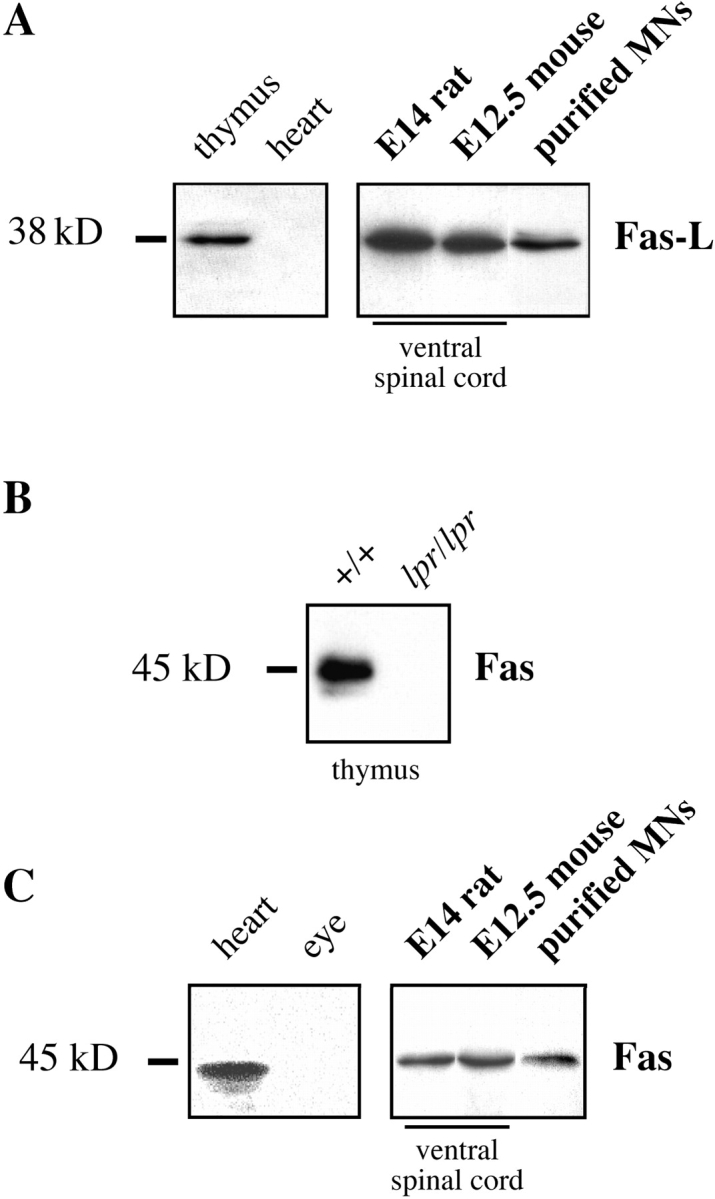
Embryonic motoneurons express Fas and FasL polypeptides in vivo. Western blot analysis was performed using specific antibodies to FasL and Fas. (A) Antibodies to FasL specifically label a 38-kD band in P17 mouse thymus, but not in heart. In ventral spinal cord from rodent embryos dissected at the stage at which motoneuron PCD is about to begin, FasL is abundant. It is also clearly present in freshly purified motoneurons from E14 rat. (B) Controls for the specificity of immunoblot-purified anti-Fas antibodies (M-20) using thymus extracts from P17 mice of the C57BL/6 strain. Wild-type (+/+) and Fas mutant (lpr/lpr) samples are shown. (C) Using immunoblot-purified antibodies, Fas is abundant in adult rat heart but not in eye. Both ventral spinal cord and freshly purified motoneurons from embryos show clear expression of a single band corresponding to Fas.
Several commercial antibodies to Fas we tested reacted with multiple bands on Western blots (data not shown). Therefore, we affinity-purified anti-Fas antibodies from polyclonal rabbit Ig (M-20) raised against the intracellular domain of Fas (see Materials and Methods). On Western blots of thymus from P17 mice, this antibody recognized a band that migrated with an apparent molecular mass of 45 kD, the expected molecular mass for Fas (Fig. 2 B). The intensity of this band was greatly reduced in thymus from P17 lpr/lpr mice (Fig. 2 B), in which levels of Fas have been reported to be at least 10-fold lower than normal (Mariani et al. 1994). Also as expected (French et al. 1996), Fas protein was detected in adult heart, but not eye (Fig. 2 C). In the same conditions, the immunopurified antibody recognized a band of identical molecular mass in extracts of ventral spinal cord from E14 rat and E12.5 mouse, and in motoneurons freshly purified from E14 rat spinal cord (Fig. 2 C). When the antibody was preincubated with the immunogenic peptide, no staining was seen (data not shown). As a final demonstration that the polypeptide present in spinal cord and motoneurons was indeed Fas, we immunopurified the Fas antibodies by incubation with the 45-kD band cut out from Western blots of E12.5 mouse spinal cord. The eluted antibodies recognized a 45-kD band in thymus that disappeared in lpr/lpr tissue (data not shown). Thus, Fas protein is expressed at significant levels by spinal motoneurons (and perhaps other spinal neurons) at the onset of motoneuron PCD. However, we were not able to immunolocalize Fas reliably on embryonic tissue sections (data not shown).
We performed immunolabeling studies on purified mouse (Fig. 3) and rat (data not shown) motoneurons cultured for 1 d with neurotrophic factors. For Fas, using the affinity-purified Ig described above, granular membrane labeling was observed in nearly all cultured motoneurons, although some were more intensely labeled than others (Fig. 3 A). Similar results (data not shown) were obtained using rat mAb RMF6 raised against the extracellular domain of Fas. The rabbit antibodies also reproducibly stained the nuclei of cultured motoneurons; both this and the membrane immunoreactivity were absent when the antibody was preincubated with the peptide used for immunization (Fig. 3 B). Antibodies to FasL also stained nearly all motoneurons (Fig. 3 C), and double-labeling experiments confirmed that most individual neurons coexpressed Fas and FasL (Fig. 3 D).
Figure 3.

Motoneurons in culture coexpress Fas and FasL. Immunolabeling of purified E12.5 mouse motoneurons cultured for 1 d in the presence of neurotrophic factors. (A) Cultured motoneurons labeled using blot-purified rabbit anti-Fas (M-20). Note punctate labeling of cell membranes and more diffuse labeling of nucleus. There was variation in intensity from cell to cell (not apparent in this field). (B) Control in which the same primary antibody was preincubated with the immunogenic peptide. Preincubation with an irrelevant peptide did not reduce staining (data not shown). (C) Immunostaining using a rat mAb against FasL (H11). Note the strong homogeneous staining. (D) Double immunolabeling for Fas (green) and FasL (red), showing the death receptor and its ligand present at the cell surface of the same motoneuron. Controls using an irrelevant rat mAb of the same class or purified rabbit antibodies gave no significant staining (data not shown).
Motoneuron Death Induced by Trophic Deprivation Involves Endogenous Activation of Fas and Caspase-8
Since motoneurons coexpressed the death receptor Fas together with its ligand, we asked whether Fas might play a role in motoneuron cell death. When purified rat motoneurons were grown in basal medium at low density (1,000 per 16-mm well) for 24 h, about half of them died (Fig. 4 A). Their death was completely prevented by BDNF (1 ng/ml). Fas-Fc, which contains the extracellular domain of Fas fused to an Ig Fc domain, is an antagonist known to block activation of Fas by FasL. Fas-Fc inhibited ∼75% of the death of trophically deprived motoneurons in a dose-dependent manner (Fig. 4 A). Higher doses of Fas-Fc were slightly less efficient (data not shown), but this may have been due to toxicity of the reagent. This suggested that endogenous FasL was activating Fas in these neurons, thereby triggering the cell death process.
Figure 4.




Activation of Fas and caspase-8 by endogenous FasL is required for death of motoneurons in the absence of trophic support. (A) Freshly purified rat motoneurons were induced to die by culturing them in basal medium only for 24 h, or were kept alive by addition of an optimal concentration of BDNF (1 ng/ml). Increasing concentrations of Fas-Fc (0.01–1 μg/ml), which inhibits Fas activation by FasL, added to basal medium promoted their survival (expressed as a percentage of the number in BDNF) in a dose-dependent manner. Addition of 1 μg/ml of Fas-Fc to motoneurons cultured in the presence of BDNF (1 ng/ml) had no effect on their survival. (B) Immunolabeling of two cultured E12.5 mouse motoneurons using a specific antibody recognizing all forms of caspase-8. Double-labeling using DAPI (data not shown) allowed for localization of nuclei (arrows). All motoneurons presented clear granular labeling of the cytoplasm. (C) After seeding, rat motoneurons were incubated with increasing doses of the caspase-8 peptide inhibitor IETD-fmk (0.01–1 μM) without or with BDNF (1 ng/ml). The number of surviving cells was measured 24 h later and expressed relative to survival in BDNF. (D) Western blotting reveals the presence of caspase-8 in ventral spinal cord from mouse embryos, and in purified mouse motoneurons. The molecular mass of the major species (43 kD) is significantly lower than in nonneural cell lines (53–55 kD).
Fas-triggered cell death in nonneuronal systems involves activation of caspase-8, which then activates downstream caspases such as caspase-3 (Nicholson and Thornberry 1997). Using a specific antibody to caspase-8 that recognizes both the inactive procaspase and the active forms (Srinivasan et al. 1998a), we found that nearly all cultured motoneurons showed a characteristic pattern of cytoplasmic labeling (Fig. 4 B). We then tested the hypothesis that caspase-8 might be an essential relay in the death of motoneurons induced by trophic deprivation. Prior application of the caspase-8–selective inhibitor peptide IETD-fmk (Garcia-Calvo et al. 1998), which alone did not affect survival in BDNF, inhibited 75% of the motoneuron loss observed in the absence of trophic support (Fig. 4 C), consistent with a requirement for caspase-8 in this process.
To determine whether caspase-8 is present in motoneurons in vivo, we performed Western blots on different tissue extracts (Fig. 4 D). As expected, nonapoptotic HeLa and Jurkat cells showed a principal doublet around 53–55 kD, which corresponds to the forms referred to as caspase-8/a and caspase-8/b (Scaffidi et al. 1997). Spinal cord extracts and freshly purified motoneurons showed a single major caspase-8–immunoreactive band at 43 kD (Fig. 4 D). It is not possible to determine from these data whether this form is identical to p43, a known intermediate in the caspase-8 activation process in nonneuronal cell types (Scaffidi et al. 1997), and it is possible that it results from other posttranslational modifications than those observed in cell lines. However, we do not believe that the 43-kD form in nervous tissue is proteolytically active, since we detected it at similar levels in normal adult spinal cord, where little or no PCD occurs (data not shown). Thus, caspase-8, a central component of the Fas signaling pathway, is present in normal spinal cord during the period of naturally occurring motoneuron death.
Fas Can Trigger Motoneuron Cell Death in a Manner Independent of Trophic Deprivation
In view of the finding that blockade of the Fas system saves motoneurons deprived of trophic support (Fig. 4), we next asked whether Fas activation could trigger motoneuron death even in the presence of trophic factors. To do this, we took advantage of reagents that have been shown to cluster Fas at the cell membrane in other systems and thereby exogenously trigger Fas-dependent cell death.
Motoneurons purified from E12.5 mouse embryos were cultured in the presence of a cocktail of trophic factors (BDNF, CNTF, and GDNF) at concentrations determined previously to ensure optimal survival (Arce et al. 1999). After 24 h to allow attachment and initial neurite outgrowth, antibody to the extracellular domain of mouse Fas was added in the continued presence of trophic factors to induce clustering of Fas. 2 d later (i.e., after 3 d in vitro [DIV]), anti-Fas antibody had caused dose-dependent motoneuron loss, up to a maximum reduction of 45% using 10 ng/ml antibody (Fig. 5 A). The half-maximal dose of antibody required to induce death of motoneurons (0.7 ng/ml) is close to that reported for lymphoid cells (Watanabe-Fukunaga et al. 1992). Control antibodies including the enhancer antibody (see below) had no effect on survival (data not shown).
Figure 5.

Activation of Fas triggers PCD of motoneurons even in the presence of optimal trophic support. Different agents known to activate Fas receptor by clustering were tested for their ability to trigger motoneuron cell death. (A) Purified E12.5 mouse motoneurons were cultured in the presence of BDNF (1 ng/ml), CT-1 (10 ng/ml), and GDNF (0.1 ng/ml) (+NTFs) for 1 d. Subsequently, the indicated concentrations of anti-Fas antibody were added in the continued presence of NTFs, and motoneuron survival was counted 2 d later, at 3 DIV. Counts were expressed relative to the value for NTFs at 3 DIV. Fas antibody induced a dose-dependent loss of 45% of the motoneurons. (B) An analogous experiment using purified motoneurons from E14 rat, cultured in the continued presence of BDNF (1 ng/ml). After 1 DIV, tagged soluble FasL was added at the indicated concentrations in the presence of 1 μg/ml of enhancer antibody, which had no effect when tested alone. Survival was counted 2 d later. (C) Comparison of the fractions of E14 rat motoneurons lost after 2 d of incubation with 10 ng/ml sFasL, in the presence of indicated neurotrophic factors used at the same concentrations as for mouse motoneurons in A. The number of motoneurons lost was expressed as a percentage of the total number of motoneurons present in the same conditions but without sFasL. All histograms are representative of at least three independent experiments. Error bars represent the mean ± range of duplicate wells.
To confirm the generality of this observation, we performed an analogous experiment on cultured rat motoneurons. Since rat Fas is not recognized by the clustering antibody, we used a recombinant form of the extracellular domain of FasL (sFasL) that is epitope-tagged. Addition of sFasL, followed by an antibody to this tag (enhancer antibody), clusters sFasL and thereby Fas, leading to Fas activation. Rat motoneurons were cultured for 1 d in the presence of optimal concentrations of BDNF (1 ng/ml). They were then treated with enhancer, either alone or in the presence of increasing concentrations of sFasL. Enhancer antibody alone (1 μg/ml) did not affect survival, but in the presence of sFasL, 40% of motoneurons were lost 2 d later in a dose-dependent fashion, with an EC50 of 1 ng/ml (Fig. 5 B). This is close to the half-maximal dose necessary to trigger death of A20 B lymphoma cells (Hahne et al. 1996). Strikingly, in this model too, motoneuron death was not enhanced by further increasing the concentrations of sFasL.
We compared the percentage of motoneurons induced to die by Fas activation in the presence of different factors. Rat motoneurons were cultured as described above, except that BDNF (1 ng/ml) was replaced by CT-1 (10 ng/ml), or GDNF (0.1 ng/ml), or CNTF (10 ng/ml; data not shown). The reduction in motoneuron number after 2 d was expressed as a percentage of the number surviving in the same conditions without sFasL (Fig. 5 C). In each case, sFasL triggered death of ∼30% of motoneurons, and similar results were obtained using FasL in basal medium alone (data not shown).
Role of Caspases-3 and -8 in Cell Death Induced by Exogenous Fas Activators
To demonstrate that the motoneurons lost after Fas activation had activated a cell death program, we studied the involvement of caspases.
We first focused on caspase-3, a downstream caspase known to be required for PCD of many neurons (Kuida et al. 1996), and of motoneurons in particular (Li et al. 1998). Using an antibody (CM1) that specifically recognizes the activated form of caspase-3 (Srinivasan et al. 1998b), we showed that many cultured motoneurons exposed to Fas activators expressed activated caspase-3, and that this reactivity was especially strong in the motoneurons with apoptotic nuclei that had not yet detached from the culture dish (Fig. 6 A). We quantified CM1-positive motoneurons 30 h after treatment (or not) with anti-Fas antibodies (Fig. 6 B). At this stage, there was not yet a significant difference in total motoneuron numbers in the presence or absence of anti-Fas (P > 0.5; data not shown). Without Fas activator, 8.0 ± 0.9% (mean ± SEM; n = 5) of motoneurons expressed intense CM1 immunoreactivity and showed fragmented chromatin using Hoechst staining. Using anti-Fas antibodies, this value was significantly increased (19.0 ± 1.6%; P = 0.001 by t test). To test the functional significance of caspase-3 activation, we used the active-site peptide DEVD, a potent cell-permeable caspase inhibitor that is selective for caspase-3 (Nicholson et al. 1995; Villa et al. 1997), but can also inhibit caspase-8 (Garcia-Calvo et al. 1998). Increasing concentrations of DEVD-fmk were added to rat motoneurons cultured in BDNF, 2 h before addition of sFasL (10 ng/ml) and enhancer antibody. Motoneuron loss was inhibited in a dose-dependent fashion (Fig. 6 C), and the concentration of DEVD required to completely prevent Fas-dependent motoneuron death (10 μM) was similar to or lower than those reported to prevent PCD in other cell types (Armstrong et al. 1997). DEVD was also able to completely inhibit the cell death of mouse motoneurons induced by sFasL or anti-Fas antibodies (Fig. 6 D). Thus, Fas most likely triggers PCD of motoneurons by a mechanism involving caspase-3.
Figure 6.

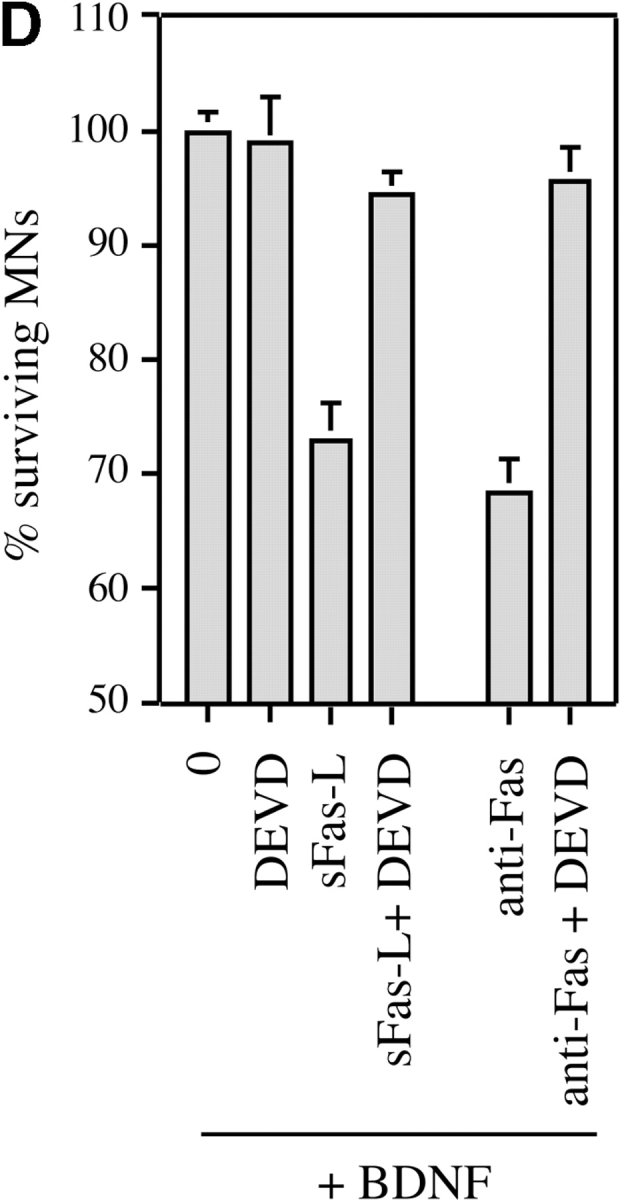
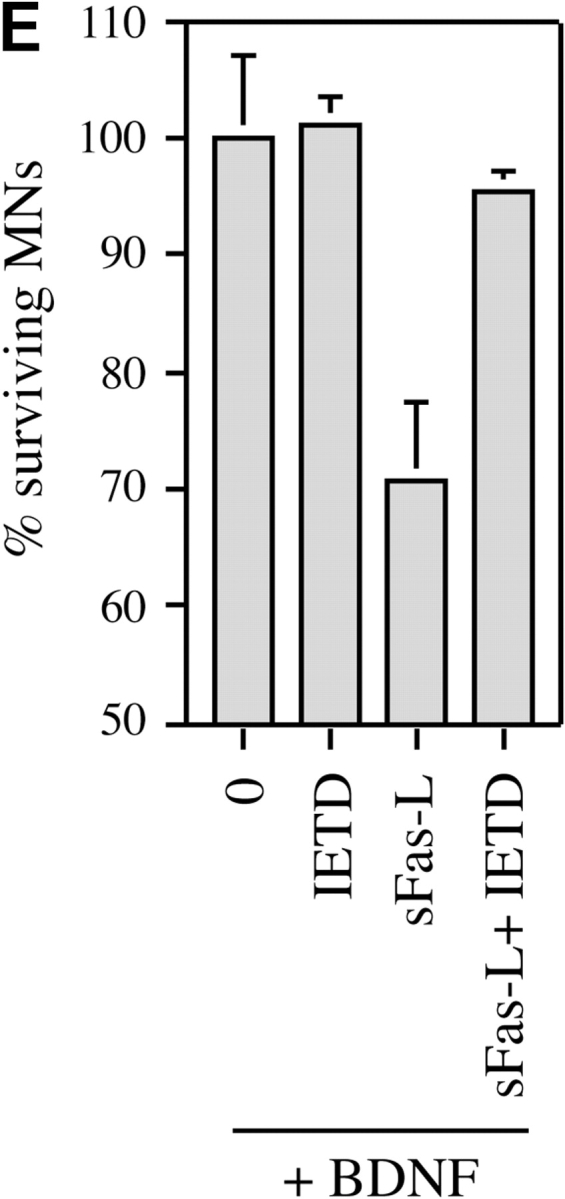
Motoneuron cell death triggered by Fas activation in the presence of trophic factors involves caspases-3 and -8. (A) Activation of caspase-3 in cultured mouse motoneurons treated with anti-Fas was visualized using the CM1 antibody, which specifically recognizes the activated form of caspase-3. The motoneuron illustrated was undergoing apoptosis, as visualized by the fragmented chromatin of the nucleus stained with DAPI (inset). Note the intense perinuclear staining for activated caspase-3. (B) Increase in the fraction of CM1-positive mouse motoneurons 30 h after treatment with anti-Fas antibodies in the presence of neurotrophic factors. After immunostaining, CM1-positive motoneurons were counted along two diameters of 14-mm coverslips (∼100 motoneurons counted for each). All CM1-positive cells showed pyknotic nuclei by DAPI staining. Values are means ± SEM of five coverslips, and are typical of two independent experiments (asterisks indicate P = 0.001 by t test). Total survival was not significantly different in the two conditions (P > 0.5; data not shown). (C) Death of E14 rat motoneurons triggered by sFasL (10 ng/ml) in the presence of BDNF (1 ng/ml) is inhibited in a dose-dependent fashion by the caspase-3 peptide inhibitor DEVD-fmk. (D) Death of mouse motoneurons triggered by sFasL (10 ng/ml) and enhancer (1 μg/ml), or anti-Fas antibodies (10 ng/ml), is blocked by DEVD-fmk (10 μM). Survival is expressed as the percentage of the number of motoneurons surviving in the presence of BDNF alone. (E) Death of rat motoneurons triggered by sFasL (10 ng/ml, in the presence of enhancer) is inhibited by the caspase-8 inhibitor IETD-fmk (1 μM). C, D, and E show combined mean values (± S.D.) from duplicate wells in two independent experiments.
In similar experiments, the caspase-8 inhibitor IETD-fmk was able to completely prevent the motoneuron loss induced by sFasL (Fig. 6 E), consistent with a requirement for caspase-8 in this process.
Resistance to Fas-induced Cell Death Is Regulated In Vitro
It was striking (Fig. 5) that ∼50% of motoneurons were resistant to the effects of exogenous Fas activation in the presence of trophic factors. We tested the survival of mouse motoneurons treated at different stages with Fas activators (Fig. 7). In a first experiment analogous to those in Fig. 5, mouse motoneurons were seeded in a cocktail of neurotrophic factors, treated with either sFasL or anti-Fas at 1 DIV, and their survival was counted 4 d later, at 5 DIV (Fig. 7 A). Some motoneurons (∼10%) died over the 5-d culture period even in the presence of trophic factors, and so values were expressed as a percentage of the value at 5 DIV with trophic factors alone. In these conditions, Fas activation led to loss of 50% of motoneurons. To rule out the possibility that incomplete motoneuron loss reflected instability of reagents in the culture dish, we then added Fas activators at both 1 and 3 DIV, and counted survival at 5 DIV (Fig. 7 B). No further motoneuron loss was observed, demonstrating that some motoneurons are truly resistant to Fas activation. We then asked whether this resistance might be regulated in vitro, by first culturing motoneurons for 3 DIV in the presence of trophic factors, then adding Fas activators, and counting survival at 5 DIV. Surprisingly, no motoneuron death at all was triggered in these conditions (Fig. 7 C).
Figure 7.

Resistance of motoneurons to the cell-killing effects of Fas activation is tightly regulated. Mouse motoneurons were cultured in the continued presence of BDNF, CNTF, and GDNF (NTFs) and then treated with the Fas activators sFasL (50 ng/ml) or anti-Fas antibodies (50 ng/ml). Motoneuron survival in all experiments was counted at 5 DIV, and expressed relative to the value in NTFs alone at that time. (A) Fas activators were added at 1 DIV only. (B) Fas activators were added at 1 and 3 DIV; no further motoneuron death was observed. (C) Fas activators were added at 3 DIV: all motoneurons became resistant to Fas activation. Histograms are representative of similar results obtained in three independent experiments. Error bars represent the mean ± range of duplicate values.
We asked whether all motoneurons might at some stage be susceptible to Fas activation. Because it was not technically possible to purify motoneurons from younger embryos, we added sFasL to E14 rat motoneurons at 0 h of culture. As at 24 h, it was not possible to trigger the death of more than half of them (data not shown). By extension, it seems likely that half the motoneurons in situ are already resistant to Fas activation as cell death begins. To exclude the possibility that the resistant neurons were intrinsically incapable of responding to Fas activation, we performed the experiment in the presence of cycloheximide (10 μM). As with many other cell types, this increased the number of motoneurons induced to die by Fas activators to 80% (two independent experiments; data not shown).
Regulation of FasL and c-Flip Expression by Neurotrophic Factors
Our results suggested: (a) that in the absence of trophic support, Fas becomes sufficiently activated by FasL to induce motoneuron cell death (Fig. 4); and (b) that in the presence of trophic factors, motoneurons become resistant to even exogenous activation of Fas (Fig. 7). We used semiquantitative RT-PCR to follow the regulation of the expression of critical molecules in cultured motoneurons. Purified motoneurons were cultured in the absence and presence of neurotrophic factors. Cells were harvested after 1 and 3 DIV and levels of indicated mRNAs were determined by semiquantitative RT-PCR.
First we asked whether trophic deprivation led to upregulation of components of the Fas system. Levels of Fas, normalized to actin, were relatively unaffected by the presence or absence of trophic factors (Fig. 8 B). In contrast, whereas levels of FasL steadily decreased in motoneurons cultured with trophic factors, they were strongly upregulated in trophically deprived motoneurons (Fig. 8 A). Thus, regulation of levels of FasL may underlie the dependence of motoneurons on external trophic support.
Figure 8.

Regulation of FasL and FLIP by neurotrophic factors. (A and B) Levels of molecules involved in Fas signaling were followed using semiquantitative RT-PCR on motoneurons cultured for 1 and 3 DIV in the presence or absence of neurotrophic factors. (A) FasL mRNA is upregulated in motoneurons cultured in the absence of trophic factors. (B) Regulation of fas and FLIP mRNAs. mRNA samples were incubated with (+) or without (−) reverse transcriptase. Fas was expressed at relatively constant levels when normalized to actin, whereas FLIP was strongly upregulated in motoneurons cultured for 3 DIV in the presence of NTFs. (C) Strong upregulation of FLIP protein in the presence of neurotrophic factors. Western blots are shown of extracts of purified motoneurons cultured for 1 and 3 DIV in the presence of NTFs; two major forms of FLIP at 55 and 52 kD are observed. The loading control was neurofilament-68. (D) Immunolabeling of Fas-resistant mouse motoneurons cultured for 3 d in the presence of NTFs, using the blot-purified polyclonal antibodies against Fas characterized in Fig. 2. Fas continues to be expressed at the cell membrane. (E) Motoneurons treated for 3 d with NTFs also continue to express caspase-8. (F) Summary diagram showing the potential involvement of the Fas system in PCD of motoneurons. The main experimental strategies adopted in our study of the role of Fas are depicted. In addition, we have indicated the three levels at which neurotrophic factors may potentially act to block Fas-related motoneuron death.
Next, we searched for molecular correlates for the state in which motoneuron death can no longer be triggered by exogenous Fas activators. As stated above, after a 3-d exposure to BDNF, motoneurons still expressed Fas mRNA (Fig. 8 B). We confirmed this by demonstrating immunoreactivity for Fas at their surface (Fig. 8 D), and further showed that caspase-8 immunoreactivity was also still present in treated motoneurons (Fig. 8 E).
Surprisingly, therefore, all major components of the Fas pathway for cell death were still present in Fas-resistant motoneurons. This suggested that trophic factor–treated motoneurons became resistant at a level downstream from Fas. Normally, Fas activates caspase-8 through the binding of the adaptor molecule FADD to the cytoplasmic domain of Fas. FADD can then recruit procaspase-8 and lead it to self-activate. FLIP is an endogenous cytoplasmic decoy that can competitively inhibit the binding of procaspase-8 to FADD, and thereby prevent caspase-8 activation (Fig. 8 E). Using semiquantitative RT-PCR, we showed that Flip mRNA was strongly upregulated after 3 DIV with neurotrophic support (Fig. 8 B). Scanning of the gels showed that the intensity of the Flip band was 6.8-fold greater at 3 DIV with NTFs than at 1 DIV with NTFs, when both were expressed relative to the intensity of the band for actin. To confirm that the change in mRNA levels was reflected in an increase of FLIP protein, we performed Western blots on cultures of motoneurons in the same conditions using a specific antibody to mouse FLIP (Fig. 8 C). As reported in other cell types (Irmler et al. 1997; Srinivasula et al. 1997), two major molecular species were labeled by this antibody in motoneuron extracts, with apparent molecular masses of 52 and 55 kD. Levels of both were markedly increased at 3 DIV with NTFs. Scanning of the blots showed that there was a mean 6.3-fold increase relative to neurofilament over the 2-d period in culture. Thus, motoneurons that survive in the presence of neurotrophic factors not only downregulate the death-inducer FasL but also upregulate the death inhibitor FLIP.
Discussion
PCD of motoneurons during development of the spinal cord has long been a classical paradigm, and its study was at the origin of the discovery of neurotrophic factors (Oppenheim 1991; Pettmann and Henderson 1998). However, we still know very little about exactly what drives the death of embryonic motoneurons. We have shown here that the death receptor Fas and its ligand FasL, which are coexpressed by motoneurons as they enter the phase of naturally occurring cell death, may potentially play an important role. Activation of Fas in cultured embryonic motoneurons is both necessary for PCD in the absence of neurotrophic factors, and sufficient to trigger death of a significant proportion of motoneurons in their presence. Levels of endogenous Fas activators and inhibitors are regulated by neurotrophic factors, suggesting that the Fas–FasL system may be a central pathway through which motoneuron numbers are controlled. Further experiments will be required to determine the role of Fas in naturally occurring and/or pathological motoneuron cell death in vivo.
The presence of Fas on developing motoneurons (and perhaps other spinal neurons) was unexpected, although Fas expression has recently been reported in both normal and lesioned adult central nervous system (D'Souza et al. 1996; Becher et al. 1998a,Becher et al. 1998b; Park et al. 1998; Sakurai et al. 1998), and a recent paper reported Fas in cerebral cortex at earlier stages (Cheema et al. 1999). Therefore, we substantiated this observation by the use of different techniques (RT-PCR, Western blotting, and immunocytochemistry using several independent antibodies) wher-ever possible in two different species (mouse and rat). Our results unequivocally demonstrate the presence of both Fas and FasL in ventral spinal cord, and freshly purified motoneurons at the stage at which motoneuron PCD is about to begin. It will be of interest to determine the pattern of expression of Fas in vivo on a cell-by-cell basis. Our preliminary data using in situ hybridization and immunohistochemistry on rodent spinal cord were not included here, since Fas and FasL signals are too close to the threshold of detection. This may also explain apparent discrepancies between our findings and those of French et al. 1996, who, although they described the presence of FasL in embryonic spinal cord, reported Fas to be absent from the embryonic nervous system.
To better understand the potential roles of Fas and FasL, we studied two potential modes of Fas activation in vitro: endogenous and exogenous. Motoneurons coexpress FasL and Fas, and both Fas-Fc and IETD-fmk can prevent their spontaneous PCD in the absence of trophic factors, demonstrating that the binding of endogenous FasL to Fas is a motor for PCD in these conditions. The fact that these results were obtained in low-density cultures suggests that this was probably an autocrine effect, as already demonstrated for T lymphocytes (Brunner et al. 1995; Dhein et al. 1995). Similar events may occur in cerebellar granule neurons and PC12 cells which, like motoneurons, upregulate FasL in the absence of trophic support (Le-Niculescu et al. 1999). Our results raise the possibility that some motoneurons die in vivo by a cell-autonomous Fas-related mechanism.
Activation of Fas by trophic deprivation is by definition blocked by neurotrophic factors. In contrast, exogenous Fas activation inhibited the survival-promoting effects of all trophic factors we tested. The loss of motoneurons observed in these conditions was very similar in both extent (50%) and time-course (3–4 d) to motoneuron PCD in the embryo, although we cannot be sure that the 50% of motoneurons that die in these conditions are the same as the 50% that die from trophic deprivation. This raises the possibility that some motoneurons in vivo may die not because they have insufficient trophic support, but because they have been induced to do so by FasL presented in a paracrine fashion by nearby cells. Both the autocrine and paracrine hypotheses will need to be tested during development in vivo. Mice bearing either hypomorphic or null mutations for Fas, or a point mutation in FasL are known (Watanabe-Fukunaga et al. 1992; Takahashi et al. 1994; Adachi et al. 1995). We have obtained these mice and are currently analyzing the detailed pattern of motoneuron PCD and lesion-induced cell death in the absence of normal Fas signaling.
It is not certain that Fas plays the same role in all motoneurons. It is striking that Fas-Fc and IETD-fmk both protected at most 75% of the motoneurons that would normally have died as a result of trophic deprivation. It is possible that higher concentrations of these reagents were simply toxic, but this suggests that other Fas-independent pathways may also be involved in triggering death of some motoneurons. Another suggestion of heterogeneity comes from the observation that 50% of motoneurons were resistant to Fas activators in all experimental conditions, even when treated immediately after purification. Strikingly, the resistance to Fas activation becomes complete after incubation for 3 d with trophic factors, and this is correlated with the upregulation of FLIP, an endogenous inhibitor of caspase-8 activation (Irmler et al. 1997; Tschopp et al. 1998; Scaffidi et al. 1999). Upregulation of FLIP over time in culture may reflect either general maturation or a specific effect of neurotrophic factors. It is not currently possible to purify rodent motoneurons at different developmental stages, but it will be interesting in the future to keep motoneurons alive by mechanisms apparently distinct from the mode of action of trophic factors, e.g., in high cAMP concentrations (Hanson et al. 1998; Meyer-Franke et al. 1998), and then test their resistance to Fas activation at different stages.
Which are the caspases that act downstream of the Fas–FasL system in inducing motoneuron death? Our results implicate both caspase-3 and caspase-8. The evidence for activation of the downstream protease caspase-3 comes both from inhibitor studies and from immunolabeling using the CM1 antibody; our results are consistent with the implication of caspase-3 in many apoptotic phenomena in neurons (Kuida et al. 1996; Li et al. 1998). In other dying neurons, activation of the caspase-3 zymogen has generally been considered to occur through the actions of caspase-9, and null mutants for this gene indeed show reduced neuronal death (Hakem et al. 1998; Kuida et al. 1998). Our results using the IETD peptide, which inhibits caspase-8 much more strongly than caspase-3 (Garcia-Calvo et al. 1998), suggest that Fas acts through caspase-8 in this system as in others (Nagata 1996). Our conclusions are not in agreement with those of a previous report (Li et al. 1998), whose authors found that caspase-8–like activity is present in chick motoneurons but is not upregulated after removal of trophic support, and that motoneurons were not saved by IETD-CHO. This may reflect a species difference and/or the different form of the peptide inhibitor used. However, it is clear that our results and those of Li et al. 1998 need to be substantiated by methods other than those based on active-site peptides, and we are currently developing other methods to study activation of procaspase-8 in the low numbers of motoneurons available.
Understanding the intracellular mechanisms that regulate sensitivity or resistance to the effects of Fas will be one of the keys to evaluating the role of Fas during normal motoneuron development, and in pathological systems. Fig. 8 E presents a possible synthesis of our current knowledge. Fas activation may occur by either endogenous or exogenous mechanisms. The fact that exogenous (but not endogenous) activation triggers PCD even in the presence of neurotrophic factors may simply reflect more intense activation of caspase-8. This could mean that the signaling through the Bcl-2 family in the mitochondrion triggered by NTFs is not sufficient to prevent subsequent release of cytochrome c and activation of caspase-3 in these conditions (Pettmann and Henderson 1998). However, in the medium term (1–3 d), neurotrophic factors can prevent PCD induced by either endogenous or exogenous Fas activation. This seems to involve transcriptional events such as downregulation of FasL or upregulation of FLIP. Unraveling the interplay between these pathways will be important for understanding the control of motoneuron survival.
The Fas-dependent mechanism of PCD we have described for motoneurons may well be relevant to the study of other developing neuronal populations, most of which undergo a quantitatively significant phase of naturally occurring cell death. If our data on spinal motoneurons can be extrapolated, it may be that sensitivity to Fas activation will not be apparent at all stages. For instance, in paradigms involving culture of neurons with neurotrophic factors for a certain period before withdrawal (Deshmukh and Johnson 1997), the neurons under study may become resistant to Fas activation before the stage at which their cell death is normally studied. An interesting possibility is that, just as different cell populations seem to depend for cell death on different members of the Bcl-2 family (Deckwerth et al. 1996) or on different caspases (Troy et al. 1996), different members of the tumor necrosis factor (TNF) receptor family such as p75 and Fas may be involved in the programmed elimination of different groups of neurons.
It will be important to determine whether triggering of motoneuron death by Fas might underlie the specific loss of motoneurons that is observed in some circumstances at later stages of development or in the adult. These include experimental or traumatic lesions, and the neurodegenerative diseases that affect motoneurons, such as the pmn or wobbler mutations in mice, and spinal muscular atrophies (SMA) or amyotrophic lateral sclerosis (ALS) in humans (Henderson 1995). If Fas expression is observed in these patients, the pathways downstream of Fas may provide a novel and specific site for therapeutic intervention in these still incurable diseases.
Acknowledgments
We thank J.R. Sanes, P. Golstein, and members of U.382, for helpful comments on the manuscript and throughout this work. We are particularly grateful to Kevin Tomaselli and A. Srinivasan of Idun Pharmaceuticals, Inc. (La Jolla, CA) for their generous gift of antibodies to caspase-8 and activated caspase-3.
This study was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Association Française contre les Myopathies (AFM), Institut pour la Recherche sur la Moelle Epinière (IRME), and European Commission BIO4 contract CT960433. C. Raoul is a recipient of a scholarship from the Ministère de l'Éducation Nationale, la Recherche et la Technologie (MENRT).
Footnotes
Abbreviations used in this paper: BDNF, brain-derived neurotrophic factor; CNTF, ciliary neurotrophic factor; CT-1, cardiotrophin-1; DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride; DIV, day(s) in vitro; FADD, Fas-associated death domain; Fas-Fc, recombinant human APO-1/Fas–Fc IgG; GDNF, glial cell line–derived neurotrophic factor; L, ligand; NTF, neurotrophic factors; PCD, programmed cell death; RT, reverse transcription; s, soluble.
References
- Adachi M., Suematsu S., Kondo T., Ogasawara J., Tanaka T., Yoshida N., Nagata S. Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat. Genet. 1995;11:294–300. doi: 10.1038/ng1195-294. [DOI] [PubMed] [Google Scholar]
- Arce V., Garcès A., de Bovis B., Filippi P., Henderson C., Pettmann B., deLapeyrière O. Cardiotrophin-1 requires LIFRβ to promote survival of mouse motoneurons purified by a novel technique. J. Neurosci. Res. 1999;55:119–126. doi: 10.1002/(SICI)1097-4547(19990101)55:1<119::AID-JNR13>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Armstrong R.C., Aja T.J., Hoang K.D., Gaur S., Bai X., Alnemri E.S., Litwack G., Karanewsky D.S., Fritz L.C., Tomaselli K.J. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J. Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A., Dixit V.M. Death receptorssignaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Becher B., Barker P.A., Owens T., Antel J.P. CD95-CD95Lcan the brain learn from the immune system? Trends Neurosci. 21 1998. 114 117a [DOI] [PubMed] [Google Scholar]
- Becher B., D'Souza S.D., Troutt A.B., Antel J.P. Fas expression on human fetal astrocytes without susceptibility to fas-mediated cytotoxicity Neuroscience. 84 1998. 627 634b [DOI] [PubMed] [Google Scholar]
- Brunner T., Mogil R.J., LaFace D., Yoo N.J., Mahboubi A., Echeverri F., Martin S.J., Force W.R., Lynch D.H., Ware C.F. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- Calderò J., Prevette D., Mei X., Oakley R.A., Li L., Milligan C., Houenou L., Burek M., Oppenheim R.W. Peripheral target regulation of the development and survival of spinal sensory and motor neurons in the chick embryo. J. Neurosci. 1998;18:356–370. doi: 10.1523/JNEUROSCI.18-01-00356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter B.D., Lewin G.R. Neurotrophins live or let diedoes p75NTR decide? Neuron. 1997;18:187–190. doi: 10.1016/s0896-6273(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Cheema Z.F., Wade S.B., Sata M., Walsh K., Sohrabji F., Miranda R.C. Fas/Apo-1 and associated proteins in the differentiating cerebral cortexinduction of caspase-dependent cell death and activation of NF-κB. J. Neurosci. 1999;19:1754–1770. doi: 10.1523/JNEUROSCI.19-05-01754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan A.M., O'Rourke K., Tewari M., Dixit V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chu-Wang I.W., Oppenheim R.W. Cell death of motoneurons in the chick embryo spinal cord. I. A light and electron microscopic study of naturally occurring and induced cell loss during development J. Comp. Neurol. 177 1978. 33 57a [DOI] [PubMed] [Google Scholar]
- Chu-Wang I.W., Oppenheim R.W. Cell death of motoneurons in the chick embryo spinal cord. II. A quantitative and qualitative analysis of degeneration in the ventral root, including evidence for axon outgrowth and limb innervation prior to cell death J. Comp. Neurol. 177 1978. 59 85b [DOI] [PubMed] [Google Scholar]
- D'Souza S.D., Bonetti B., Balasingam V., Cashman N.R., Barker P.A., Troutt A.B., Raine C.S., Antel J.P. Multiple sclerosisFas signaling in oligodendrocyte cell death. J. Exp. Med. 1996;184:2361–2370. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth T.L., Elliott J.L., Knudson C.M., Johnson E.M., Jr., Snider W.D., Korsmeyer S.J. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M., Jr. Programmed cell death in neuronsfocus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol. Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- Dhein J., Walczak H., Baumler C., Debatin K.M., Krammer P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- Ellis R.E., Horvitz H.R. Two C. elegans genes control the programmed deaths of specific cells in the pharynx. Development. 1991;112:591–603. doi: 10.1242/dev.112.2.591. [DOI] [PubMed] [Google Scholar]
- Frade J.M., Barde Y.A. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 1998;20:35–41. doi: 10.1016/s0896-6273(00)80432-8. [DOI] [PubMed] [Google Scholar]
- Frade J.M., Barde Y.A. Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development. 1999;126:683–690. doi: 10.1242/dev.126.4.683. [DOI] [PubMed] [Google Scholar]
- French L.E., Tschopp J. Constitutive Fas ligand expression in several non-lymphoid mouse tissuesimplications for immune-protection and cell turnover. Behring Inst. Mitt. 1996;97:156–160. [PubMed] [Google Scholar]
- French L.E., Hahne M., Viard I., Radlgruber G., Zanone R., Becker K., Muller C., Tschopp J. Fas and Fas ligand in embryos and adult miceligand expression in several immune-privileged tissues and coexpression in adult tissues characterized by apoptotic cell turnover. J. Cell Biol. 1996;133:335–343. doi: 10.1083/jcb.133.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Hahne M., Rimoldi D., Schroter M., Romero P., Schreier M., French L.E., Schneider P., Bornand T., Fontana A., Lienard D. Melanoma cell expression of Fas(Apo-1/CD95) ligandimplications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- Hakem R., Hakem A., Duncan G.S., Henderson J.T., Woo M., Soengas M.S., Elia A., de la Pompa J.L., Kagi D., Khoo W. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Hamburger V. The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J. Exp. Zool. 1934;68:449–494. [Google Scholar]
- Hamburger V. History of the discovery of neuronal death in embryos. J. Neurobiol. 1992;23:1116–1123. doi: 10.1002/neu.480230904. [DOI] [PubMed] [Google Scholar]
- Hanson M.G., Jr., Shen S., Wiemelt A.P., McMorris F.A., Barres B.A. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J. Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson C.E. Neurotrophic factors as therapeutic agents in amyotrophic lateral sclerosispotential and pitfalls. Adv. Neurol. 1995;68:235–240. [PubMed] [Google Scholar]
- Henderson C.E., Phillips H.S., Pollock R.A., Davies A.M., Lemeulle C., Armanini M., Simpson L.C., Moffet B., Vandlen R.A., Koliatsos V.E., Rosenthal A. GDNFa potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- Henderson C.E., Bloch-Gallego E., Camu W. Purified embryonic motoneurons. In: Cohen J., Wilkin G., editors. Nerve Cell CultureA Practical Approach. Oxford University Press; London: 1995. pp. 69–81. [Google Scholar]
- Hollyday M., Hamburger V. Reduction of the naturally occurring motor neuron loss by enlargement of the periphery. J. Comp. Neurol. 1976;170:311–320. doi: 10.1002/cne.901700304. [DOI] [PubMed] [Google Scholar]
- Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J.L., Schroter M., Burns K., Mattmann C. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Kuida K., Zheng T.S., Na S., Kuan C., Yang D., Karasuyama H., Rakic P., Flavell R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K., Haydar T.F., Kuan C.Y., Gu Y., Taya C., Karasuyama H., Su M.S., Rakic P., Flavell R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- La Bella V., Cisterni C., Salaun D., Pettmann B. Survival motor neuron (SMN) protein in rat is expressed as different molecular forms and is developmentally regulated. Eur. J. Neurosci. 1998;10:2913–2923. doi: 10.1111/j.1460-9568.1998.00298.x. [DOI] [PubMed] [Google Scholar]
- Le-Niculescu H., Bonfoco E., Kasuya Y., Claret F.X., Green D.R., Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Prevette D., Oppenheim R.W., Milligan C.E. Involvement of specific caspases in motoneuron cell death in vivo and in vitro following trophic factor deprivation. Mol. Cell. Neurosci. 1998;12:157–167. doi: 10.1006/mcne.1998.0709. [DOI] [PubMed] [Google Scholar]
- Mariani S.M., Matiba B., Armandola E.A., Krammer P.H. The APO-1/Fas (CD95) receptor is expressed in homozygous MRL/lpr mice. Eur. J. Immunol. 1994;24:3119–3123. doi: 10.1002/eji.1830241231. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A., Herr I., Jeremias I., Hahne M., Brandt R., Vogel J., Schenkel J., Herdegen T., Debatin K.M. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J. Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein M.M., Hengartner M.O., Tsung N., Ellis R.E., Horvitz H.R. Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans gene ces-2. Nature. 1996;382:545–547. doi: 10.1038/382545a0. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A., Wilkinson G.A., Kruttgen A., Hu M., Munro E., Hanson M.G., Jr., Reichardt L.F., Barres B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan C.E., Prevette D., Yaginuma H., Homma S., Cardwell C., Fritz L.C., Tomaselli K.J., Oppenheim R.W., Schwartz L.M. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosistelling cells their time is up. Curr. Biol. 1996;6:1241–1243. doi: 10.1016/s0960-9822(02)70706-9. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nagata S., Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Nguyen Q.T., Parsadanian A.S., Snider W.D., Lichtman J.W. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science. 1998;279:1725–1729. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W., Thornberry N.A. Caspaseskiller proteases. Trends Biochem. Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W., Ali A., Thornberry N.A., Vaillancourt J.P., Ding C.K., Gallant M., Gareau Y., Griffin P.R., Labelle M., Lazebnik Y.A. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W., Prevette D., Yin Q.W., Collins F., MacDonald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science. 1991;251:1616–1618. doi: 10.1126/science.2011743. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W., Houenou L.J., Johnson J.E., Lin L.F., Li L., Lo A.C., Newsome A.L., Prevette D.M., Wang S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;3730:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- Park C., Sakamaki K., Tachibana O., Yamashima T., Yamashita J., Yonehara S. Expression of fas antigen in the normal mouse brain. Biochem. Biophys. Res. Commun. 1998;252:623–628. doi: 10.1006/bbrc.1998.9572. [DOI] [PubMed] [Google Scholar]
- Pennica D., Arce V., Swanson T.A., Vejsada R., Pollock R.A., Armanini M., Dudley K., Phillips H.S., Rosenthal A., Kato A.C., Henderson C.E. Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron. 1996;17:63–74. doi: 10.1016/s0896-6273(00)80281-0. [DOI] [PubMed] [Google Scholar]
- Pettmann B., Henderson C.E. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- Saas P., Walker P.R., Hahne M., Quiquerez A.L., Schnuriger V., Perrin G., French L., Van Meir E.G., de Tribolet N. Fas ligand expression by astrocytoma in vivomaintaining immune privilege in the brain? J. Clin. Invest. 1997;99:1173–1178. doi: 10.1172/JCI119273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M., Hayashi T., Abe K., Sadahiro M., Tabayashi K. Delayed selective motor neuron death and fas antigen induction after spinal cord ischemia in rabbits. Brain Res. 1998;797:23–28. doi: 10.1016/s0006-8993(98)00290-x. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Medema J.P., Krammer P.H., Peter M.E. FLICE is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J. Biol. Chem. 1997;272:26953–26958. doi: 10.1074/jbc.272.43.26953. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz I., Krammer P.H., Peter M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999;274:1541–1548. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- Srinivasan A., Li F., Wong A., Kodandapani L., Smidt R., Jr., Krebs J.F., Fritz L.C., Wu J.C., Tomaselli K.J. Bcl-xL functions downstream of caspase-8 to inhibit Fas- and tumor necrosis factor receptor 1-induced apoptosis of MCF7 breast carcinoma cells J. Biol. Chem. 273 1998. 4523 4529a [DOI] [PubMed] [Google Scholar]
- Srinivasan A., Roth K.A., Sayers R.O., Shindler K.S., Wong A.M., Fritz L.C., Tomaselli K.J. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system Cell Death Differ. 5 1998. 1004 1016b [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Ahmad M., Ottilie S., Bullrich F., Banks S., Wang Y., Fernandes-Alnemri T., Croce C.M., Litwack G., Tomaselli K.J. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- Suda T., Takahashi T., Golstein P., Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Tanaka M., Brannan C.I., Jenkins N.A., Copeland N.G., Suda T., Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- Troy C.M., Derossi D., Prochiantz A., Greene L.A., Shelanski M.L. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J. Neurosci. 1996;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J., Irmler M., Thome M. Inhibition of fas death signals by FLIPs. Curr. Opin. Immunol. 1998;10:552–558. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- Villa P., Kaufmann S.H., Earnshaw W.C. Caspases and caspase inhibitors. Trends Biochem. Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R., Brannan C.I., Copeland N.G., Jenkins N.A., Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- White F.A., Keller-Peck C.R., Knudson C.M., Korsmeyer S.J., Snider W.D. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J. Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]