

SUMMARY
All reported GnRH receptor mutants (causing human hypogonadotropic hypogonadism) are misfolded proteins that cannot traffic to the plasma membrane. Pharmacoperones correct mis-folding and rescue mutants, routing them to the plasma membrane where they regain function. Because pharmacoperones are often peptidomimetic antagonists, these must be removed for receptor function after rescue; in vivo this necessitates pulsatile pharmacoperone administration. As an antecedent to in vivo studies, we determined whether pharmacoperones need to be present at the time of synthesis or whether previously misfolded proteins could be refolded and rescued. Accordingly, we blocked either protein synthesis or intra-cellular transport. Biochemical and morphological studies using 12 mutants and 10 pharmacoperones representing three different chemical classes show that previously synthesized mutant proteins, retained by the quality control system (QCS), are rescued by pharmacoperones, showing that pharmacoperone administration in vivo likely need not consider whether the target protein is being synthesized at the time of drug administration.
Keywords: G protein coupled receptor, hypogonadotropic hypogonadism, protein folding, protein trafficking
INTRODUCTION
Because of interest in the GnRHR as a therapeutic target (Conn and Crowley, 1991; Ulloa-Aguirre et al., 2003) and the small size of this receptor (328 amino acids in the human sequence, which makes site-directed mutation more facile than for larger proteins), it has become an important model for understanding the folding of G protein coupled receptors (Conn et al., 2002; Ulloa-Aguirre et al., 2004; Castro-Fernandez et al., 2005; Janovick et al., 2002). Seventeen GnRHR point mutants have been identified from patients with human hypogonadotropic hypogonadism (HH; Leaños-Miranda et al., 2003; Leaños-Miranda et al., 2002; Ulloa-Aguirre et al., 2003; Ulloa-Aguirre et al., 2004) and all appear to exert their effect (i.e. loss or diminution of GnRH efficacy) by producing misfolded and misrouted receptors rather than by loss of the ability to bind ligand or couple to effector. Accordingly, 15 of the 17 HH-associated mutants can be rescued to some degree by pharmacological chaperones (“pharmacoperones”), small molecules (usually peptidomimetic antagonists) that enter cells and correct folding errors (Conn et al., 2002; Bernier et al., 2004a; Bernier et al., 2004b). In the two remaining cases (hGnRHR(Ser168Arg) and hGnRHR(Ser217Arg)), thermodynamically unfavorable substitutions cause severe twisting of transmembrane segment 4 (TMS4) or TMS5, respectively, irreversibly preventing the alignment of extracellular loop 2 (ECL2) and the amino terminal, needed for proper positioning of amino acids Cys14 and Cys200, a bridge which is requisite for creation of a properly folded human GnRHR that passes the cellular quality control system (Knollman et al., 2005; Janovick et al., 2006).
When coexpressed with wild type (WT) hGnRHR, many of the mutants isolated from HH patients, show a dominant-negative effect on WT expression, resulting in loss of WT plasma membrane binding and effector coupling (Leaños-Miranda et al., 2002; Leaños-Miranda et al., 2003), an effect that is explained by recognition of the WT-mutant oligomer by the QCS and retention of the oligomer in the endoplasmic reticulum (ER; Brothers et al., 2004). As in the case of mutants expressed alone, pharmacoperones rescue both the mutant and the WT receptor from ER retention of this oligomer by this dominant-negative effect.
In making the jump from cell culture to an in vivo model, it is necessary to address the fact that many pharmacoperones are actually antagonists of the receptors that they rescue. Accordingly, this will likely mean that drugs based on these agents will have to be administered in a pulsatile fashion to enable them to be washed out and avoid blocking the receptor from activation by agonist; this is independent of the route of administration. In principle, the need to administer pharmacoperones periodically could conflict with the need to maintain their presence if they are required at the time of synthesis of the target molecule. A key question for moving from cell culture to in vivo work is then, “can a misfolded/misrouted protein that is already retained by the QCS be rescued” or, alternatively, “is it necessary that the pharmacoperone be present at the time of synthesis?” For this reason experiments were designed to determine if it is necessary for pharmacoperones to be present at the time of (mutant) receptor synthesis in order to be functional.
METHODS
Materials
The GnRH analog, D-tert-butyl-Ser6-des-Gly10-Pro9-ethylamide-GnRH (Buserelin, Hoechst-Roussel Pharmaceuticals, Somerville, NJ), myo-[2-3H(N)]-inositol (Perkin Elmer, Boston, MA; NET-114A), competent cells (Promega, Madison, WI), PCR primers, DMEM, OPTI-MEM, lipofectamine, phosphate buffered saline, and pcDNA3.1 (Invitrogen, San Diego, CA), endofree maxi-prep kits (Qiagen, Valencia, CA), were obtained as indicated. The following chemical structures (collectively referenced as “pharmacoperones”) were utilized; those of the quinolone class are prefaced by the letter “Q” and those of the indole class by the letters “IN” and were produced by Merck and Company (Ashton et al., 2001a; Ashton et al., 2001b; Ashton et al., 2001c; DeVita et al., 1999a; DeVita et al., 1999b; DeVita et al., 2001; Walsh et al., 2000): Q89, (7-chloro-2-oxo-4-{2-[(2S)-piperidin-2-yl]ethoxy}-N-pyrimidin-4-yl-3-(3,4,5-trimethylphenyl)-1,2-dihydroquinoline-6-carboxamide); Q76, (N-(7-chloro-3-(3,5-dimethylphenyl)-2-oxo-4-{2-[(2S)-piperidin-2-yl]ethoxy}-1,2-dihydroquinolin-6-yl)-N′-cyclopropylurea); Q08, ((2S)-2-(2-{[7-chloro-6-[(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]-3-(3,5-dimethylphenyl)-2-oxo-1,2-dihydroquinolin-4-yl]oxy}ethyl)piperidinium trifluoroacetate); IN30, ((2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl]-2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-{2-[4-(methylsulfinyl)phenyl]ethyl}propan-1-amine); IN31b, ((2S)-N-[2-(4-carboxyphenyl)ethyl]-2-[5-[1,1-dimethyl-2-(4-methylpiperazin-1-yl)-2-oxoethyl]-2-(3,5-dimethylphenyl)-1H-indol-3-yl]propan-1-aminium trifluoroacetate); IN3, ((2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl]-2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-(2-pyridin-4-ylethyl)propan-1-amine. Erythromycin-derived macrolides were prepared by Abbott Laboratories (Bush et al., 1999; Diaz et al., 1999) and are prefaced by the letter “A.” A-7662.0, (Erythromycin A); A-64755.0 (11-deoxy-11-[carboxy-phenylethylamino]-6-O-methyl-erythromycin A 11,12-(cyclic carbamate)); A-177775.0, (3′-N-desmethyl-3′-N-cyclopentyl-11-deoxy-11-[carboxy-(3,4-dichlorophenylethylamino)]-6-O-methyl-erythromycin A 11,12-(cyclic carbamate)); A-222509.0, 3′,3′-N-desmethyl-3′,3′-N-cyclopropylmethyl-11-deoxy-11-[carboxy-(3-chloro,4-fluoro-phenylethylamino)]-6-O-methyl-erythromycin A 11,12-(cyclic carbamate)). Cycloheximide and monensin (Sigma, St. Louis, MO) were obtained as indicated. ER tracker™ dye and Wheat Germ Agglutinin-AlexaFluor633 were obtained from Molecular Probes (Eugene, OR).
Other reagents were obtained from commercial sources and were of the highest degree of purity available. GnRHR WT and mutant cDNAs were prepared using site directed mutagenesis, as reported (Janovick et al., 2002). The identity of all cDNA mutants and the correctness of all PCR-derived coding sequences were verified by ABI PRISM 3130 Genetic Analyzer, according to manufacturer’s instructions (Applied Biosystems, Foster City, CA).
Creation of Stable (tTA + Mutant GnRH Receptor) HeLa Cells
The stable HeLa (tTA; tetracycline-controlled transactivator; Krestel et al., 2001) cell line was a kind gift from Dr. Peter Seeburg (Max-Planck-Institute for Medical Research, Heidelberg). The cells were maintained in growth medium (DMEM/10%FCS/20μg/ml Gentamicin) and grown at 37 C, 5% CO2 in a humidified atmosphere until the cell density reached about 90%.
The human (hGnRHR(E90K) (“E90K”)) mutant GnRHR was cloned into pTRE2-Hygromycin vector (the response vector) and then transfected into the stable HeLa (tTA; tTA binds the TRE and activates transcription in the absence of Tetracycline or Doxycycline) cell line. Selection antibiotics were used at 400 μg/ml G418 + 200 μg/ml Hygromycin. Single colonies were selected and screened for expression of the mutant hGnRHR(E90K). E90K was selected for these studies since it is normally expressed at unmeasurably low levels at the plasma membrane (assessed by radioligand binding or by Buserelin-stimulated IP, Janovick et al., 2002) and, once rescued turns over at a rate that is indistinguishable for the WT receptor.
Transient Transfection and Co-Transfection
Cos-7 cells were cultured, plated and transfected as previously reported (Janovick et al., 2002). Cells were transiently transfected with 20 ng DNA of WT human or mutant GnRHR (as indicated) and 80 ng pcDNA3.1 without insert (“empty vector”), and 1 μl lipofectamine in 0.125 ml OPTI-MEM (room temperature), according to manufacturer’s instructions. Empty vector (pcDNA3.1, without insert) was included to bring the total cDNA to 100 ng/well. 100 ng/well is a concentration of cDNA that does not interfere with the transfection efficiency (Brothers et al., 2004). Twenty-two and 22.5 hours after transfection, the medium was removed and replaced with DMEM/10%FCS/20 μg/ml Gentamicin containing cycloheximide (20μg/ml), monensin (10 μM), or medium alone and then incubated for 30 or 60 minutes as a “preincubation” period; cycloheximide and monensin were continuously present thereafter for a total of 24.5 or 25 hours. After the preincubation time, the fluid was removed and the indicated pharmacoperone was added and incubated for 4 hours.
Confocal Experiments
Two-well glass coverslip bottom culture slides (Costar) were soaked in 12 N HCl for 1 h to facilitate cell attachment. The slides were then rinsed extensively with sterile water and once with growth medium prior to use. 105 cells in 1 ml DMEM/10% fetal calf serum/20 μg/ml Gentamicin were plated per well in chambered slides and co-transfected with hGnRHR(E90K) and the GFP-tagged E90K mutant as described above. Twenty-two hours after transfection, the cells were pretreated with 20 μg/ml cycloheximide, then IN3 (1 μg/ml) as described above. One micromolar ER tracker™ dye and 5 μg/ml Wheat Germ Agglutinin-AlexaFluor633 (both from Molecular Probes, Eugene, OR) were diluted in DMEM/0.1% BSA, supplemented with 10 mM HEPES pH 7.4, and added to the cells at room temperature. After approximately 10 minutes, cells were imaged with a Leica TCS SP confocal microscope (Leica Microsystems, Exton, PA) using a 40x NA1.25 Pl Apo objective. ER tracker was excited at 361nm and emission was detected in the 400nm–470nm interval. GFP was excited at 488nm and detected in the 500nm–570nm interval and Alexa633 excited at 633nm and detected at 650–720nm. GFP and Alexa 633 were imaged simultaneously; ER tracker was imaged sequentially to eliminate the possibility of bleed through into the GFP channel. Images of single confocal planes were contrast enhanced in Photoshop 7.0 (Adobe Systems Incorporated, San Jose, CA). Translocation of the mutant receptor was measured using MetaMorph (Molecular Devices, Union City, CA) in single confocal sections, approximately 1 μM thick, from 6 cells per group. GFP, WGA and ER Tracker, displayed in individual channels, were segmented by intensity using the auto-threshold function. The percentage of total integrated intensity of GFP per section that is co-localized with ER Tracker and WGA-AlexaFluor633 respectively, were measured using the “Measure Co-localization” function. Data shown are mean values per group ± S.E.M.
Inositol Phosphate (IP) Assays
Cells were treated with pharmacoperones as previously reported (Janovick et al., 2001; Janovick et al., 2003), where indicated. Cells were washed then “pre-loaded” for 6 hours (unless otherwise indicated) with 4 μCi/ml myo-[2-3H(N)]-inositol in 0.25 ml DMEM (prepared without inositol; Brothers et al., 2004). Then, cells were washed twice with 0.3 ml DMEM (without inositol) containing 5 mM LiCl (LiCl prevents inositol phosphate (IP) degradation), then treated for 2 h with in 0.25 ml of 100 nM Buserelin in the same medium. Total IPs were determined as previously described (Huckle and Conn, 1987).
Radioligand binding
Cells were cultured and plated in growth medium as described above, except 105 cells in 0.5 ml growth medium were added to 24-well Costar cell culture plates (cell transfection and medium volumes were doubled accordingly). Twenty-two and 22.5 hours after transfection, respectively, the medium was removed and replaced with 0.5 ml fresh growth medium, containing cycloheximide (20 μg/ml) for a 30 or 60 min preincubation as shown. Twenty-seven hours after transfection, cells were washed twice with 0.5 ml DMEM containing 0.1% BSA with 20 μg/ml gentamicin and 20 μg/ml cycloheximide, then 0.5 ml of DMEM containing cycloheximide was added in order to duplicate the conditions in the IP assay. After 18 h, cells were washed twice with 0.5 ml DMEM/0.1% BSA/10 mM HEPES, then a range of concentrations of [125I]-Buserelin prepared in our laboratory (as modified from Janovick et al., 2006, for use in cell cultures, specific activity is 700–800 μCi/μg, from 1.25 × 105 to 4 × 106 cpm/ml) in 0.5 ml of the same medium was added to the cells and allowed to incubate at room temperature for 90 min (Janovick et al., 2006). This time was selected in order to achieve maximal binding. The amount of new receptor synthesis in this period is negligible at room temperature. After 90 min, the media was removed and radioactivity was measured as described previously (Janovick et al., 2006). To determine nonspecific binding, the same concentrations of radioligand were added to similarly transfected cells in the presence of 10 μM unlabeled GnRH. Saturation binding curve fits and calculations (Bmax and Kd) were computed with Sigma Plot 8.02 (Jandel Scientific Software, Chicago, IL, USA), using a nonlinear one-site binding model.
Statistics
Each experiment was repeated a minimum of three times. Replicates of at least 6 data points for each treatment group within an experiment were analyzed with one-way ANOVA, followed by paired Student’s t-test for individual comparisons (SigmaStat 3.0, Jandel Scientific Software, Chicago, IL; P < 0.05 was considered significant).
RESULTS
Figure 1 shows the effect of varying the time of rescue and time of preloading with tritiated inositol on total IP production (CPM) following stimulation with a saturating concentration of the GnRH agonist, Buserelin (100 nM). The number of CPM recovered is a linear function of the amount of the time allowed for radiolabeled inositol incorporation into the cells. Presumably, this reflects an increase in specific activity of the intracellular inositol pool over time.
Figure 1.
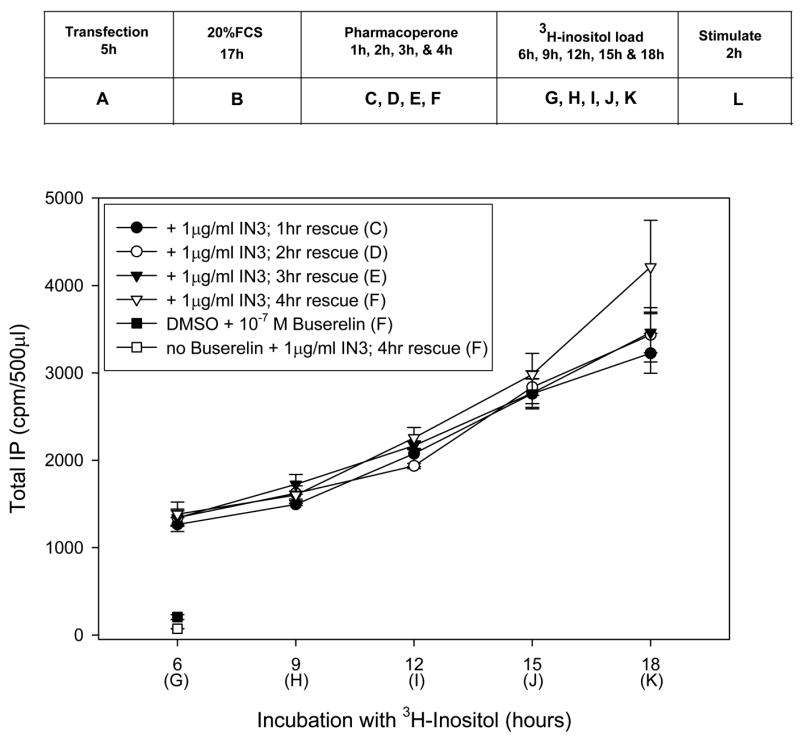
Effects of IN3 and 3H-inositol exposure times on total IP production (CPMs) in stably transfected HeLa cells containing the hGnRHR (E90K) mutant in response to 100 nM Buserelin (except for the single point noted). Cells were treated with 1 μg/ml IN3 for 1–4 hours (“C–F”) then preloaded with 3H-inositol for 6–18 hours (“G–K”). Averages and SEMs were calculated from at least 3 independent experiments performed in replicates of 6.
More surprisingly however, was the observation that the amount of time for rescue suggested that anywhere from 1–4 hours exposure to the pharmacoperone IN3 (“C–F” shown in the box) resulted in similar amounts of rescue of hGnRHR (E90K). It is important to notice that the rescue period may be viewed as including the period of tritiated inositol loading (6–18 h; “G–K,” shown in the box), since the IN3 had been removed from the medium prior to the preloading period, “G–K,” This period allows time for the mutant to traffic to the plasma membrane and residual IN3 is likely present in the cells. For this reason it would be incorrect to conclude that a 1 hour rescue time (alone) is sufficient time to effect rescue without this additional time.
Table 1 shows the effect of the protein synthesis inhibitor, cycloheximide, on receptor Bmax, which is diminished by 26% with a 30 min preincubation; as expected Kd was unchanged. A 60 min preincubation does not increase the effect on Bmax.
Table 1. Effect of Cycloheximide on Kd and Bmax of GnRHR.
Table shows binding characteristics of human WT GnRHR. Cos-7 cells were transiently transfected with 20 ng hWT GnRHR + 80 ng empty vector as described in Methods. Twenty-two hours after transfection, the cells were pretreated with or without cycloheximide (20 μg/ml) for 30 or 60 min. Freshly prepared cycloheximide was then added for 4 hours, cells were washed +/− cycloheximide in DMEM/BSA, then DMEM was added +/− cycloheximide for 18 hours. Receptor Kd and Bmax was determined by Scatchard analysis as described in Methods. Averages and SEMs were calculated from at least 3 independent experiments performed in replicates of 4.
| Medium | Cycloheximide | |||
|---|---|---|---|---|
| 30min | 60min | 30min | 60min | |
| Kd (pMol) | 996 ± 310 | 721 ± 270 | 742 ± 150 | 779 ± 280 |
| Bmax (fMol/105 cells) | 5.03 ± 1.31 | 4.29 ± 1.06 | 2.81 ± 0.39 | 2.49 ± 0.42 |
In Figure 2, the box shows a time line for the incubation time(s) for transfection, protein inhibition preincubation, rescue and 3H preload and stimulation. We selected (Figure 2A) the “longest” condition from Fig. 1 (4 h rescue with IN3, “E” in the box, 18 h loading with tritiated inositol, “G” in the box) and, separately, the “shortest” condition (Fig. 2B) from the same Figure (4 h rescue with IN3, “E” in the box, 6 h loading with tritiated inositol; “F” in the box) in order to compare the effect of inhibition of protein synthesis (by cycloheximide) or protein trafficking (monensin inhibits trafficking from the Golgi to the plasma membrane) in the stably transfected HeLa or transiently transfected Cos-7 cells. Inhibitors were added either 30 “C” or 60 minutes “D” in the box, prior to addition of IN3. Each inhibitor (i.e. cycloheximide and monensin) was then present through the end of the experiment (i.e. during the periods of time when the rescue drug was present, the tritiated inositol loading period and during the Buserelin challenge and all washes). Although the cells remained responsive the Buserelin at all time points and excluded trypan blue (not shown, an indicator of viability), some cell debris (yellow) was visible in micrographs (Fig. 3).
Figure 2.

Effect of inhibiting protein synthesis or protein trafficking on the ability of IN3 to rescue the receptors. Stably transfected HeLa cells or Cos-7 cells transiently transfected with the E90K mutant GnRHR were treated with either the protein synthesis inhibitor cycloheximide (20 μg/ml) or monensin (10 μM) for 30 or 60 minutes prior to addition of 1 μg/ml IN3 for 4 hours (“E”). Inhibitors were present up until the end of the agonist challenge, at which point total IP production was assessed. (A): cells were pre-loaded for 18 hours with 3H-inositol (“G”). (B): cells were pre-loaded for 6 hours with 3H-inositol (“F”). Cells were stimulated with 100 nM Buserelin for 2 hours (“H”). Averages and SEMs were calculated from at least 3 independent experiments performed in replicates of 6.
Figure 3.
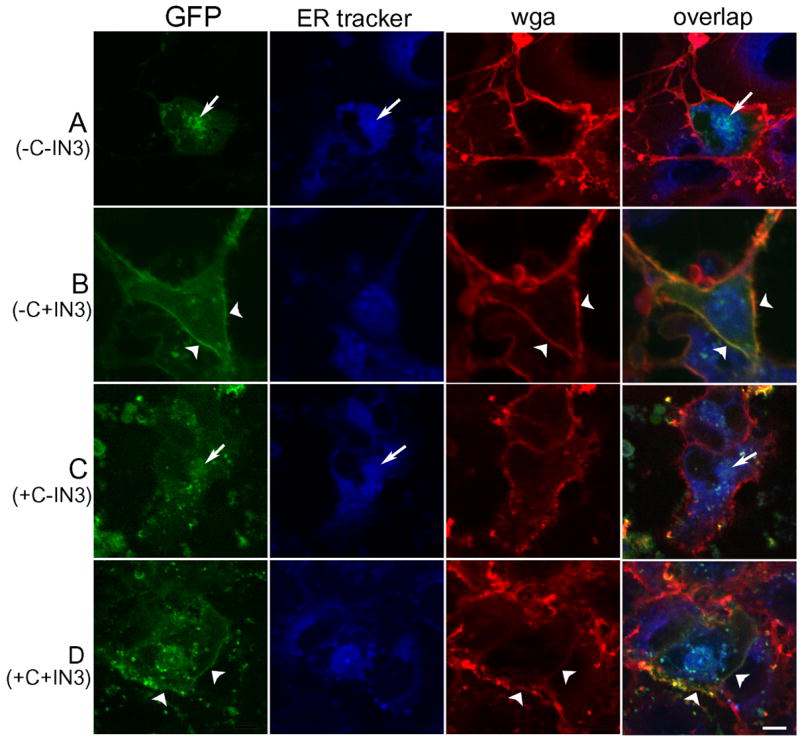
Confocal images of Cos-7 cells transiently co-transfected with the GFP-tagged hGnRHR(E90K) in green and the dominant-negative hGnRHR (E90K) and labeled with ER tracker™ (blue) and wheat-germ agglutinin Alexa-633 (red). A and C: Mutant receptor is trapped in the ER where it co-localizes with ER Tracker™ (arrows). B and D: Treatment with 1 μg/ml IN3 for 4 hours allows the mutant receptor to leave the ER and move to the plasma membrane (arrowheads). Treatment with 20 μg/ml cycloheximide for 25 hours to inhibit new protein synthesis (C and D) did not affect the ability of IN3 to rescue the mutant phenotype. The bar in D shows a distance of 10 μM in all images.
The data from the addition of 30 or 60 minute exposure to inhibitor prior to Buserelin are very similar; this indicates that a preincubation period longer than 30 minutes does not result in further inhibition and the period of time (and the dose) appears sufficient to inhibit production of new protein. This is consonant with the observations in Table 1. This time was selected to be substantially longer than mean transit time of ribosomes on a mammalian mRNA, about 2–8 minutes (Roper and Wicks, 1978).
In the longest condition of exposure to inhibitor, both Cos-7 and HeLa cells expressing hGnRHR(E90K) showed diminished total IP production in response to Buserelin, reflecting the diminution of protein synthesis itself by cycloheximide or the diminished trafficking due to monensin; there was less absolute receptor protein in the cell. Nonetheless, in the presence of cycloheximide or monensin, IN3 resulted in measurable rescue of protein during a period of time when protein synthesis is shut off by cycloheximide. This suggests that, of the mutant misfolded protein synthesized before the inhibitors and retained, that rescue of this previously synthesized mutant could still occur. Accordingly the pharmacoperone serves to aid in receptor refolding at the post-translational level.
In Figure 2B the net preloading time with tritiated inositol was decreased to 6 hours. This was done to decrease the total time of exposure to inhibitor, yet provide a condition in which the effect of rescue could be measured. As expected the decreased labeling time produced fewer total CPM (see CPM values in Figure 1), yet the decreased total exposure to inhibitor increased the total percentage of rescue compared with cells that had never been exposed to inhibitor.
Figure 3 shows images of single confocal sections, approximately 1 μm thick, where the endoplasmic reticulum, labeled by ER tracker™, is shown in blue, and plasma membrane, labeled with wheat-germ agglutinin, is shown in red. GFP tagged-GnRHR(E90K) is shown in green. Cells were pre-treated with cycloheximide (20 μg/ml for 25 hours, “C and D”) and IN3 (1 μg/ml for 4 hours, “B and D”) prior to imaging. Cells with extremely high levels of expression show protein retention in the ER and abnormal morphology and therefore only cells with weak to moderate expression level were taken into account. In A and C, the mutant receptor is trapped in the ER (arrows). In B and D, after treatment with IN3, most of the receptor has moved out of the ER and into the plasma membrane (arrow heads). Translocation was unaffected by the inhibition of new protein synthesis by cycloheximide. This observation supports the biochemical data in suggesting that a misfolded-misrouted mutant which has been previously synthesized and retained in the ER can be rescued by the pharmacoperone, IN3.
Measurements of the integrated intensity of GFP co-localized with ER tracker in single confocal sections, approximately 1 μm thick, show that ER localization decreased from 76 ± 5% to 44 ± 8% of total GFP fluorescence, after IN3 treatment, in the absence of cycloheximide. When new protein synthesis was inhibited by cycloheximide, ER localization decreased from 81 ± 8% to 27 ± 4% of total GFP. In the same conditions, the integrated intensity of GFP co-localized with the membrane marker WGA-Alexa Fluor 633 increased from 3 ± 1% to 23 ± 7% of total GFP fluorescence in the absence of cycloheximide, and from 19 ± 2% to 29 ± 5% after cycloheximide treatment. These observations suggest that most of the hGnRHR(E90K) transfected in Cos7 cells is present in the endoplasmic reticulum. Treatment with IN3 causes the receptor to move out of the ER and, after 4 hours, part of it appears on the plasma membrane. The effect of IN3 on receptor location is independent of new protein synthesis. This suggests that IN3 can act on previously mislocated receptors and release them from the ER. P values for a non-paired t-test comparing percentage of total GFP fluorescence in the ER before and after IN3 treatment were 0.013 with no cycloheximide and 0.0007 after cycloheximide treatment. The lack of new receptor synthesis and mandatory temporary sequestration in the ER allows a higher percentage of the total to be rescued after IN3 treatment. P values comparing membrane distribution of the receptor mutant with IN3 treatment were 0.03 and 0.13, without and with cycloheximide treatment, respectively. The lower significance for the cycloheximide group is probably due to the increased non-specific signal given by cell debris brightly fluorescent in both GFP and AlexaFluor633 channels. Even though the large majority of cells appeared normal after treatment, and a propidium iodide test failed to show significantly increased cell death (data not shown), there are visibly more cell debris after cycloheximide treatment (Figure 3, C and D, extracellular yellow bright spots).
Figure 4 shows that a selection of naturally occurring mutants behaves similarly to E90K, in that they are rescued even when protein synthesis has been inhibited by cycloheximide or when monensin was used to block the Golgi apparatus. Blockade of protein synthesis results in less total protein that is rescued compared to uninhibited cells, an observation that is likely due to the decreased total amount available for rescue.
Figure 4.
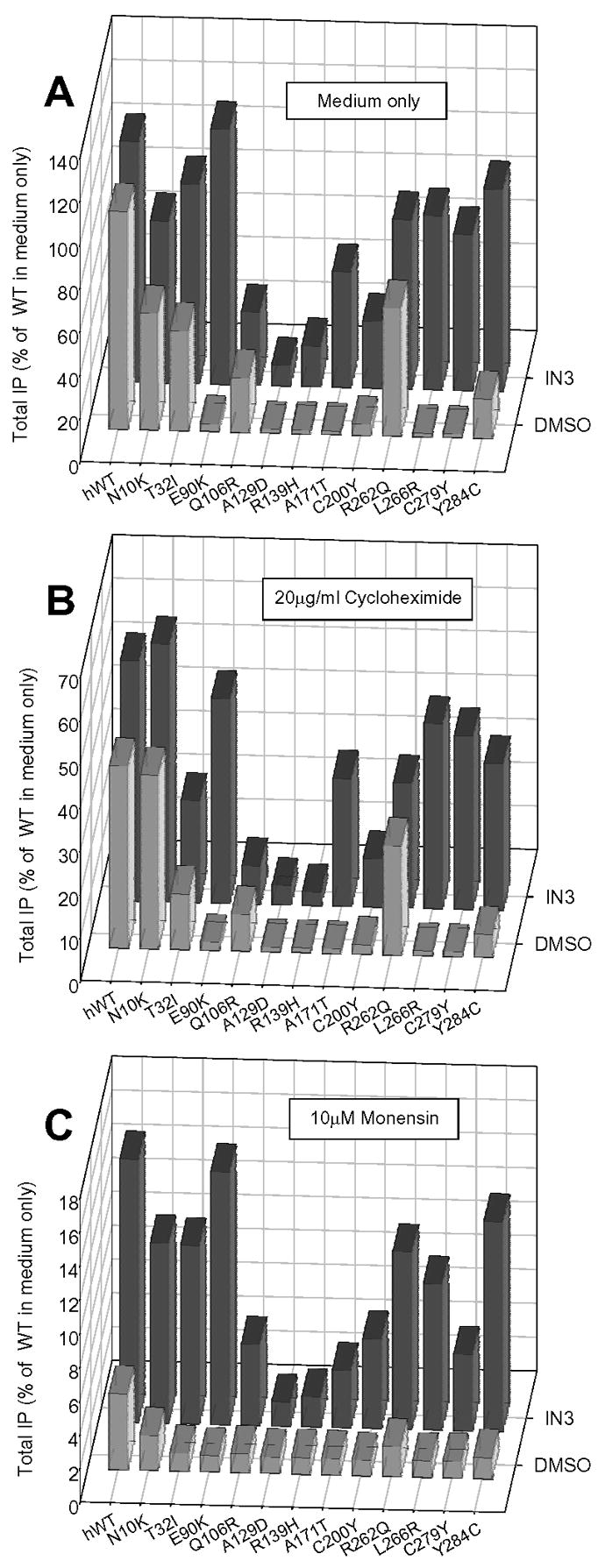
Effect of inhibiting protein synthesis or protein trafficking on the ability of IN3 to rescue the HH mutant GnRHRs. Cos-7 cells transiently transfected with mutant GnRHRs were treated with either medium only (A) the protein synthesis inhibitor (20 μg/ml) cycloheximide (B) or (10 μM) monensin (C) for 60 minutes prior to addition of IN3. Cells were pre-loaded for 6 hours with 3H-inositol. Inhibitors were present up until the end of the agonist challenge, at which point total IP production was assessed. Averages and SEMs were calculated from at least 3 independent experiments performed in replicates of 6.
Figure 5 shows that a range of rescue agents from three different chemical classes, indoles, quinolones and erythromycin macrolides continue to function even when protein synthesis is blocked by cycloheximide. Chemically related pharmacoperones are arranged in the order listed from lowest affinity (left) to highest affinity (right). For some of the lowest affinity agents, rescue was not measurable. This is not surprising, based on earlier studies (Janovick et al., 2003).
Figure 5.

Effect of inhibiting protein synthesis on the ability of three different chemical classes of pharmacoperones to rescue the hGnRHR E90K mutant. Cos-7 cells transiently transfected with mutant GnRHR were treated with either medium alone or the protein synthesis inhibitor (20 μg/ml) cycloheximide for 60 minutes prior to addition of pharmacoperones. Cells were pre-loaded for 6 hours with 3H-inositol. Cycloheximide was present up until the end of the agonist challenge, at which point total IP production was assessed. (A): Indoles (1 μg/ml each). (B): Quinolones (1 μg/ml each). (C): Erythromycin macrolides (1 μg/ml each). Averages and SEMs were calculated from at least 3 independent experiments performed in replicates of 6.
DISCUSSION
Our prior cell culture studies have relied on transient transfection in Cos-7 cells in order to show that pharmacoperones rescue many misfolded mutants of the GnRHR. While Cos-7 cells cannot be stably transfected, they are well-known for their ability to aggressively synthesize proteins. In the current studies, we used both Cos-7 cells and a stably transfected HeLa cell line expressing hGnRHR (E90K) mutant. The stable cells were included in these studies because the transcription and translation products may be presumed to be at equilibrium and because we wished to consider a second system to determine if primate cells derived from different tissues behaved similarly to the Cos-7 cells. HeLa cells were selected as they are human derived while Cos-7 cells are non-human primate derived. Both Cos (from monkey renal tissue) and HeLa (from a human cervical cancer) cells are primate-derived.
The recent studies rely on inhibitors of protein synthesis (cycloheximide; Roper and Wicks, 1978), or of vesicular movement from the Golgi to the membrane (monensin; Ellinger and Pavelka, 1984). The data indicate that previously synthesized mutant GnRHRs that are misfolded and retained by the ER QCS can be rescued even when the pharmacoperone is not present and after the synthesis is complete.
The classic studies of Anfinsen (Kresge et al., 2006; Haber and Anfinsen, 1962) in which RNase A, an enzyme of 124 residues and four disulfide bonds, was denatured in urea and with a sulfhydral reducing agent, then subsequently renatured with restoration of 90% activity, first suggested that the native conformation of some proteins is adopted spontaneously. Our view of protein maturation and routing has grown more complex as we have come to understand the roles that pro-proteins, post-translational modifications, and the quality control system play along with the endogenous chaperone proteins that assist in folding and serve to identify and retain misfolded proteins (Schrag et al., 2003; Kleizen and Braakman, 2004; Castro-Fernandez et al., 2005). One notable feature is that the recognition of “defective” mutant proteins is based on common features of misfolded proteins (i.e. an exposed hydrophobic plate on a soluble protein for example) rather than failure of function such as inability to bind substrate or ligand. This consideration explains why proteins that are retained by the QCS, frequently retain intrinsic function, but are misrouted.
The observation that many protein mutants are actually misfolded proteins and can be rescued by low molecular weight pharmacoperones which correct folding errors presented the possibility that such molecules might serve as therapeutic agents. Such pharmacoperones are often receptor antagonists, selected because of their known ability to bind to the receptor. Accordingly, once the rescued molecule is stabilized in the lateral plane of the plasma membrane, pharmacoperones must be removed so that they do not block the active site of the receptor from agonist.
Determining the period and duration for such pulsatile administration is also a function of whether a preexisting (i.e. ER-retained) mutant can be rescued or whether the pharmacoperone must be present at the time of synthesis in order to serve as a template for the nascent receptor. If the latter condition occurs and the pharmacoperone need not be present at the moment of synthesis, this means that the flexibility in determination of the pulse pattern may be optimized to serve only the requirement for its removal, so as not to act as an antagonist. In this case the pharmacoperone drug could, in principle, have to be present less often and for shorter periods.
A recent observation (Janovick et al., 2006; Conn et al., 2006a; Conn et al., 2006b) is that some WT proteins are routed with less than 100% efficiency to the plasma membrane, the balance being misfolded and retained in the ER. This presents the possibility that it might be possible to utilize pharmacoperone to mediate (enhance) the levels of WT proteins in order to obtain therapeutic benefits.
One way to consider this is that pharmacoperones alter the molecules of a dynamic protein to increase the percentage that is in the “correct” state at any time. These molecules then are able to pass the standards of the cellular QCS.
Although there is not general agreement as to its exact mechanism of action (Dinter and Berger, 1998), there is a widespread view that monensin, an ionophore that is commonly used as a feed additive, in some way disrupts Golgi mediated protein trafficking. Morphological studies suggest that this agent causes substantial swelling of this organelle in a wide range of tissues and it is not clear how or if pharmacoperone-mediated rescue allows the GnRHR to move through a grossly swollen Golgi apparatus. In that regard, the degree of rescue observed in the present work in the presence of monensin is quite modest. The physical disruption may manifest its action by inhibiting a site specific post-translational modification (glycosylation) that may be involved in the chaperone mediated processing. In principle pharmacoperones may allow the proper configuration to be achieved, absent these modifications.
A very limited number of studies (Bernier et al., 2006) have shown that pharmacoperones function in vivo, although there has not yet been an effort in non-human primates or rodents to optimize the pattern of administration. The present study is a needed antecedent to selection of the appropriate time for pulsatile administration of pharmacoperones in vivo and suggests that, at least in the case of GnRH receptor mutants with three chemical classes of pharmacoperones, the drug need not be present at the time of synthesis, as appears to be the case for other vasopressin receptor subtypes (Hawtin, 2006; Morello et al., 2000; Wuller et al., 2004). The present model suggests that pharmacoperones act post-translationally to aid in receptor folding.
Acknowledgments
This work has been supported by NIH grants HD-19899, RR-00163, and HD-18185.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashton WT, Sisco RM, Kieczykowski GR, Yang YT, Yudkovitz JB, Cui J, Mount GR, Ren RN, Wu TJ, Shen X, Lyons KA, Mao AH, Carlin JR, Karanam BV, Vincent SH, Cheng K, Goulet MT. Orally bioavailable, indole-based nonpeptide GnRH receptor antagonists with high potency and functional activity. Bioorg Med Chem Lett. 2001a;11:2597–2602. doi: 10.1016/s0960-894x(01)00512-1. [DOI] [PubMed] [Google Scholar]
- Ashton WT, Sisco RM, Yang YT, Lo JL, Yudkovitz JB, Gibbons PH, Mount GR, Ren RN, Butler BS, Cheng K, Goulet MT. Potent nonpeptide GnRH receptor antagonists derived from substituted indole-5-carboxamides and –acetamides bearing a pyridine side-chain terminus. Bioorg Med Chem Lett. 2001b;11:1727–1731. doi: 10.1016/s0960-894x(01)00275-x. [DOI] [PubMed] [Google Scholar]
- Ashton WT, Sisco RM, Yang YT, Lo JL, Yudkovitz JB, Cheng K, Goulet MT. Substituted indole-5-carboxamides and –acetamides as potent nonpeptide GnRH receptor antagonists. Bioorg Med Chem Lett. 2001c;11:723–1726. doi: 10.1016/s0960-894x(01)00274-8. [DOI] [PubMed] [Google Scholar]
- Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol. 2004a;4:528–533. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Bernier V, Lagace M, Bichet D, Bouvier M. Pharamcological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004b;15:222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Bernier V, Morello JP, Zarruk A, Debrand N, Salahpour A, Longergan M, Arthus MF, Laperriere A, Brouard R, Bouvier M, Bichet DG. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232–243. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Cornea A, Janovick JA, Conn PM. Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol. 2004;18:1787–1797. doi: 10.1210/me.2004-0091. [DOI] [PubMed] [Google Scholar]
- Bush EN, Kramer DL, Cybulski VA, Mohning KM, Besecke LM, Diaz GJ, Bammert GF, Sauer D, Haviv F, Borre A. Activity and pharmacokinetics of A-177775, a non-peptide GnRH antagonist, in rats and dogs. Program and Abstracts of the 81st Annual Meeting of the Endocrine Society; San Diego, CA. 1989. pp. Abstract P3–225. [Google Scholar]
- Castro-Fernandez C, Maya-Nunez G, Conn PM. Beyond the signal sequence: protein routing in health and disease. Endocr Rev. 2005;26:479–503. doi: 10.1210/er.2004-0010. [DOI] [PubMed] [Google Scholar]
- Conn PM, Crowley WF. Gonadotropin-releasing hormone and its analogues. N Engl J Med. 1991;324:93–103. doi: 10.1056/NEJM199101103240205. [DOI] [PubMed] [Google Scholar]
- Conn PM, Leaños-Miranda A, Janovick JA. Protein origami: therapeutic rescue of misfolded gene products. Mol Inter. 2002;2:308–316. doi: 10.1124/mi.2.5.308. [DOI] [PubMed] [Google Scholar]
- Conn PM, Janovick JA, Brothers SP, Knollman PE. “Effective Inefficiency:” cellular control of protein trafficking as a mechanism of post-translational regulation. J Endocrinol. 2006a;190:13–16. doi: 10.1677/joe.1.06771. [DOI] [PubMed] [Google Scholar]
- Conn PM, Knollman PE, Brothers SP, Janovick JA. Protein folding as post-translational regulation: evolution of a mechanism for controlled plasma membrane expression of a GPCR. Mol Endocrinol. 2006b;20:3035–3041. doi: 10.1210/me.2006-0066. [DOI] [PubMed] [Google Scholar]
- Diaz GJ, Besecke LM, Rao M, Cybulski VA, Segreti JA, Mohning KM, Bush EN, Sauer D, Dalton C, Haviv F. Improved oral activity of a novel non-peptide GnRH antagonist, when co-administered with the protease inhibitor ritonavir. Program and Abstracts of the 81st Annual Meeting of the Endocrine Society; San Diego, CA. 1999. pp. Abstract P3–226. [Google Scholar]
- DeVita RJ, Hollings DD, Goulet MT, Wyvratt MJ, Fisher MH, Lo JL, Yang YT, Cheng K, Smith RG. Identification and Initial Structure-Activity Relationships of a Novel Non-Peptide Quinolone GnRH Receptor Antagonist. Bioorg Med Chem Lett. 1999a;9:2615–2620. doi: 10.1016/s0960-894x(99)00446-1. [DOI] [PubMed] [Google Scholar]
- DeVita RJ, Goulet MT, Wyvratt MJ, Fisher MH, Lo JL, Yang YT, Cheng K, Smith RG. Investigation of the 4-Alkylamino Substituent of Non-Peptide Quinolone GnRH Receptor Antagonists. Bioorg Med Chem Lett. 1999b;9:2621–2624. doi: 10.1016/s0960-894x(99)00447-3. [DOI] [PubMed] [Google Scholar]
- DeVita RJ, Walsh TF, Young JR, Jiang J, Ujjainwalla F, Toupence RB, Parikh M, Huang SX, Fair JA, Goulet MT, Wyvratt MJ, Lo JL, Ren N, Yudkovitz JB, Yang YT, Cheng K, Cui J, Mount G, Rohrer SP, Schaeffer JM, Rhodes L, Drisko JE, McGowan E, MacIntyre DE, Vincent S, Carlin JR, Cameron J, Smith RG. A Potent, Nonpeptidyl 1H-Quinolone Antagonist for the Gonadotropin-Releasing Hormone Receptor. J Med Chem. 2001;44:917–922. doi: 10.1021/jm000275p. [DOI] [PubMed] [Google Scholar]
- Dinter A, Berger EG. Golgi-disturbing agents. Histochem Cell Biol. 1998;109:571–590. doi: 10.1007/s004180050256. [DOI] [PubMed] [Google Scholar]
- Ellinger A, Pavelka M. Effect of monensin on the Golgi apparatus of absorptive cells in the small intestine of the rat. Morphological and cytochemical studies. Cell Tissue Res. 1984;235:187–194. doi: 10.1007/BF00213739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber E, Anfinsen CB. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J Biol Chem. 1962;237:1839–1844. [PubMed] [Google Scholar]
- Hawtin SR. Pharmacological chaperone activity of SR49059 to functionally recover misfolded mutations of the vasopressin V1a receptor. J Biol Chem. 2006;281:14604–14614. doi: 10.1074/jbc.M511610200. [DOI] [PubMed] [Google Scholar]
- Huckle WR, Conn PM. Use of lithium ion in measurement of stimulated pituitary inositol phospholipid turnover. Methods Enzymo. 1987;141:149–155. doi: 10.1016/0076-6879(87)41063-x. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87:3255–3262. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003;305:608–614. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Knollman PE, Brothers SP, Ayala-Yanez R, Aziz AS, Conn PM. Regulation of G protein-coupled receptor trafficking by inefficient plasma membrane expression: molecular basis of an evolved strategy. J Biol Chem. 2006;281:8417–8425. doi: 10.1074/jbc.M510601200. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippinicott-Schwartz J, Brefeldin A. Insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Klotz IM. Numbers of receptor sites from Scatchard graphs: facts and fantasies. Science. 1982;217:1247–1249. doi: 10.1126/science.6287580. [DOI] [PubMed] [Google Scholar]
- Knollman PE, Janovick JA, Brothers SP, Conn PM. Parallel regulation of membrane trafficking and dominant-negative effects by misrouted gonadotropin-releasing hormone receptor mutants. J Biol Chem. 2005;280:24506–24514. doi: 10.1074/jbc.M501978200. [DOI] [PubMed] [Google Scholar]
- Kresge N, Simoni RD, Hill RL. The thermodynamic hypothesis of protein folding: the work of Christian Anfinsen. J Biol Chem. 2006;281:e11–e13. [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel R. A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res. 2001;29:E39. doi: 10.1093/nar/29.7.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaños-Miranda A, Janovick JA, Conn PM. Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002;87:4825–4828. doi: 10.1210/jc.2002-020961. [DOI] [PubMed] [Google Scholar]
- Leaños-Miranda A, Ulloa-Aguirre A, Ji TH, Janovick JA, Conn PM. Dominant-negative action of disease-causing gonadotropin-releasing hormone receptor (GnRHR) mutants: a trait that potentially coevolved with decreased plasma membrane expression of GnRHR in humans. J Clin Endocrinol Metab. 2003;88:3360–3367. doi: 10.1210/jc.2003-030084. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper MD, Wicks WD. Evidence for acceleration of the rate of elongation of tyrosi aminotransferase nascent chains by dibutyryl cyclic AMP. Proc Natl Acad Sci USA. 1978;75:140–144. doi: 10.1073/pnas.75.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag JD, Procopio DO, Cygler M, Thomas DY, Bergeron JJ. Lectin control of protein folding and sorting in the secretory pathway. Trends Biochem Sci. 2003;28:49–57. doi: 10.1016/s0968-0004(02)00004-x. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leaños-Miranda A, Conn PM. Misrouted cell surface receptors as a novel disease aetiology and potential therapeutic target: the case of hypogonadotropic hypogonadism due to gonadotropin-releasing hormone resistance. Expert Opin Ther Targets. 2003;7:175–185. doi: 10.1517/14728222.7.2.175. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- Walsh TF, Toupence RB, Young JR, Huang SX, Ujjainwalla F, DeVita RJ, Goulet MT, Wyvratt MJ, Fisher MH, Lo JL, Ren N, Yudkovitz JB, Yang YT, Cheng K, Smith RG. Potent Antagnists of Gonadotropin Releasing Hormone Receptors Derived from Quinolone-6-Carboxamides Bioorg. Med Chem Lett. 2000;10:443–447. doi: 10.1016/s0960-894x(00)00024-x. [DOI] [PubMed] [Google Scholar]
- Wuller S, Wiesner B, Loffler A, Furkert J, Krause G, Hermosilla R, Schaefer M, Schulein R, Rosenthal W, Oksche A. Pharmacochaperones post-translationally enhance cell surface espression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279:47254–47263. doi: 10.1074/jbc.M408154200. [DOI] [PubMed] [Google Scholar]
- Zhu BT. The competitive and noncompetitive antagonism of receptor-mediated drug actions in the presence of spare receptors. J Pharmacol Toxicol Methods. 1993;29:85–91. doi: 10.1016/1056-8719(93)90055-j. [DOI] [PubMed] [Google Scholar]