

Abstract
The protein cross-linking enzyme tissue transglutaminase binds in vitro with high affinity to fibronectin via its 42-kD gelatin-binding domain. Here we report that cell surface transglutaminase mediates adhesion and spreading of cells on the 42-kD fibronectin fragment, which lacks integrin-binding motifs. Overexpression of tissue transglutaminase increases its amount on the cell surface, enhances adhesion and spreading on fibronectin and its 42-kD fragment, enlarges focal adhesions, and amplifies adhesion-dependent phosphorylation of focal adhesion kinase. These effects are specific for tissue transglutaminase and are not shared by its functional homologue, a catalytic subunit of factor XIII. Adhesive function of tissue transglutaminase does not require its cross-linking activity but depends on its stable noncovalent association with integrins. Transglutaminase interacts directly with multiple integrins of β1 and β3 subfamilies, but not with β2 integrins. Complexes of transglutaminase with integrins are formed inside the cell during biosynthesis and accumulate on the surface and in focal adhesions. Together our results demonstrate that tissue transglutaminase mediates the interaction of integrins with fibronectin, thereby acting as an integrin-associated coreceptor to promote cell adhesion and spreading.
Keywords: adhesion, integrins, tissue transglutaminase, fibronectin, focal adhesions
Introduction
Transglutaminases are a family of Ca++-dependent enzymes that mediate covalent cross-linking of proteins by forming amide bonds between glutamines and ε-amino groups of lysine residues (Folk 1980). At least five transglutaminase genes were identified in vertebrates. Among them, tissue transglutaminase (tTG) is the only family member expressed in a wide variety of tissues and cell types (Thomazy and Fesus 1989). Although tTG cross-links several intracellular and extracellular proteins in vitro, its function(s) remain poorly understood. Previous work implicated tTG in programmed cell death (Oliverio et al. 1997). Elevated tTG expression and enzymatic activity in a number of cell types inhibits proliferation and accompanies cell differentiation and senescence (Aeschlimann et al. 1993). Some data suggest tTG involvement in regulation of tumor growth and metastasis (Johnson et al. 1994; van Groningen et al. 1995). A variety of growth factors, cytokines, and retinoids upregulate tTG expression in many cell lines (Davies et al. 1985; George et al. 1990; Nagy et al. 1996; Ritter and Davies 1998). The GTP-binding protein Gh, which mediates intracellular signaling by the α1B adrenergic receptor, was identified as tTG, indicating that tTG has signaling functions apart from its enzymatic activity (Nakaoka et al. 1994). A potential significance of tTG in neurodegenerative diseases stems from the fact that this enzyme polymerizes proteins with polyglutamine expansions such as huntingtin (Kahlem et al. 1996, Kahlem et al. 1998), and binds to and cross-links β-amyloid peptides in vitro (Dudek and Johnson 1994). Transglutaminase activity has been shown to increase in Alzheimer's disease brain (Johnson et al. 1997).
tTG localizes primarily in the cytoplasm, with a small part of its intracellular pool present in the nucleus (Lesort et al. 1998). In addition, some amounts of the enzyme are present on the cell surface and in the extracellular matrix (ECM) (Upchurch et al. 1991; Aeschlimann et al. 1995; Verderio et al. 1998). Since there is no leader sequence in tTG, it remains unknown how it is exported to the cell surface. tTG was shown to bind several ECM proteins, including fibronectin (Fn) (Bowness et al. 1987; Achyuthan et al. 1988; Turner and Lorand 1989). tTG interacts in vitro with a 42-kD gelatin-binding domain of Fn that consists of modules I6II1,2I7-9 and lacks integrin-binding motifs (see Fig. 1 A; Radek et al. 1993). One well-documented property of cell surface tTG is the ability to cross-link ECM proteins, such as Fn, fibrinogen, osteopontin, and laminin–nidogen complexes (Aeschlimann and Paulsson 1991; Martinez et al. 1994; Kleman et al. 1995; Kaartinen et al. 1997). It was suggested that processing of Fn by surface tTG on endothelial cells serves to stabilize the ECM and anchor cells to the basement membranes (Martinez et al. 1994).
Figure 1.
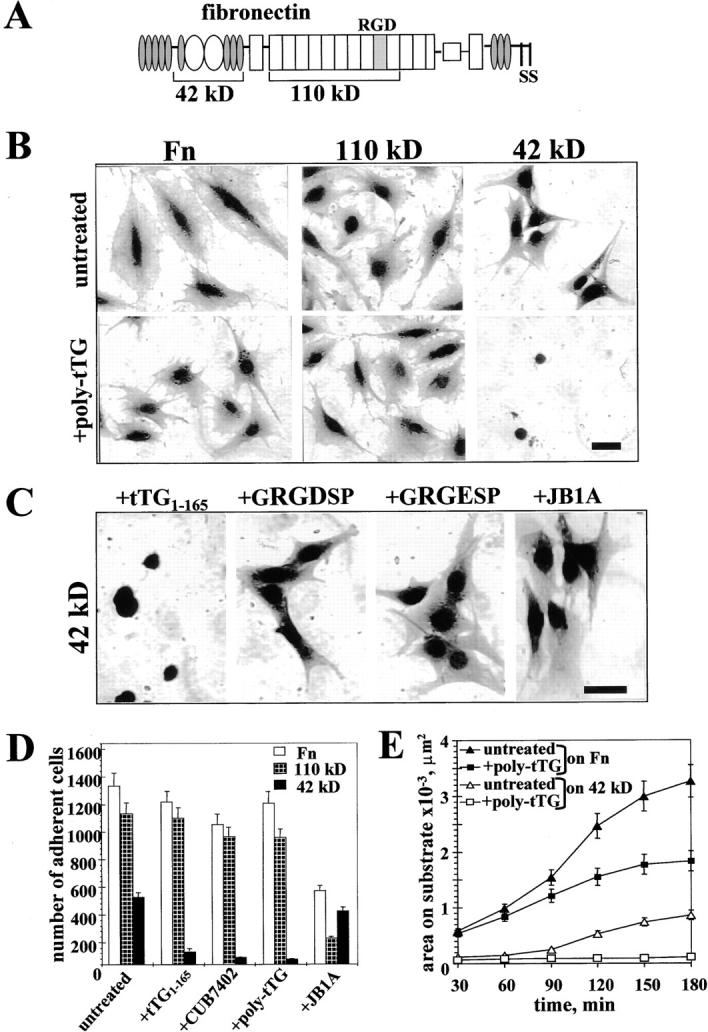
Surface tTG mediates adhesion and spreading of WI-38 fibroblasts on 42-kD Fn fragment. (A) A scheme of modular structure of Fn and its 42-kD and 110-kD proteolytic fragments. (B and C) Spreading assays with WI-38 fibroblasts. (B) Cells were plated for 4 h on Fn, 110-kD, or 42-kD Fn fragments either untreated or in the presence of 10 μg/ml polyclonal anti-tTG antibody. (C) Cells were plated for 4 h on 42-kD Fn fragment in the presence of 10 μM recombinant NH2-terminal tTG domain tTG1-165, 200 μM GRGDSP or GRGESP peptides, or 10 μg/ml function-blocking mAb JB1A against human β1 integrins. Bar, 20 μM. (D) Quantitative adhesion assays with WI-38 fibroblasts plated for 1 h on Fn (open bars), 110-kD (crossed bars), and 42-kD (filled bars) fragments without treatment or in the presence of 10 μM tTG1-165, 10 μg/ml mAb CUB7402, 10 μg/ml polyclonal anti-tTG antibody, or 10 μg/ml blocking anti-β1 integrin mAb JB1A. Shown are the means of quadruplicate measurements. (E) Quantitative spreading assays with WI-38 fibroblasts plated on Fn or 42-kD Fn fragment for 30–180 min either without treatment or in the presence of 10 μg/ml polyclonal anti-tTG antibody. Shown are the average areas on substrate for 120 cells. (B–E) Adhesion and spreading assays were performed with cells in serum-free medium in the presence of cycloheximide.
Emerging evidence suggests a role for tTG in cell adhesion. Overexpression of tTG in fibroblasts increased cell spreading and reduced susceptibility to detachment with trypsin (Gentile et al. 1992). Reduced expression of tTG led to diminished cell adhesion and spreading of endothelial cells, and a tTG-inactivating mAb reduced cell adhesion and spreading on Fn (Jones et al. 1997; Verderio et al. 1998). Additionally, tTG was shown to stabilize melanoma cell adhesion under laminar flow (Menter et al. 1991). So far, there is no explanation for the apparent involvement of tTG in cell adhesion. Here we show that cell surface tTG (a) interacts with integrins of the β1 and β3 subfamilies, (b) mediates their association with 42-kD gelatin-binding domain of Fn that lacks integrin-binding sites, and (c) potentiates integrin signaling. tTG-dependent stimulation of cell adhesion and spreading does not require its enzymatic activity and is not shared by its functional homologue, a catalytic subunit of blood coagulation factor XIII (FXIIIa), indicating an unconventional role of this enzyme in cell adhesion. Together, our data demonstrate a novel function of cell surface tTG as an integrin-associated adhesion coreceptor for Fn.
Materials and Methods
cDNAs, Antibodies, and Fn Fragments
A cDNA encoding human endothelial tTG (Gentile et al. 1991) was provided by Dr. P. Davies (University of Texas, Houston, TX). A cDNA for human placental FXIIIa (Grundmann et al. 1986) was obtained from Dr. E. Davie (University of Washington, Seattle, WA). Rabbit polyclonal antibodies against guinea pig liver tTG and human red blood cell tTG were provided by Dr. P. Birckbichler (University of Oklahoma, Oklahoma City, OK) and Dr. L. Lorand (Northwestern University, Chicago, IL), respectively, and were described previously (Gentile et al. 1991; Jeong et al. 1995; Jones et al. 1997; Verderio et al. 1998). mAbs CUB7402 and TG100 against tTG were obtained from NeoMarkers. An NH2-terminal tTG domain tTG1-165 was expressed in pGEX-2T plasmid (Amersham Pharmacia Biotech) in E. coli as glutathione S-transferase fusion protein and purified on glutathione–Sepharose, followed by cleavage with thrombin. 42-kD gelatin-binding (I6II1-2I7-9) and 110-kD cell-binding (III2-11) proteolytic fragments of Fn were generated and purified as described (Isaacs et al. 1989). Rabbit polyclonal antibody against FXIIIa was from Calbiochem and antibody against FGF receptor FGFR-1 Flg (H-76) was from Santa Cruz Biotechnology. Antivinculin mAb 7F9 was described earlier (Belkin et al. 1996). Rhodamine-phalloidin was from Molecular Probes. mAb 25E11 against human β3 integrin and mAb YFC118.3 against human β2 integrin were from Chemicon.
PCR-based Mutagenesis
C277→S tTG mutant was generated by PCR-based mutagenesis and confirmed by sequencing.
Cell Culture and Transfections
WI-38 fibroblasts and human erythroleukemia (HEL) cells were obtained from ATCC and cultured by standard methods. Rat embryonic fibroblasts REF52 and rat smooth muscle cells PAC-1 were kindly provided by Dr. K. Burridge (University of North Carolina, Chapel Hill, NC) and Dr. V.E. Koteliansky (Biogen Inc., Cambridge, MA). REF52 cells were transfected with wild-type or mutant tTG cDNAs or FXIIIa cDNA in pcDNA3.1-zeo vector (Invitrogen) using Superfect™ (Quiagen) and selected with 100 μg/ml zeocin™ (Invitrogen). β1 integrin cDNAs, CHO cell lines expressing β1 integrin cytoplasmic domain variants, and antibodies against integrin subunits were described previously (Belkin et al. 1996, Belkin et al. 1997; Retta et al. 1998).
Assays for Cell Adhesion and Spreading on Fn and Fn Fragments
Glass coverslips or 24-well plastic wells were coated with 10 μg/ml Fn, 110-kD cell-binding, or 42-kD gelatin-binding Fn fragment for 1 h at 37°C, blocked with 10 mg/ml BSA, and then used for cell plating. Since trypsin rapidly degrades cell surface tTG, we used EDTA for cell detachment in adhesion and spreading experiments. 35S-labeled WI-38 fibroblasts, REF52 transfectants, and untreated or 12-O-tetradecanoylphorbol 13-acetate (TPA)-treated HEL cells were preincubated with 20 μg/ml cycloheximide for 2 h in serum-free medium, detached with EDTA, and plated on protein-coated wells (1.5 × 106 cpm/well) for 1 h in serum-free medium with 10 mg/ml BSA and cycloheximide. In some cases, WI-38 fibroblasts or HEL cells were preincubated for 30 min on ice with 10 μg/ml anti-tTG mAbs TG100, CUB7402, or polyclonal antibody, or blocking anti–β1 integrin mAb JB1A (Chemicon). WI-38 cells were also plated on 42-kD Fn fragment in the presence of 10 μM tTG1-165 or 200 μM GRGDSP or GRGESP peptides. After 1 h, cells were washed three times with PBS, lysed in 1% SDS, and bound radioactivity was determined by scintillation counting. Adhesion on ovalbumin was used as a negative control. The number of adherent cells in each experiment was determined based on incorporation of 35S radioactivity per 103 cells.
For spreading assays, unlabeled WI-38, REF52 fibroblasts, or HEL cells were plated for 4 h on protein-coated glass coverslips using the conditions of adhesion experiments. Coverslips were washed with PBS, fixed with 3% formaldehyde, stained with Coomassie blue, destained, and photographed. To quantitate the degree of spreading of WI-38 fibroblasts and REF52 transfectants on Fn and Fn fragments over a time course, the cells were plated on the substrates for 30–180 min using the above mentioned conditions. The cells were fixed at certain timepoints, stained with Coomassie blue, and the average spreading areas were determined for 90–120 cells not contacting each other using a video monitor to trace the perimeters of adherent cells.
Analysis of Integrin Association with tTG by Coimmunoprecipitation and Affinity Isolation on Fn and Fn Fragments
Adherent or suspended cells were washed with PBS and lysed in ice-cold RIPA buffer, containing 1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM TrisCl, pH 7.5, with 0.5 mM PMSF, 0.5 mM benzamidine, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Cell lysates were precleared by centrifugation (14,000 rpm for 30 min at 4°C). 1 mg of total cellular protein or 3 × 108 cpm of protein-incorporated 35S radioactivity was used for each sample in immunoprecipitation or affinity binding experiments. RIPA cell lysates were used for immunoprecipitation with antibodies (2–8 μg per sample) against integrins or tTG, followed by protein G–Sepharose, or used for binding assays with 100 μl of immobilized Fn and Fn fragments (1 mg protein/ml Sepharose).
Cell surface biotinylation of 2 × 107 EDTA-detached TPA-differentiated HEL cells was performed in PBS containing 0.1 mg/ml membrane-impermeable biotinylation agent Sulfo-NHS-Biotin (Pierce) for 20 min, and the reaction was quenched with 20 mM Tris in PBS. Biotinylated proteins were visualized on the blots with neutravidin-peroxidase (Pierce).
Immunodepletion analysis and reprecipitation were performed as described (Berditchevski et al. 1996). Pulse-chase analysis of integrin biosynthesis was described earlier (Akiyama and Yamada 1987). In brief, 5 × 106 TPA-treated HEL cells were pulse-labeled for 1 h with 200 μCi/ml 35S[Translabel] (a mixture of [35S]methionine and [35S]cysteine; ICN Radiochemicals). Then it was replaced with the normal growth medium for 0–18 h. The cells were washed with PBS, lysed in RIPA buffer, and cell lysates were used to immunoprecipitate β1 integrins. After immunoprecipitation, the resulting immune complexes were split into halves and half of each sample was boiled in SDS sample buffer. The other half of each sample was boiled in 1% SDS, the eluate was reconstituted with 1% Triton X-100, 150 mM NaCl, 50 mM TrisCl, pH 7.5, to a final SDS concentration of 0.1%, and subjected to reprecipitation with polyclonal antibody to tTG. The resulting β1 integrin immune complexes and tTG immunoprecipitates were analyzed by SDS-PAGE and autoradiography.
To analyze association of tTG and Fn with β1 integrins in WI-38 fibroblasts, cells were labeled overnight with 50 μCi/ml [35S]Translabel. 35S-labeled RIPA lysates were subjected to immunoprecipitation with mAb CUB7402 against tTG or mAb 9EG7 against human β1 integrin. An excess (1 μM) of unlabeled Fn, 110-kD Fn fragment, 42-kD Fn fragment, or 5 μM unlabeled tTG fragment tTG1-165 was added to some immunoprecipitation samples during incubations with primary antibodies and protein G–Sepharose. After immunoprecipitation, the immune complexes were split into halves and half of each sample was boiled in SDS sample buffer. The other half was boiled in 1% SDS, the eluate was reconstituted with 1% Triton X-100, 150 mM NaCl, 50 mM TrisCl, pH 7.5, to a final SDS concentration of 0.1%, and subjected to reprecipitation with polyclonal antibody to Fn. Immunoprecipitates were washed several times with RIPA buffer, and analyzed by SDS-PAGE and autoradiography.
For coimmunoprecipitation and affinity isolation experiments with HEL cells, they were labeled overnight with 50 μCi/ml [35S]Translabel. To increase tTG synthesis, HEL cells were plated on Fn and treated with 150 ng/ml TPA for 24–48 h. After the treatment, 85–90% of cells in the population became adherent and spread. Both untreated and TPA-treated 35S-labeled cells were lysed in RIPA buffer, and precleared lysates were used for immunoprecipitation or affinity binding to Fn and Fn fragments.
To visualize 35S-labeled proteins, SDS-PAGE in 10% acrylamide/0.25% bis-acrylamide gels was followed by treatment of gels with Autofluor (Amersham Pharmacia Biotech) and fluorography. tTG was detected on blots with 0.4 μg/ml mAb TG100 and FXIIIa with 0.5 μg/ml polyclonal antibody. For immunoblotting of β1 and β3 integrins, antibodies to β1A cytodomain or β3 integrin subunit (Chemicon) were used at 0.5 μg/ml. Blots were developed with peroxidase-conjugated secondary IgG (Chemicon) and ECL reagents (Pierce).
Cross-Linking Experiments
To analyze association of cell surface tTG with integrins, cross-linking experiments were performed as follows. 2 × 107 TPA-treated HEL cells were detached with EDTA and live cells were incubated for 20 min at 20°C in PBS containing 0.5 mM membrane-impermeable cross-linker Dithiobis (sulfosuccinimidylpropionate) (DTSSP; Pierce). The reaction was stopped by adding 20 mM TrisCl, pH 7.5. The cells were pelleted, washed twice with PBS, and lysed in ice-cold RIPA buffer with protease inhibitors. β1, β2, and β3 integrins and tTG were immunoprecipitated from RIPA lysates of DTSSP-treated cells as described above. Immunoprecipitation samples were divided into halves and boiled in SDS-PAGE sample buffer either without or with reducing agents, and analyzed on 10% gels. To avoid an appearance of large IgG bands on the blot with the nonreduced immunoprecipitates, it was developed with 0.3 μg/ml biotinylated mAb TG100 against tTG, followed by neutravidin-peroxidase.
Immunofluorescence
Live nonpermeabilized REF52 cells transfected either with vector alone or with tTG cDNA were double stained with 10 μg/ml anti-tTG mAb CUB7402 and 20 μg/ml hamster mAb HM β1-1 to rat β1 integrin. To localize tTG and β1 integrins during spreading of tTG transfectants on 42-kD Fn fragment, the cells were fixed with 3% formaldehyde in PBS 1 or 2 h after plating and then double stained without cell permeabilization. A combination of rhodamine-conjugated donkey anti–mouse IgG and fluorescein-conjugated goat anti–hamster IgG (Chemicon) were used as secondary antibodies. To visualize focal adhesions and actin stress fibers, cells were fixed with 3% formaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and then costained with 10 μg/ml antivinculin mAb 7F9 and rhodamine-phalloidin (1:500), followed by fluorescein-conjugated donkey anti–mouse IgG (Chemicon).
Flow Cytometry
For flow cytometry, live REF52 transfectants were stained for cell surface tTG with 10 μg/ml polyclonal anti-tTG antibody. Staining for cell surface FXIIIa was performed with 10 μg/ml polyclonal antibody. After incubation with secondary fluorescein-labeled IgG, the cells were analyzed in FACScan™ flow cytometer (Becton Dickinson).
Measurements of Transglutaminase Activity
Transglutaminase activity in cell lysates (cytosolic fractions) of REF52 transfectants was measured by incorporation of [3H]putrescine into N,N-dimethylcaseine as described previously (Lorand et al. 1972; Jones et al. 1997; Verderio et al. 1998). Transglutaminase activity on the surface of live REF52 transfectants or HEL cells was determined as reported earlier (Verderio et al. 1998), with some modifications. 2 × 106 live EDTA-detached cells in 0.5 ml PBS containing 2 mM Ca++ and 10 mg/ml N,N-dimethylcaseine, were incubated with 10 μCi [3H]putrescine for 1 h at 37°C on the rotator. Supernatants were precipitated with ice-cold 10% TCA, the pellets were washed sequentially with 5% TCA, ice-cold ethanol, acetone, then dried and redissolved in 100 μl 1% SDS. Protein-incorporated 3H radioactivity was determined by scintillation counting.
Adhesion-dependent Focal Adhesion Kinase Phosphorylation
For focal adhesion kinase (FAK) phosphorylation experiments, 2 × 105 REF52 transfectants were either kept in suspension or plated for 3 h in serum-free medium containing 20 μg/ml cycloheximide on dishes precoated with Fn, Fn fragments, laminin, or anti-tTG antibodies. To perform a time course analysis of FAK phosphorylation with the transfectants, the cells were plated on Fn for 45, 90, and 180 min. Immunoprecipitation of FAK from cell lysates with polyclonal antibody 5158 and immunoblotting for phosphotyrosine were describred earlier (Belkin et al. 1996).
Results
Cell Surface tTG Is an Adhesion Receptor for the 42-kD Fragment of Fn
tTG binds in vitro with high affinity to the 42-kD gelatin-binding domain of Fn (Turner and Lorand 1989; Radek et al. 1993). Since this enzyme is present on the surface of some cells (Upchurch et al. 1991; Aeschlimann et al. 1995; Verderio et al. 1998), its ability to bind Fn might contribute to cell adhesion. Therefore, we examined the effects of anti-tTG antibodies on adhesion and spreading of WI-38 fibroblasts that express tTG on their surface (Upchurch et al. 1991). These cells adhered and spread on surfaces coated with Fn and its 110-kD fragment as well as its 42-kD fragment, although spreading was somewhat less on the latter (Fig. 1 B, upper panels). Treatment with the polyclonal anti-tTG antibody moderately decreased spreading of WI-38 fibroblasts on Fn, had no effect on 110-kD Fn fragment, but completely abolished spreading of these cells on the 42-kD Fn fragment (Fig. 1 B, lower panels). A recombinant NH2-terminal tTG domain, tTG1-165, which binds Fn in vitro (Jeong et al. 1995), strongly decreased spreading of WI-38 fibroblasts on the 42-kD fragment, whereas GRGDSP peptide and function-blocking anti–β1 integrin mAb JB1A had no effect (Fig. 1 C). In quantitative adhesion assays, tTG1-165 and two antibodies against tTG markedly inhibited adhesion of WI-38 fibroblasts to the 42-kD fragment, while having little or no effect on adhesion to Fn or its 110-kD fragment (Fig. 1 D). In contrast, the function-blocking anti–β1 integrin mAb JB1A, which strongly decreased adhesion of WI-38 fibroblasts to Fn and even more to the 110-kD fragment, had very little effect on adhesion to the 42-kD fragment (Fig. 1 D). A time course analysis demonstrated a potent inhibition of spreading of WI-38 fibroblasts on Fn and its 42-kD fragment by polyclonal anti-tTG antibody (Fig. 1 E). Together, these results show that surface tTG mediates adhesion and spreading of fibroblasts on the 42-kD gelatin-binding domain of Fn, which does not contain integrin-binding motifs.
It was reported earlier that HEL cells that normally grow in suspension adhere to Fn after treatment with TPA (Jarvinen et al. 1987; Ylanne et al. 1990). Using flow cytometry with live HEL cells, we found that TPA treatment increased the level of surface tTG (Fig. 2 A). In parallel, transglutaminase activity on the surface of live TPA-treated HEL cells rose ∼10-fold compared with untreated cells (Fig. 2 B). Quantitative adhesion experiments demonstrated that TPA treatment increased adhesion to Fn, and even more so to the 42-kD Fn fragment, without affecting adhesion to the 110-kD fragment of Fn (Fig. 2 C). Importantly, the antibody against tTG negated the effects of TPA on cell adhesion to Fn and to the 42-kD Fn fragment. Similarly, spreading of HEL cells on Fn and its 42-kD fragment was enhanced by TPA, and this effect was abolished by anti-tTG antibody (Fig. 2 D). These observations indicate that the induction of cell surface tTG can play a major role in stimulation of cell adhesion and spreading due to its interaction with the 42-kD gelatin-binding domain of Fn.
Figure 2.

TPA-induced adhesion and spreading of HEL cells on 42-kD Fn fragment is mediated by surface tTG. (A) Expression levels of tTG on the surface of live untreated and TPA-treated cells were determined by immunostaining with 10 μg/ml polyclonal anti-tTG antibody and flow cytometry. (B) Transglutaminase activity on the surface of live untreated and TPA-treated cells was quantified by measuring cell-mediated incorporation of [3H]putrescine into N,N-dimethylcaseine. Bars show the means of triplicate measurements. (C) Quantitative adhesion assays with untreated and TPA-treated cells plated for 1 h on Fn (open bars), 110-kD (crossed bars), and 42-kD (filled bars) fragments either without or in the presence of 10 μg/ml polyclonal anti-tTG antibody. (D) Spreading assays with untreated and TPA-treated cells. Cells were plated for 4 h on Fn, 110-kD, or 42-kD Fn fragments without any treatment or after TPA treatment either in the absence or with 10 μg/ml polyclonal anti-tTG antibody. Bar, 20 μM. (C and D) For adhesion and spreading assays HEL cells were plated in serum-free medium in the presence of cycloheximide.
Stimulation of Cell Adhesion and Spreading by tTG Is Not Shared by its Homologue FXIIIa and Does Not Require tTG Cross-Linking Activity
To explore further the role of tTG in cell adhesion, rat REF52 fibroblasts were transfected with human tTG or its enzymatically inactive mutant C277→S. Flow cytometry of live REF52 transfectants was performed with a polyclonal anti-tTG antibody that recognizes both rat and human tTG. The cells expressing human tTG or tTG[C277→S] had increased amounts of tTG on the surface compared with vector controls, showing that both proteins were exported to the cell surface (Fig. 3 A). However, transfection of REF52 cells with human FXIIIa cDNA did not result in surface expression or secretion of this protein, indicating that mechanisms of externalization of these two transglutaminases are different (Fig. 3 B). Expression of tTG or FXIIIa, but not of the catalytic mutant tTG[C277→S], increased transglutaminase activity in the cytosolic fractions of the transfectants compared with vector-expressing cells (Fig. 3 C). Measurements of transglutaminase activity on the surface of live transfectants revealed a sharp increase after transfection with tTG, a reduction after transfection with tTG[C277→S], and no change in cells expressing FXIIIa (Fig. 3 D). In agreement with previous reports (Gentile et al. 1992; Verderio et al. 1998), overexpression of tTG strongly promoted cell spreading (Fig. 3 E). Interestingly, REF52 cells expressing tTG[C277→S] also appeared significantly more spread in regular culture than vector-transfected controls or FXIIIa transfectants (Fig. 3 E).
Figure 3.

Expression of exogenous tTG or its enzymatically inactive mutant tTG[C277→S], but not of FXIIIa promotes cell spreading. (A–E) REF52 cells were transfected with vector (vect.), wild-type tTG (tTG), tTG noncatalytic mutant C277→S (tTG[C277-S]), or FXIIIa (FXIIIa). (A) Expression levels of tTG on the surface of live transfectants were determined by immunostaining with 10 μg/ml polyclonal anti-tTG antibody and flow cytometry. (B) Expression levels of FXIIIa on the surface of live transfectants were determined by immunostaining with 10 μg/ml polyclonal antibody against FXIIIa and flow cytometry. (C) Transglutaminase activity in the cytosolic fractions of the transfectants was quantified by incorporation of [3H]putrescine into N,N-dimethylcaseine by transamidating enzymes present in cell lysates (Lorand et al. 1972). Shown are the results of triplicate determinations. (D) Transglutaminase activity on the surface of live transfectants was quantified by measuring cell-mediated incorporation of [3H]putrescine into N,N-dimethylcaseine. Bars represent the means of triplicate measurements. (E) Cells expressing vector, tTG, tTG[C277→S], or FXIIIa were photographed in regular culture. Bar, 20 μM.
When plated on Fn in the presence of cycloheximide, tTG transfectants displayed increased spreading compared with control cells, as did the tTG[C277→S] mutant transfectants (Fig. 4 A). Spreading on 110-kD Fn fragment was unaffected by transfection with tTG or tTG[C277→S] (Fig. 4 A). Most notably, cells expressing exogenous tTG or tTG[C277→S] spread much better than vector-transfected cells on the 42-kD Fn fragment (Fig. 4 A). Again, the observed stimulation of cell spreading appeared to be specific for cell surface tTG, since no changes were seen for FXIIIa transfectants (Fig. 4 A).
Figure 4.
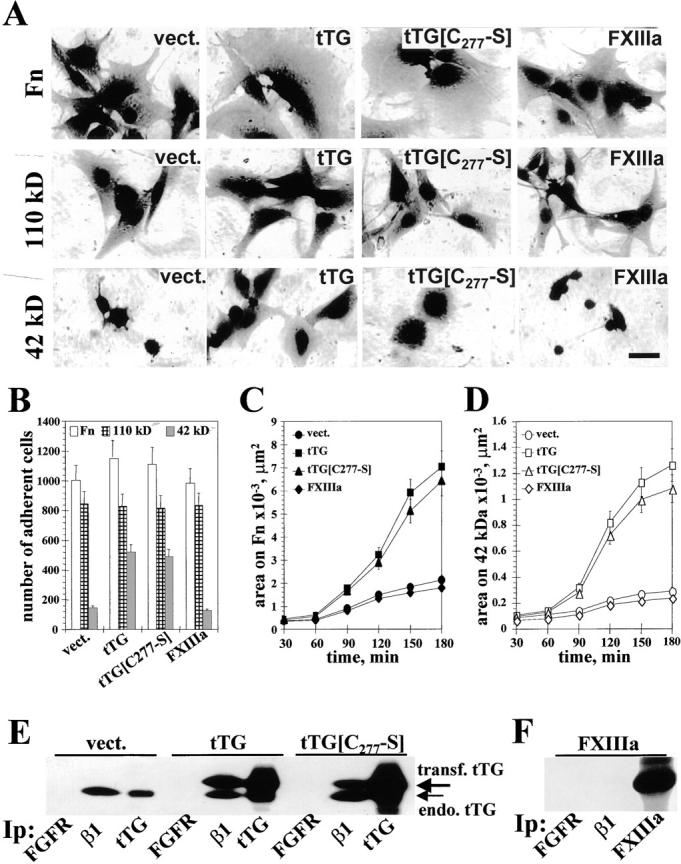
tTG-dependent stimulation of cell adhesion and spreading on Fn and its 42-kD fragment does not require the cross-linking activity. (A) Spreading assays with REF52 transfectants. Cells expressing vector, tTG, tTG[C277→S], or FXIIIa were plated for 4 h on Fn, 110-kD, or 42-kD Fn fragments. Bar, 20 μM. (B) Quantitative adhesion assays with REF52 transfectants plated for 1 h on Fn (open bars), 110-kD (crossed bars), and 42-kD (filled bars) fragments. Shown are the means of quadruplicate measurements. (C and D) Quantitative spreading assays with REF52 transfectants. Cells expressing vector, tTG, tTG[C277→S], or FXIIIa were plated for 30–180 min on Fn (C) or the 42-kD Fn fragment (D). Shown are the average areas on the substrates for 90 cells. (A–D) Cells were plated for adhesion and spreading assays in serum-free cycloheximide-containing medium. (E) Analysis of tTG association with β1 integrins. FGF receptor, β1 integrins, and tTG were immunoprecipitated with polyclonal antibody Flg(H-76) or HMβ1-1 and CUB7402 mAbs, respectively, from REF52 cells expressing vector, tTG or tTG[C277→S]. The resulting immunoprecipitates were blotted for tTG. Large arrow marks the transfected human tTG and small arrow indicates the endogenous rat tTG. (F) FXIIIa is not associated with β1 integrins. FGF receptor, β1 integrins, and FXIIIa were immunoprecipitated from FXIIIa-expressing REF52 cells and the resulting immunoprecipitates were probed for FXIIIa with a polyclonal antibody.
Quantitative adhesion assays with REF52 cells overexpressing tTG or its enzymatically inactive mutant C277→S showed that their adhesion on Fn or its 110-kD fragment was not significantly altered, but adhesion on the 42-kD Fn fragment was strongly promoted (Fig. 4 B). Expression of exogenous tTG or tTG[C277→S] also drastically enhanced spreading over time in cells plated on Fn (Fig. 4 C) or the 42-kD Fn fragment (Fig. 4 D). No significant changes were detected in quantitative adhesion and spreading assays for FXIIIa transfectants (Fig. 4, B–D). Collectively, these results show that the enzymatic activity of tTG is not required for the enhancement of cell adhesion and spreading.
tTG Interacts Directly with Multiple β1 and β3 Integrins
Cell spreading on ECM is an active process that requires outside-in signaling. Surprisingly, surface tTG is capable of promoting cell spreading on Fn and its 42-kD fragment, even though it lacks a transmembrane domain (Gentile et al. 1991). Therefore, we hypothesized that tTG might interact and collaborate with some transmembrane adhesion receptors, possibly integrins. Immunoprecipitation of β1 integrins from REF52 transfectants, followed by immunoblotting for tTG, showed that endogenous rat tTG as well as transfected human tTG and tTG[C277→S], both of which migrate slower on gels, all coprecipitated with endogenous β1 integrins (Fig. 4 E). No tTG could be detected in the immunoprecipitates with antibody against FGF receptor FGFR-1, proving specificity of immunoprecipitation (Fig. 4 E). Although a large amount of FXIIIa was expressed in REF52 transfectants, no association of FXIIIa with β1 integrins was detected by coimmunoprecipitation in this cell line (Fig. 4 F). These data suggested that tTG might specifically associate with integrins.
Immunoprecipitation in RIPA buffer, which disrupts integrin–ligand interactions (data not shown), was used to isolate integrin–tTG complexes from cell lysates. We initiated the analysis with HEL cells because they do not synthesize any detectable Fn (Jarvinen et al. 1987). A predominant 80-kD protein that comigrated with tTG and coprecipitated with β1 integrins from lysates of 35S-labeled TPA-treated HEL cells was immunodepleted with polyclonal antibody against tTG, strongly suggesting that it is tTG (Fig. 5 A, arrow). Additionally, after preadsoption of tTG, the intensity of the β1 integrin band slightly decreased, whereas a detectable amount of β1 integrin appeared in tTG immunoprecipitates (Fig. 5 A). The identity of the 80-kD protein was further proved by reprecipitation experiments (Berditchevski et al. 1996). β1 integrins were first immunoprecipitated from 35S-labeled HEL cells using mAb 9EG7 (Fig. 5 B). The resulting immune complexes were then eluted with either 0.4% SDS at 25°C or 1% SDS with boiling and reprecipitated with antibody against the cytoplasmic domain of β1A integrin (Fig. 5 B, lane 1) or three anti-tTG antibodies (Fig. 5 B, lanes 2–4). The 80-kD protein appeared in all cases, proving its identity as tTG (Fig. 5 B, arrow). Notably, β1 integrins coprecipitated with tTG even after treatment with 0.4% SDS, showing extremely stable association between these proteins (Fig. 5 B, left panel). However, after boiling in 1% SDS, integrins no longer coprecipitated with tTG (Fig. 5 B, right panel), demonstrating that their association is noncovalent. The absence of other 35S-labeled proteins in β1 integrin or tTG immunoprecipitates after reprecipitation proves that this association is highly specific and is not mediated by a third protein.
Figure 5.

tTG associates with multiple β1 and β3 integrins in different cell types. (A) Immunodepletion. An 80-kD protein associated with β1 integrins is immunodepleted with anti-tTG antibody. β1 integrins and tTG were immunoprecipitated from RIPA lysates of TPA-treated 35S-labeled HEL cells with mAb 9EG7 or polyclonal anti-tTG antibody, respectively. Note a comigration of 80-kD protein coprecipitating with β1 integrins, with tTG (arrow). Preadsorbtion of 35S-labeled RIPA lysates with polyclonal anti-tTG antibody followed by immunoprecipitation of β1 integrins with mAb 9EG7 caused a disappearance of 80-kD protein from the β1 integrin immunoprecipitates. Arrows in A–E point to tTG bands. Brackets in A and B mark α5β1 integrin. (A–C) Molecular weight markers are shown to the right of the gels. (B) Reprecipitation. An 80-kD protein associated with β1 integrins is reprecipitated by three antibodies against tTG. β1 integrins were immunoprecipitated from RIPA lysates of TPA-treated 35S-labeled HEL cells with 9EG7 mAb. The 35S-labeled β1 integrin immune complexes were treated with 0.4% SDS (left panel) or boiled in 1% SDS (right panel). 35S-labeled eluates from the β1 integrin immune complexes were divided into four equal aliquots and subjected to reprecipitation in RIPA buffer with antibody against β1A integrin cytodomain (lane 1), anti-tTG polyclonal antibody (lane 2), mAb CUB7402 (lane 3), or mAb TG100 (lane 4) against tTG. (C) tTG interacts with β1 integrins inside the cell during biosynthesis. TPA-treated HEL cells were labeled with [35S]Translabel for 1 h, then chased with regular medium for 0, 2, 6, or 18 h. β1 integrins were immunoprecipitated from 35S-labeled RIPA lysates with 9EG7 mAb. The resulting immunoprecipitates were divided into halves. Half of each sample was run on the gel (upper panel), whereas another half was boiled in 1% SDS and tTG was reprecipitated from these samples using anti-tTG polyclonal antibody (lower panel). Large and small arrowheads point to mature β1 integrin and its underglycosylated precursor, respectively. (D) Association of tTG with multiple integrins. Immunoprecipitates from PAC-1 smooth muscle cells with antibodies against α1, α3, α5, αv, β1, and β3 integrins, FGF receptor, tTG, or without primary antibody (cont.) were blotted for tTG. (E) β1 integrin cytodomain is not required for binding tTG. Transfected human and endogenous hamster β1 integrins were immunoprecipitated with mAbs TS2/16 and 7E2, respectively, from CHO cells expressing exogenous β1A, β1D, or β1 integrin with deleted cytodomain. No primary antibody was used in control immunoprecipitations (cont.). The immunoprecipitates were blotted for tTG.
Pulse-chase analysis and immunoprecipitation of β1 integrins from TPA-induced HEL cells showed that immediately after 1 h labeling, an 80-kD protein was complexed with β1 integrins (Fig. 5 C, upper panel, arrow). At this timepoint, de novo synthesized β1 integrins exist as underglycosylated precursors which have yet to be exported to the cell surface (Akiyama and Yamada 1987). Reprecipitation of tTG from β1 integrin immune complexes demonstrated that the 80-kD protein represents tTG (Fig. 5 C, lower panel). Early association of tTG with β1 integrins indicated that this interaction occurs during integrin biosynthesis and maturation inside the cell.
Analysis of complex formation between αβ integrin heterodimers and tTG in PAC-1 rat smooth muscle cells by coimmunoprecipitation and immunoblotting for tTG showed that tTG associates with α1β1, α3β1, α5β1, as well as αvβ3 integrins (Fig. 5 D). We also tested CHO cells transfected with human integrins β1A, β1D, and β1Δcyto (β1 subunit with a deleted cytodomain) (Belkin et al. 1996, Belkin et al. 1997; Retta et al. 1998), and found all of these integrins to associate with tTG, indicating that β subunit cytoplasmic domains are not required for integrin complex formation with tTG (Fig. 5 E).
Detection of tTG Complexes with β1 and β3 Integrins on the Cell Surface
To test association of tTG with other integrins on the cell surface, we biotinylated cell surface proteins on live TPA-differentiated HEL cells that express high levels of β1, β2, and β3 integrins. Immunoprecipitation with antiintegrin antibodies followed by blotting with avidin-peroxidase revealed an 80-kD protein that comigrated with tTG and was associated with β1 and β3, but not with β2 integrins (Fig. 6 A, upper panel, arrow). Furthermore, proteins that comigrated with β1 and β3, but not with β2 integrins, could be detected in tTG immunoprecipitates (Fig. 6 A, upper panel). The identity of the 80-kD protein as tTG and its lack of association with β2 integrins was confirmed by blotting the corresponding immunoprecipitates for tTG (Fig. 6 A, lower panel).
Figure 6.
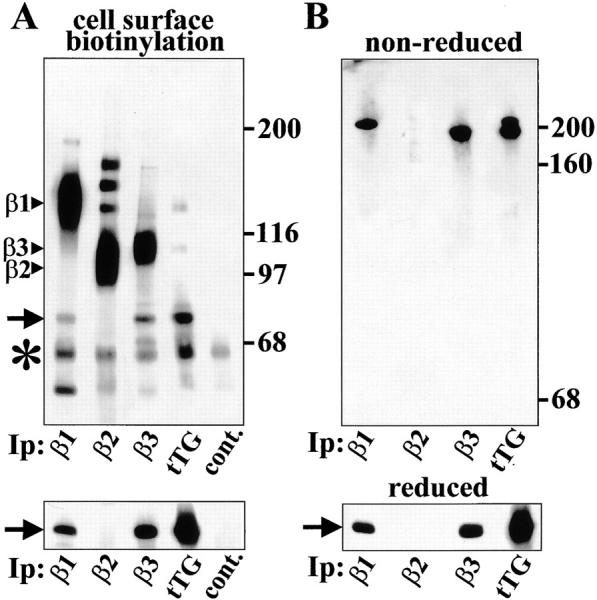
tTG is associated with β1 and β3, but not with β2 integrins on the cell surface. (A) Interaction of tTG with β1 and β3 integrins on the cell surface. (Upper panel) TPA-differentiated HEL cells were biotinylated in suspension and β1, β2, and β3 integrins and tTG were immunoprecipitated from RIPA lysates of surface-biotinylated cells. Anti–mouse IgG was used in control immunoprecipitations (cont.). Biotinylated proteins in the immunoprecipitates were visualized on blots by neutravidin-peroxidase. (Lower panel) The same immunoprecipitates as in the top panel were blotted for tTG. Note association of tTG with β1 and β3 but not with β2 integrins. β1, β2, and β3 integrin bands are marked by arrowheads. Asterisks mark IgG heavy chains. Molecular weight markers are shown to the right of the blot. (B) tTG can be cross-linked to β1 and β3 integrins on the cell surface. TPA-differentiated HEL cells were treated for 20 min with 0.5 mM membrane-impermeable reducible cross-linker DTSSP in suspension and β1, β2, and β3 integrins and tTG were immunoprecipitated from RIPA lysates. After immunoprecipitation samples were run on 8% gels under nonreducing (upper panel) or reducing (lower panel) conditions. To avoid the appearance of IgG bands, the blots were probed for tTG using biotinylated anti-tTG mAb TG100 followed by neutravidin-peroxidase. Arrow in A and B points to tTG bands. Molecular weight markers (nonreduced: myosin heavy chain, 200 kD; IgG, 160 kD; and BSA, 68 kD) are shown to the right of the blots.
We also tested whether tTG can be chemically cross-linked to cell surface integrins (Fig. 6 B). After treatment of live TPA-induced HEL cells with membrane-impermeable reducible cross-linker DTSSP, we immunoprecipitated β1, β2, and β3 integrins and tTG from RIPA lysates and subjected the resulting immune complexes to SDS-PAGE and immunoblotting for tTG. Under nonreducing conditions, tTG appeared exclusively as a high molecular weight complex immunoprecipitated with antibodies against β1 and β3 integrins, but not with antibody to β2 integrin (Fig. 6 B, upper panel). The sample immunoprecipitated with antibody against tTG revealed two bands corresponding in mobility to those obtained with anti-β1 and anti-β3 antibodies. Under reducing conditions, all of the tTG migrated as an 80-kD protein (Fig. 6 B, lower panel). The apparent molecular weight of the complexes with β1 (M r ∼210,000) and β3 (M r ∼195,000) integrins, strongly suggests a stoichiometry of 1:1 of integrin–tTG complexes. The absence of any other tTG-containing bands in the nonreduced samples indicates that all the cell surface tTG is associated with β1 or β3 integrins.
tTG Mediates the Formation of Ternary Complexes of β1 and β3 Integrins with Fn via Its Interaction with the 42-kD Fn Fragment
The foregoing data demonstrate that tTG functions as a cell adhesion receptor for the gelatin-binding region of Fn and interacts with β1 and β3 integrins. We next examined biochemically whether tTG by itself can mediate association of integrins with this part of Fn, which lacks integrin-binding motifs. First, a protein corresponding to Fn was observed in both tTG and β1 integrin immunoprecipitates from 35S-labeled WI-38 human fibroblasts (Fig. 7 A, arrowhead). Addition of unlabeled Fn or its 42-kD fragment to the 35S-labeled RIPA lysates effectively displaced 35S-labeled Fn from the β1 integrin immunoprecipitates, whereas unlabeled 110-kD fragment was unable to do so. None of these treatments decreased the amounts of tTG associated with β1 integrins (Fig. 7 A, arrow). In parallel experiments, an NH2-terminal fragment tTG1-165 strongly inhibited association of Fn with tTG and completely displaced 35S-labeled Fn from both the tTG and β1 integrin immune complexes (Fig. 7 B, arrowhead). The presence of Fn in the immunoprecipitates was confirmed by using half of each sample for reprecipitation with anti-Fn antibody after boiling the immune complexes in 1% SDS (Fig. 7C and Fig. D). The use of GRGDSP peptide in coimmunoprecipitation assays did not cause any decrease in the amounts of 35S-labeled Fn (data not shown). These data indicate that in the RIPA lysates Fn interacts indirectly with β1 integrins due to association of its 42-kD fragment with integrin-bound tTG.
Figure 7.
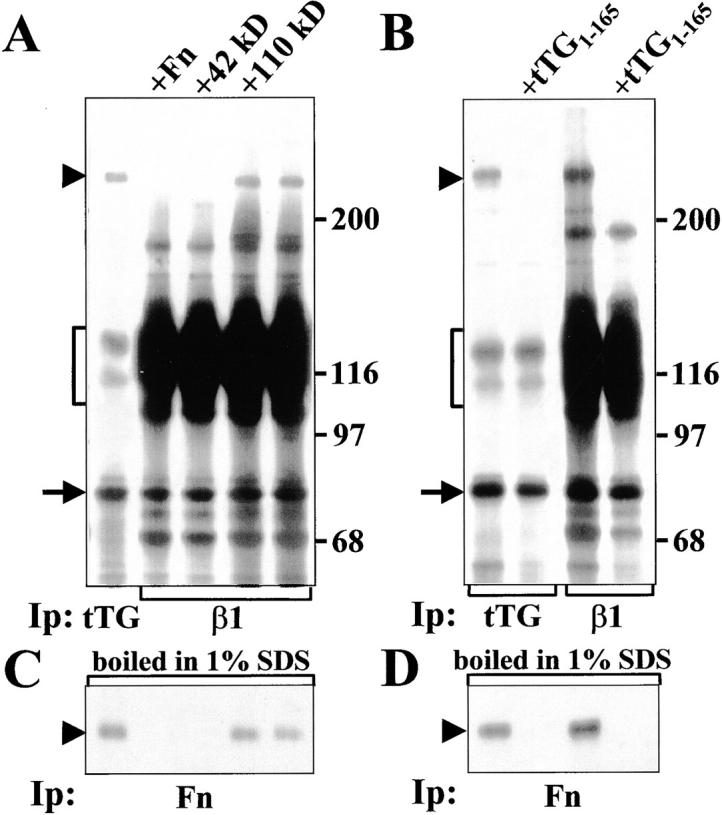
Interaction of tTG with β1 integrins allows formation of ternary complexes with Fn. (A) tTG (left lane) or β1 integrins (all other lanes) were immunoprecipitated from RIPA lysates of 35S-labeled WI-38 fibroblasts either in the presence of 1 μM unlabeled Fn, its 42-kD fragment, its 110-kD fragment or without any of these proteins added. (B) tTG (left two lanes) or β1 integrins (right two lanes) were immunoprecipitated from RIPA lysates of 35S-labeled WI-38 fibroblasts either in the absence or with 5 μM unlabeled NH2-terminal tTG fragment tTG1-165. After immunoprecipitation half of each sample shown in A and B was boiled in 1% SDS, reconstituted with 10 volumes of 1% Triton X-100 in TBS and subjected to reprecipitation with polyclonal antibody against Fn (C and D). Note a disappearance of 35S-labeled Fn bands in the samples treated with excess unlabeled Fn, 42-kD Fn fragment, or tTG1-165, but not with excess unlabeled 110-kD Fn fragment. Arrowheads indicate Fn bands. Brackets mark α5β1 integrin. Arrows point to tTG bands. Molecular weight markers are shown to the right of the gels.
An even more convincing proof of this concept came from experiments with HEL cells, which synthesize essentially no endogenous Fn even when treated with TPA, and express large amounts of α5β1 and αIIbβ3 integrins, which serve as Fn receptors (Jarvinen et al. 1987; data not shown). We found that stimulation of these cells with TPA sharply increased the overall expression of tTG while having no effect on the levels of α5β1 and αIIbβ3 (see below). This provided a convenient inducible model with which to test the ability of tTG to mediate the interaction of integrins with immobilized Fn and its fragments. Affinity chromatography of 35S-labeled RIPA lysates of both untreated and TPA-treated cells (Fig. 8A and Fig. B) showed that tTG was the predominant protein in the eluates from immobilized Fn and its 42-kD fragment, but did not bind the 110-kD Fn fragment (Fig. 8A and Fig. B, lanes 2–4). The identity of tTG was confirmed by immunoprecipitation (Fig. 8A and Fig. B, lanes 5) and by blotting the unlabeled eluates and immunoprecipitates for tTG (Fig. 8G and Fig. H). Consistent with the drastic enhancement of tTG synthesis in HEL cells by TPA, this treatment also greatly elevated the amounts of α5β1 and αIIbβ3 integrins coprecipitating with tTG and vice versa (Fig. 8, A–F, lanes 5; Fig. 8G and Fig. H, lanes 6 and 7). Notably, α5β1 and αIIbβ3 integrins bound to immobilized Fn and its 42-kD fragment, but not to the 110-kD fragment, indicating that binding of these integrins to Fn and its 42-kD fragment in RIPA buffer is indirect and mediated by tTG (Fig. 8, A–F, lanes 2–4). As a consequence of larger amounts of integrin–tTG complexes, more α5β1 and αIIbβ3 integrins were isolated on Fn and the 42-kD fragment, from the TPA-treated than from the untreated cells (Fig. 8, A–F, lanes 2 and 4). Taken together, these data establish that tTG serves as a bridge to link α5β1 and αIIbβ3 integrins with the gelatin-binding domain of Fn.
Figure 8.
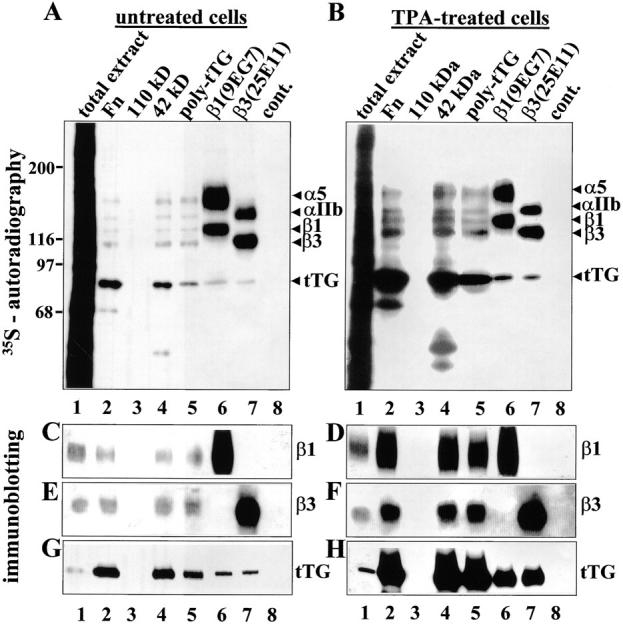
tTG mediates association of α5β1 and αIIbβ3 integrins with Fn via its 42-kD fragment. 35S-labeled untreated (A) and TPA-treated (B) HEL cells were extracted with RIPA buffer (lanes 1). Cell extracts were incubated with Sepharose-immobilized Fn (lanes 2), 110-kD (lanes 3), or 42-kD (lanes 4) Fn fragments, or immunoprecipitated with polyclonal anti-tTG antibody (lanes 5), anti–β1 integrin mAb 9EG7 (lanes 6), anti–β3 integrin mAb 25E11 (lanes 7) or control Sepharose beads (lanes 8). 35S-labeled eluates from immobilized Fn and Fn fragments, and immunoprecipitates were analyzed by SDS-PAGE and autoradiography. Unlabeled RIPA extracts of untreated (C, E, and G) or TPA-treated (D, F, and H) HEL cells, analogous to those in A and B, respectively, were incubated with immobilized Fn and Fn fragments or immunoprecipitated with antibodies against tTG, β1, and β3 integrins. Eluates from immobilized Fn and Fn fragments and immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with polyclonal antibody to β1A integrin cytodomain (C and D), polyclonal antibody to β3 integrin (E and F), or anti-tTG mAb tTG100 (G and H). (A and B) Protein bands corresponding to α5, αIIb, β1, and β3 integrins and tTG are marked to the right of each gel. Molecular weight markers are shown to the left of the gels.
tTG Amplifies Integrin Signaling, Colocalizes with β1 Integrins on the Cell Surface, and Causes Enlargement of Focal Adhesions
Since tTG mediates binding of integrins to Fn, we next analyzed whether tTG functionally collaborates with integrins in adhesion-dependent signal transduction. Analysis of integrin-mediated tyrosine phosphorylation of FAK was performed with REF52 cells overexpressing tTG (Fig. 9A and Fig. B). Tyrosine phosphorylation of FAK was very low in cells in suspension regardless of the levels of tTG expression (Fig. 9 A). A gradual time-dependent increase in FAK phosphorylation was observed in both types of transfectants during adhesion and spreading on Fn, but the phosphotyrosine content of FAK was consistently higher in the tTG-transfected cells (Fig. 9 A). We also compared the extent of FAK phosphorylation in the transfectants plated on different substrata for 3 h (Fig. 9 B). Unlike in the case of Fn, no difference was seen for the two cell lines adhering to 110-kD Fn fragment, despite the substantial levels of FAK phosphorylation in the transfectants. However, adhesion on 42-kD Fn fragment or anti-tTG antibody caused a much greater increase in FAK phosphorylation in cells overexpressing tTG compared with vector-transfected controls. In contrast, the levels of FAK phosphorylation were similar in these cell lines after adhesion on laminin, an ECM protein that does not bind tTG (Aeschlimann and Paulsson 1991). Enzymatically inactive mutant tTG[C277→S] had similar effects on integrin signaling (data not shown). These results demonstrate that association of integrin-bound tTG with the 42-kD Fn fragment is sufficient to trigger tyrosine phosphorylation of FAK even in the absence of direct integrin–ligand interaction. tTG also potentiates integrin-mediated FAK phosphorylation in cells adhering on Fn, showing cooperativity with integrins in outside-in signal transduction.
Figure 9.
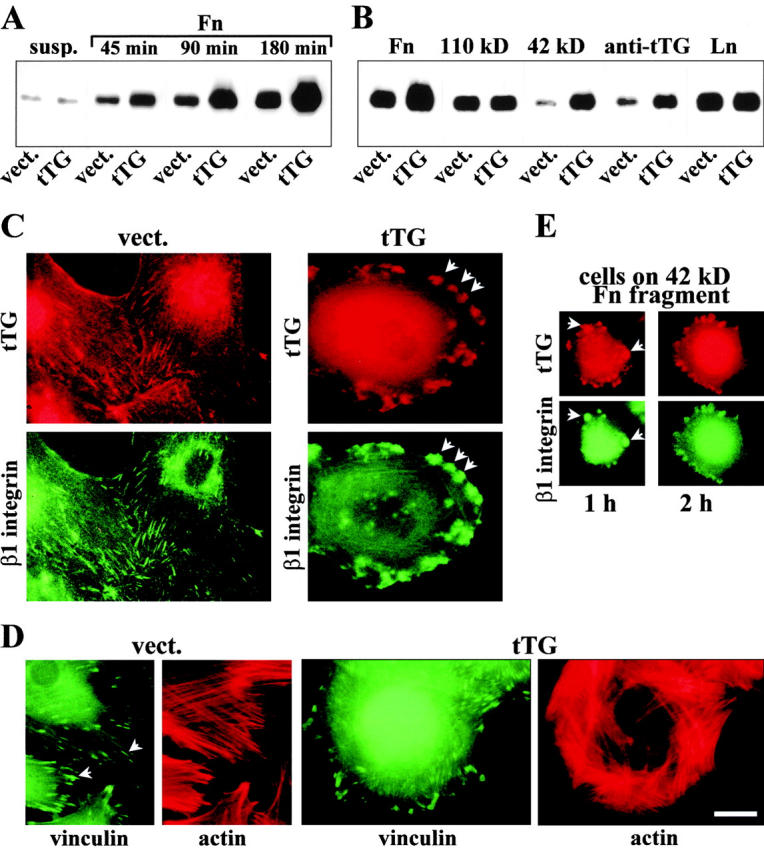
tTG amplifies integrin-mediated tyrosine phosphorylation of FAK and colocalizes with β1 integrins at focal adhesions. (A and B) tTG potentiates FAK phosphorylation. (A) Time course of FAK phosphorylation in REF52 cells expressing vector (vect.) or wild-type tTG (tTG), plated on Fn. Cells were either kept in suspension (susp.) or plated on Fn for 45, 90, or 180 min. (B) REF52 cells expressing vector (vect.) or wild-type tTG (tTG), were plated for 3 h on dishes coated with Fn, 110-kD, or 42-kD Fn fragments, polyclonal anti-tTG antibody or laminin (Ln). (A and B) The transfectants were plated on ECM proteins or anti-tTG antibody in serum-free medium in the presence of cycloheximide. The cells were lysed and FAK was immunoprecipitated from cell lysates followed by SDS-PAGE and immunoblotting of the immune complexes for phosphotyrosine with PY20 mAb (Belkin et al. 1996). (C–E) tTG colocalizes with β1 integrins on the cell surface of REF52 fibroblasts and causes enlargement of focal adhesions. (C) Live, nonpermeabilized cells transfected with vector (vect.) or tTG (tTG) were double stained for cell surface tTG with mAb CUB7402 and β1 integrins with hamster mAb HMβ1-1. Note codistribution of these proteins at focal adhesions and much larger size of these structures in tTG transfectants. (D) Formaldehyde-fixed, permeabilized cells transfected with vector or tTG, were double stained for vinculin and actin with mAb 7F9 and rhodamine-phalloidin. Note the increased size of focal adhesions and altered organization of actin bundles in the tTG transfectants. (E) Cells overexpressing tTG were plated for 1 or 2 h on 42-kD Fn fragment, fixed, and then double stained for surface tTG with mAb CUB7402 and β1 integrins with hamster mAb HMβ1-1. Arrows indicate focal adhesion sites. Bar, 20 μM.
Since tTG is physically and functionally associated with integrins, we compared their localization on the cell surface. Immunostaining of live nonpermeabilized REF52 cells on Fn revealed codistribution of tTG and β1 integrins at focal adhesions (Fig. 9 C, left panels). Overexpression of tTG showed its accumulation on the surface of REF52 cells in distinct clusters that colocalized with β1 integrins and appeared much larger in size than focal adhesions in vector-transfected cells (Fig. 9 C, arrows). Double staining for vinculin and actin showed an enlargement of focal adhesions in cells overexpressing tTG (Fig. 9 D, arrows). Also, the overall organization of the actin cytoskeleton in these cells was altered with a number of thick parallel bundles of actin filaments arranged peripherally (Fig. 9 D). Notably, β1 integrins were colocalized with tTG in REF52 transfectants spreading on the 42-kD Fn fragment, suggesting that aggregation of surface tTG leads to integrin clustering (Fig. 9 E, arrows).
Discussion
In this study we describe a novel function of cell surface tTG as an integrin-associated adhesion coreceptor for Fn. tTG exerts this function by associating with several β1 and β3 integrins while simultaneously binding to Fn via its NH2-terminal domain. The latter interaction involves a previously unrecognized adhesive site on Fn that is located within the 42-kD gelatin-binding domain (modules I6II1,2I7-9) and is distinct from its classical RGD-containing and all other known cell-binding motifs. Experiments presented in this study prove that cells can adhere, spread, and form focal adhesions on the isolated 42-kD Fn fragment, and that surface tTG is critical for these processes. tTG also promotes adhesion and enhances spreading, focal adhesion formation, and adhesion-triggered signaling of cells adhering to whole Fn.
A recent work by Hocking et al. 1998 reported that the 70-kD NH2-terminal Fn fragment, which includes the 42-kD gelatin-binding domain, binds directly to α5β1 integrin and elicits integrin-mediated signals. These effects were attributed to the 29-kD Fn fragment (modules I1-5), and not to the adjacent 42-kD domain. In addition, another recent study showed that numerous isolated type III modules of Fn can support cell adhesion by interacting with integrins, therefore greatly expanding the number of potential integrin-binding adhesive sites in Fn (Chi-Rosso et al. 1997). In our work, we demonstrate that another part of the Fn molecule, its 42-kD gelatin-binding domain that lacks integrin-binding motifs and does not interact with purified integrins in vitro, contributes to cell adhesion. In contrast to other reports on integrin-Fn association, we show that integrins can interact with Fn indirectly, and cell surface tTG mediates the interaction of integrins with the gelatin-binding domain of Fn.
The adhesive function of tTG strictly depends on its interaction with integrins. We estimate that in different cell types, 5–40% of β1 integrins are complexed with tTG. In contrast, only 0.5–10% of the total tTG cellular pool is associated with integrins; the great majority of tTG resides in the cytoplasm, making it often difficult to detect integrins in tTG immunoprecipitates from whole cell lysates. On the other hand, the chemical cross-linking experiments indicate that all the tTG on the cell surface is present as 1:1 complexes with integrins. The interaction of tTG with integrins occurs primarily via the extracellular domains of integrin β subunits. Among several integrins containing homologous β1, β2, and β3 subunits, only members of the β1 and β3 subfamilies interact with tTG, whereas β2 integrins do not. This emphasizes the specificity of this interaction and implies new functional differences between integrin subgroups.
In this study, we show that integrin–tTG complexes are formed inside the cell early during biosynthesis. We were unable to reconstitute integrin interaction with tTG in vitro using purified proteins, most likely because the formation of these complexes involves some as-yet unidentified intracellular intermediate(s) (data not shown). These facts might explain some previous data showing inability of purified tTG added to cells to influence cell adhesion and spreading (Jones et al. 1997). We also generated a truncated tTG mutant tTG[Δ592-687] with the deleted fourth (most COOH-terminal) domain and found that although it retained enzymatic and Fn-binding activities, it did not associate with integrins and was not transported to the cell surface (data not shown). These results suggest that integrins might be involved in transporting tTG to the cell surface. Thus, the integrin–tTG interaction established in this work may provide an explanation for the surface localization of tTG reported previously (Martinez et al. 1994; Aeschlimann et al. 1995; Verderio et al. 1998), and consequently, for its effects on cell adhesion and spreading (Gentile et al. 1992; Jones et al. 1997; Verderio et al. 1998).
How does the integrin–tTG interaction promote cell adhesion? Integrins are relatively low affinity receptors for ECM proteins, including Fn. In contrast, tTG binds with high affinity to Fn and its 42-kD fragment (Turner and Lorand 1989; Radek et al. 1993), and as shown here, forms stable complexes with integrins. The presence of integrin-bound tTG on the surface creates a possibility for cells to use an additional binding site within Fn for the interaction with integrins. This potentially doubles the number of sites in the Fn matrix that cells can access in the process of adhesion and spreading. If the Fn chains are fully extended, as in the case of the fibrillar matrix (Dzamba and Peters 1991), the tTG-binding site would be separated from the RGD motif by >20 nm, minimizing steric restrictions against their simultaneous occupation. Furthermore, the tTG-binding site on integrins most likely involves sequences outside the integrin ligand-binding pocket, consistent with the fact that their association is not perturbed by 110-kD cell-binding Fn fragment or RGD-containing peptides and function-blocking anti–β1 integrin antibodies (data not shown). Therefore, two types of ternary adhesion complexes can be envisioned as shown schematically in Fig. 10. Cells can adhere to Fn via integrins in the absence of tTG (Fig. 10 A). In the simplest case, tTG serves merely as a bridge between integrin and Fn (Fig. 10 B). This alone could strengthen adhesion because of the higher affinity and by allowing a second integrin molecule to access the RGD site in the same Fn chain. However, beyond this is the possibility for even more stable ternary complexes, where each protein interacts with two others (Fig. 10 C). Either scenario should increase cell adhesion and spreading on Fn. In agreement with the proposed model, tTG promotes integrin clustering in the membrane, and consequently, amplifies integrin outside-in signaling (Fig. 9). Integrin clustering could be further facilitated if tTG forms stable bivalent dimers, as does its homologue in FXIIIa (Yee et al. 1994). It is also possible that binding to tTG could affect the activation state of integrins.
Figure 10.
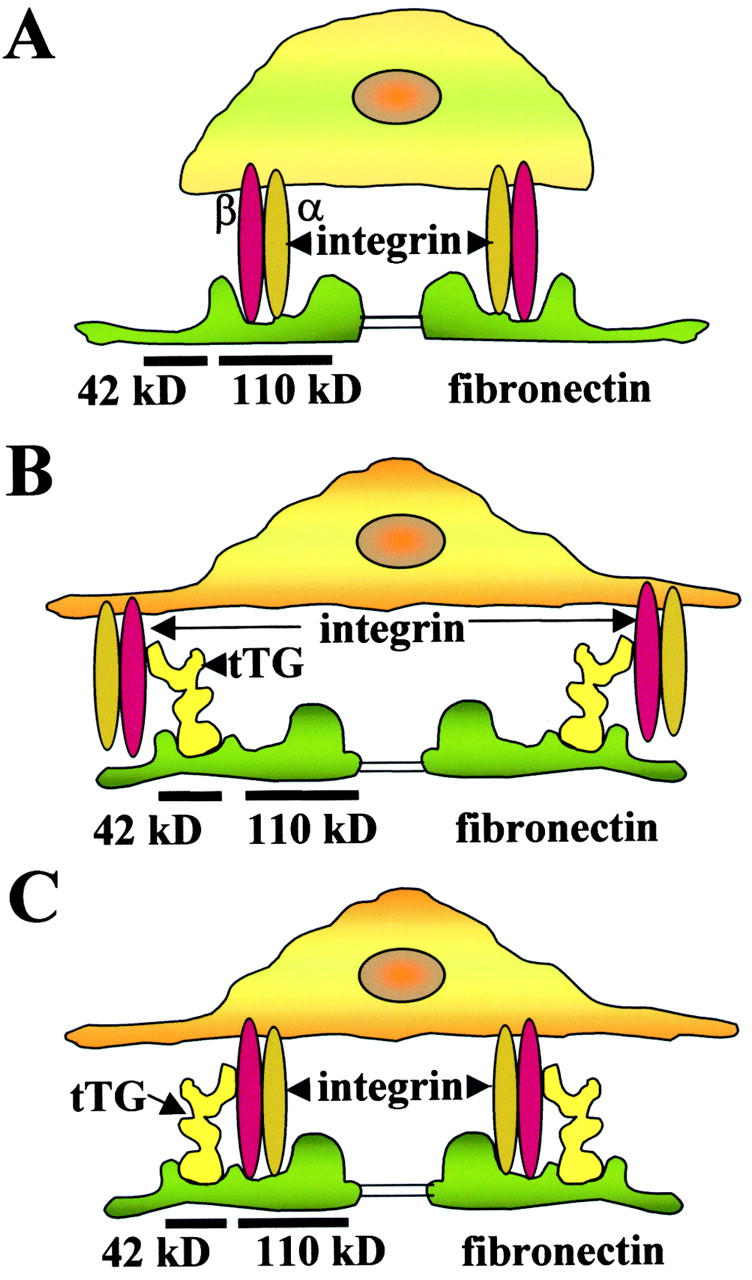
A model proposing the role of tTG in cell adhesion. Association of integrins with tTG promotes cell adhesion and spreading due to formation of ternary adhesion complexes with Fn. (A) Integrin-mediated adhesion to Fn in the absence of tTG. (B) tTG enhances adhesion acting as a bridge between integrins and Fn. (C) tTG enhances adhesion by mediating the formation of ternary complexes where all three proteins interact with each other.
Based on the interaction of tTG with several ECM proteins reported previously (Bowness et al. 1987; Achyuthan et al. 1988; Aeschlimann and Paulsson 1991; Aeschlimann et al. 1995) and the evidence presented here that tTG associates with multiple integrins, the proposed model is likely applicable to ECM proteins other than Fn. tTG is widely expressed in vivo, but its physiological role remains poorly understood. This study defines a function for tTG in cell adhesion that is distinct from and independent of its enzymatic (cross-linking) activity. However, this newly identified function could serve to concentrate transglutaminase activity at sites of adhesion and to strengthen locally the ECM. Upregulation of integrin–tTG association might be expected to occur when cells require enhanced adhesion and anchoring to the underlying substrate. This could take place during normal cell differentiation and under certain pathological conditions involving abnormal adhesion, such as tumor metastasis. Cell-modulated association of tTG with integrins represents a novel way to modify integrin adhesive function, in addition to regulation of integrin expression and ligand-binding affinity. Further studies will help to elucidate the physiological significance of this interaction and mechanisms of its regulation on the cellular level.
Acknowledgments
The authors wish to thank P. Davies, E. Davie, P. Birckbichler, L. Lorand, L. Romer, K. Burridge, V.E. Koteliansky, R. Fässler, S.F. Retta, and G. Tarone for valuable reagents and cell lines used in this study. We thank L. Zaritskaya for help with flow cytometry experiments. We are particularly grateful to K. Ingham for many stimulating discussions, providing Fn fragments, and critical reading of the manuscript.
This study was supported by National Institutes of Health grants R29 CA 77697 to A.M. Belkin and HL 21791 to K. Ingham.
Footnotes
Abbreviations used in this paper: ECM, extracellular matrix; FAK, focal adhesion kinase; Fn, fibronectin; FXIIIa, factor XIII; HEL, human erythroleukemia; TPA, 12-O-tetradecanoylphorbol 13-acetate; tTG, tissue transglutaminase.
References
- Achyuthan K.E., Mary A., Greenberg C.S. The binding sites on fibrin for guinea pig transglutaminase are similar to those of blood coagulation factor XIII. J. Biol. Chem. 1988;26:14296–14301. [PubMed] [Google Scholar]
- Aeschlimann D., Paulsson M. Cross-linking of laminin-nidogen complexes by tissue transglutaminase. A novel mechanism for basement membrane stabilization. J. Biol. Chem. 1991;266:15308–15317. [PubMed] [Google Scholar]
- Aeschlimann D., Wetterward A., Fleisch H., Paulsson M. Expression of tissue transglutaminase in skeletal tissue correlates with events of terminal differentiation of chondrocytes. J. Cell Biol. 1993;120:1461–1470. doi: 10.1083/jcb.120.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschlimann D., Kaupp O., Paulsson M. Transglutaminase-catalyzed matrix cross-linking in differentiating cartilageidentification of osteonectin as a major glutaminyl substrate. J. Cell Biol. 1995;129:881–892. doi: 10.1083/jcb.129.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama S.K., Yamada K.M. Biosynthesis and acquisition of biological activity of the fibronectin receptor. J. Biol. Chem. 1987;262:17536–17542. [PubMed] [Google Scholar]
- Belkin A.M., Zhidkova N.I., Balzac F., Altruda F., Tomatis D., Maier A., Tarone G., Koteliansky V.E., Burridge K. β1D integrin displaces the β1A isoform in striated muscleslocalization at junctional structures and signaling potential in nonmuscle cells. J. Cell Biol. 1996;132:211–226. doi: 10.1083/jcb.132.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin A.M., Retta F.S., Pletjushkina O.Y., Balzac F., Silengo L., Fassler F., Koteliansky V.E., Burridge K., Tarone G. Muscle β1D integrin reinforces the cytoskeleton-matrix linkmodulation of integrin adhesive function by alternative splicing. J. Cell Biol. 1997;139:1583–1595. doi: 10.1083/jcb.139.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berditchevski F., Zutter M.M., Hemler M.E. Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins) Mol. Biol. Cell. 1996;7:193–207. doi: 10.1091/mbc.7.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowness J.M., Folk J.E., Timpl R. Identification of a substrate site for liver transglutaminase on the aminopeptide of type III collagen. J. Biol. Chem. 1987;262:1022–1024. [PubMed] [Google Scholar]
- Chi-Rosso G., Gotwals P.J., Yang Y., Ling L., Jiang K., Chao B., Baker D.P., Burkly L.C., Fawell S.E., Koteliansky V.E. Fibronectin type III repeats mediate RGD-independent adhesion and signaling through activated β1 integrins. J. Biol. Chem. 1997;272:31447–31452. doi: 10.1074/jbc.272.50.31447. [DOI] [PubMed] [Google Scholar]
- Davies P.J.A., Murtaugh M.P., Moore W.T., Jr., Johnson G.S., Lucas D. Retinoic acid-induced expression of tissue transglutaminase in human promyelocytic leukemia (HL-60) cells. J. Biol. Chem. 1985;260:5166–5174. [PubMed] [Google Scholar]
- Dudek S.M., Johnson G.V. Transglutaminase facilitates the formation of polymers of the beta-amyloid peptide. Brain Res. 1994;651:129–133. doi: 10.1016/0006-8993(94)90688-2. [DOI] [PubMed] [Google Scholar]
- Dzamba B.J., Peters D.M.P. Arrangement of cellular fibronectin in noncollagenous fibrils in human fibroblast cultures. J. Cell Sci. 1991;100:605–612. doi: 10.1242/jcs.100.3.605. [DOI] [PubMed] [Google Scholar]
- Folk J.E. Transglutaminases. Annu. Rev. Biochem. 1980;49:517–531. doi: 10.1146/annurev.bi.49.070180.002505. [DOI] [PubMed] [Google Scholar]
- Gentile V., Saydak M., Chiocca E.A., Akande O., Birckbichler P.J., Lee K.N., Stein J.P., Davies P.J. Isolation and characterization of cDNA clones to mouse macrophage and human endothelial cell tissue transglutaminase. J. Biol. Chem. 1991;266:478–483. [PubMed] [Google Scholar]
- Gentile V., Thomazy V., Piacentini M., Fesus L., Davies P.J. Expression of tissue transglutaminase in Balb-C 3T3 fibroblastseffects on cell morphology and adhesion. J. Cell Biol. 1992;119:463–474. doi: 10.1083/jcb.119.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George M.D., Vollberg T.M., Floyd E.E., Stein J.P., Jetten A.M. Regulation of transglutaminase type II by transforming growth factor-beta 1 in normal and transformed human epidermal keratinocytes. J. Biol. Chem. 1990;265:11098–11104. [PubMed] [Google Scholar]
- Grundmann U., Amann E., Zettlmeissl G., Kupper H.A. Characterization of cDNA coding for human factor XIII. Proc. Natl. Acad. Sci. USA. 1986;83:8024–8028. doi: 10.1073/pnas.83.21.8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking D.C., Sottile J., McKeown-Longo P.J. Activation of distinct α5β1-mediated signaling pathways by fibronectin's cell adhesion and matrix assembly domains. J. Cell Biol. 1998;141:241–253. doi: 10.1083/jcb.141.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs B.S., Brew S.A., Ingham K.C. Reversible unfolding of the gelatin-binding domain of fibronectinstructural stability in relation to function. Biochemistry. 1989;28:842–850. doi: 10.1021/bi00428a065. [DOI] [PubMed] [Google Scholar]
- Jarvinen M., Ylanne J., Vartio T., Virtanen I. Tumor promoter and fibronectin induce actin stress fibers and focal adhesion sites in spreading human erythroleukemia (HEL) cells. Eur. J. Cell Biol. 1987;44:238–246. [PubMed] [Google Scholar]
- Jeong J.M., Murthy S.N., Radek J.T., Lorand L. The fibronectin-binding domain of transglutaminase. J. Biol. Chem. 1995;270:5654–5658. doi: 10.1074/jbc.270.10.5654. [DOI] [PubMed] [Google Scholar]
- Johnson G.V., Cox T.M., Lockhart J.P., Zimmerman M.D., Miller M.L., Powers R.E. Transglutaminase activity is increased in Alzheimer's disease brain. Brain Res. 1997;751:323–329. doi: 10.1016/s0006-8993(96)01431-x. [DOI] [PubMed] [Google Scholar]
- Johnson T.S., Knight C.R., el-Alaoui S., Mian S., Rees R.C., Gentile V., Davies P.J., Griffin M. Transfection of tissue transglutaminase into a highly malignant hamster fibrosarcoma leads to a reduced incidence of primary tumour growth. Oncogene. 1994;9:2935–2942. [PubMed] [Google Scholar]
- Jones R.A., Nicholas B., Mian S., Davies P.J., Griffin M. Reduced expression of tissue transglutaminase in a human endothelial cell line leads to changes in cell spreading, cell adhesion and reduced polymerization of fibronectin. J. Cell Sci. 1997;110:2461–2472. doi: 10.1242/jcs.110.19.2461. [DOI] [PubMed] [Google Scholar]
- Kaartinen M.T., Pirhonen A., Linnala-Kankunen A., Maenpaa P.H. Transglutaminase-catalyzed cross-linking of osteopontin is inhibited by osteocalcin. J. Biol. Chem. 1997;272:22736–22741. doi: 10.1074/jbc.272.36.22736. [DOI] [PubMed] [Google Scholar]
- Kahlem P., Terre C., Green H., Dijan P. Peptides containing glutamine repeats as substrates for transglutaminase-catalyzed cross-linkingrelevance to diseases of the nervous system. Proc. Natl. Acad. Sci. USA. 1996;93:14580–14585. doi: 10.1073/pnas.93.25.14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlem P., Green H., Dijan P. Transglutaminase action imitates Huntington's diseaseselective polymerization of huntingtin containing expanded polyglutamine. Mol. Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- Kleman J.P., Aeschlimann D., Paulsson M., van der Rest M. Transglutaminase-catalyzed crosslinking of fibrils of collagen V/XI in A204 rhabdomyosarcoma cells. Biochemistry. 1995;34:13768–13775. doi: 10.1021/bi00042a007. [DOI] [PubMed] [Google Scholar]
- Lesort M., Attanavanich K., Zhang J., Johnson G.V. Distinct nuclear localization and activity of tissue transglutaminase. J. Biol. Chem. 1998;273:11991–11994. doi: 10.1074/jbc.273.20.11991. [DOI] [PubMed] [Google Scholar]
- Lorand L., Campbell-Wilkes L.K., Cooperstein L. A filter paper assay for transamidating enzymes using radioactive amine substrates. Anal. Biochem. 1972;50:623–631. doi: 10.1016/0003-2697(72)90074-7. [DOI] [PubMed] [Google Scholar]
- Martinez J., Chalupowicz D.G., Roush R.K., Sheth A., Barsigian C. Transglutaminase-mediated processing of fibronectin by endothelial cell monolayers. Biochemistry. 1994;33:2538–2545. doi: 10.1021/bi00175a024. [DOI] [PubMed] [Google Scholar]
- Menter D.G., Patton J.T., Updyke T.V., Kerbel R.S., McIntire L.V., Nicolson G.L. Transglutaminase stabilizes melanoma adhesion under laminar flow. Cell Biophys. 1991;18:123–143. doi: 10.1007/BF02989810. [DOI] [PubMed] [Google Scholar]
- Nagy L., Saydak M., Shipley N., Lu S., Basilion J.P., Yan Z.H., Syka P., Chandraratna R.A., Stein J.P., Heyman R.A., Davies P.J. Identification and characterization of a versatile retinoid response element in the mouse transglutaminase gene promoter. J. Biol. Chem. 1996;271:4355–4365. doi: 10.1074/jbc.271.8.4355. [DOI] [PubMed] [Google Scholar]
- Nakaoka H., Perez D.M., Baek K.J., Das T., Husain A., Misono K., Im M.J., Graham R.M. Gha GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264:1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- Oliverio S., Amendola A., Di Sano F., Farrace M.G., Fesus L., Nemes Z., Piredda L., Spinedi A., Piacentini M. Tissue transglutaminase-dependent posttranslational modification of the retinoblastoma gene product in promonocytic cells undergoing apoptosis. Mol. Cell. Biol. 1997;17:6040–6048. doi: 10.1128/mcb.17.10.6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radek J.T., Jeong J.M., Murthy S.N., Ingham K.C., Lorand L. Affinity of human erythrocyte transglutaminase for a 42-kDa gelatin-binding fragment of human plasma fibronectin. Proc. Natl. Acad. Sci. USA. 1993;90:3152–3156. doi: 10.1073/pnas.90.8.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta S.F., Balzac F., Ferraris P., Belkin A.M., Fassler R., Humphries M.J., De Leo G., Silengo L., Tarone G. β1-Integrin cytoplasmic subdomains involved in dominant negative function. Mol. Biol. Cell. 1998;9:715–731. doi: 10.1091/mbc.9.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter S.J., Davies P.J. Identification of a transforming growth factor-beta1/bone morphogenetic protein 4 (TGF-beta/BMP4) response element within the mouse tissue transglutaminase gene promoter. J. Biol. Chem. 1998;273:12798–12806. doi: 10.1074/jbc.273.21.12798. [DOI] [PubMed] [Google Scholar]
- Thomazy V., Fesus L. Differential expression of tissue transglutaminase in human cells. An immunohistochemical study. Cell Tissue Res. 1989;255:215–224. doi: 10.1007/BF00229084. [DOI] [PubMed] [Google Scholar]
- Turner P.M., Lorand L. Complexation of fibronectin with tissue transglutaminase. Biochemistry. 1989;28:628–635. doi: 10.1021/bi00428a032. [DOI] [PubMed] [Google Scholar]
- Upchurch H.F., Conway E., Maxwell M.D. Localization of cellular transglutaminase on the extracellular matrix after woundingcharacteristics of the matrix-bound enzyme. J. Cell. Physiol. 1991;149:375–382. doi: 10.1002/jcp.1041490304. [DOI] [PubMed] [Google Scholar]
- van Groningen J.J., Klink S.L., Bloemers H.P., Swart G.W. Expression of tissue-type transglutaminase correlates positively with metastatic properties of human melanoma cell lines. Int. J. Cancer. 1995;60:383–387. doi: 10.1002/ijc.2910600319. [DOI] [PubMed] [Google Scholar]
- Verderio E., Nicholas B., Gross S., Griffin M. Regulated expression of tissue transglutaminase in Swiss 3T3 fibroblastseffects on the processing of fibronectin, cell attachment and cell death. Exp. Cell Res. 1998;239:119–138. doi: 10.1006/excr.1997.3874. [DOI] [PubMed] [Google Scholar]
- Yee V.C., Pedersen L.C., Le Trong I., Bishop P.D., Stenkamp R.E., Teller D.C. Three-dimensional structure of a transglutaminasehuman blood coagulation factor XIII. Proc. Natl. Acad. Sci. USA. 1994;91:7296–7300. doi: 10.1073/pnas.91.15.7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylanne J., Cheresh D.A., Virtanen I. Localization of beta 1, beta 3, alpha 5, alpha v, and alpha IIb subunits of the integrin family in spreading erythroleukemia cells. Blood. 1990;76:570–577. [PubMed] [Google Scholar]