

Abstract
Perlecan is a heparan sulfate proteoglycan that is expressed in all basement membranes (BMs), in cartilage, and several other mesenchymal tissues during development. Perlecan binds growth factors and interacts with various extracellular matrix proteins and cell adhesion molecules. Homozygous mice with a null mutation in the perlecan gene exhibit normal formation of BMs. However, BMs deteriorate in regions with increased mechanical stress such as the contracting myocardium and the expanding brain vesicles showing that perlecan is crucial for maintaining BM integrity. As a consequence, small clefts are formed in the cardiac muscle leading to blood leakage into the pericardial cavity and an arrest of heart function. The defects in the BM separating the brain from the adjacent mesenchyme caused invasion of brain tissue into the overlaying ectoderm leading to abnormal expansion of neuroepithelium, neuronal ectopias, and exencephaly. Finally, homozygotes developed a severe defect in cartilage, a tissue that lacks BMs. The chondrodysplasia is characterized by a reduction of the fibrillar collagen network, shortened collagen fibers, and elevated expression of cartilage extracellular matrix genes, suggesting that perlecan protects cartilage extracellular matrix from degradation.
Keywords: perlecan, basement membrane, cardiac muscle, exencephaly, chondrodysplasia
Perlecan is a heparan sulfate proteoglycan and a major component of basement membranes (BMs) and cartilage (Hassell et al. 1980; French et al. 1999). Although several biochemical and structural properties of perlecan have been reported, its in vivo function remains largely unknown. The core protein is composed of several different sequence motifs that are also found in the low density lipoprotein receptor and in other extracellular matrix (ECM) proteins such as laminin and the neural cell adhesion molecule (Murdoch et al. 1992). The NH2 terminus of perlecan contains three glycosaminoglycan attachment sites (Costell et al. 1997) that are usually modified by the addition of heparan sulfate side chains.
During mouse development perlecan is first expressed in two-cell embryos (Dziadek and Timpl 1985). The expression of perlecan increases on the external surface of trophectodermal cells of blastocysts (Smith et al. 1997), which correlates with the acquisition of attachment competence of blastocysts in vitro. Since perlecan expression is attenuated during delayed implantation (Smith et al. 1997) a role for perlecan in the attachment of the embryo to the uterine epithelium was suggested. During postimplantation development, perlecan is detected in blood vessel walls and in the developing heart and skeletal muscle (Handler et al. 1997). In Caenorhabditis elegans, mutations in the unc-52 gene, the orthologue of the mammalian perlecan gene, lead to disruptions of sarcomeres and cause detachment of body wall muscle (Rogalski et al. 1993), indicating an important role of perlecan for muscle function. A significant increase in perlecan expression occurs during organogenesis of the kidney, lung, liver, spleen, gastrointestinal tract, and cartilage (Handler et al. 1997). The levels of perlecan are low in precartilaginous tissues (French et al. 1999), but are high in mature cartilage. Recent in vitro findings have shown that perlecan supports chondrocyte differentiation (French et al. 1999), which together with its expression pattern, suggests a role for this molecule in skeletogenesis.
In contrast to the well characterized expression pattern, only a few functional properties of perlecan are known. The presence of perlecan in BMs and its ability to interact with other BM components such as collagen type IV, laminin, and nidogen/entactin in vitro suggested that it is involved in BM assembly (Reinhardt et al. 1993; Hopf et al. 1999). It also binds cell adhesion molecules, such as β1 and β3 integrins (Hayashi et al. 1992; Brown et al. 1997) and α-dystroglycan (Peng et al. 1998; Talts et al. 1999), and several of these components are also known to participate in BM assembly (Bloch et al. 1997; Henry and Campbell 1998; Sasaki et al. 1998).
One property that perlecan shares with several other proteoglycans is its ability to bind and store growth factors. The heparan sulfate side chains bind FGF-2 and may serve as a low affinity coreceptor, thus, playing a role in FGF-2–mediated mitogenesis and angiogenesis (Aviezer et al. 1994). The observation that high levels of perlecan in metastatic melanomas correlate with a more aggressive phenotype (Timar et al. 1992) supports the latter hypothesis. The core protein is also capable of binding different growth factors including PDGF-B and FGF-7 (Göhring et al. 1998; Sharma et al. 1998).
Heparan sulfate proteoglycans are thought to be essential for the glomerular filtration apparatus. Antibodies against perlecan core protein destroy the filtering properties of the glomerular BM and cause proteinuria (Miettinen et al. 1986). Moreover, in long-term diabetes mellitus, the content of heparan sulfate proteoglycans is decreased (Comper et al. 1996), which is believed to contribute to the development of diabetic nephropathy with characteristic proteinuria and ultimately renal failure. Perlecan has also been implicated in the pathogenesis of Alzheimer's disease (AD) amyloidosis. A common feature of AD amyloids is the presence of perlecan within the deposits (Snow and Wight 1989) where it interacts with the β-amyloid (Aβ) protein and its precursor (Castillo et al. 1997). It seems that this interaction enhances the formation of Aβ fibrils and protects Aβ from protease degradation (Gupta-Bansal et al. 1995).
To directly test the function of perlecan in vivo, we have generated mice lacking perlecan gene expression. We demonstrate that perlecan is essential for maintaining the integrity of cartilage ECM and BMs of contracting cardiac muscle cells and expanding brain vesicles.
Materials and Methods
Generation of Perlecan-deficient Mice
A 700-bp DNA fragment from the 5′ region of the mouse perlecan cDNA was used to screen a genomic library derived from a mouse D3/129 embryonic stem (ES) cell line (a gift from J.S. Mudgett, Merck Sharp & Dohme, NJ) to isolate perlecan genomic clones. The targeting construct (see Fig. 1 A) consisted of an 8-kb fragment containing exon 5, an expression cassette flanked by loxP sites in which the phosphoglycerate kinase promoter controls the expression of the neomycin (neo) gene and the Herpes simplex virus thymidine kinase (HSV-tk) gene, respectively, an 0.8-kb fragment containing exon 6 followed by a single loxP site and a 1.5-kb fragment containing exon 7 (for more detailed information contact: reinhard. fassler@pat.lu.se).
Figure 1.

Targeting strategy, Southern blots, PCR, and RIA analysis of ES cells and mice lacking perlecan. (A) Structure of the wild-type perlecan allele, targeting construct, and targeted perlecan allele before and after cre-loxP–mediated deletion. The dark boxes show exons of the perlecan gene, the open box shows the neo-tk cassette, and the loxP sites are indicated as triangles. Probe 1 was used to detect homologous recombination, and probe 2 was used to identify the null allele. Restriction sites are as follows: E, EcoRI; B, BamHI; and H, HindIII. (B) Southern blot analysis of EcoRI-digested genomic DNA derived from wild-type, heterozygous, and homozygous mutant mice hybridized with probe 1. (C) Detection of mutant mRNA lacking exon 6 by RT-PCR using the total RNA prepared from wild-type, heterozygous, and homozygous ES cells. Primers from exons 5 and 8 amplified 640 bp from the wild-type and 479 bp from the mutant allele. PCR products were probed with exon 7–specific radiolabeled oligonucleotide. (D) Radioimmunoassay measurement of laminin-1, nidogen-1, and perlecan in the serum-free medium derived from wild-type and mutant ES cells. Values are means ± SD of triplicate culture dishes and are expressed as micrograms per milliliter of supernatant.
The targeting construct was electroporated into R1 ES cells. ES cells culture, electroporation, isolation, and analysis of G418-resistant ES cell clones was carried out as described (Fässler and Meyer 1995). Genomic DNAs were digested with EcoRI, and probed with an external 3-kb BamHI-EcoRI genomic DNA fragment (see Fig. 1, probe 1). ES cell clones that had undergone homologous recombination were transiently transfected with a Cre expression plasmid (a gift from Dr. Werner Müller, University of Cologne, Germany) and selected in the presence of FIAU. Surviving ES cell clones were isolated and analyzed by Southern blot by cleaving genomic DNA with EcoRI, BamHI, or HindIII, respectively, and probed with an internal genomic DNA fragment (see Fig. 1, probe 2). The generation of germline chimeras and breeding were carried out as described (Fässler and Meyer 1995).
Antibodies
The following primary antibodies were used for immunohistochemistry: rat anti-collagen type II (diluted 1:400; obtained from Dr. Rikard Holmdahl, Lund University, Sweden); rabbit anti-collagen type X (1:500; obtained from Dr. Bjorn Olsen, Harvard Medical School, Boston, MA); rabbit anti–laminin-1 (1:200); rabbit anti–perlecan antibodies against domain I (1:1,000), domain III-3 (1:500) and domain V (1:1,000); rat anti–nidogen-1 (1:30); mouse anti–β-tubulin isotype III (1:100; Sigma Chemical Co.); rabbit anti-nestin (1:2,000; obtained from Dr. Zaal Kokaia, Lund University, Sweden); mouse anti-reelin (1:1,000; obtained from Dr. André Goffinet, University of Namur, Belgium); and Ki-67 (1:50; Dianova, Hamburg, Germany).
The following secondary antibodies were used: biotinylated goat anti–rabbit Ig, biotinylated goat anti–rat Ig (obtained from Vector Laboratories Inc.); FITC-conjugated donkey anti–mouse; and Cy3-conjugated goat anti–rat (both obtained from Jackson ImmunoResearch Laboratories, Inc.).
Reverse Transcriptase (RT) –PCR Assays, Radioimmunoassays, and Northern Blot
Total RNA from E12.5 embryos was isolated as described (Fässler and Meyer 1995). RT-PCR was carried out with a commercial kit (Titan RT-PCR; Boehringer Mannheim). The primer sequences were as follows: 5′-CCCGAATACAGGAAGATCCC-3′ specific for exon 5, and 5′-TCACAGGCGAACTCGTTAGGCTCA-3′ specific for exon 8. The amplification products were separated on 2% agarose gel, blotted onto nylon filters, and hybridized with a γ-[32P]ATP–labeled oligonucleotide (5′-CAGACATGTCCCTGCAGTCAGGCC-3′) specific for exon 7.
Confluent ES cells were grown overnight in serum-free medium. The medium was collected and the cells were lysed with RIPA buffer supplemented with proteinase inhibitors (5 mM NEM, 2 mM PMSF, 5 mM EDTA). Radioimmunoassays were performed with the medium and cell extract using either tissue-derived (laminin, perlecan) or recombinant proteins (nidogen-1) as reference inhibitors. Two assays specific for perlecan domains I and III-3 were used and gave similar results (Costell et al. 1997).
For Northern blotting, cartilage samples were dissected from the limbs of normal and perlecan-deficient E17.5 embryos, and total RNA was prepared, separated, blotted, and probed as described (Fässler and Meyer 1995). The oligo-labeled probes represented ∼500 bp of the mouse Col2a1, matrilin-3, and cartilage matrix protein (COMP) cDNAs, respectively. Signal intensities were measured from nonsaturated films using a Gel-ProAnalyzer (Media Cybernetics).
Histological Analysis, Immunohistochemistry, and Staining of Skeletons
For histological analyses, staged embryos were fixed in 4% fresh paraformaldehyde in PBS overnight, pH 7.2, dehydrated in a graded alcohol series, and embedded in paraffin (Paraplast X-tra; Sigma Chemical Co.). Sections were cut at 6–8 μm and stained with hematoxylin/eosin or cresyl violet (Nissl staining). To detect proteoglycans and calcification, bone sections were stained with Safranin orange and van Kossa (Aszódi et al. 1998). To detect proliferating cells, 5-bromo-2′-deoxyuridine (BrdU; Sigma Chemical Co.) and anti-BrdU mAb (Boehringer Mannheim) were used as described (Aszódi et al. 1998). Apoptosis was analyzed by the terminal deoxynucleotidyl transferase (TdT)–mediated dUTP nick end labeling (TUNEL) method using a commercial in situ cell death detection kit (Boehringer Mannheim) according to the manufacturer's instructions.
For immunohistochemistry, embryos were either fixed in 4% buffered paraformaldehyde or in 95% ethanol/5% acetic acid overnight at 4°C, embedded in paraffin, and sectioned at 6–8 μm. Immunostaining was performed either by using the Vectastain ABC Elite kit (Vector Laboratories) or by using an immunofluorescence method described earlier (Aszódi et al. 1998). Skeletons of E17.5 embryos were prepared and stained as described by Aszódi et al. 1998 and photographed on a dissecting microscope.
Plastic Embedding and Electron Microscopy
For light microscopy of heart tissue, 4% paraformaldehyde immersion-fixed embryos were postfixed with 2% osmium tetroxide in PBS for 2 h at 4°C. After a thorough wash in PBS for 3× 10 min, the embryos were block-stained with 1% uranyl acetate in 70% ethanol for 8 h. Afterwards the specimens were dehydrated, infiltrated with and embedded in araldite, cut with a glass knife (1–2 μm) on a Reichert ultramicrotome (Leica), and stained with methylene blue. Ultrastructural analysis of basement membranes and cartilage tissue was performed as described earlier (Bloch et al. 1997; Aszódi et al. 1998).
For scanning electron microscopy, E10.5 embryos were prepared as previously described (Walter and Müller 1985) with the modification that samples were critical point dried in a Balzer critical point dryer using 100% ethanol as the intermediate solvent. Embryos were mounted on aluminum holders, sputtered with 60 nm palladium/gold, and examined in a Jeol JSM-T330 scanning electron microscope. Electron microscopy after negative staining with anti–laminin-1 antibody with protein A coupled to colloidal gold was done as described by Roth et al. 1980.
Results
Targeted Disruption of the Perlecan Gene
The perlecan gene was inactivated in ES cells by homologous recombination using the cre/loxP technique. A neomycin/thymidine kinase (neo/tk) cassette flanked by single loxP sites was inserted into intron 5 and a single loxP site was inserted into intron 6 (Fig. 1 A). Eight independently targeted ES cell lines were obtained. Two of them (clones 243 and 310) were transiently transfected with a cre recombinase expression plasmid, selected in FIAU, and analyzed for cre-mediated deletion. In three clones derived from 243 and 1 clone derived from 310, respectively, the sequence between the outermost loxP sites (including the neo/tk cassette, part of intron 5, exon 6, and part of intron 6) was deleted (Fig. 1 A). One clone carrying the constitutive null allele from 243 and from 310, respectively, was injected into C57BL/6 blastocysts and transferred to pseudopregnant females. Chimeric males from both clones gave germline transmission of the mutated perlecan gene.
ES Cells Carrying a Homozygous Mutation in the Perlecan Gene Do Not Secrete Perlecan
To verify that the deletion of exon 6 in the perlecan gene leads to a constitutive null mutation, ES cells carrying a homozygous mutation were generated. Heterozygous ES cells were retransfected with the original targeting construct and again selected for neo expression. Afterwards, one of the clones with a recombination event on the wild-type allele was transiently transfected with the cre expression plasmid to obtain ES clones with a homozygous deletion of exon 6 (Fig. 1 B).
To test whether the mutant allele was transcribed, RT-PCR was performed with total RNA extracted from normal, heterozygous, and homozygous ES cells. The amplified fragment encompasses sequences in exons 5–8. The size of the amplified DNA fragment was 640 bp for the wild-type allele and 479 bp for the mutant allele, indicating that splicing occurs between exon 5 and 7 (Fig. 1 C). Splicing of exon 5–7 leads to a reading frame shift and to the formation of a truncated perlecan protein consisting of the 116–NH2-terminal amino acids that make approximately half of domain I.
To determine whether the mutant mRNA was translated, normal and homozygous ES cells were grown in serum-free culture medium, and the supernatant as well as the cell extract were subjected to a highly sensitive RIA using polyclonal antibodies against laminin-1, nidogen-1, or the NH2-terminal domain I of perlecan. Whereas normal and mutant ES cells produce similar amounts of laminin and nidogen-1, only normal but not mutant ES cells produce perlecan (Fig. 1 D).
These data show that although the mutant perlecan allele is transcribed, no truncated protein could be detected with a domain I–specific polyclonal antibody. Most likely, the truncated form of domain I is not properly folded and is consequently degraded intracellularly as soon as it is translated. This was confirmed by episomal transfection of human EBNA-293 cells with domain I lacking 38 COOH-terminal residues that did not produce any recombinant fragment (data not shown). Therefore, the mutation in the perlecan gene is designated as a loss of function mutation.
Mice Homozygous for the Perlecan-Null Mutation Die at Two Developmental Stages: between E10 and E12 and Perinatally
Mice heterozygous for the mutation appeared normal and did not display any overt anatomical or behavioral abnormalities. No mice homozygous for the mutation were detected among 728 weaned progeny from heterozygous intercrosses (Table ). Analysis of newborn offspring detected three homozygotes and several cannibalized mice among 98 neonates (Table ). The three homozygotes showed exencephaly, chondrodysplasia, hemorrhage in several organs (see below), severe cleft palates, and died around birth.
Table 1.
Progeny of Perlecan+/− × Perlecan+/− Crosses
| Stage | No. of litters | Perlecan genotype (%) | −/− Viable | Percentage of expected −/− progeny | ||
|---|---|---|---|---|---|---|
| +/+ | +/− | −/− | ||||
| P21 | 91 | 246 (34) | 482 (66) | 0 | 0 | 0 |
| P0 | 13 | 32 (33) | 63 (64) | 3 (3) | 0 | 0 |
| E18.5 | 4 | 12 (40) | 18 (60) | 0 (0) | 0 | 0 |
| E17.5 | 15 | 39 (35) | 67 (59) | 7 (6) | 7 | 20 |
| E16.5 | 2 | 6 (40) | 8 (53) | 1 (7) | 1 | 20 |
| E15.5 | 6 | 12 (29) | 25 (60) | 5 (12) | 5 | 41 |
| E14.5 | 8 | 12 (20) | 43 (72) | 5 (8) | 5 | 28 |
| E13.5 | 11 | 23 (26) | 60 (68) | 5 (6) | 5 | 18 |
| E12.5 | 15 | 28 (24) | 63 (55) | 24 (21) | 19 | 63 |
| E11.5 | 34 | 74 (28) | 151 (57) | 40 (15) | 17 | 23 |
| E10.5 | 34 | 78 (29) | 138 (50) | 56 (21) | 53 | 74 |
| E9.5 | 11 | 17 (21) | 43 (53) | 21 (26) | 21 | 105 |
E, embryonic day; and P, postnatal day.
To determine when the remaining homozygotes die, embryos were examined from E9.5 to birth (Table ). At E9.5, wild-type, heterozygous, and homozygous mutant embryos were represented in a normal Mendelian ratio (Table ). Perlecan-null embryos at E9.5 were indistinguishable from wild-type and heterozygous littermates. E9.5 perlecan-null embryos formed brain vesicles, optic vesicles, branchial arches, otic pits, limb buds, a beating heart, and 20–25 somites (not shown). Therefore, development to E9.5 proceeds normally in the absence of perlecan.
By E10.5, defects began to appear in perlecan-null embryos. Although they were still present in the expected percentage (Table ) and were of normal size, ∼70–80% were dead as demonstrated by the absence of the heart beating and the presence of severe hemopericardium (Fig. 2 B, arrow). 20–30% of homozygotes were alive and of normal appearance (not shown). By E11.5 and E12.5 the percentage of dead embryos with hemopericardium increased further, whereas some of the living embryos developed abnormally in the head region and survived to the perinatal period. At all stages analyzed, the placental development was unaffected by the loss of perlecan. The placental size, architecture, and the blood content were similar between normal and perlecan-null embryos (not shown). In addition, PECAM whole mount stainings of E9.5 and E10.5 embryos revealed that homozygotes had no defects in angiogenesis, sprouting, and remodeling to generate vessels of different sizes (not shown). At later stages of development (E13–E17), we could observe the formation of microaneurysms associated with bleedings in several tissues including lung, skin, and brain (not shown).
Figure 2.
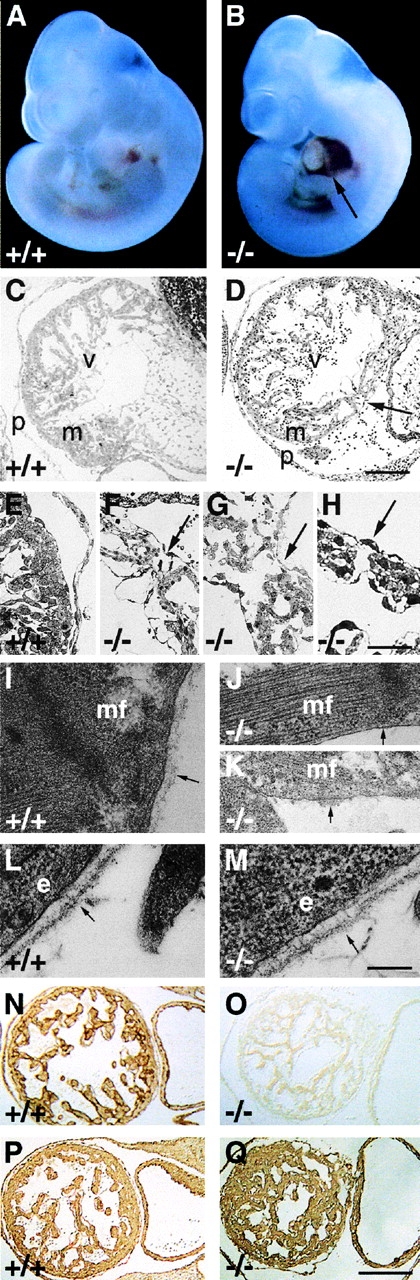
Development of the hemopericard in E10.5 perlecan-null embryos. (A and B) Whole mount pictures of E10.5 wild-type (A) and perlecan-deficient embryos showing blood leakage into the pericardial cavity (B, arrow). (C–H) Light microscopy of semi-thin sections of wild-type (C and E) and perlecan-null hearts (D and F–H) stained with methylene blue. (C) Myocardial wall with well developed trabeculation separated from the thin pericardium by a cell-free cavity. (E) Higher magnification shows that cardiomyocytes are covered by an endo- and epicardial cell layer. (D) Myocardial wall of homozygotes is disrupted (arrow), whereas the endo- and epicardial cell layers are intact. The pericardial cavity contains blood cells and is surrounded by a thickened pericardium. (F–H) Three different perlecan-null hearts with small ruptures in the myocardial wall (arrows). (I–K) Electron microscopy of a normal (I) cardiac muscle cell with BM composed of a lamina rara and densa (arrow). Mutant cardiac muscle cell shows a typical myofilament organization but lacks a BM (arrow in J) or with BM-like material (arrow in K) on the cell surface. (L and M) Typical BM with a lamina rara and densa (arrow) beneath the dermal epithelium of wild-type (L) and perlecan-null (M) embryos. (N-Q) Immunostaining for perlecan (N and O) and laminin-1 (P and Q) of a wild-type (N and P) and perlecan-null (O and Q) heart. Perlecan is absent but laminin-1 is expressed in homozygous hearts. Abbreviations: v, ventricle; m, myocardium; p, pericardium; mf, muscle filament; and e, epithelial cell. Bars: (C and D) 250 μm; (E–H) 100 μm; (I–M) 250 nm; and (N–Q) 250 μm.
These results indicate that a null mutation in the perlecan gene leads to two patterns of development. Many of the perlecan-null embryos die during an early crisis (E10.5–E12.5) characterized by hemopericardium and heart arrest. The few remaining mutants survive to the perinatal period (Table ), but develop severe brain and skeletal defects (see below).
Early Lethality in Perlecan-deficient Embryos Is Due to Myocardial Defects
At E10–11, many perlecan-null embryos manifested several signs of cardiac insufficiency characterized by intrapericardial hemorrhages (Fig. 2 B), weak heartbeats, or cardiac arrest. To investigate these defects, wild-type and alive perlecan-null embryos were analyzed by immunohistochemical and ultrastructural methods. The high resolution analysis of plastic-embedded hearts derived from E10.5 wild-type embryos showed a continuous wall of several layers of cardiomyocytes covered on both sides by a single cell layer of endothelial cells forming the endocardium and epicardium (Fig. 2C and Fig. E). In a large number of perlecan-null embryos, the compact layer of cardiomyocytes was interrupted by small intercellular clefts that were often covered by an intact layer of endo- and epicardium (Fig. 2D and F–H). In a few embryos, the clefts in the myocardium were filled with endocardial cells (Fig. 2D and Fig. F). The defects were associated with blood cell leakage into the pericardial cavity (Fig. 2D and Fig. F). Adjacent to the myocardial defects, the pericardial tissue was thickened because of an increase in cell number and matrix deposition (Fig. 2 D). No defects were observed in the blood vessels of affected hearts (not shown).
At E9.5, thin and normally appearing BMs were observed around perlecan-null cardiomyocytes (not shown). However, at E10.5 normal cardiomyocytes were covered by a continuous BM (Fig. 2 I), whereas perlecan-null cardiomyocytes lacked BM deposition (Fig. 2 J) or showed small patches of electron dense material on their cell surface (Fig. 2 K). The formation of sarcomeres (Fig. 2 J) and tight junctions were normal in perlecan-null cardiomyocytes. Ultrastructural analysis of BMs in other tissues including skin (Fig. 2L and Fig. M) and gut (not shown) revealed no abnormalities, suggesting that at E10.5 the BM defects are restricted to the heart where they are exposed to mechanical stress. To test whether the BM defects in the perlecan-null myocardium are associated with abnormal expression of BM proteins, immunostaining for perlecan (Fig. 2N and Fig. O), laminin-1 (Fig. 2P and Fig. Q), and collagen IV (not shown) was performed on normal and perlecan-null cardiac tissue. With the exception of perlecan, all BM components were similarly expressed in mutant tissue (Fig. 2P and Fig. Q). These data demonstrate that perlecan-deficient cardiac muscle cells lack BM or are covered by abnormal BM, which disrupts the integrity of the myocardium and leads to the formation of small clefts in myocardial tissue and finally to blood leakage into the pericardial cavity.
Development of Exencephaly and Neuronal Ectopias
About 80% of the perlecan-null embryos surviving the first crisis developed an exencephalic malformation that was first visible between E10.5 and E11.5. To test whether the absence of perlecan results in an abnormal closure of the neural tube, scanning electron microscopy was performed with normal and perlecan-null E10.5 embryos. All normal (n = 3) and perlecan-null embryos (n = 11) analyzed, displayed properly closed neuropores (Fig. 3, A–C). This was also confirmed by histological studies on E9.5 and E10.5 cephalic regions derived from mutant embryos (not shown). Some perlecan-null embryos had holes in the fore- and midbrain and showed collapsed brain vesicles (Fig. 3 C). The cephalic region of normal embryos was covered by an intact layer of ectodermal cells (Fig. 3 D). However, at higher magnification, the surface ectoderm of seven out of nine homozygotes showed small clefts that were 20–30 μm in width that contained round cells with small extensions (Fig. 3 E). In nullizygotes with severe defects, round cells with small extensions traversed the cephalic mesenchyme to reach the surface ectoderm (Fig. 3 F). To exclude that the clefts were caused artificially during embryo handling, heads were incubated with colloidal gold-conjugated antibodies binding to laminin-1 (Fig. 3 G) and analyzed by scanning electron microscopy. Preparation artefacts occurring after antibody incubation were devoid of staining (Fig. 3H and Fig. J, and arrow in I and K), whereas clefts already present in homozygotes before handling showed colloidal gold staining of the exposed BM (Fig. 3I and Fig. K).
Figure 3.
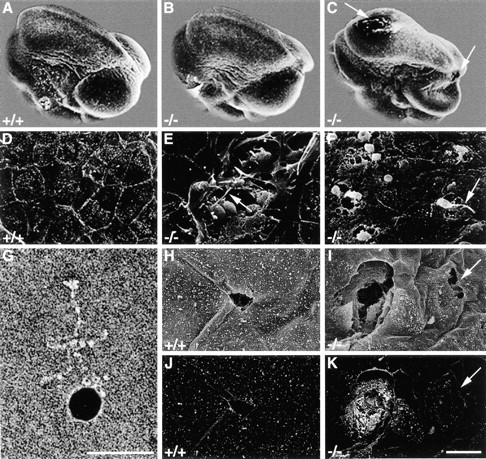
Scanning electron microscopy revealed brain defects in perlecan-null embryos. (A–C) Scanning electron microscopy shows that the neural tube is closed in wild-type (A) and perlecan-deficient (B and C) E10.5 embryos. Some perlecan-null embryos show holes (C, arrows) in the fore- and midbrain and collapsed brain vesicles. (D–F) High magnification of the surface ectoderm. In wild-type embryos (D), the cephalic region is covered with an intact layer of ectodermal cells. Perlecan-null embryos (E) show small clefts that are 20–30 μm in width and contain round cells with small extensions (arrow). In other perlecan-deficient embryos (F), round cells with extensions burst through the surface ectoderm (arrow). G shows an antibody coupled to colloidal gold reacting with purified laminin. (H–K) Backscattered electron analysis shows that small defects in the ectoderm of normal mice caused during the preparation are not labeled with the gold-conjugated antibody (secondary electron image H and backscattered electron image J), while the clefts in perlecan-null embryos are labeled (secondary electron image I, backscattered electron image K). Arrow in K indicates an ectodermal defect caused during the preparation. Bars: (D–F and H–K) 20 μm; and (G) 50 nm.
Histological analysis of brain sections from normal and perlecan-null E9.5 embryos revealed normal BM between neural tissue and mesenchyme (not shown). At E11.5, 70% of homozygotes showed areas in which the BM surrounding the telencephalic vesicles was disrupted (Fig. 4D and Fig. F), and the brain tissue had invaded into the cephalic mesenchyme and fused with the overlaying ectoderm (Fig. 4 H). Immunostaining revealed that the ectopias contained many nestin-positive cells (a marker for neuroepithelial cells; Fig. 4I and Fig. J), but lacked βIII isotype tubulin-positive cells (a marker for committed neurons; Fig. 4K and Fig. L). In the ectopic region, the neuroepithelium appeared thickened and the cells in the ventricular zone region were round instead of elongated as observed in the neocortex of normal embryos (Fig. 4G and Fig. H). At E11.5, clusters of neuroepithelial cells were exposed to the amniotic cavity and formed small disruptions of 5–10 μm (Fig. 4 M). Immunostaining for proliferative cells with Ki-67 antibodies and for apoptotic cells with TUNEL labeling revealed no abnormalities in E10.5 and E11.5 perlecan-null brain tissues, neither in ectopias nor in normal appearing areas of the neocortex (not shown). At later stages, several homozygotes without obvious exencephaly showed a ruffled brain surface because the marginal zone of the neocortex was studded with large ectopias associated with a severe distortion of the laminar architecture of the cortex (Fig. 4O and Fig. Q).
Figure 4.
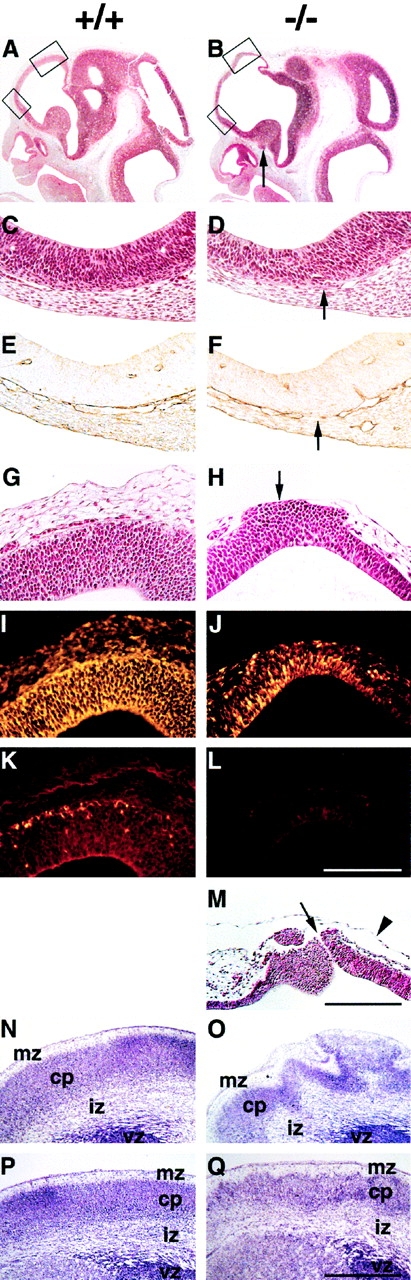
Exencephaly and neuronal ectopias develop in the anterior region of the forebrain. (A and B) Hematoxylin/eosin staining of sagittal brain sections from wild-type (A) and perlecan-null (B) E11.5 embryos. Note the extension and the thinning of the anterior part of the forebrain in the perlecan-null embryo (upper box in B). Ectopias are visible ventral of the medial ganglionic eminence (arrow) in the homozygous embryo. (C–F) Higher magnification of the lower boxes indicated in A and B. In a normal brain, the neuroepithelium and the underlying mesenchyme are separated by a BM (C) that contains laminin-1 (E). In the perlecan-null brain, the BM is discontinuous (D, arrow) and shows interrupted laminin-1 staining (F, arrow). (G and H) Higher magnification of the upper boxes indicated in A and B. In a normal brain, the neuroepithelium and the overlying mesenchyme are separated by a BM (G). In the perlecan-null brain, neuroepithelial cells have invaded the overlying ectoderm (H, arrow). (I–L) Immunohistochemical localization of nestin and β-tubulin type III in the neocortex of normal and perlecan-null embryos. The neocortex of the wild-type embryo contains nestin-positive cells (I) and a few β-tubulin type III–positive cells in the cortical subplate (K). Ectopic cells in the perlecan-null embryo are positive for nestin (J) but negative for β-tubulin type III (L). (M) Hematoxylin/eosin staining of the forebrain region of an E11.5 homozygote showing a defect in the ectoderm and a small hole of 5–10 μm (arrow). Note that the amniotic membrane is still intact (arrowhead). (N–Q) Nissl staining of coronal sections of wild-type (N and P) and perlecan-null (O and Q) E17.5 forebrain regions. The perlecan-null brain shows a ruffled surface, large ectopias, and abnormal lamination (O). Posterior to this area, the ruffles and defects in lamination are less severe (Q). Abbreviations: mz, marginal zone; cp, cortical plate; iz, intermediate zone; and vz, ventricular zone. Bars: (C–L) 125 μm; (M) 250 μm; and (N–Q) 500 μm.
All perlecan-null embryos analyzed so far, including those without exencephaly, exhibited neuronal ectopias in the ventral telencephalic region of the brain, when examined at E11.5 and later stages (Fig. 5). The ectopias appeared as small, compact clusters of βIII isotype tubulin-positive cells (Fig. 5, A–F) that had invaded the mesenchyme (Fig. 5B, Fig. D, Fig. F, Fig. H, Fig. J, and Fig. L) at areas where the basement membrane is disrupted as shown by immunostaining for laminin-1 (Fig. 5I and Fig. J). Immunostaining for laminin-1 and perlecan at E13.5 revealed that both molecules were expressed around brain vessels and in the leptomeninges surrounding the brain tissue (Fig. 5I and Fig. K). No perlecan expression was found in the brain parenchyma of normal mice (Fig. 5 K).
Figure 5.
Neuronal ectopias in the ventral forebrain. (A–D) Hematoxylin/eosin staining of coronal brain sections from wild-type (A and C) and perlecan-null (B and D) E12.5 embryos. Ectopias (B and D, arrows) are visible in the ventral forebrain of homozygotes. (E and F) Cells within the ectopias are β-tubulin type III–positive (F, arrow). (G–L) Hematoxylin/eosin and immunostaining of sagittal brain sections from wild-type (G, I, and K) and perlecan-null (H, J, and L) E13.5 embryos. Ectopias (H, J, and L, arrows) are visible in the ventral forebrain. Laminin-1 is expressed in the leptomeninges and around the capillaries in both the wild-type (I) and perlecan-null (J) brain. In homozygotes, laminin-1 staining is disrupted around the ectopias (arrows). Perlecan is expressed around capillaries and in the leptomeninges but not in brain of normal embryos (K). No perlecan staining is detectable in homozygotes (L). Bars: (C–F and I–L) 250 μm.
Chondrodysplasia Associated with Abnormal Endochondral Ossification
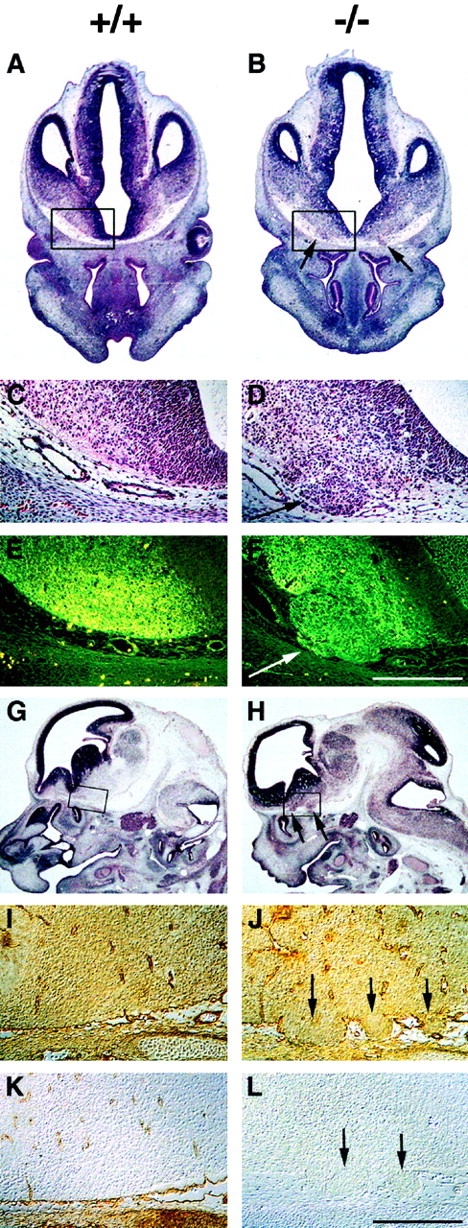
All perlecan-null embryos that did not exhibit apparent heart defects continued their intrauterine development but died perinatally. Between E15 and the newborn stage, these animals developed a severe osteochondrodysplasia characterized by dwarfism, cleft palate, short limbs, and a short and abnormally bended vertebral column (Fig. 6 A and not shown). Approximately 80% of homozygotes had exencephaly and lacked calvarial bones (Fig. 6 A, −/−b). Homozygotes without exencephaly had a domed skull (Fig. 6 A, −/−a). Whole mount skeletons of E17.5 embryos showed that all bones were present in perlecan-null embryos, except in exencephalic embryos, which lacked frontal and parietal bones (Fig. 6 B, −/−a and −/−b). Detailed inspection of the mutant skeleton showed that long bones were approximately half of the size of wild-types. In addition, the cortical bone was thickened. In the skull, the mandible and nasal bone were shortened, and the structures of the middle and inner ear were poorly developed (Fig. 6 B). The bones of chondrocranium (occipital, sphenoidal, and ethmoidal) were shortened and undermineralized (data not shown).
Figure 6.
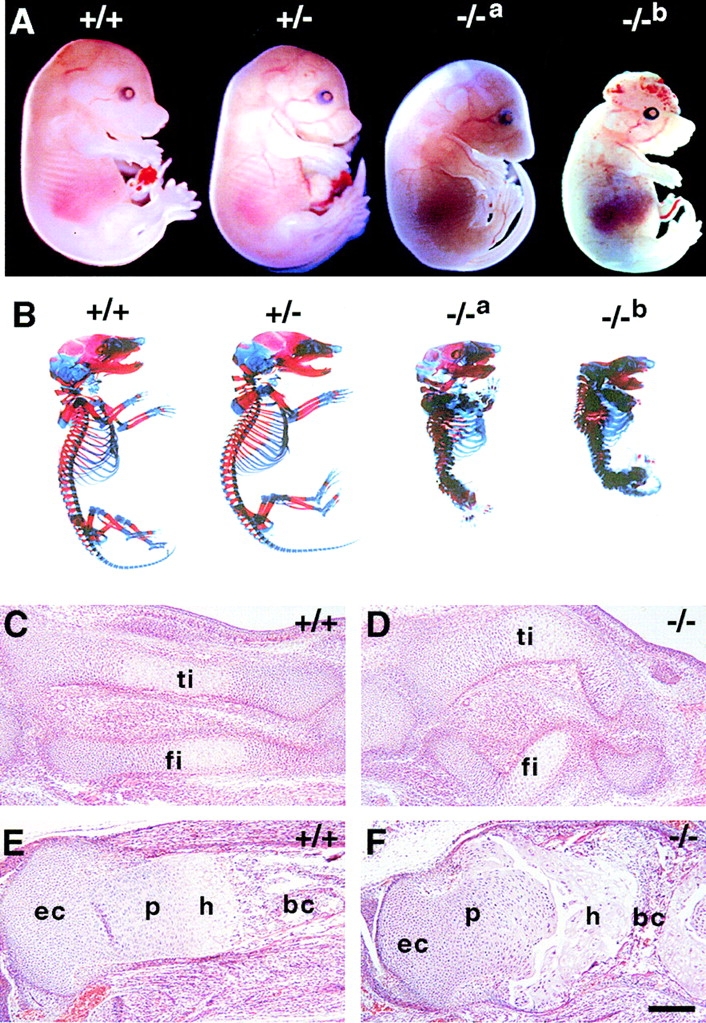
Skeletal abnormalities in perlecan-null embryos. (A) Lateral view of E16.5 normal, heterozygous, and perlecan-null (−/−a and −/−b) embryos. Loss of perlecan results in disproportionate dwarfism with short limbs, neck, and snout. Some homozygotes have domed skull (−/−a), others lack the roof of skull and exhibit exencephaly (−/−b). (B) Alcian blue/alizarin red stained skeletons of normal, heterozygous, and perlecan-null (−/−a and −/−b) E17.5 embryos. The skeleton of heterozygotes is normal. The spine of homozygous mutants is short and displays severe kyphoscoliosis, the thorax is narrow, and the ribs, vertebrae, and long bones are malformed. Embryos with exencephaly (−/−b) lack frontal and parietal bones of the skull. (C–F) Comparative histological analysis of developing hindlimbs from normal (C and E) and homozygotes (D and F). Sagittal sections from E14 (C and D) and E16.5 (E and F) embryos stained with hematoxylin/eosin. At E14, the mutant tibia (ti) and fibula (fi) are short, thick, and curved (D). At E16.5, the mutant tibia (F) is short and thick, the metaphyseal bone is extremely reduced, and the growth plate is disorganized. Abbreviations: ec, epiphyseal cartilage; p, zone of proliferative chondrocytes; h, zone of hypertrophic chondrocytes; and bc, bone cavity. Bar: (C–F) 100 μm.
Histological analysis at various stages of development revealed that the cartilage anlage of all long bones occurred normally in the mutant mice (not shown). Between E13 and 14, the first alterations in size and shape of the perlecan-null long bones became apparent (Fig. 6C and Fig. D). Mutant bones had disorganized growth plates characterized by the absence of the typical columnar arrangement of hypertrophic chondrocytes. In addition, hypertrophic chondrocytes showed an atypical morphology, and growth plates were always dissociated from their epiphyses (Fig. 6 F). Such gaps in the tissue were never observed in normal bones and, therefore, suggest a mechanical weakness of the mutant cartilage leading to damage during tissue processing. Perlecan-null bones had small marrow cavities.
Impaired Mineralization and Reduced Collagen Content
Safranin-O staining was reduced in perlecan-null cartilage, suggesting a decreased proteoglycan content (Fig. 7A and Fig. B). van Kossa staining revealed that normal bones showed clear mineralization in the longitudinal septa of the late hypertrophic zone (Fig. 7 A), perlecan-null tissue had minimal or no mineral deposits in the matrix around hypertrophic chondrocytes, and the calcified trabecula were transversely oriented in perlecan-null bones (Fig. 7 B). Immunohistochemistry showed expression of perlecan in cartilage as well as the surrounding mesenchyme of normal but not mutant mice (Fig. 7C and Fig. D). Matrix proteins including collagen types II, IX, X, and XI, aggrecan, matrilin-1 and -3, and COMP were expressed in homozygotes (Fig. 7F and Fig. H, and not shown).
Figure 7.
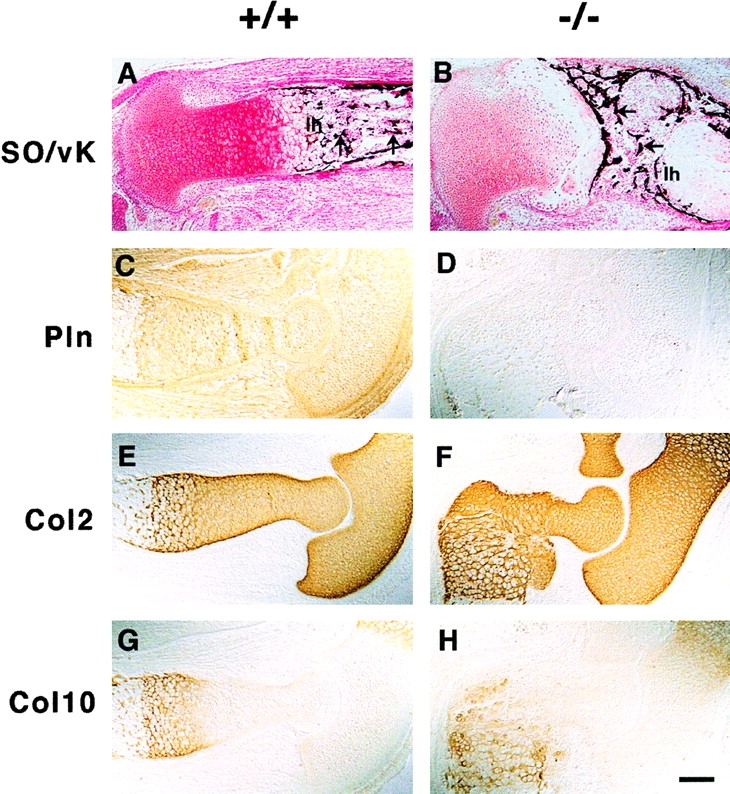
ECM expression in long bones. (A and B) Safranin orange (SO) and van Kossa (vK) double staining show reduced proteoglycan content in mutant (B) as compared with wild-type (A) cartilage and the absence of mineralization of longitudinal septa in the lower hypertrophic zone (1 h) of the mutant growth plate (A and B). Also, note the transversally oriented trabecular bones in the mutant (arrows). (C–H) Immunostaining of perlecan and collagen types II (Col2) and X (Col10) on consecutive sections of elbows from normal and perlecan-null E15.5 embryos. Perlecan is present in normal cartilage, in the periosteum/perichondrium, and in the surrounding connective tissues (C). In mutant embryos, perlecan staining is absent (D). The distribution of collagen types II (E and F) and X (G and H) is similar in normal (E and G) and perlecan-null (F and H) cartilage. Bar: (A–D and G and H) 100 μm; 50 μm in E and F.
The ultrastructure of cartilage tissue derived from an E17.5 limb showed that wild-type hypertrophic chondrocytes were electron lucent with a paucity of organelles in the cytosol (Fig. 8A and Fig. C). The wild-type chondrocytes also showed contacts with the surrounding matrix that was homogeneously filled with fibrillar collagen (Fig. 8 A). In contrast, hypertrophic chondrocytes of perlecan-null mice displayed an increased density of organelles and distended cisternae of ER (Fig. 8B and Fig. D). The cytosol was enriched with free ribosomes and polysomes (Fig. 8 D, arrowheads). The collagen fibrils in wild-type growth plate cartilage showed a random distribution, had uniform length and diameter, and formed a typical network (Fig. 8 E). The perlecan-null growth plate cartilage lacked such collagen network and the fibrils were shorter in length (Fig. 8 F).
Figure 8.
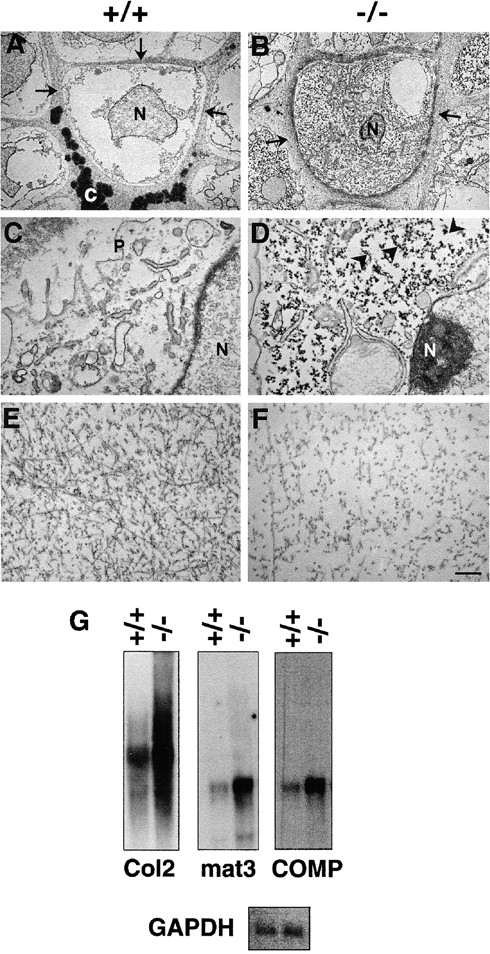
Ultrastructure of hypertrophic chondrocytes and territorial matrix. (A and C) The hypertrophic chondrocyte in a normal mouse femur has low organelle density. The septa are homogeneously filled with fibrillar collagen (arrows) and calcified material (c). (B and D) The hypertrophic chondrocyte in a perlecan-null femur displays increased density of organelles, and the cytosol is highly enriched with free ribosomes and polysomes (arrowheads). The adjacent pericellular matrix compartment is filled with fibrillar collagen but lacks calcification. (E and F) The territorial zone in normal cartilage shows random distribution of collagen fibrils. The collagen fibrils are of uniform diameter and length (E). The territorial zone in homozygotes lacks a well organized collagen fibrillar network. The collagen fibrils are shorter in length, lower in contrast, and the density is reduced. (G) The expression of Col2a1, matrilin-3, and COMP mRNA is increased in perlecan-deficient cartilage. Abbreviations: P, plasma membrane; N, cell nucleus; and C, calcification. Bars: (A and B) 5 μm; 1 μm (C and D); and (E and F) 0.5 μm.
To test whether an increased expression of cartilage ECM genes was responsible for the high metabolic activity in perlecan-null chondrocytes, Northern assays were performed with the total RNA derived from cartilage of normal and homozygous E18 limbs. Analysis of optical densities of mRNA signals such as shown in Fig. 8 G revealed that Col2a1 expression was increased threefold, and matrilin-3 and COMP expression was increased fivefold in mutant cartilage.
Discussion
We have generated perlecan-deficient mice, and we demonstrate that perlecan is essential for embryonic development. It plays a major role for maintaining the integrity of basement membranes and cartilage matrix, but has no apparently critical function in basement membrane assembly, organogenesis, and mesenchymal cell migration.
Early Embryonic Development Is Not Affected by the Loss of Perlecan
Perlecan is expressed throughout development. In the preimplantation period, it is found between blastomeres and on the external trophectodermal cell surface of blastocysts just before they become attachment-competent, suggesting that embryo-derived perlecan initiates implantation by attaching embryos to the uterine epithelium (Smith et al. 1997). Our results do not confirm such a role for perlecan. We found neither a delayed nor a reduced implantation rate of perlecan-null embryos, and the Mendelian distribution of genotypes was normal at all stages analyzed between E5.5 and E9.5. Perlecan was also thought to be important for BM assembly and function (Timpl 1993; Timpl and Brown 1996). These complex and highly ordered BM structures are composed of many constituents and serve as barriers, substratum for epithelial sheets, and sinks for growth factor storage and release (Timpl and Brown 1996). Despite the ability of perlecan to interact with several BM components, adhesion molecules involved in BM assembly, and growth factors (Timpl 1993; Timpl and Brown 1996), all BMs form in the absence of perlecan and appear morphologically normal. A likely explanation for this finding is that other heparan sulfate proteoglycans substitute for the loss of perlecan and the glycosaminoglycan chains attached to it. A possible candidate is agrin, which is present in most if not all BMs and can also bind growth factors, α-dystroglycan and BM components (Ruegg and Bixby 1998). Mice lacking the neuronal splice variant of agrin have defects in neuromuscular synaptogenesis but are otherwise apparently normal (Gautam et al. 1996). The lack of a general phenotype in these mutants could be due to the very low expression of nonneuronal agrin or the functional substitution by perlecan.
Perlecan is Essential for Maintaining Myocardial Basement Membranes and Heart Function
At E9.5 all perlecan-null embryos analyzed were of normal size, had similar pulse rates to their wild-type and heterozygous littermates, and showed no histological abnormalities. Between E10.5 and E12.5, ∼70–80% of the perlecan-null embryos died. When alive homozygotes were dissected, their hearts were of normal size and shaped with a well developed myocardium that was lined by endocardium internally and epicardium externally. However, the ventricles were suffused with blood leakage into the pericardial cavity. Cardiac muscle cells that normally differentiate at around E7.5–E8, form intercalated discs (Challice and Viragh 1973) and an immature BM on the free cell surface (Kitten et al. 1987). Their contractions are initially arrhythmic, but at around E9 they contract regularly and with high frequency. The intraventricular pressure significantly rises in the developing chicken heart between embryonic day 5 and 6 (Faber 1968), which corresponds to E10–12 in mouse. At around E9, a thin but distinct BM had formed on cardiac muscle cells in normal and perlecan-null mice. In all perlecan-null embryos with hemopericardium, however, this BM had deteriorated. At E10.5, we observed small clefts in the myocardium that were often still lined by endocardium and epicardium. Ultrastructural analysis revealed striking abnormalities in their BMs. Either the lamina densa was completely absent or the cell surface was sparsely covered by densities of irregular shape and size. These alterations could be observed throughout the mutant hearts and were not restricted to the defects in the myocardium. Furthermore, some cardiomyocytes were lined by partly normal and partly abnormal BMs. Interestingly, the homozygotes that survived the E10.5 crisis also showed the same BM defect in their hearts. We further confirmed this defect by crossing homozygotes with a transgenic strain expressing the green fluorescence protein under the control of the cardiac actin promoter (Fleischmann et al. 1998). When rhodamine-labeled dextran was injected into the atrium of such embryos using the patch clamp technique to control volume and pressure, the cardiac muscle wall of ∼70% of the homozygous embryos showed two to five leaking transmural channels, and the remaining 30% began to leak when the injection pressure was slightly increased to a level tolerated in normal or heterozygous control hearts (Bloch, N., unpublished observation). At present, we do not know why the heart of these surviving homozygotes does not develop holes in the myocardium. A likely explanation is that cell–cell contacts such as intercalated discs, which were normal in affected and unaffected homozygotes, sufficiently compensate for the BM defects. Altogether, our findings suggest that loss of perlecan is not crucial for the assembly of BMs on early contracting cardiomyocytes but for the maintenance of their structural and functional integrity when subjected to mechanical stress.
Perlecan Is Required for Normal Brain Development
All perlecan-null embryos that survive the first crisis develop brain anomalies. This was unexpected since we and others found no perlecan expression in the central nervous system (Handler et al. 1997). We used different polyclonal antibodies raised against domains I, III, and V of perlecan that revealed expression in the choroid plexus, the subendothelial BMs, and in the BM surrounding the brain.
The brain defects were first visible at E10.5 in the anterior region of the expanding brain vesicle. At this stage, two well developed BMs separate the ectoderm and the brain tissue from the mesenchyme. The disruption of both BMs caused aberrant fusion of brain tissue with the overlying ectoderm. Using EM scanning, the BM defects appeared as small clefts, usually not >20–30 μm in width, which contained small round cells. This type of defect exposed brain tissue to amniotic fluid, which led to the destruction of the tissue and the development of exencephaly.
Exencephaly can result from defects in neural tube closure, abnormal neuronal migration, or altered proliferation and/or apoptosis of neurons or neuronal precursor cells. We did not observe any of these three defects in perlecan-null embryos. All embryos analyzed by electron or light microscopy showed normal closure of the neural tube, and we could not find abnormal rates of neuronal proliferation and apoptosis in the expanding brain vesicles and other regions of the developing brain before, or at the time of, the onset of defects. When the exencephaly was fully developed we saw increased proliferation of neuronal and glial cells in some of the mutant brains that, however, may be a secondary effect of the amniotic fluid on brain cells. Finally, we found no evidence of abnormal neuronal migration as the cause of the exencephaly. The earliest neuronal cells born in the forebrain are Cajal-Retzius neurons and the subplate neurons that start to migrate at around E9.5-E10 (Derer and Derer 1990). Most other neurons start to migrate around E12 from the ventricular zone along the radial glia to form the typical laminae of the cortex (Rakic 1995). The cells in the affected brain areas of perlecan-null embryos expressed neither reelin nor other neuronal markers such as βIII tubulin but stained strongly for nestin, suggesting that they are neuroepithelial cells.
It was shown in earlier studies that the intraventricular pressure in the developing brain is much higher than the pressure in the amniotic cavity (Jelinek and Pexieder 1968). In chick embryos, it was demonstrated that the release of the intraventricular pressure leads to the collapse of the brain vesicles and to the formation of ruffles (Desmond and Jacobson 1977) similar to that seen in the perlecan-null brains. We observed that the disruptions in BMs occurred always in the polar area of the brain vesicles where the brain tissue is thin and, therefore, most vulnerable to the vesicular pressure and before the expansion of the neuroepithelium. Therefore, these data suggest that perlecan maintains BM stability by withstanding the tensile force exerted by the expanding brain vesicles. Since the BM has an important barrier function, particularly during development, where tissues are constantly remodeled, small gaps in the BM can lead to an abnormal expansion of neuroepithelial cells and eventually to tissue fusion. A similar process has been observed in mice lacking the laminin α5 chain, which develop small gaps in the BM of the developing hind buds, allowing mesenchymal cells to invade the overlaying ectoderm and eventuating in syndactyly (Miner et al. 1998).
In addition to the exencephaly, all perlecan-null embryos developed small neuronal ectopias ventral to the medial ganglionic eminence in the developing forebrain. These ectopias that were first visible at E11.5, maintained a constant size and contained postmitotic neuronal cells. Similarly, as on the surface of the brain vesicle, the cells had migrated through the disrupted BM and invaded the surrounding mesenchyme. At present, we are not sure why these neurons fail to stop their migration. It is probable that the migrating neurons exert mechanical stress on the perlecan-null BM, which leads to small gaps allowing cells to invade the mesenchyme. The BM had small gaps that were very similar to those found on the surface of the brain vesicles and the ectopias were always found at the same location, i.e., ventral to the ganglion eminence where active remodeling, extensive migration, and expansion of cells is occurring. The surrounding mesenchyme, being a mechanically resilient tissue, would physically prevent the further aberrant migration of these neurons.
Perlecan Supports Endochondral Ossification by Maintaining the Cartilage ECM
All perlecan-null embryos surviving the first critical period between E10 and E12.5 develop chondrodysplasia characterized by disproportionate dwarfism, disorganization of the growth plate, cleft palate, and perinatal lethality most probably because of respiratory failure. During mouse development, perlecan mRNA is expressed in the precartilaginous tissues, accumulates until E14–E15 in all cartilage primordia, and then sharply decreases to background level (Handler et al. 1997, French et al. 1999). However, perlecan expression is not detectable in membranous bones (French et al. 1999). Despite the abundant deposition of perlecan in cartilage, its function during skeletal development is speculative and mainly based on in vitro experiments. French et al. 1999 demonstrated recently that 10T1/2 cells efficiently aggregate into dense cartilaginous nodules when cultured on perlecan, suggesting that perlecan promotes chondrogenic differentiation. We find that the cartilaginous analogue forms normally in perlecan-null mice, indicating that perlecan is not essential for the condensation and differentiation of prechondrogenic mesenchyme into cartilaginous blastema.
During endochondral ossification, the formation of the cartilage model is followed by a complex process that includes the further differentiation of chondrocytes, matrix calcification, vascularization, and the replacement of cartilage by bone (Horton 1993). In long bones, endochondral ossification takes place mainly in the epiphyseal growth plate. The perlecan-null bones showed mild changes in epiphyseal cartilage but severe abnormalities in the growth plate, suggesting that perlecan is important for its proper organization and function. Since the perlecan-null chondrocytes can undergo an apparently normal differentiation cascade characterized by the synthesis of lineage-specific matrix components such as type X collagen and of other differentiation genes such as BMP2, Sox9, and indian hedgehog (Aszódi, A., unpublished observation), these abnormalities are apparently not associated with an altered chondrocyte differentiation or maturation. In addition to the growth plate abnormalities, perlecan-null long bones exhibit a bended shape. The shape deformation begins shortly after the differentiation of hypertrophic chondrocytes. This finding might be explained by the abnormal hypertrophic matrix, which has a decreased resistance against mechanical forces arising from muscle contraction.
Interestingly, the skeletal abnormalities observed in the perlecan-null mice resemble the phenotype of Col2a1-deficient mice (Aszódi et al. 1998) and Dmm (disproportionate micromelia) mice that carry a three-nucleotide deletion in the C-propeptide coding region of the Col2a1 gene, resulting in a severe reduction of type II collagen fiber network in cartilage (Pace et al. 1997). Like in Col2a1-null or Dmm mice, the cartilage of perlecan-null mice is very soft and structurally disorganized. The long bones are shortened and thickened and the bony trabecula are oriented transversely to the long axis. The reduced Safranin-O staining of cartilage and the almost complete lack of mineralization in the late hypertrophic zone is observed in both perlecan-null and Col2a1-null mice. Surprisingly, the ultrastructural analysis of perlecan-null cartilage revealed a lack of the typical collagen network. The reduced amount and the shortening of collagen fibrils suggests that perlecan plays a pivotal role in maintaining the collagen network. In contrast to the reduced collagen fibrils, the EM analysis revealed a dilated ER and a tremendous increase in free ribosomes and polysomes in the cytosol of chondrocytes, which indicates an unusually high biosynthetic activity in perlecan-null chondrocytes. This was further supported by Northern analysis showing that cartilage tissue derived from perlecan-null embryos expressed three to five times more mRNA coding for Col2a1, matrilin-3, and COMP. Apparently, perlecan-null chondrocytes try to compensate for the loss of cartilage ECM by increasing its synthesis. Since the de novo synthesis does not compensate for the disintegrated matrix, the growth plates deteriorate and are unable to lay down a calcified matrix in the hypertrophic zone leading to insufficient endochondral ossification (template for endochondral bone). These experimental findings revealed a new function of perlecan during endochondral bone formation. A mechanistical explanation could be that perlecan protects the cartilage ECM possibly by binding and inactivating tissue proteases or by masking the ECM proteins and, hence, protecting them from protease attack. Interestingly, it has been shown that perlecan inhibits the proteolytic degradation of fibrillar Aβ (Gupta-Bansal et al. 1995), which is thought to play an important role for the accumulation and persistence of neurotoxic plaques in Alzheimer's disease. It is conceivable that similar mechanisms are responsible for the inhibition of proteolysis by perlecan in developing cartilage and Alzheimer's disease.
Acknowledgments
We thank Drs. Ray Boot-Handford (University of Lund), Luis Puelles (University of Vigo), Markus Ruegg (University of Basel), Mike Dictor, Kenny Campbell, and Peter Ekblom (all three from University of Lund) for discussion and Drs. Masaharu Ogawa (Kochi Medical School), Zaal Kokaia (University of Lund), André Goffinet (University of Namur School of Medicine), and Bjorn Olsen (Harvard Medical School) for antibody gifts.
M. Costell was supported by the European Community and an EMBO short-term fellowship, and E.G. by the Inflammation Network of the stiftelse för strategisk forskning. This work was also funded by the Swedish Cancer Foundation. Additional support was obtained from the Kocks as well as Österlund Foundations.
Footnotes
Abbreviations used in this paper: Aβ, β amyloid; AD, Alzheimer's disease; BMs, basement membranes; ECM, extracellular matrix; ES, embryonic stem; RT-PCR, reverse transcriptase–PCR.
References
- Aszódi A., Chan D., Hunziker E., Bateman J.F., Fässler R. Collagen II is essential for the removal of the notochord and the formation of invertebral discs. J. Cell Biol. 1998;30:1399–1412. doi: 10.1083/jcb.143.5.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviezer D., Hecht D., Safran M., Eisinger M., David G., Yayon A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell. 1994;79:1005–1013. doi: 10.1016/0092-8674(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Bloch W., Forsberg E., Lentini S., Brakebusch C., Martin K., Krell H.W., Weidle U.H., Addicks K., Fässler R. β1 integrin is essential for teratoma growth and angiogenesis. J. Cell Biol. 1997;139:265–278. doi: 10.1083/jcb.139.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.C., Sasaki T., Göhring W., Yamada Y., Timpl R. The C-terminal domain V of perlecan promotes β1 integrin-mediated cell adhesion, binds heparin, nidogen and fibulin-2 and can be modified by glycosaminoglycans. Eur. J. Biochem. 1997;250:39–46. doi: 10.1111/j.1432-1033.1997.t01-1-00039.x. [DOI] [PubMed] [Google Scholar]
- Castillo G.M., Ngo C., Cummings J., Wight T.N., Snow A.D. Perlecan binds to the β-amyloid proteins (Aβ) of Alzheimer's disease, accelerates Aβ fibril formation, and maintains Aβ fibril stability. J. Neurochem. 1997;69:2452–2465. doi: 10.1046/j.1471-4159.1997.69062452.x. [DOI] [PubMed] [Google Scholar]
- Challice C.E., Viragh S. The architectural development of the early mammalian heart. Tissue Cell. 1973;6:447–462. doi: 10.1016/0040-8166(74)90037-8. [DOI] [PubMed] [Google Scholar]
- Comper W.D., Glasgow E.F., Singh A.K. The glomerulus. In: Comper W.D., editor. Extracellular Matrix. Harwood Academic Publishers; Amsterdam: 1996. pp. 410–442. [Google Scholar]
- Costell M., Mann K., Yamada Y., Timpl R. Characterization of recombinant perlecan domain I and its substitution by glycosaminoglycans and oligosaccharides. Eur. J. Biochem. 1997;243:115–121. doi: 10.1111/j.1432-1033.1997.t01-1-00115.x. [DOI] [PubMed] [Google Scholar]
- Derer P., Derer M. Cajal-Retzius cell ontogenesis and death in mouse brain visualized with horseradish peroxidase and electron microscopy. Neuroscience. 1990;36:839–856. doi: 10.1016/0306-4522(90)90027-2. [DOI] [PubMed] [Google Scholar]
- Desmond M.E., Jacobson A.G. Embryonic brain enlargement requires cerebrospinal fluid pressure. Dev. Biol. 1977;57:188–198. doi: 10.1016/0012-1606(77)90364-5. [DOI] [PubMed] [Google Scholar]
- Dziadek M., Timpl R. Expression of nidogen and laminin in basement membranes during mouse embryogenesis and in teratocarcinoma cells. Dev. Biol. 1985;111:372–382. doi: 10.1016/0012-1606(85)90491-9. [DOI] [PubMed] [Google Scholar]
- Faber J.J. Mechanical function of the septating embryonic heart. Am. J. Physiol. 1968;214:475–481. doi: 10.1152/ajplegacy.1968.214.3.475. [DOI] [PubMed] [Google Scholar]
- Fässler R., Meyer M. Consequences of lack of β1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- Fleischmann M., Bloch W., Kolossov E., Andressen C., Muller M., Brem G., Hescheler J., Addicks K., Fleischmann B.K. Cardiac specific expression of the green fluorescent protein during early murine embryonic development. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1998;440:370–376. doi: 10.1016/s0014-5793(98)01476-8. [DOI] [PubMed] [Google Scholar]
- French M.M., Smith S.E., Akanbi K., Sanford T., Hecht J., Farach-Carson M.C., Carson D.D. Expression of the heparan sulfate proteoglycan, perlecan, during mouse embryogenesis and perlecan chondrogenic activity in vitro. J. Cell Biol. 1999;145:1103–1115. doi: 10.1083/jcb.145.5.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M., Noakes P.G., Moscoso L., Rupp F., Scheller R.H., Merlie J.P., Sanes J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Göhring W., Sasaki T., Heldin C.H., Timpl R. Mapping of the binding of platelet-derived growth factor to distinct domains of the basement membrane proteins BM-40 and perlecan and distinction from the BM-40 collagen-binding epitope. Eur. J. Biochem. 1998;255:60–66. doi: 10.1046/j.1432-1327.1998.2550060.x. [DOI] [PubMed] [Google Scholar]
- Gupta-Bansal R., Frederickson R.C., Brunden K.R. Proteoglycan-mediated inhibition of Aβ proteolysis. A potential cause of senile plaque accumulation. J. Biol. Chem. 1995;270:18666–18671. doi: 10.1074/jbc.270.31.18666. [DOI] [PubMed] [Google Scholar]
- Handler M., Yurchenco P.D., Iozzo R.V. Developmental expression of perlecan during murine embryogenesis. Dev. Dyn. 1997;210:130–145. doi: 10.1002/(SICI)1097-0177(199710)210:2<130::AID-AJA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hassell J.R., Gehron-Robey P., Barrach H.-J., Wilczek J., Rennard S.I., Martin G.R. Isolation of a heparan sulfate-containing proteoglycan from basement membrane. Proc. Natl. Acad. Sci. USA. 1980;77:4494–4498. doi: 10.1073/pnas.77.8.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Madri J.A., Yurchenco P.D. Endothelial cells interact with the core protein of basement membrane perlecan through β1 and β3 integrinsan adhesion modulated by glycosaminoglycan. J. Cell. Biol. 1992;119:945–959. doi: 10.1083/jcb.119.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry M.D., Campbell K.P. A role for dystroglycan in basement membrane assembly. Cell. 1998;11:859–870. doi: 10.1016/s0092-8674(00)81708-0. [DOI] [PubMed] [Google Scholar]
- Hopf M., Göhring W., Kohfeldt E., Yamada Y., Timpl R. Recombinant domain IV of perlecan binds to nidogens, laminin-nidogen complex, fibronectin, fibulin-2 and heparin. Eur. J. Biochem. 1999;259:917–925. doi: 10.1046/j.1432-1327.1999.00127.x. [DOI] [PubMed] [Google Scholar]
- Horton W.A. Morphology of connective tissuecartilage. In: Royce P.N., Steinmann B., editors. Connective Tissue and Its Heritable Disorders. Molecular Genetic, and Medical Aspects. Wiley-Liss; New York: 1993. pp. 73–84. [Google Scholar]
- Jelinek R., Pexieder T. The pressure of encephalic fluid in chick embryos between the 2nd and 6th day of incubation. Physiol. Bohemoslov. 1968;17:297–305. [PubMed] [Google Scholar]
- Kitten G.T., Markwald R.R., Bolender D.L. Distribution of basement membrane antigens in cryopreserved early embryonic hearts. Anat. Rec. 1987;217:379–390. doi: 10.1002/ar.1092170409. [DOI] [PubMed] [Google Scholar]
- Miettinen A., Stow J.L., Mentone S., Farquhar M.G. Antibodies to basement membrane heparan sulfate proteoglycans bind to the laminae rarae of the glomerular basement membrane (GMB) and induce subepithelial GMB thickening. J. Exp. Med. 1986;163:1064–1084. doi: 10.1084/jem.163.5.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner J.H., Cunningham J., Sanes J.R. Roles for laminin in embryogenesisexencephaly, syndactyly, and placentopathy in mice lacking the laminin α5 chain. J. Cell. Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch A.D., Dodge G.R., Cohen I., Tuan R.S., Iozzo R.V. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 1992;267:8544–8557. [PubMed] [Google Scholar]
- Pace J.M., Li Y., Seegmiller R.E., Teuscher C., Taylor B.A., Olsen B.R. Disproportionate micromelia (Dmm) in mice caused by a mutation in the C-propeptide coding region of Col2a1. Dev. Dyn. 1997;208:25–33. doi: 10.1002/(SICI)1097-0177(199701)208:1<25::AID-AJA3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Peng H.B., Ali A.A., Dagett D.F., Rauvala H., Hassell J.R., Smalheiser N.R. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell. Adhes. Commun. 1998;5:475–489. doi: 10.3109/15419069809005605. [DOI] [PubMed] [Google Scholar]
- Rakic P. Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc. Natl. Acad. Sci. USA. 1995;92:11323–11327. doi: 10.1073/pnas.92.25.11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt D, Nischt R., Fox J.W., Chu M.-L., Krieg T., Timpl R. Mapping of nidogen binding sites for collagen type IV, heparan sulfate proteoglycan, and zinc. J. Biol. Chem. 1993;268:10881–10887. [PubMed] [Google Scholar]
- Rogalski T.M., Williams B.D., Mullen G.P., Moerman D.G. Products of the unc-52 gene in Caenorhabditis elegans are homologous to the core protein of the mammalian basement membrane heparan sulfate proteoglycan. Genes Dev. 1993;7:1471–1484. doi: 10.1101/gad.7.8.1471. [DOI] [PubMed] [Google Scholar]
- Roth J., Bendayan M., Orci L. FITC-protein A-gold complex for light and electron microscopic immunocytochemistry. J. Histochem. Cytochem. 1980;26:1074–1081. doi: 10.1177/28.1.6153194. [DOI] [PubMed] [Google Scholar]
- Ruegg M.A., Bixby J.L. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–27. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- Sasaki T., Forsberg E., Bloch W., Addicks K., Fässler R., Timpl R. Deficiency of β1 integrins in teratoma interferes with basement membrane assembly and laminin-1 expression. Exp. Cell Res. 1998;238:70–81. doi: 10.1006/excr.1997.3837. [DOI] [PubMed] [Google Scholar]
- Sharma B., Handler M., Eichstetter I., Whitelock J.M., Nugent M.A., Iozzo R.V. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Investig. 1998;102:1599–1608. doi: 10.1172/JCI3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.E., French M.E., Julian J., Paria B.C., Dey S.K., Carson D.D. Expression of heparan sulfate proteoglycan (perlecan) in the mouse blastocyst is regulated during normal and delayed implantation. Dev. Biol. 1997;184:38–47. doi: 10.1006/dbio.1997.8521. [DOI] [PubMed] [Google Scholar]
- Snow A.D., Wight T.N. Proteoglycan in the pathogenesis of Alzheimer's disease and other amyloidoses. Neurobiol. Aging. 1989;10:481–497. doi: 10.1016/0197-4580(89)90108-5. [DOI] [PubMed] [Google Scholar]
- Talts J.F., Andac Z., Göhring W., Brancaccio A., Timpl R. Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO (Eur. Mol. Biol. Org.) J. 1999;18:863–870. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timar J., Ladanyi A., Lapis K., Moczar M. Differential expression of proteoglycans on the surface of human melanoma cells characterized by altered metastatic potential. Am. J. Pathol. 1992;141:467–474. [PMC free article] [PubMed] [Google Scholar]
- Timpl R. Proteoglycans of basement membranes. Experientia. 1993;49:417–428. doi: 10.1007/BF01923586. [DOI] [PubMed] [Google Scholar]
- Timpl R., Brown J. Supramolecular assembly of basement membranes. Bioessays. 1996;18:123–132. doi: 10.1002/bies.950180208. [DOI] [PubMed] [Google Scholar]
- Walter, P., and M. Müller. 1985. The science of biological specimen preparation for microscopy and microanalysis. Proc. Pfefferkorn Conf. 4th, O'Hare, IL, 195–201.