

Abstract
Smooth muscle myosin in the dephosphorylated state does not form filaments in vitro. However, thick filaments, which are composed of myosin and myosin-binding protein(s), persist in smooth muscle cells, even if myosin is subjected to the phosphorylation– dephosphorylation cycle. The characterization of telokin as a myosin-assembling protein successfully explained the discrepancy. However, smooth muscle cells that are devoid of telokin have been observed. We expected to find another ubiquitous protein with a similar role, and attempted to purify it from chicken gizzard. The 38k protein bound to both phosphorylated and dephosphorylated myosin to a similar extent. The effect of the myosin-binding activity was to assemble dephosphorylated myosin into filaments, although it had no effect on the phosphorylated myosin. The 38k protein bound to myosin with both COOH-terminal 20 and NH2-terminal 28 residues of the 38k protein being essential for myosin binding. The amino acid sequence of the 38k protein was not homologous to telokin, but to human p32, which was originally found in nuclei as a subunit of pre-mRNA splicing factor-2. Western blotting showed that the protein was expressed in various smooth muscles. Immunofluorescence microscopy with cultured smooth muscle cells revealed colocalization of the 38k protein with myosin and with other cytoskeletal elements. The absence of nuclear immunostaining was discussed in relation to smooth muscle differentiation.
Keywords: myosin assembly, myosin binding, myosin filament, human p32, smooth muscle
Introduction
The thick filaments observed in various muscle cells are composed of myosin filaments that are associated with myosin-binding proteins. Assembly into filaments is an inherent property of myosin (Huxley 1963). Myosin-binding proteins, such as myosin-binding protein-C (C-protein), myosin-binding protein-H (H-protein), M-protein, titin, and myomesin are thought to stabilize the filaments (for review, see Epstein and Fischman 1991; Furst and Gautel 1995).
In the case of skeletal muscle myosin, assembly is observed regardless of the phosphorylation state of myosin. However, smooth muscle myosin does not form filaments unless its regulatory light chain (RLC) is phosphorylated (for review, see Stull et al. 1991; Trybus 1991; Gallagher et al. 1997). Smooth muscle myosin molecules, if not phosphorylated, are in a folded structure called the 10S form, and hardly assemble filaments. When the myosin is phosphorylated, its shape changes to the extended 6S form and filaments are assembled (Onishi and Wakabayashi 1982; Trybus et al. 1982; Craig et al. 1983). Thus, the filaments formed from the phosphorylated myosin become unstable upon dephosphorylation of the RLC and, as a result, easily fall apart.
These in vitro properties of smooth muscle myosin filaments are of particular interest when we observe the persistent nature of thick filaments in smooth muscle cells. Their myosin is repeatedly subject to cycles of phosphorylation and dephosphorylation in response to contraction, followed by relaxation (for review, see Stull et al. 1991; Somlyo and Somlyo 1994). However, there is no sign of assembly and disassembly of thick filaments, indicating that they are stable (Somlyo et al. 1981; Cooke et al. 1989; Xu et al. 1996). By raising an mAb to the 10S form, Horowitz et al. 1994 examined the intracellular form of myosin. The concentration of 10S myosin was very low, regardless of contraction and relaxation states, most of the myosin being in the 6S form. Therefore, myosin filaments are expected to be stabilized as thick filaments by myosin-binding proteins in the cell. The first examination of this suggestion was by Shirinsky et al. 1993, who observed that telokin binds to dephosphorylated smooth muscle myosin to stimulate myosin assembly. However, telokin is not present in all smooth muscle cells (Gallagher and Herring 1991), indicating the presence of another myosin-binding protein(s) with a similar role in the cell.
We were interested in this indication and adopted a strategy to purify myosin-binding proteins from skeletal muscles (Starr and Offer 1971). We chose a crude preparation of smooth muscle myosin as a starting material and purified a protein of 38 kD that bound to myosin, causing filament assembly.
Materials and Methods
Preparation of Proteins
Fresh chicken gizzards were minced within 2 h of killing, and kept frozen at −80°C until use. Proteins other than myosin were purified from the frozen mince, unless otherwise specified. Dephosphorylated myosin was purified from chicken gizzard according to Ebashi 1976 with modifications (Ikebe and Hartshorne 1985a) and used as unphosphorylated myosin. We checked that myosin preparations did not contain the 38k myosin-assembling protein by SDS-PAGE or Western blotting. When required, myosin was phosphorylated with myosin light chain kinase (MLCK) in the presence of Ca2+ and calmodulin (Okagaki et al. 1991). Rod, light meromyosin (LMM), and heavy meromyosin (HMM) of gizzard myosin were prepared proteolytically as described (Ikebe and Hartshorne 1985b; King et al. 1995). Myosin was also purified from chicken breast muscle and bovine heart and used as skeletal and cardiac myosin, respectively (Okagaki et al. 1993). In this paper, myosin denotes smooth muscle myosin unless otherwise mentioned. MLCK, telokin, and tropomyosin were purified from chicken gizzard according to Hayakawa et al. 1994, Ito et al. 1989, and Ishikawa et al. 1989, respectively. Monomeric actin was purified from chicken breast muscle and used as actin filaments after polymerization (Okagaki et al. 1991). The concentration of actin filaments was expressed as that of monomeric actin.
Preparation of the 38k Protein
Following our experience with C-protein, a myosin-binding protein of skeletal muscle (Okagaki et al. 1993), we used a crude myosin preparation (see below) as the starting material. From this, we purified a protein of 38 kD in SDS-PAGE using myosin-assembling activity under dark-field microscopy (see below) as a probe. All columns (see below) were incorporated into a high performance liquid chromatography system (model L-6200; Hitachi).
The mince of chicken gizzard was thawed, suspended in 3 vol of buffer containing 30 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl, pH 7.5, and 1 mM DTT, and precipitated by centrifugation at 1,000 g for 10 min. This washing was repeated four times to remove soluble proteins. The washed mince was homogenized in 3 vol of extraction buffer (0.1 M NaCl, 5 mM ATP, 2 mM EDTA, 20 mM Tris, pH 7.5, 1 mM DTT, 0.5 mM PMSF, and 0.5 μg/ml leupeptin) using a cooking mixer. From the homogenate, the 38k protein was extracted together with myosin for 40 min with occasional mixing on ice. The homogenized muscle was centrifuged at 1,000 g for 40 min. After centrifugation, supernatants were pooled and the extraction procedure was repeated on the precipitate. The second supernatant was mixed with the pool (for SDS-PAGE, see Fig. 1, lane 2). The 38k protein could be extracted from the muscle with 2 mM ATP, but higher concentrations of ATP increased the yield of the protein. Higher salt solutions (0.2–0.3 M NaCl) also extracted more of the 38k protein from the muscle, but also increased contamination. Thus, we adopted the above extraction buffers. NaCl was added to the pooled extracts to increase its concentration to 0.3 M. The mixture was further centrifuged at 100,000 g for 2 h to precipitate actin filaments and actin-associated proteins. We denote this supernatant as partially purified myosin in this paper (Fig. 1, lane 3). The supernatant was subjected to stepwise ammonium sulfate fractionation. Aliquots of each step of the fractionation were desalted with Biogel P-6 (BioRad Laboratories), equilibrated with buffer A (1 mM ATP, 2 mM MgCl2, 0.1 M NaCl, 20 mM Tris-HCl, pH 7.5, 1 mM DTT, and 0.5 mM PMSF) and mixed with unphosphorylated myosin at a final concentration of 0.2 μM in buffer A. The mixtures were subjected to the dark-field microscopy to examine myosin-assembling activity (see Centrifugation Assay). The fractions precipitated at between 55 and 80% saturation (Fig. 1, lane 4) contained the myosin-assembling activity. The fractions were dissolved in buffer B (20 mM Tris-HCl, pH 7.5, 1 mM DTT, 0.5 mM PMSF, and 0.5 μg/ml leupeptin), the volume of which was selected to adjust its conductivity to be as low as that of 0.2 M NaCl. Then the solution was clarified by centrifugation at 100,000 g for 2 h. The supernatant was applied to DEAE Toyopearl 650M (Tosoh), equilibrated with buffer B supplemented with 0.2 M NaCl, followed by elution with a linear gradient of NaCl from 0.2–0.5 M. Aliquots of each eluate were desalted and tested for myosin-assembling activity as described. The activity was detected in the fractions eluted with 0.3–0.4 M NaCl. The fractions containing activity were composed of polypeptides of 38 kD, 17 kD, and minor contaminants (Fig. 1, lane 5). They were pooled, concentrated with Centricon-30 (Millipore), and applied to a Superdex HR75 column (Amersham-Pharmacia) equilibrated with buffer B supplemented with 0.3 M NaCl. Fractions corresponding to 15–30 kD, as estimated by a molecular weight marker for gel filtration (Amersham-Pharmacia), showed the myosin-assembling activity. The major protein band of these fractions was 38 kD on SDS-PAGE (Fig. 1, lane 6). We used this fraction for the most of biochemical experiments. During column chromatography, the 38k protein often degraded to polypeptides of 20 and 17 kD if leupeptin was absent. We routinely obtained 0.8–1.0 mg of the 38k protein from 200 g of gizzard.
Figure 1.

Purification of the 38k protein. Aliquots of samples from the following purification steps were applied to SDS-PAGE: 1, Total homogenate of gizzard; 2, myosin extracted from gizzard; 3, crude myosin preparation (supernatant in the presence of 0.3 M NaCl and 5 mM ATP); 4, ammonium sulfate precipitate of partially purified myosin preparation at 55–80% saturation; 5, the fraction of DEAE Toypearl 650M with the highest myosin-assembling activity; and 6, the active principle eluted from Superdex HR75. The 38k to the right of lane 6 denotes the band of the 38k protein. For details, see Materials and Methods.
Observation of Myosin Filament Assembly by the 38k Protein
Myosin at 0.2 μM was mixed with the 38k protein or the 38k protein-containing fractions in buffer A and incubated at room temperature for 30 min. A drop of the mixture was mounted on a glass slide, sealed with a coverslip, and observed with a dark-field microscope (Okagaki et al. 1991). Myosin filaments were detected as small spots of uniform size. The fine structure of myosin filaments was confirmed by EM. A drop of the above mixture was placed on a grid coated with collodion and carbon, stained with l% uranyl acetate, and observed with JEM 100C electron microscope operated at 80 kV.
Centrifugation Assay for Myosin-assembling, Myosin-binding, and Actin-binding Activities
Myosin (1.1 μM), either in phosphorylated or unphosphorylated form, was mixed with various concentrations of the 38k protein in buffer A, and the mixture was incubated at room temperature for 30 min, followed by centrifugation at 100,000 g for 40 min. The supernatant and precipitate were separately subjected to SDS-PAGE. The SDS-PAGE gels were stained with Coomassie brilliant blue and the protein bands were densitometrically scanned. The density of the protein band was calculated by NIH image (version 3.0). The densities of myosin heavy chain (MHC), the 38k protein, and telokin were considered, respectively, as assembled myosin, the 38k protein bound to myosin, and telokin bound to myosin.
The relationship of the amount of the 38k protein bound to unphosphorylated myosin is shown (see Fig. 3 e). Unphosphorylated myosin remains in the supernatant if insufficient 38k protein is added. Therefore, it is possible that part of the 38k protein remains in the supernatant, despite its binding to myosin. However, we believe that this is unlikely and that the 38k protein bound to unphosphorylated myosin is mostly recovered in the precipitate (compare open and closed circles in Fig. 3 e). The binding of 5.4 μM of 38k protein to 10 μM actin filaments in the presence and absence of tropomyosin was examined by a similar centrifugation assay (Ishikawa et al. 1989).
Figure 3.

Sedimentation assay of myosin assembly. Myosin (1.1 μM) and the 38k protein were mixed in buffer A, and the mixture was allowed to assemble. The assembled myosin was precipitated by centrifugation and quantified. a, SDS-PAGE pattern of supernatants (1, 3, and 5) and precipitates (2, 4, and 6) after centrifugation of the mixture of unphosphorylated myosin and the 38k protein. Concentrations of the 38k protein were 0 μM (1 and 2), 1.4 μM (3 and 4), and 5.4 μM (5 and 6). b, SDS-PAGE pattern of supernatants (1, 3, and 5) and precipitates (2, 4, and 6) from the sedimentation assay with phosphorylated myosin. Concentrations of the 38k protein were 0 μM (1 and 2), 1.4 μM (3 and 4), and 5.4 μM (5 and 6). c, SDS-PAGE pattern of supernatants (1, 3, and 5) and precipitates (2, 4, and 6) from the binding assay of the 38k protein to actin filaments. Actin filaments alone at 10 μM (1 and 2); 10 μM actin filaments and 5.4 μM of the 38k protein (3 and 4); and 10 μM actin filaments, 1.4 μM tropomyosin, and 5.4 μM of the 38k protein (5 and 6). d, The amount of assembled myosin was plotted against concentration of the 38k protein mixed with 1.1 μM myosin. Open and closed circles indicate unphosphorylated and phosphorylated myosin, respectively. e, The amount of the 38k protein bound to myosin was plotted against the 38k protein mixed with 1.1 μM myosin. Open and closed circles denote the unphosphorylated and phosphorylated myosin, respectively. MHC, 38k, A, α-TM, and β-TM denote the bands of MHC, the 38k protein, actin, and α- and β-tropomyosins, respectively.
The centrifugation assay of the 38k protein bound to insoluble myosin fragments was also performed in a similar way. Various concentrations of the 38k protein were mixed with 1.1 μM rod or LMM in buffer A, centrifuged, and analyzed as above. The interaction of the 38k protein to soluble myosin-fragment of HMM was examined by the gel filtration (see Fig. 5, inset), i.e., 8.5 μM HMM and 40 μM 38k protein in 300 μl buffer A was applied to the column Superose 6 (Amersham-Pharmacia).
Figure 5.

The amounts of the 38k protein coprecipitated by binding to myosin and its fragments, and striated muscle myosins. The assembly of unphosphorylated myosins was observed only in the presence of the 38k protein (see Fig. 3). The assembly of rod, LMM, skeletal, and cardiac myosins do not require the 38k protein. About 95% of total amount of rod, 85% of that of LMM, 90% of that of skeletal and cardiac myosins could be precipitatable under our assay conditions. The 38k protein was mixed with 1.1 μM of various types of myosin and its fragments and centrifuged to coprecipitate with them. The amounts of the 38k protein recovered in the precipitate were quantified by the densitometry. Rod (open circles), LMM (closed circles), unphosphorylated myosin (open triangles), skeletal (open squares), and cardiac myosins (closed squares). Data are from the average of two sets of independent experiments. Inset, Gel filtration pattern of the mixture of HMM and the 38k protein. The mixture of 8.5 μM HMM and 40 μM 38k protein in 300 μl buffer A was applied to Superose 6 equilibrated with buffer A and eluted with buffer A in a flow rate of 0.5 ml/min. A280 in arbitrary unit (ordinate) was plotted against elution time (abscissa). HMM and the 38k protein was separately eluted in the peak at 31 and 35 min, respectively. SDS-PAGE patterns of peak fractions of HMM (left) and the 38k protein (right) were also shown. Arrowheads indicate HMM (H) and the 38k protein (38).
The molecular weights used for calculation based upon their primary structure are 440,000 for MHC, 24,000 for the 38k protein (or human p32, its human isoform, see below), 18,000 for telokin, 42,000 for actin, and 35,000 for tropomyosin. The molecular weights of myosin fragments used for calculation are: 250,000, 150,000, and 260,000 for rod, LMM, and HMM, respectively.
Determination of Partial Amino Acid Sequence
The partial amino acid sequence of the 38k protein was determined after cleavage with Staphylococcus V8 protease (Pierce Chemical Co.). The weight ratio of the 38k protein to V8 protease was 100:1 and the digestion was for 30 min at room temperature at the boundary between the stacking and running gels of SDS-PAGE. The 38k protein was degraded to only two bands of 20 and 17 kD, and smaller fragments were not obtained by prolonged digestion of up to 1 h. The cleaved protein was subjected to SDS-PAGE and then electrotransferred to poly vinyl demethyl fluorate (PVDF) membrane, the bands of 38 (uncleaved protein), 20, and 17 kD were excised and sequenced (476A; Applied Biosystems). Two sequences were obtained; LHTEGDKAFAQFLTDEI from the 38- and the 20-kD bands, and REVSFQPTGESD from the 17-kD band.
cDNA Cloning of the 38k Protein
Total RNA was extracted from fresh chicken gizzard by homogenizing the tissue in TRIZOL reagent (Life Technologies) using a polytron homogenizer. mRNA was then purified with Oligotex-dT30 <super> (Japan Roche) according to the manufacturer's protocol. cDNA was synthesized by First-Strand cDNA-synthesis kit (Amersham-Pharmacia) using oligo-dT adapter primer (RR012A; Takara). Based on the above two partial amino acid sequences, we designed two primers: p1, 5′-CA(CT)AC (ATC)GA(AG)GG(ATC)GA(CT)AA(AG)GC(ATC)TT(CT)GC(ATC) CA(AG)TT(CT)-3′; and p2R, 5′-(AG)TC(CT)TC(ACGT)TC(GAT) CC(GAT)GT(ACGT)(GT)(AT)(CT)TG(AG)(GC)(AC)(GAT)(AT)(GC)(ACGT)(GC)C-3′. PCR was performed with these primers (30 cycles of 95°C [1 min], 55°C [2 min], 72°C [3 min]) using the above cDNA as template, and a product of 400 bp as examined by agarose gel electrophoresis was obtained. This PCR product, denoted as S1, was subcloned into the pGEM-T Easy vector system (Promega) and the nucleotide sequence of the cDNA clones were determined by the dideoxy chain termination method with Dye Primer Cycle Sequencing Kit (Applied-Biosystems) using an automated DNA sequencer (Prism 377A; Applied Biosystems). To extend cDNA sequence in the 3′-direction, we performed 3′-RACE (Frohman et al. 1988). Based on the 3′-end sequence of S1, we designed the primer p4, 5′-GAGGAGGAAAGTGACATTTTCAC-3′. Using p4 and the oligo-dT adapter as primers, and the above cDNA as template, PCR was performed as described above. A 700-bp product, denoted as S2, containing a termination codon and poly-A sequence, was obtained. The PCR product was subcloned into the pGEM-T Easy vector and its nucleotide sequence was determined as described above. The cDNA fragment of the 38k protein, denoted as S3, was obtained by PCR using primers corresponding to the sequences of the 5′-end of S1, 5′-GATTCACACGGAGGGGGATAAAGCG-3′; and the 3′-end of the coding region of S2, 5′-GAGCACTGCTTTCCTAACCTACTGACATTTGAC-3′. The above cDNA was used as template. The PCR product was subcloned to pGEM-T Easy vector.
The gizzard cDNA library, synthesized by priming with oligo-dT using TimeSaver cDNA synthesis kit (Amersham-Pharmacia), was constructed in λZAPII (Stratagene) vectors. To confirm sequence of the 5′-end of S3, cDNA clone of the 38k protein was obtained by screening the gizzard cDNA library (2 × 105 plaques) by plaque hybridization with the direct labeling of the above PCR product of S3 with alkaline phosphatase by AlkPhos Direct labeling kit (Amersham-Pharmacia). The detection was performed with CDP-Star chemiluminescent (Tropix Inc). pBluescript SK(−) (Stratagene) carrying cDNA inserts was prepared by in vivo excision from the λZAPII vectors. Both strands were completely sequenced by the same method as described above. The nucleotide sequence data reported in this paper will appear in the GenBank/EMBL/DDBJ nucleotide sequence databases with accession number AB029946.
Expression of the Human p32 and the 38k Protein
As will be described, the 38k protein had a high sequence homology to human p32. pT7 plasmid containing cDNA encoding human p32, a gift from Dr. A. Krainer (Cold Spring Harbor Laboratory, NY), was transformed into Escherichia coli strain BL21(DE3) (Krainer et al. 1991). Cells expressing human p32 after induction with 1 mM isopropylthio-β-d-galactoside (IPTG) were collected by centrifugation at 10,000 g for 10 min and resuspended in 0.3 M NaCl, 20 mM Tris, pH 7.5, 1 mM DTT, 0.5 mM PMSF, and 0.5 μg/ml leupeptin. The cells were frozen, thawed, sonicated (SONIFIER 200; Branson) and centrifuged at 10,000 g for 30 min. Human p32 was purified from the supernatant by ammonium sulfate fractionation, followed by DEAE chromatography as described for the purification of the 38k protein from chicken gizzard. The recombinant protein was used in the present experiments as human p32.
Constructs of truncated forms of the 38k protein were made by PCR reactions using primers containing a 5′ NcoI restriction site including ATG and a 3′ EcoRI site. The primers used were: M1, 5′-GCATGCCATGGACACGGAGGGGGATAAAGC-3′; M2, 5′-GCATGCCATGGGCATCCCCCCCGCGGTGG-3′; M3, 5′-CGGAATTCCTATTCTCCGGTGGGCTGAAAGC-3′; M4, 5′-CGGAATTCCTACTGACATT-TGACAAAGC-3′; M5, 5′-GCATGCCATGGCGACTCCGAACTTC-GTTGTGGAGG-3′; M6, 5′-GCATGCCATGGATTGGAAGGACACCAACTACACC-3′; M7, 5′-GCATGCCATGGCCAAAGTGTCCGGC-3′; and M8, 5′-CCGGAATTCCTATGCAGTGCTGAGCTCAAT-3′.
The cDNA fragment of S3 (see above) was used as a template for the PCR reactions. The primers were used in the following combinations: full-length protein (1–207 residues), M1 and M4; ΔN28 (29–207 residues), M7 and M4; ΔN63 (64–207 residues), M2 and M4; ΔN88 (89–207 residues), M6 and M4; ΔNC (64–141 residues), M2 and M3; ΔC66 (1–141 residues), M1 and M3; and ΔC20 (1–187 residues), M1 and M8. PCR was as described, PCR products were cut with NcoI and EcoRI, and subcloned to the NcoI/EcoRI site of pET21d (Novagen). These plasmids were transformed into BL21(DE3) and the truncated and full-length forms of the 38k protein were expressed as described. The expressed proteins were purified as described above and used as the recombinant 38k protein and the truncated 38k protein. The 38k protein denotes native protein purified from smooth muscle.
Immunochemical Experiments
Rabbits and guinea pigs were injected with human p32. Their sera were used as anti-38k protein antibodies after affinity purification through NHS-sepharose (Amersham-Pharmacia) conjugated with human p32. For Western blotting, samples after SDS-PAGE were electrotransferred onto PVDF membrane to react with the anti-38k protein antibody. Immunoreaction was detected using goat anti–rabbit IgG conjugated with HRP (55689; Cappel) as secondary antibody, coupled with the ECL system (Amersham-Pharmacia).
To estimate the content of the 38k protein in various smooth muscle tissues, they were excised from a freshly killed adult rat, frozen in liquid nitrogen, broken into small chips with a frozen air hammer, and then mixed with an SDS-containing buffer for SDS-PAGE. The mixture was subjected to Western blotting, together with specified amounts of human p32 as a standard. Intensities of the immunoreacted bands were quantified by densitometry as described above.
For indirect immunofluorescence microscopy, we used the smooth muscle cell line, AC01, recently established from mouse aorta (Ohmi et al. 1997). This cell line appears in the Japanese Collection of Research Biosources Cell Bank. Cells were fixed with 3.5% formaldehyde, 1% glutaraldehyde, and 0.1% Triton X-100 on ice for 30 min, washed with PBS several times, and reacted with the anti-38k protein antibody or the following commercial antibodies: smooth muscle MHC polyclonal, BTI564 (Biochemical Technologies Inc.); desmin polyclonal, PS031 (SANBIO); anti–α-actin monoclonal, A2547 (Sigma Chemical Co.); and anti–β-actin mAbs, A5441 (Sigma Chemical Co.). To visualize the localization of proteins, the specimen was treated with the following secondary antibodies; goat anti–rabbit IgG conjugated with FITC (55641; Cappel), goat anti–rabbit IgG conjugated with rhodamine (55674; Cappel), goat anti–mouse IgG conjugated with rhodamine (55539; Cappel), and goat anti–guinea pig IgG conjugated with FITC (57000; Cappel). For visualization of mitochondria, cells were stained with MitoTracker Red CMXRos (Molecular Probes) before fixation according to the manufacture's protocol.
To demonstrate the colocalization of the 38k protein/myosin and that of the 38k protein/mitochondria in the smooth muscle cells (see Fig. 7), they were subjected to the double staining with anti-38k protein antibody, and with anti-MHC antibody or with MitoTracker Red CMXRos. Each pair of images were incorporated into Adobe Photoshop (version 4.0) and then processed to create product images (see Fig. 7c and Fig. f).
Figure 7.

Myosin assembly by human p32. a, Electron micrographs of 0.2 μM unphosphorylated myosin alone (1) or mixed with 0.6 μM human p32 (2). b, Length distribution of myosin filaments formed by human p32. The histogram was obtained by measurement of 150 filaments. An arrow indicates average length. c, Quantification of myosin assembly by the sedimentation assay. Myosin at 1.1 μM was mixed with human p32 and subjected to the centrifugation assay of assembly. Amounts of assembled myosin were plotted against concentration of human p32. Open and closed circles denote unphosphorylated and phosphorylated myosins, respectively.
Results
Myosin Filaments Formed by the 38k Protein
Purified myosin hardly assembled to form filaments when unphosphorylated (Fig. 2 a). However, when the 38k protein was present, unphosphorylated myosin could form filaments (Fig. 2 c). The preparation of the 38k protein did not show kinase activity for myosin when incubated with myosin in the presence of ATP for 1 h (data not shown). Thus, the myosin assembling activity could not be due to an elevation of phosphorylation level of myosin, but to an inherent property of the 38k protein.
Figure 2.
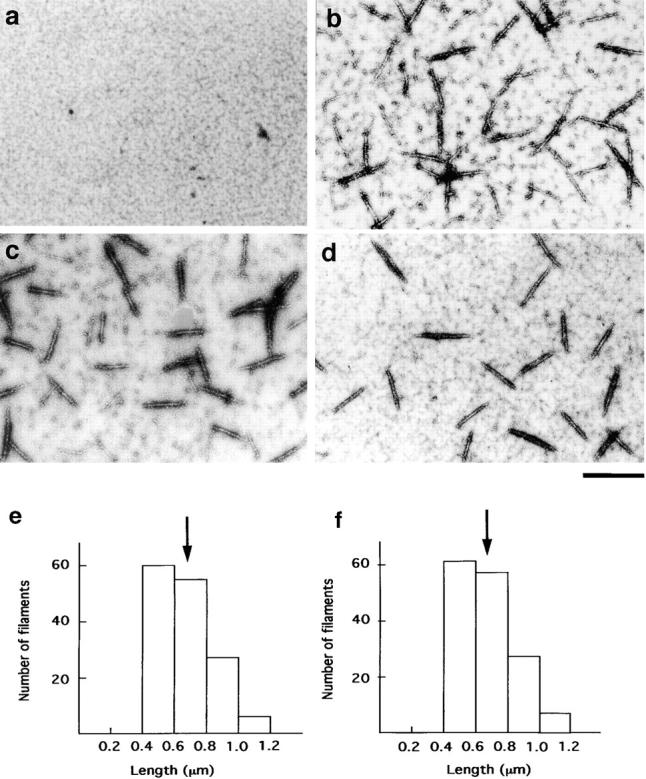
Electron micrographs of myosin filaments assembled with the 38k protein. In the absence of the 38k protein, unphosphorylated myosin (0.2 μM) did not form filaments (a). However, they were formed from phosphorylated myosin in the absence of the 38k protein (b). Unphosphorylated myosin (0.2 μM) was assembled by 0.55 μM of the 38k protein (c). Filaments formed by 0.2 μM unphosphorylated myosin with 0.69 μM telokin (d). Length distribution of filaments formed by 0.2 μM unphosphorylated myosin with 0.55 μM of the 38k protein (e) and that of filaments formed from phosphorylated myosin in the absence of the 38k protein (f). These histograms were obtained by measurement of the length of 150 filaments. Arrows in e and f indicate average length.
As shown in the histograms (Fig. 2e and Fig. f), the length of assembled filament was from 0.4–1.2 μm and the diameter was ∼20 nm. The length and diameter of these filaments were almost identical to those of filaments formed by phosphorylated myosin without the 38k protein (Fig. 2 b). The average length of the filaments formed with the 38k protein was 0.65 ± 0.15 μm, SEM (n = 150; Fig. 2 e, arrow), whereas that of phosphorylated myosin was 0.65 ± 0.16 μm, SEM (n = 150; Fig. 2 F, arrow).
Thus, the 38k protein keeps myosin in filaments, even though myosin is unphosphorylated. As will be discussed later, we expect that the 38k protein could prevent disassembly of thick filaments when myosin is dephosphorylated by phosphatase in vivo.
Quantification of Myosin-binding and Myosin-assembling Activities of the 38k Protein
To quantify the myosin filament assembling activity of the 38k protein, unphosphorylated myosin was incubated in the presence of various concentrations of the 38k protein and the assembled unphosphorylated myosin was precipitated by centrifugation. In the absence of the 38k protein, >90% of unphosphorylated myosin remained in the supernatant after centrifugation (Fig. 3 a, lanes 1 and 2). The amount of unphosphorylated myosin recovered in the precipitate was increased by increasing concentration of the 38k protein, as shown by the open circles in Fig. 3 d. Concentration to give half-maximal assembly was 0.8 ± 0.2 μM, SEM (n = 3), with the maximal effect being observed in the presence of ∼3 μM of the 38k protein. The amount of the 38k protein precipitated together with unphosphorylated myosin also increased, as is shown by open circles in Fig. 3 e. The maximum amount of the 38k protein bound to 1.1 μM myosin was 1.2 ± 0.1 μM, SEM (n = 3) and the concentration giving half-maximal binding was 1.7 ± 0.2 μM, SEM (n = 3).
The binding experiment was also performed using phosphorylated myosin. Phosphorylated myosin could assemble into filaments without the 38k protein, as was seen by microscopic observation (Fig. 2 b). When quantified by the centrifugation assay (Fig. 3 b), >90% of the myosin was sedimented (Fig. 3 b, lane 2). There was no obvious effect on the assembly when the 38k protein was added (Fig. 3 d, closed circles).
The closed circles in Fig. 3 e show the binding of the 38k protein to phosphorylated myosin. Similar to unphosphorylated myosin, the half-maximal binding was observed at 1.4 ± 0.2 μM, SEM (n = 3). The binding to phosphorylated myosin is distinct from telokin, which does not show any binding to phosphorylated myosin (Shirinsky et al. 1993). The distinction may derive from the difference in the binding site of myosin (see Fig. 5).
The binding of the 38k protein to skeletal muscle myosin was much weaker than that of smooth muscle myosin (see Fig. 5). The same was observed with cardiac myosin (see Fig. 5). These observations indicate that its binding is specific for smooth muscle myosin and agree with the observation that it is absent from skeletal muscle cells (see Fig. 6 d).
Figure 6.

Amino acid sequence of the 38k protein deduced from cDNA sequencing. a, The sequences of the 38k protein as denoted by chicken 38k (GenBank/EMBL/DDBJ accession number AB029946) are aligned with human p32 (GenBank/EMBL/DDBJ identification number, g338043) and mouse YL2 (GenBank/EMBL/DDBJ identification number, g743485). Two sequences of the 38k protein determined by direct amino acid sequencing are underlined. Hatched sequences indicate identical amino acids. b, Expression of human p32 in E. coli. SDS-PAGE pattern of total cell homogenate before (1) and after (2) IPTG induction. Purified human p32 (3) and the 38k protein (4). c, Immunoblotting with anti-38k protein antibody. Total homogenate of chicken gizzard (1, 2, and 3) and the purified 38k protein (4, 5, and 6). Protein staining (1 and 4) and immunostaining with anti-38k protein antibody (2 and 5), and with preimmune serum (3 and 6). 38k and h-p32 denote the bands of the 38k protein and human p32, respectively. d, Expression of the 38k protein in various muscles. The 38k protein was detected in the homogenate of various smooth muscle tissues of rat. Lanes: esophagus (1), stomach (2), duodenum (3), small intestine (4), cecum (5), rectum (6), uterus (7), aorta (8), air way (9), skeletal muscle (10), and cardiac muscle (11). 10 μg of each homogenate were applied.
To examine whether the protein can bind to myofibrillar components other than myosin, we studied its binding to actin filaments in the presence and absence of tropomyosin. The 38k protein bound to neither actin filaments (Fig. 3 c, lanes 3 and 4) nor actin filaments associated with tropomyosin (Fig. 3 c, lanes 5 and 6).
Absence of Cross-Talk between the 38k Protein and Telokin
To determine whether the 38k protein and telokin share the same binding site(s) in the myosin molecule, we allowed the 38k protein to compete with telokin for myosin binding. We initially confirmed that telokin induced assembly of unphosphorylated myosin under our experimental conditions (Fig. 2 d and 4 a). The effect was saturated when >2 μM of telokin was mixed with 1 μM of myosin. To this mixture, various amounts of the 38k protein were added. As shown in Fig. 4 c, even when the assembly was maximally induced by telokin, the addition of the 38k protein further stimulated assembly. The maximal binding of the 38k protein to myosin did not change whether or not telokin was present (Fig. 4 d, open circles). Accordingly, the maximal binding of telokin to myosin was not affected by the 38k protein (Fig. 4 d, closed circles).
Figure 4.

Relationship in myosin-assembling activity between the 38k protein and telokin. a, Effect of telokin on 1.1 μM myosin assembly. The amount of assembled myosin was plotted against telokin concentration. Open and closed circles indicate unphosphorylated and phosphorylated myosin, respectively. b, Amount of telokin coprecipitated with 1.1 μM myosin. The amount of telokin bound to myosin was plotted against telokin concentration. Open and closed circles indicate unphosphorylated and phosphorylated myosin, respectively. c, Myosin assembly in the presence of telokin and the 38k protein. In the presence of a saturation of telokin (3.5 μM, as judged from a), various concentrations of the 38k protein were mixed with 1.1 μM unphosphorylated myosin and then the mixture was centrifuged to precipitate myosin filaments. The amount of assembled myosin was plotted against concentration of the 38k protein. d, In the experiment of c, the amount of the 38k protein (open circles) and that of telokin (closed circles) bound to myosin filaments was plotted against concentration of the 38k protein.
The possibility that the 38k protein binds directly to telokin without affecting its myosin-binding activity can be rejected because telokin did not interact with the 38k protein when the interaction was examined by gel filtration (data not shown).
Binding of the 38k Protein to Myosin Fragments
To examine which portion of the myosin molecule the 38k protein binds to, we allowed it to bind to rod, LMM, or intact myosin. The fragments themselves are insoluble and easy to precipitate under the conditions for myosin binding. As shown in Fig. 5, the 38k protein could bind to LMM and rod, as well as myosin, although the maximum extent of binding to LMM was lower than those to rod and to myosin.
We then mixed the 38k protein with HMM of a soluble myosin fragment, and the mixture was subjected to the gel filtration. Both proteins were eluted in the different fractions, and their interaction was not detected (Fig. 5, inset).
Taken together, the above data indicate that the major site of the binding of the 38k protein should be in LMM. The indication is compatible with the absence of cross-talk between the 38k protein and telokin, because telokin binds to myosin at subfragment-2 (Shirinsky et al. 1993)
Both NH2-terminal and COOH-terminal Parts of the 38k Protein Are Required for Myosin Assembly
The myosin-assembling activity of the recombinant 38k protein was confirmed by observing that myosin filaments formed in its presence were the same as those formed by the native 38k protein under EM (data not shown). To test which part of the 38k protein contributes to the myosin-assembling activity, we produced several constructs of deletion mutants comparable to those of telokin (Silver et al. 1997).
As shown in Table , when full-length, recombinant 38k protein at 4 μM was mixed with 1.1 μM myosin in either phosphorylated or unphosphorylated form, the amount of the 38k protein bound to the myosin was ∼1.0 μM. However, the NH2-terminal–deleted mutants ΔN28, ΔN63, and ΔN88 did not bind to phosphorylated or unphosphorylated myosin. Similarly, the COOH-terminal–deleted mutants, ΔC20 and ΔC66, could not bind to either type of myosin. Thus, deletion of the NH2-terminal 28 or COOH-terminal 20 amino acid residues of the 38k protein completely abolished the interaction with myosin. The significance of these data will be discussed below with reference to the crystal structure of human p32, which was published recently by Jiang et al. 1999.
Table 1.
Binding of the 38k Protein and its Deletion Mutants to Myosin
| Unphosphorylatedmyosin | Phosphorylatedmyosin | |
|---|---|---|
| Full-length (1–207) | 1.00 | 1.09 |
| ΔN28 (29–207) | 0.02 | 0.02 |
| ΔN63 (64–207) | 0.01 | 0.02 |
| ΔN88 (89–207) | 0.01 | 0.01 |
| ΔNC (64–141) | 0.00 | 0.00 |
| ΔC66 (1–141) | 0.00 | 0.01 |
| ΔC29 (1–187) | 0.02 | 0.03 |
Mutants of the 38k protein (4.0 μM) were mixed with myosin (1.1 μM) in buffer A and precipitated with myosin as described in Materials and Methods. Amounts of the 38k protein or its mutants recovered in the precipitate were quantified and expressed in micromoles (μM). Data are the average of two sets of independent experiments. The numbers in parentheses refer to the number of the amino acid residue from the NH2 terminus of the full-length 38k protein.
Comparison of the Amino Acid Sequences of the 38k Protein and Human p32
Two partial amino acid sequences (LHTEGDKAFAQFLTDEI and REVSFQPTGESD) were obtained by cleavage with V8 protease. The former sequence was also obtained from undigested 38k protein, suggesting that the sequence is at the NH2 terminus of the protein. The amino acid sequence deduced from the cDNA sequence of the 38k protein (see Materials and Methods) is shown in Fig. 6 a. The calculated molecular weight was 23,681. Comparison with the GenBank/EMBL/DDBJ data base revealed that the sequence is highly homologous to human p32, which was originally identified as a protein copurified with human splicing factor-2 (Krainer et al. 1991; Honore et al. 1993). Alignment of the 38k protein to p32 showed that 78% of the amino acid sequence of chicken 38k protein was identical to that of human p32. Mouse YL2 (Luo et al. 1994), a homologue of p32, was also highly homologous (74%) with the 38k protein.
Characterization of Expressed Human p32 as a Homologue of the 38k Protein
Human p32 is known to contribute to various cellular functions, such as splicing of pre-mRNA (Krainer et al. 1991; Mayeda et al. 1992; Honore et al. 1993), binding of rev protein of the HIV virus to enhance duplication of the genomic DNA of the virus (Luo et al. 1994; Tange et al. 1996) and cell surface receptors for complements (Ghebrehiwet et al. 1994; Lim et al. 1996), and for high molecular weight kininogen (Joseph et al. 1996). However, to our knowledge, it has not yet been characterized as a cytoskeletal protein.
To clarify any functional difference between the 38k protein and human p32, we examined assembly of myosin by human p32. As shown in Fig. 7 a, it induced formation of filaments of unphosphorylated myosin, as well as the 38k protein did. The length distribution of the myosin filaments (Fig. 7 b) was similar to that of the 38k protein (Fig. 2 e), the average length of the filaments being 0.69 ± 0.16 μm, SEM (n = 150; Fig. 7 b, arrow). The myosin-assembling activity was also quantified by the centrifugation assay. The concentration of human p32 required to give half-maximal assembly estimated from Fig. 7 c was 0.9 ± 0.2 μM, SEM (n = 3), with the concentration giving half-maximal binding at 1.8 ± 0.2 μM, SEM (n = 3). These figures are similar to those of the 38k protein (compare Fig. 7 c with Fig. 3 d). Thus, the human p32 appears to possess similar activity to that of the 38k protein.
We stained the blot of SDS-PAGE of total homogenate of chicken gizzard and the purified 38k protein with anti-38k protein antibody. As shown in Fig. 6 c, the sole immunoreacting band is 38 kD. Because the antibody was raised against human p32 (see Materials and Methods), we conclude that the human p32 is a homologue of the 38k protein.
Distribution of the 38k Protein in Smooth Muscle Tissues
To examine the expression of the 38k protein in various smooth muscle tissues, their homogenates were subjected to Western blots. The 38k protein was detected in almost every smooth muscle tissue, including esophagus, stomach, small intestine, cecum, uterus, aorta, and airway as shown in Fig. 6 d. It was especially highly expressed in stomach and digestive tracts. Thus, the 38k protein can be thought of as a universal component of smooth muscle. It was also detected in cardiac muscle, but not clearly in skeletal muscle (Fig. 6 d, lanes 10 and 11). It could also be detected in nonmuscle tissues, such as liver, lung, spleen, and brain (data not shown), giving good conformity with the presence of human p32 in a variety of cells (Simon and Georgatos 1994; Deb and Datta 1996; Dedia and Muller-Esterl 1996; Joseph et al. 1996; Tange et al. 1996), but we cannot exclude the possibility that the reaction was due to contamination by smooth muscle cells from blood vessels.
Colocalization of the 38k Protein with Myosin in Cultured Smooth Muscle Cells
To determine whether the 38k protein is associated with myosin in smooth muscle cells, we examined its localization in the cultured smooth muscle cell line, AC01, by indirect immunofluorescence. Double staining (Fig. 8, a and b), followed by the image analysis (Fig. 8 c), indicates that the localization of the 38k protein coincides with myosin. Together with the same staining as shown in Fig. 8, a and b, the analysis indicates that the 38k protein is associated with myosin in the whole cells. In the case of the double staining of the 38k protein (Fig. 8 d) and mitochondria (Fig. 8 e, see below), the overlapping staining is limited to the central part of the cell (Fig. 8 f).
Figure 8.

Localization of the 38k protein in the cultured smooth muscle cell line AC01. a–c, Staining pattern with anti–38k protein (a) and that with anti-MHC (b) antibodies. To amplify colocalized region of the 38k protein and MHC, images of a and b were multiplied in c, i.e., c = a × b. d–f, The cell was stained with MitoTracker Red CMXRos to visualize position of mitochondria. Staining pattern with anti-38k protein (d) and that with MitoTracker Red CMXRos (e). To demonstrate colocalized region of the 38k protein with mitochondria, images of d and e were multiplied in f, i.e., f = d × e. Magnification is the same in all pictures. Bar, 30 μm.
To examine whether the 38k protein is localized in the contractile apparatus composed of myosin and/or actin, we performed further double staining with antibodies against components of the contractile apparatus other than myosin. At least two types of actin isoforms are detected in AC01 cells by Western blotting, α- and β-actin (data not shown). Localization of the 38k protein in the cells is close to that of α-actin (Fig. 9c and Fig. d), but does not necessarily match that of β-actin (Fig. 9e and Fig. f). The localization of the 38k protein overlaps that of desmin, a major component of dense bodies (Fig. 9g and Fig. h).
Figure 9.

Colocalization of the 38k protein with components of contractile apparatus in the cultured smooth muscle cell line, AC01. a–h, Double staining of AC01 cell with anti-38k protein antibody, as detected by FITC-labeled second antibody and with various antibodies as detected by rhodamine-labeled second antibody. Staining pattern with anti-38k protein antibody (a, c, e, and g) and with antibodies against MHC (b), α-actin (d), β-actin (f), or desmin (h). Magnification is the same in all pictures. Bar, 30 μm.
Human p32 has been found to localize in nuclei (Krainer et al. 1991; Mayeda et al. 1992; Honore et al. 1993; Luo et al. 1994; Simon and Georgatos 1994; Nikolankaki et al. 1996; Tange et al. 1996), mitochondria (Muta et al. 1997, Jiang et al. 1999), the vesicular fraction (Dedia and Muller-Esterl 1996), and at the cell surface (Ghebrehiwet et al. 1994; Deb and Datta 1996; Joseph et al. 1996; Lim et al. 1996). However, in our hands, with cultured smooth muscle cell, such localization, except in mitochondria, could not be confirmed clearly (Fig. 8, d–f).
Discussion
In this report, we identified and characterized a novel myosin-binding protein of smooth muscle, a protein of 38 kD that assembles myosin into filaments. The myosin-assembling activity is similar to the activity reported for telokin (Shirinsky et al. 1993). Telokin binds at the NH2-terminal end of subfragment-2 of unphosphorylated myosin to change myosin from the 10S form to the 6S form (Masato et al. 1997; Silver et al. 1997). Thus, unphosphorylated myosin can be assembled into filaments. However, the binding site for the 38k protein in the myosin molecule is different from that of telokin (Fig. 4 and Fig. 5).
It must be noted that telokin binds to only unphosphorylated myosin (Shirinsky et al. 1993). Therefore, it is expected in vitro that telokin may dissociate from myosin filaments to the cytosol when smooth muscle myosin is phosphorylated upon excitation of smooth muscle cells. However, because the 38k protein binds to both phosphorylated and unphosphorylated myosin, the 38k protein is thought to remain as a component of myosin filaments, whether or not smooth muscle cells are excited. The other distinction is that expression of telokin is limited to only a few types of smooth muscles (Gallagher and Herring 1991; Herring and Smith 1996), but the 38k protein is expressed in various types of smooth muscle cells, as shown in Fig. 6 d.
We compared the amino acid sequence of the 38k protein to other myosin-binding proteins. The similarity of the amino acid sequence of the 38k protein to telokin (Gallagher and Herring 1991; Yoshikai and Ikebe 1992) is unexpectedly low, <5%. The 38k protein also showed little sequence homology with myosin-binding proteins of striated muscle, such as C-protein (Furst et al. 1992; Okagaki et al. 1993; Weber et al. 1993), H-protein (Vaughan et al. 1993), M-protein (Noguchi et al. 1992), and titin (Labeit et al. 1990), or other components of smooth muscle that interact with myosin, such as the 130 kD subunit of myosin phosphatase (Shimizu et al. 1994) and MLCK (Olson et al. 1990). Thus, the primary structure of the 38k protein is unique amongst myosin-binding proteins, suggesting that its myosin-binding, and hence myosin-bundling, activities are by a novel mode.
Recent analysis of the crystal structure of human p32 showed that it consists of seven consecutive antiparallel β-sheets flanked by one NH2-terminal and two COOH-terminal α helices, and that the α helix at the NH2 terminus has extensive contact with the COOH terminus (Jiang et al. 1999). We expect that the myosin-binding site of the 38k protein is at the contact, because deletion of either NH2-terminal or COOH-terminal residues (Table ) leads to the loss of myosin-binding activity of the 38k protein.
We quantified the intracellular concentration of the 38k protein with an antibody as described in Materials and Methods. Its concentration in gizzard was 6.4 ± 0.5 μM, SD (n = 3). Since the concentration of myosin is reported as 46 μM (Yamazaki et al. 1987), the molar ratio of myosin to the 38k protein is ∼1 to 0.14. Cellular myosin in the relaxed state is mostly unphosphorylated (for review, see Somlyo and Somlyo 1994). From the kinetic profile shown in Fig. 3 d, the assembly of 1.1 μM unphosphorylated myosin was maximal in the presence of ∼3 μM of the 38k protein, indicating that 2.7-fold molar excess of the protein over myosin was required for full assembly. Therefore, the cytoplasmic concentration of the 38k protein is too low to explain myosin assembly in vivo. The Prosite database predicts that the 38k protein has residues that can be phosphorylated with protein kinase C at Thr130 and with casein kinase-2 at Thr2, Thr176, Ser185, and Thr86. It is possible that the myosin-assembling activity may be enhanced upon phosphorylation of the 38k protein. Another explanation is that, like thick filaments of skeletal muscle, a few myosin-binding proteins, including telokin (Shirinsky et al. 1993) and the 38k protein, are working at the same time to assemble myosin into thick filaments of smooth muscle.
As explained in relation to Fig. 9, the 38k protein has been reported to be present in a variety of cellular organelles, including mitochondria, nuclei, vesicles, and cell surfaces. The present study describes additional localization of cytoskeleton. We realized that the variety may be attributable to that of the cells/organs used for the respective experiments and speculated that all of them may not function at once. Rather, they might change according to the cellular states, e.g., differentiation. The first step in testing our speculation, we stained smooth muscle cell (AC01) under two distinct types, i.e., differentiated and dedifferentiated, using an antibody against the 38k protein in a similar way described for Fig. 8 and Fig. 9. Our unpublished data shows that the antibody densely stained the nucleoli of the dedifferentiated type. However, in the differentiated type, as shown in Fig. 8 and Fig. 9, the staining of nucleoli was obscured with the increase in the cytoskeletal staining. The results are compatible with our speculation, but require additional experiments to demonstrate it.
Acknowledgments
We wish to thank Mrs. Yuki Hanyuda, Mrs. Keiko Arai, and Mrs. Yukie Roppongi for excellent technical assistance. We also thank all our colleagues in our department for critical discussion and advice.
This work was supported in part by grants-in-aid for Scientific Research from the Ministry of Education, Science and Culture of Japan, and grants from the Ichiro Kanehara Foundation, the Yamanouchi Foundation for Research of Metabolic Disorders, and The Smoking Research Foundation.
Footnotes
T. Okagaki's present address is Laboratories of Marine Food Science, Faculty of Bioresources, Mie University, Tsu-city, Mie 514-0102, Japan.
Abbreviations used in this paper: C-protein, myosin-binding protein-C; HMM, heavy meromyosin; LMM, light meromyosin; MHC, myosin heavy chain; MLCK, myosin light chain kinase.
References
- Cooke P.H., Fay F.S., Craig R. Myosin filaments isolated from skinned amphibian smooth muscle cells are side-polar. J. Muscle Res. Cell Motil. 1989;10:206–220. doi: 10.1007/BF01739811. [DOI] [PubMed] [Google Scholar]
- Craig R., Smith R., Kendrick-Jones J. Light chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature. 1983;302:436–439. doi: 10.1038/302436a0. [DOI] [PubMed] [Google Scholar]
- Deb T.B., Datta K. Molecular cloning of human fibroblast hyaluronic acid-binding protein confirms its identity with P-32, a protein copurified with splicing factor SF2. J. Biol. Chem. 1996;271:2206–2212. doi: 10.1074/jbc.271.4.2206. [DOI] [PubMed] [Google Scholar]
- Dedia J., Muller-Esterl W. Kininogen binding protein p33/gC1qR is localized in the vesicular fraction of endothelial cells. FEBS Lett. 1996;399:255–258. doi: 10.1016/s0014-5793(96)01339-7. [DOI] [PubMed] [Google Scholar]
- Ebashi S. A simple method of preparing actin-free myosin from smooth muscle. J. Biochem. 1976;79:229–231. doi: 10.1093/oxfordjournals.jbchem.a131052. [DOI] [PubMed] [Google Scholar]
- Epstein H.F., Fischman D.A. Molecular anatomy of protein assembly in muscle development. Science. 1991;251:1039–1044. doi: 10.1126/science.1998120. [DOI] [PubMed] [Google Scholar]
- Frohman M.A., Dush M.K., Martin G.R. Rapid production of full-length cDNAs from rare transcriptsamplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst D.O., Gautel M. The anatomy of a molecular gianthow the sarcomere cytoskeleton is assembled from immunoglobulin superfamily molecules. J. Mol. Cell. Cardiol. 1995;27:951–959. doi: 10.1016/0022-2828(95)90064-0. [DOI] [PubMed] [Google Scholar]
- Furst D.O., Vinkemeier U., Weber K. Mammalian skeletal muscle C-proteinpurification from bovine muscle, binding to titin and the characterization of a full-length human cDNA. J. Cell Sci. 1992;102:769–778. doi: 10.1242/jcs.102.4.769. [DOI] [PubMed] [Google Scholar]
- Gallagher P.J., Herring B.P. The carboxyl terminus of the smooth muscle myosin light chain kinase is expressed as an independent protein, telokin. J. Biol. Chem. 1991;266:23945–23952. [PMC free article] [PubMed] [Google Scholar]
- Gallagher P.J., Herring B.J., Stull J.T. Myosin light chain kinases. J. Muscle Res. Cell Motil. 1997;18:1–6. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- Ghebrehiwet B., Lim B.-L., Peerschke E.I.B., Willis A.C., Reid K.B.M. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J. Exp. Med. 1994;179:1809–1821. doi: 10.1084/jem.179.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K., Okagaki T., Higashi-Fujime S., Kohama K. Bundling of actin filaments by myosin light chain kinase from smooth muscle. Biochem. Biophys. Res. Commun. 1994;199:788–791. doi: 10.1006/bbrc.1994.1298. [DOI] [PubMed] [Google Scholar]
- Herring B.P., Smith A.F. Telokin expression is mediated by a smooth muscle cell-specific promoter. Am. J. Physiol. 1996;270:C1656–C1665. doi: 10.1152/ajpcell.1996.270.6.C1656. [DOI] [PubMed] [Google Scholar]
- Honore B., Madsen P., Rasmussen H.H., Vandekerckhove J., Celis J.E., Leffers H. Cloning and expression of a cDNA covering the complete coding region of the P32 subunit of human pre-mRNA splicing factor SF2. Gene. 1993;134:283–287. doi: 10.1016/0378-1119(93)90108-f. [DOI] [PubMed] [Google Scholar]
- Horowitz A., Trybus K.M., Bowman D.S., Fay F.S. Antibodies probe for folded monomeric myosin in relaxed and contracted smooth muscle. J. Cell Biol. 1994;126:1195–1200. doi: 10.1083/jcb.126.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley H.E. Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. J. Mol. Biol. 1963;7:281–308. doi: 10.1016/s0022-2836(63)80008-x. [DOI] [PubMed] [Google Scholar]
- Ikebe M., Hartshorne D.J. Effects of Ca2+ on the conformation and enzymatic activity of smooth muscle myosin J. Biol. Chem. 260 1985. 13146 13153a [PubMed] [Google Scholar]
- Ikebe M., Hartshorne D.J. Proteolysis of smooth muscle myosin by Staphylococcus aureus proteasepreparation of heavy meromyosin and subfragment 1 with intact 20000-Dalton light chains Biochemistry. 24 1985. 2380 2387b [DOI] [PubMed] [Google Scholar]
- Ishikawa R., Yamashiro S., Matsumura F. Differential modulation of actin-severing activity of gelsolin by multiple isoforms of cultured rat cell tropomyosin. J. Biol. Chem. 1989;264:7490–7497. [PubMed] [Google Scholar]
- Ito M., Dabrowska R., Guerriero V., Hartshorne D.J. Identification of turkey gizzard of an acidic protein related to the C-terminal portion of smooth muscle myosin light chain kinase. J. Biol. Chem. 1989;264:13971–13974. [PubMed] [Google Scholar]
- Jiang J., Zhang Y., Krainer A.R., Xu R.-M. Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl. Acad. Sci. USA. 1999;96:3572–3577. doi: 10.1073/pnas.96.7.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph K., Ghebrehiwet B., Peerschke E.I.B., Reid K.B.M., Kaplan A.P. Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XIIidentity with the receptor that binds to the globular “heads” of C1q (gC1q-R) Proc. Natl. Acad. Sci. USA. 1996;93:8552–8557. doi: 10.1073/pnas.93.16.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King L., Jiang M.J., Huang T.-S., Sheu G.-C. Protease-susceptible sites and properties of fragments of aortic smooth-muscle myosin. Biochem. J. 1995;312:511–518. doi: 10.1042/bj3120511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainer A.R., Mayeda A., Kozak D., Binns G. Functional expression of cloned human splicing factor SF2homology to RNA binding proteins, U1 70k, and Drosophila splicing regulators. Cell. 1991;66:383–394. doi: 10.1016/0092-8674(91)90627-b. [DOI] [PubMed] [Google Scholar]
- Labeit S., Barlow D., Gautel M., Gibson T., Holt J., Hsieh C., Francke U., Leonard K., Wardale J., Whiting A., Trinick J. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature. 1990;345:273–276. doi: 10.1038/345273a0. [DOI] [PubMed] [Google Scholar]
- Lim B.-L., Reid K.B.M., Ghebrehiwet B., Peerschke E.I.B., Leigh L.A.E., Preissner K.T. The binding protein for globular heads of complement C1q, gC1qR. J. Biol. Chem. 1996;271:26739–26744. doi: 10.1074/jbc.271.43.26739. [DOI] [PubMed] [Google Scholar]
- Luo Y., Yu H., Peterlin B.M. Cellular protein modulated effects of human immunodeficiency virus type 1 rev. J. Virol. 1994;68:3850–3856. doi: 10.1128/jvi.68.6.3850-3856.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masato T., Numata T., Katoh T., Morita F., Yazawa M. Crosslinking of telokin to chicken gizzard smooth muscle myosin. J. Biochem. 1997;121:225–230. [PubMed] [Google Scholar]
- Mayeda A., Zahler A.M., Krainer A.R., Roth M.B. Two members of a conserved family of nuclear phosphoproteins are involved in pre-mRNA splicing. Proc. Natl. Acad. Sci. USA. 1992;89:1301–1304. doi: 10.1073/pnas.89.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muta T., Kang D., Kitajima S., Fujiwara T., Hamasaki N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997;272:24363–24370. doi: 10.1074/jbc.272.39.24363. [DOI] [PubMed] [Google Scholar]
- Nikolankaki K., Simons G., Georgatos S.D., Giannakouros T. A nuclear envelope-associated kinase phosphorylates arginine serine motifs and modulates interactions between the lamin B receptor and other nuclear proteins. J. Biol. Chem. 1996;271:8365–8372. doi: 10.1074/jbc.271.14.8365. [DOI] [PubMed] [Google Scholar]
- Noguchi J., Yanagisawa M., Imamura M., Kasuya Y., Sakurai T., Tanaka T., Masaki T. Complete primary structure and tissue expression of chicken pectoralis M-protein. J. Biol. Chem. 1992;267:20302–20310. [PubMed] [Google Scholar]
- Ohmi K., Masuda T., Yamaguchi H., Sakurai T., Kudo Y., Katsuki M., Nonomura Y. A novel aortic smooth muscle cell line obtained from p53 knock out mice expressed several differentiation characteristics. Biochem. Biophys. Res. Commun. 1997;238:154–158. doi: 10.1006/bbrc.1997.7218. [DOI] [PubMed] [Google Scholar]
- Okagaki T., Higashi-Fujime S., Ishikawa R., Takano-Ohmuro H., Kohama K. In vitro movement of actin filaments on gizzard smooth muscle myosinrequirement of phosphorylation of myosin light chain and effects of tropomyosin and caldesmon. J. Biochem. 1991;109:858–866. doi: 10.1093/oxfordjournals.jbchem.a123471. [DOI] [PubMed] [Google Scholar]
- Okagaki T., Weber F.E., Fischman D.A., Vaughan K.T., Mikawa T., Reinach F.C. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J. Cell Biol. 1993;123:619–626. doi: 10.1083/jcb.123.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson N.J., Pearson R.B., Needleman D.S., Hurwitz M.Y., Kemp B.E., Means A.R. Regulatory and structural motifs of chicken gizzard myosin light chain kinase. Proc. Natl. Acad. Sci. USA. 1990;87:2284–2288. doi: 10.1073/pnas.87.6.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi H., Wakabayashi T. Electron microscopic studies of myosin molecules from chicken gizzard muscle Ithe formation of the intramolecular loop in the myosin tail. J. Biochem. 1982;92:871–879. doi: 10.1093/oxfordjournals.jbchem.a134001. [DOI] [PubMed] [Google Scholar]
- Shimizu H., Itoh M., Miyahara H., Ichikawa K., Okubo S., Konishi T., Naka M., Tanaka T., Hirano K., Hartshorne D.J. Characterization of myosin-binding subunit of smooth muscle myosin phosphatase. J. Biol. Chem. 1994;269:30407–30411. [PubMed] [Google Scholar]
- Shirinsky V.P., Vorotnikov A.V., Birukov K.G., Nanaev A.K., Collonge M., Lukas T.J., Sellers J.R., Watterson D.M. A kinase-related protein stabilized unphosphorylated smooth muscle myosin minifilaments in the presence of ATP. J. Biol. Chem. 1993;268:16578–16583. [PubMed] [Google Scholar]
- Silver D.L., Vorotnikov A.V., Watterson D.M., Shirinsky V.P., Sellers J.R. Sites of interaction between kinase-related protein and smooth muscle myosin. J. Biol. Chem. 1997;272:25353–25359. doi: 10.1074/jbc.272.40.25353. [DOI] [PubMed] [Google Scholar]
- Simon G., Georgatos S.D. The lamin B receptor-associated protein p34 shares sequence homology and antigenic determinants with the splicing factor 2-associated protein p32. FEBS Lett. 1994;346:225–228. doi: 10.1016/0014-5793(94)00479-x. [DOI] [PubMed] [Google Scholar]
- Somlyo A.V., Somlyo A.P. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Somlyo A.V., Butler T.M., Bond M., Somlyo A.P. Myosin filaments have non-phosphorylated light chains in relaxed smooth muscle. Science. 1981;294:567–569. doi: 10.1038/294567a0. [DOI] [PubMed] [Google Scholar]
- Starr R., Offer G. Polypeptide chains of intermediate molecular weight in myosin preparations. FEBS Lett. 1971;15:40–44. doi: 10.1016/0014-5793(71)80075-3. [DOI] [PubMed] [Google Scholar]
- Stull J.T., Gallagher P.J., Herring B.P., Kamm K.E. Vascular smooth muscle contractile elements. Cellular regulation. Hypertension. 1991;17:723–732. doi: 10.1161/01.hyp.17.6.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange T.O., Jensen T.H., Kjems J. In vitro interaction between human immunodeficiency virus type 1 rev protein and splicing factor ASF/SF2-associated protein, p32. J. Biol. Chem. 1996;271:10066–10072. doi: 10.1074/jbc.271.17.10066. [DOI] [PubMed] [Google Scholar]
- Trybus K.M. Assembly of cytoplasmic and smooth muscle myosins. Curr. Opin. Cell Biol. 1991;3:105–111. doi: 10.1016/0955-0674(91)90172-u. [DOI] [PubMed] [Google Scholar]
- Trybus K.M., Huiatt T.W., Lowey S. A bent monomeric conformation of myosin from smooth muscle. Proc. Natl. Acad. Sci. USA. 1982;79:6151–6155. doi: 10.1073/pnas.79.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan K.T., Weber F.E., Einheber S., Fischman D.A. Molecular cloning of chicken MyBP-H (86kDa protein) reveals conserved protein structure in the myosin-binding protein (MyBP) family containing immunoglobulin C2 and fibronectin type III motifs. J. Biol. Chem. 1993;268:3670–3676. [PubMed] [Google Scholar]
- Weber F.E., Vaughan K.T., Reinach F.C., Fischman D.A. Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment. Eur. J. Biochem. 1993;216:661–669. doi: 10.1111/j.1432-1033.1993.tb18186.x. [DOI] [PubMed] [Google Scholar]
- Xu J.-Q., Harder B.A., Uman P., Craig R. Myosin filament structure in vertebrate smooth muscle. J. Cell Biol. 1996;134:53–66. doi: 10.1083/jcb.134.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki K., Itoh K., Sobue K., Mori T., Shibata N. Purification of caldesmon and myosin light chain (MLC) kinase from arterial smooth musclecomparisons with gizzard caldesmon and MLC kinase. J. Biochem. 1987;101:1–9. doi: 10.1093/oxfordjournals.jbchem.a121879. [DOI] [PubMed] [Google Scholar]
- Yoshikai S., Ikebe M. Molecular cloning of the chicken gizzard telokin gene and cDNA. Arch. Biochem. Biophys. 1992;299:242–247. doi: 10.1016/0003-9861(92)90270-7. [DOI] [PubMed] [Google Scholar]