

SUMMARY
Inflammatory bowel disease (IBD) has been attributed to over-exuberant host immunity or the emergence of harmful intestinal flora. The transcription factor T-bet orchestrates inflammatory genetic programs in both adaptive and innate immunity. We describe a profound and unexpected function for T-bet in influencing the behavior of host inflammatory activity and commensal bacteria. T-bet deficiency in the innate immune system results in spontaneous and communicable ulcerative colitis in the absence of adaptive immunity and increased susceptibility to colitis in immunologically intact hosts. T-bet controls the response of the mucosal immune system to commensal bacteria by regulating TNF-α production in colonic dendritic cells, critical for colonic epithelial barrier maintenance. Loss of T-bet influences bacterial populations to become colitogenic, and this colitis is communicable to genetically intact hosts. These findings reveal a novel function for T-bet as a peacekeeper of host-commensal relationships and provide new perspectives on the pathophysiology of IBD.
INTRODUCTION
The intestinal lumen is massively colonized by bacteria and for most metazoans this relationship is beneficial (Ley et al., 2006). Prokaryotes partner with their eukaryotic hosts to aid in the extraction of energy and nutrients from food, and non-pathogenic bacteria suppress pathogenic species (Backhed et al., 2005). An epithelial cell barrier is essential for this symbiosis as it creates a boundary necessary for coexistence by preventing mucosal inflammation in response to bacterial or other luminal stimuli (Magalhaes et al., 2007). However, in some individuals this balance is upset, resulting in persistent intestinal inflammation, that manifests as the 2 distinct clinical entities of IBD, Crohn’s disease (CD) and ulcerative colitis (UC) (Rakoff-Nahoum et al., 2006). Determining the factors that regulate these complex host-commensal relationships and promote the development of colitis is of great clinical and scientific interest.
T-bet (Tbx21) is a T-box transcription factor family that regulates the differentiation and function of immune system cells. Although initially described to direct the development of a major subset of lymphocytes called T helper 1 cells, recent work has firmly established T-bet as a regulator of the Type 1 proinflammatory immune response in cells of both the adaptive and innate immune systems (Glimcher, 2007). It has been implicated as a key effector protein in autoimmune, allergic, and neoplastic diseases; nevertheless, its role in balancing immunity and autoimmunity is complex, and it is not well-understood if T-bet protects against or promotes chronic intestinal inflammation (Weigmann and Neurath, 2002).
Here we reveal an unexpected and powerful role for T-bet in host-commensal interactions. Loss of T-bet in mice lacking an adaptive immune system results in a spontaneous, highly penetrant, aggressive, and communicable colitis that resembles human UC. Both the T-bet deficient genetic background and the microbiota its absence engenders are required for disease initiation; but once established, the microbiota from the afflicted mice is vertically transmissible and causes intestinal inflammation in wild type (WT) mice. Thus we have identified a novel function for T-bet in moderating host-commensal relationships. This new mouse model of spontaneous UC provides exciting opportunities to further our understanding of the etiology and pathogenesis of UC, to interrogate the complex nature of commensal microbial communities, and to serve as a robust host for the evaluation of new therapeutics.
RESULTS
T-bet expression protects against colitis and T-bet-/- x RAG2-/- mice develop spontaneous colitis
The sulfated polysaccharide dextran sodium sulfate (DSS) induces colitis via direct toxicity on mucosal epithelium. T-bet -/- mice developed more severe colitis upon DSS administration than WT mice (Figure 1 A and B), as evidenced by a more extensive and severe inflammatory infiltrate, containing neutrophils, mononuclear cells, and more lymphoid aggregates, more edema, extensive ulceration, and crypt loss (Figure 1B). This observation was unexpected as DSS colitis arises from overabundant Type 1 cytokines, IFNγ, IL-12, IL-1 and TNF-α. T-bet-/- T cells produce a Type 2 cytokine profile that is protective for most T cell-driven colitis models. Indeed, T-bet deficiency is protective against the T cell adoptive transfer SCID colitis model (Neurath et al., 2002). To clarify whether T-bet’s protective function resided in innate immunity, we examined T-bet-/- mice bred onto the RAG2-/- background. A comparative survey of the gastrointestinal (GI) tracts of T-bet-/-x RAG2-/- mice, RAG2-/-, T-bet-/-, and WT mice in our colony revealed that T-bet-/-x RAG2-/- mice spontaneously developed a highly penetrant and severe colitis (Figure 1C and 1D). This colitis was apparent by 4 wks of age and increased in severity over time (Figure 1E). Of note, our mice are housed in a barrier facility, documented to be free of known colitogenic pathogens such as Helicobacter hepaticus, bilis, and muridarum.
Figure 1. T-bet expression protects against colitis and T-bet-/-x RAG2-/- (TRUC) mice develop spontaneous colitis.
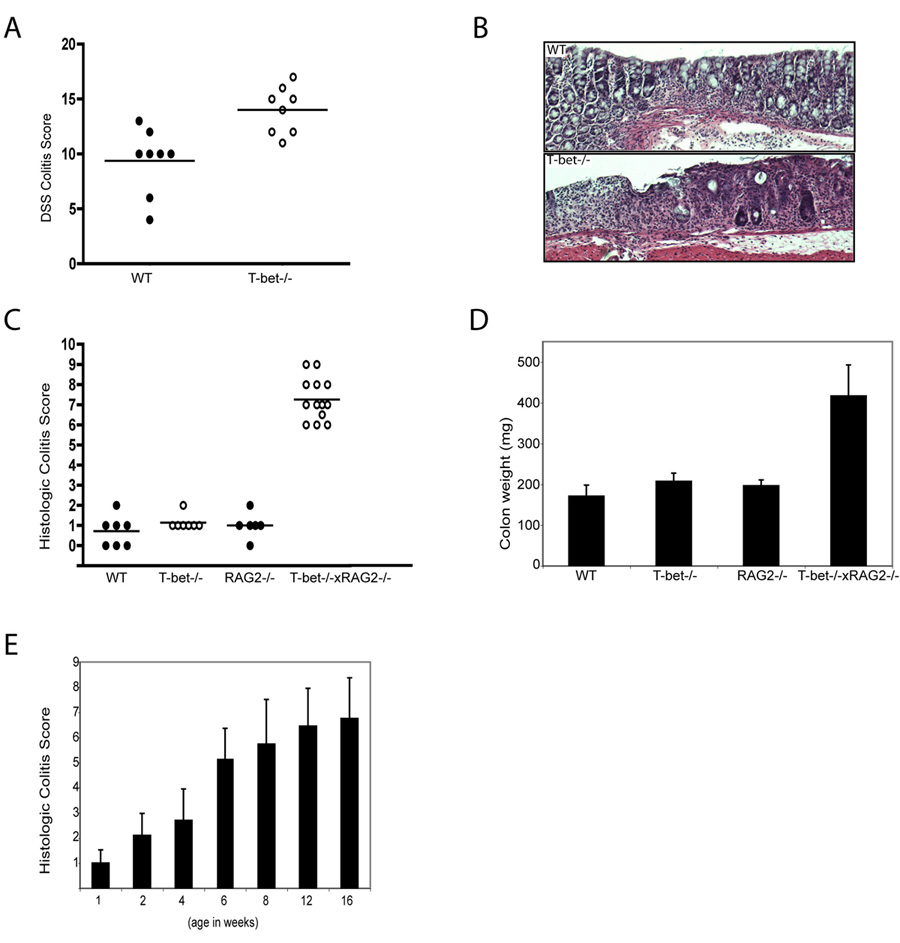
(A) WT and T-bet-/- mice, age 8 wks, treated with 4% DSS for 8 days. One representative exp of 3, n=8 per group, p = .0031. (B) Photomicrographs of DSS treated WT (upper panel) and T-bet-/- (lower panel) colons. (C) TRUC mice develop a spontaneous, highly penetrant colitis. Colitis score for each mouse denoted by a dot, the mean for each group shown as a horizontal bar. n=7 per group, all 12 wks old. (D) Mean colon weights (mg) for the mice in 1C. (E) TRUC colitis severity increases with time. Mean colitis score ± stdev, n=10 per group except for 1 week old group, n=8.
Spontaneous colitis in TRUC mice phenocopies human UC and is characterized by an early colonic epithelial barrier breach
T-bet-/-x RAG2-/- (herein referred to as TRUC [T-bet-/-x RAG-/- Ulcerative Colitis] mice develop colitis that bears a striking resemblance to human UC (Figure 2A). notable for anorectal prolapse (Figure 2A 1), from rectal inflammation. TRUC colons demonstrated continuous inflammation of the rectum and left colon by 4 wks of age (Figure 2A 2b) and by 8 wks, there was marked inflammation and colonic thickening (Figure 2A 2d) not seen in RAG2-/- controls (Figure 2A 2c). Microscopically, TRUC colitis phenocopies many aspects of human UC (Figure 2A 4) with a mixed inflammatory infiltrate in the lamina propria containing both mononuclear and polymorphonuclear cells, neutrophil infiltration of the crypt and surface epithelium, and epithelial injury with surface denudations and ulcerations associated with crypt loss and epithelial mucodepletion. The stomach and small intestine appeared microscopically normal, similar to UC but in contrast with CD and most spontaneous mouse models of colitis. Examination of mice from one week postnatally revealed no evidence of colitis until 4 wks (Figure 1E). However, intrarectal instillation of fluorescently labeled dextran, to test the integrity of the colonic epithelial barrier in 3.5 week-old mice (Karhausen et al., 2004) demonstrated significantly increased permeability in TRUC compared to RAG2-/- and T-bet-/- mice (Figure 2B) with 2-fold higher serum fluorescence (p=.0002 TRUC vs RAG2-/-). Colonic permeability increased with age, with a 3-fold increase between 4–5 wks and an 8.6 fold increase between 5–6wks (p=.00036) (Figure 2C). Hence, a decrease in colonic epithelial integrity precedes histologically detectable colitis and worsens with progression.
Figure 2. Spontaneous colitis in TRUC mice phenocopies human UC and is characterized by a colonic epithelial barrier breach.
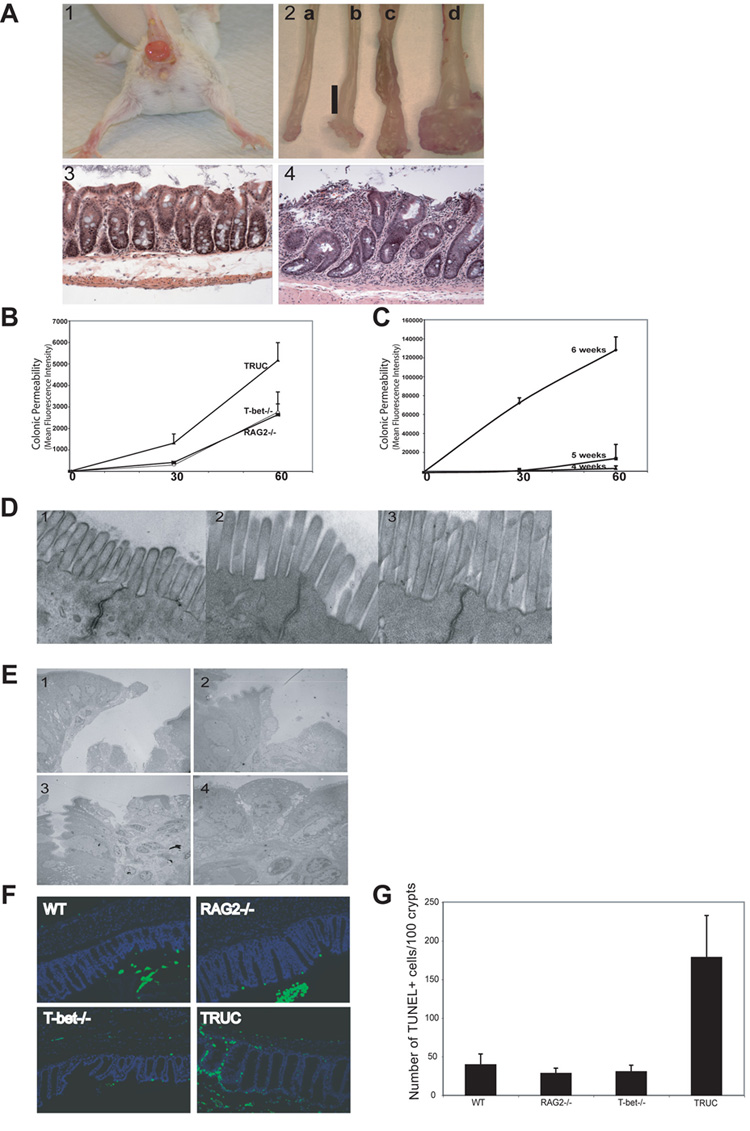
(A)(1) TRUC mouse (8 wks) with anorectal prolapse. (2) Photograph distal colons RAG2-/- 4 wks (a) 8 wks (c) and TRUC 4 wks (b) 8 wks (d) (anorectal junction at bottom). Vertical bar delineates the inflamed, thickened colonic wall. Colonic wall thickening with prolapsed rectal mucosa in 8 wk TRUC. (3) Normal colonic mucosa 6 wk RAG2-/- mouse, 100x. (4) Representative disease 6 wk TRUC. Note mucosal thickening, surface ulceration, crypt distortion and hyperplasia, and dense mixed inflammatory cell infiltrate in the lamina propria (compare to panel 3), 100x. (B)Intra-rectal FITC-dextran was administered to 3.5 wk T-bet-/-, RAG2-/-, and TRUC. Serum fluorescence measured at the indicated time points. One representative exp of 3, n=4–6 per group, [p =.0002, 60 min TRUC vs RAG2-/-]. (C) Intra-rectal FITC-dextran administered to TRUC at 4, 5, and 6 wks. Serum fluorescence at the indicated time points, [p=.0036, 60 min 6 vs 5 wks]. One representative exp of 3, n= 5 per group.
(D) TRUC colonic tight junctions surveyed by EM: representative images prior to 2 wks (1), 3 wks (2), and 4 wks (3), 25,000x. (E) Colonic epithelial discontinuities present in 3.5 wk TRUC. Representative EM images, (1) 800x (2) 1500x (3) 1000x (4) 3000x. (F) Increased apoptosis in TRUC colonic epithelium. WT, T-bet-/-, RAG2-/-, and TRUC colonic epithelium at 5 wks stained with DAPI (blue) and TUNEL (green), 200x. (G) Epithelial crypt and TUNEL+ cell counts. 5 slides generated from each genotype group (2–3 mice per genotype). 500 crypts per genotype were scored.
Although, abnormal tight junctions may contribute to IBD pathogenesis (Bruewer et al., 2006), EM (electron micrographs) of TRUC and control colonic mucosa from mice aged 2–12 wks showed intact epithelial tight junctions (Fig 2D, 2 wks (Panel 1), 3 wks (Panel 2), and 4 wks (Panel 3)). However, numerous, large epithelial discontinuities were observable on EM by 3.5 wks of age (Figure 2E 1–4), that likely accounted for the increased permeability. Further, the degree of cell death was well above the intrinsic rate of epithelial turnover. We quantified colonic epithelial apoptosis by TUNEL(Terminal transferase dUTP nick end labeling) in 5 wk old mice when significant colonic permeability exists (Figure 2C). While TUNEL+ cells were noted at the top of crypts in all genotypes, TUNEL+ cells were distributed throughout the crypt in TRUC mice and in many cases the entire crypt consisted of TUNEL+ cells (Figure 2F). Quantitation of TUNEL+ cells/100 crypts for all 4 genotypes showed a 4–5 fold increase in TUNEL+ cells in TRUC mice, (Figure 2G), accentuated if TUNEL+ cells/epithelial cells/crypt is plotted (Supp. Data Panel B).
TNF-α drives tissue injury in TRUC colitis and TNF-α over-production maps to colonic DCs
We analyzed the TRUC versus RAG2-/- colonic cytokine milieu in colon explant cultures. TNF-α was significantly elevated in TRUC explant cultures at 4 wks and increased over time (Figure 3A). There were no marked differences in the levels of other IBD associated cytokines, IFNγ, IL-1α, IL-1β, IL-6, IL-10, IL-12, IL-13, or IL-23 at 4 wks (Figure 3B). TNF-α is a key effector cytokine in IBD thought to lead to an increased inflammatory tone of the intestinal epithelium. TNF-α neutralizing Ab therapy is widely used in IBD treatment. Anti-TNF-α Ab therapy suppressed TRUC colitis as evidenced by colon weight (Supplemental Figure A) and colitis score (Figure 3C). The microscopic appearance of TNF-α Ab treated TRUC colons was indistinguishable from control mice. Of interest, anti-TNF-α treatment decreased the number of apoptotic epithelial cells to normal levels in mice treated from 4–8 wks of age (Figure 3D).
Figure 3. TNF-α drives tissue injury in TRUC colitis and TNF-α overproduction maps to colonic DCs.
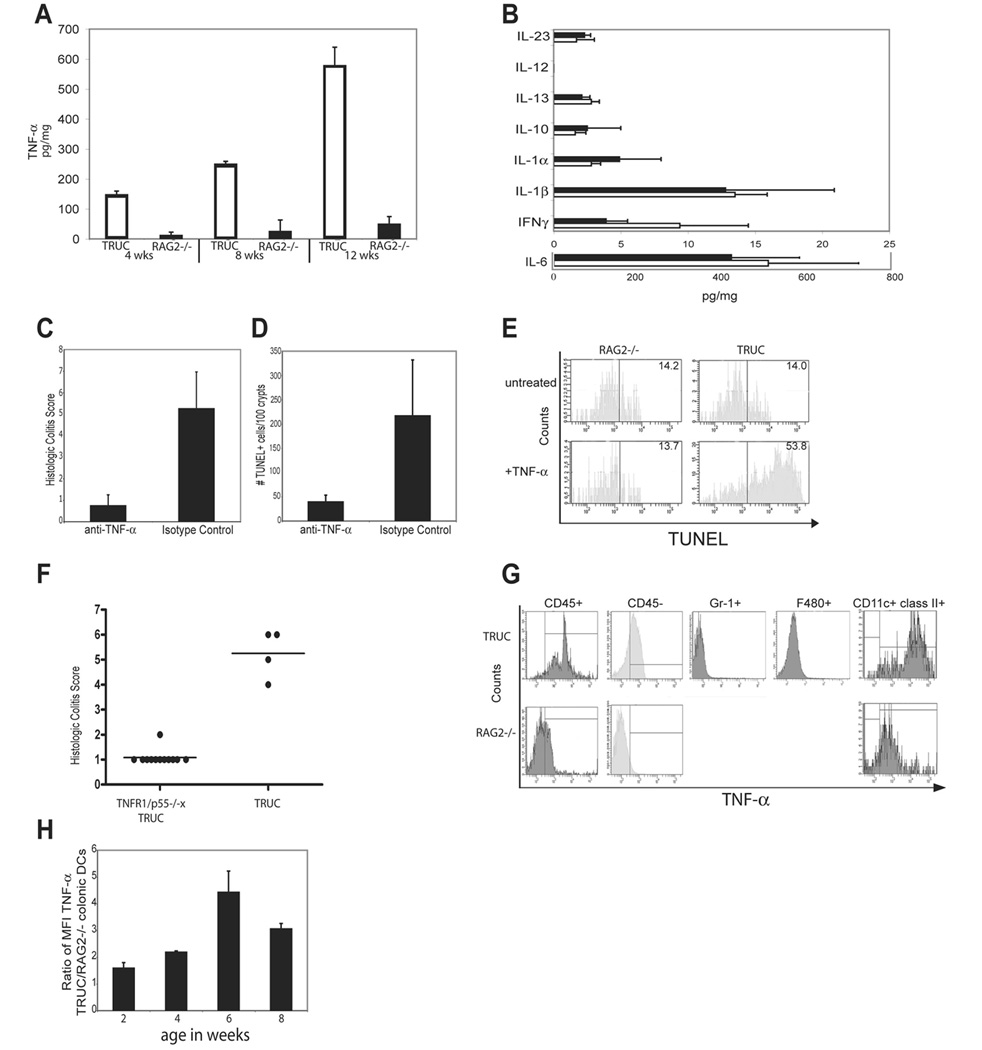
(A) TNF-α ELISAs on colon explant cultures from 4, 8, and 12 wk old TRUC (open) and RAG2-/- (shaded). One representative exp of 3 is shown, samples pooled, n=4 per group. (B) IL-23, IL-13, IL-12p40, IL-10, IL-6, IL-1α, IL-1β, and IFNγ levels of colon explant cultures from TRUC (open) and RAG2-/- (shaded), samples pooled, n= 4 per group. Data represent the mean of 3 independent exp, error bars denote stdev. (C) 4 wk TRUC were treated with anti-TNF-α Ab or an isotype control for 4 wks. Colitis scores for treated vs control, p=.0023. One representative exp of 3 is shown, n=4 per group. (D) Apoptotic (TUNEL+) epithelial cells counts; anti-TNF-α vs control, p= .0016. (E) TUNEL staining of cultured TRUC colonic epithelial cells (CD45- cytokeratin 5/8+) treated with TNF-α compared to TNF-α treated RAG2-/- colonic epithelial cells (53.8% vs 13.7%). One representative exp of 4 is shown. (F) Colitis scores for 8 wk TRUC TNFR1/p55-/- and TRUC, p<.0001. Colonic cell suspensions were, stained with Abs directed against cell surface markers, and permeabilized to allow for detection of intracytoplasmic TNF-α by flow cytometry. (G) TNF-α production by immunocytes (CD45+). RAG2-/- cells as controls. Abs against Gr-1, F4/80, and CD11c paired with class II were used to identify the immunocytes with the highest TNF-α production. (H) Colonic DC production of TNF-α during the disease course, 2–8 wks. The mean and stdev of 3 independent exp are plotted. Vertical bars denote the right-sided tail of the isotype control staining. Data are representative of 3 independent exp; samples were pooled, n=10–20 per group.
TNF-α could modulate either pro-survival or pro-death signaling pathways in cells. We examined the effects of TNF-α on the apoptosis of colonic epithelial cells (CEC), isolated from RAG2-/- and TRUC colons cultured with or without TNF-α. After 6 days, the untreated RAG2-/- and TRUC CECs had quite similar percentages (%) of apoptosis (14.2 vs 14.0 %: RAG2-/- vs TRUC); however, TNF-α treated cells displayed a marked difference in apoptosis (13.7 vs 53.8%: RAG2-/- vs TRUC) (Figure 3E). Since CECs do not express T-bet, their exposure to the TRUC colonic environment likely sensitized them to the effects of TNF-α. Cell surface TNFR1 does differ between TRUC and RAG2-/- CECs even from 2 week old mice (data not shown). To discern whether CEC death is a direct or indirect effect of TNF-α, we generated TRUC mice that were also deficient in TNF-α receptor by crossing them onto TNFR1/p55-/- mice. 8 wk old triple-deficient mice showed no evidence of colitis, suggesting that TNF-α signaling through TNFR1/p55 is a central event in disease pathophysiology (Figure 3F). These experiments established the centrality of TNF-α in this model, and led to the question of what cell type(s) was responsible for TNF-α production in the colon.
We performed intracellular cytokine analysis on cells isolated from TRUC and RAG2-/- colons. Leukocyte (CD45+) populations had higher staining for TNF-α than epithelial and other non-leukocyte cells (Figure 3G). Among the leukocyte subsets, there was no observable staining for NK cells by flow cytometry (FC) or IHC in TRUC colons (data not shown). TNF-α in macrophages, granulocytes, and neutrophils could not account for the levels of TNF-α observed (Figure 3G). However, there was significant accumulation of TNF-α in TRUC colonic DCs (Figure 3G) suggesting that colonic DCs are the principal TNF-α producing cell type. A time course of TNF-α production of TRUC and RAG2-/- colonic DCs from mice aged 2–8 wks (Figure 3H) showed increased TNFα as early as 2 wks of age, prior to any observed epithelial discontinuities by microscopy. Thus over-production of TNF-α by colonic DCs precedes the development of epithelial barrier abnormalities and colitis.
T-bet regulates production of TNF-α in DCs
To ask if T-bet was expressed in colonic DCs as in DCs in the peripheral lymphoid system, RAG2-/- colons were stained with CD11c to identify DCs and adjacent serial sections were stained for T-bet (Figure 4A 1 and 2). While the required serial sectioning prevents precise co-localization, there were many T-bet+ cells in areas of dense CD11c staining. We confirmed this finding, by staining FACS isolated colonic DCs from RAG2-/- mice with α-T-bet Ab and DAPI. T-bet staining (red) is evident and, there is co-localization with the DAPI (blue) stain in the nucleus (Figure 4A 3).
Figure 4. T-bet regulates production of TNF-α in DCs.
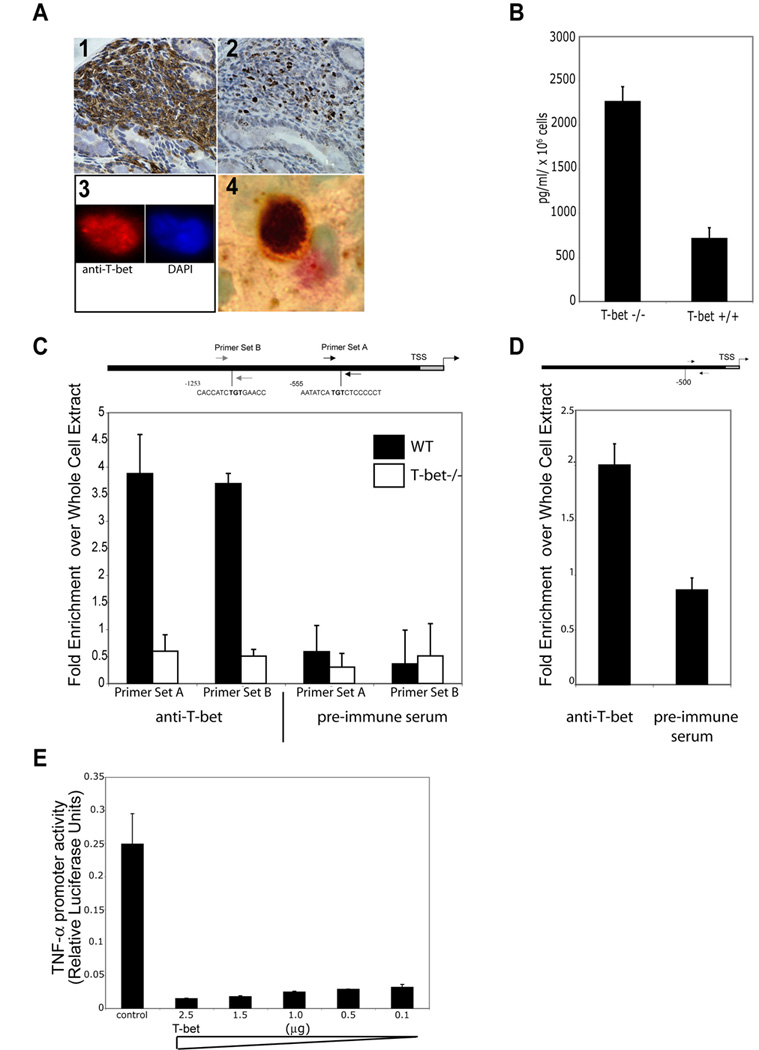
T-bet is expressed in colonic DCs. (1) CD11c staining RAG2-/- colonic mucosa, 200x. (2) Serial section of (1) stained with anti-T-bet Ab, 4B10, 200x. (3) Sorted colonic, mouse DC stained with anti-T-bet Ab (red) and DAPI (blue), 1000x. (4) Human colonic DC, S100 (pink), anti-T-bet (brown), 1000x. (B) Loss of T-bet expression in BMDCs results in increased production of TNF-α.
T-bet binds the TNF-α promoter. (C) qPCR of mouse bone BMDCs ChIP samples. Primer sets A and B, approximately 500 and 1200 bps upstream of the TSS. A schema of the promoter showing T-box consensus sites and the location of primers used. Data are the mean of 3 exp (D) qPCR of human myeloid DC ChIP samples, one representative exp of 3. (E) T-bet negatively regulates TNF-α gene transcription. RAW cells transiently cotransfected with a 3kb TNF-α promoter luciferase construct, a constitutively active Iκκβ mutant construct to activate TNF-α transcription, a Renilla construct for normalization of transfection efficiency, and varying concentrations of a full length T-bet cDNA construct or control plasmid. TNF-α promoter activity is shown as a function of relative luciferase activity. Data are representative of 3 independent exp.
We next examined T-bet expression in human colonic biopsies. Specimens were stained with S100 (DCs red) and T-bet (brown). A representative DC with T-bet staining is shown (Figure 5A 4). Demonstration of T-bet expression in human colonic DCs is a novel observation as T-bet expression had only previously been demonstrated in human colonic T cells (Neurath et al., 2002). There was no T-bet expression in CECs (Figure 4A 2) by IHC nor has T-bet been detectable in CEC lysates (data not shown). T-bet is expressed in both mouse and human colonic DCs.
Figure 5. CD4+CD25+ T-regulatory cells control TRUC.
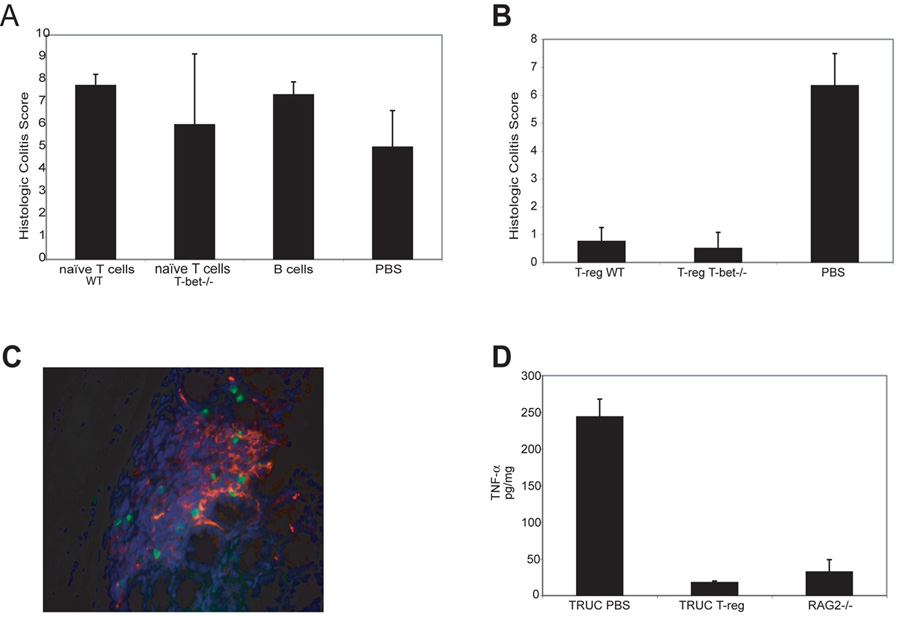
(A) 4 week TRUC injected with PBS, 1×106 B cells, or 1×106 naïve T cells, WT or T-bet-/-. Colitis scores at 2 wks post-injection. Representative data from one of 3 exp, n=4 per group. (B). 4 week TRUC mice injected with 75,000 T-regs (CD4+CD25+CD62L+), WT or T-bet-/-, or PBS. Colitis scores at 4 wks post-injection. One representative exp of 3 is shown, n=6–8 per group. (C) Photomicrograph of TRUC colon (T-reg treated). T-regs (CD3, green) and DCs (CD11c, red), 400x. (D) Adoptive transfer of T-regs normalizes TRUC TNF-α levels. 4 week TRUC injected with 75,000 T-regs (CD4+CD25+CD62L+) or PBS. TNF-α levels from colon explant cultures 4 wks post-injection, RAG2-/- colon explants as a control. Samples pooled, n=4–5 per group.
To explore T-bet’s role in DC TNF-α production, we used mouse bone marrow DCs (BMDCs) as colonic DC yields are insufficient for biochemical evaluation. LPS matured T-bet-/- BMDCs, similar to colonic DCs, overproduce TNF-α compared to WT BMDCs (Figure 4B). The observations that loss of T-bet expression in colonic and BMDCs results in increased TNF-α production suggested that T-bet may directly regulate TNF-α in DCs.
Chromatin immunoprecipitation (ChIP) and quantitative real timer PCR (qPCR) (Figure 4C) assays revealed that T-bet bound the mouse TNF-α promoter at two T-box consensus sites at approximately −1200 and −500 relative to the transcription start site (TSS) but not at regions examined upstream of −1200 (data not shown), using T-bet-/- BMDCs and pre-immune sera as controls. We also performed ChIP assays in human myeloid DCs. Although there is little sequence homology between the 5’ UTR of the mouse and human TNF-α promoters and T-box consensus sites can be rather degenerate, T-bet bound the human TNF-α promoter as demonstrated by ChIP and qPCR (Figure 4D) at approximately −400 relative to the TSS and did not bind more upstream.
To investigate whether T-bet is a negative regulator of TNF-α transcription as would be predicted from the TNF-α overproduction phenotype of T-bet-/- DCs. RAW cells were cotransfected with a TNF-α promoter luciferase construct, a constitutively active Iκκβ mutant to activate TNF-α transcription, and T-bet. Transfection with T-bet reduced NFκB-driven TNF-α promoter activity in RAW cells in a dose dependent fashion suggesting that T-bet functions as a negative regulator of TNF-α transcription (Figure 4E).
CD4+CD25+ T-regulatory cells control TRUC colitis
T-bet-/- immunosufficient mice do not develop spontaneous colitis although they display increased susceptibility to DSS and have been shown to have increased susceptibility to oxazolone-induced colitis, a Th2 model of colitis (Neurath et al., 2002). T-bet-/- and WT mice possess an adaptive immune system as do most IBD patients. To test the hypothesis that a component of the adaptive immune system might act as a repressor of colitis in immunosufficient hosts, we performed adoptive transfer (AT) experiments in TRUC. As evidenced by colitis scores, the protection we observed could not be attributed to conventional T helper or B cells since transfer of naïve CD4 T cells (T-bet-/- or WT) or B cells did not protect against disease (Figure 5A). The CD4 T-regulatory cell (T-reg) is a potent repressor of effector T cell function that might have prevented the development of colitis in the T-bet-/- host. AT of T-regs reverses intestinal inflammation in H. hepaticus infected RAG2-/- mice, an innate immune driven colitis model (Maloy et al., 2005). AT of either WT or T-bet-/- T-regs controlled TRUC colitis (Figure 5B). Colons from mice infused with T-regs were free of any signs of colitis. Imaging TRUC colons to document that the transferred cells had trafficked to the colon using CD3 staining revealed that the CD3+ cells were in close contact with DCs as shown by co-staining with CD11c (Figure 5C) suggesting that cross-talk between T-regs and DCs may suppress the T-bet deficient DC pro-inflammatory phenotype. Colon explant cultures from TRUC mice injected with T-regs 4 wks earlier showed markedly reduced TNF-α levels similar to control RAG-/- colonic explants (Figure 5D). We hypothesized that colonic DC T-bet deficiency might confer a genetic susceptibility to aberrant TNF-α inflammatory responses to the microbial environment.
The TRUC colonic environment is a niche supporting a colitogenic microbial community whose suppression prevents colitis across generations
The colonic luminal contents are abundant in microbiota (Hooper and Gordon, 2001) and intestinal DCs constantly sample these microbes (Chieppa et al., 2006; Niess et al 2006). Most mouse models of colitis are microbe-dependent and patients with IBD often have favorable if not durable responses to antibiotic therapy. To determine if TRUC colitis was driven by the gut microbiota, mice were treated with broad spectrum antibiotics: vancomycin (V), metronidazole (M), neomycin (N), and ampicillin (A)—a combination previously shown to deplete enteric microbial communities (Fagarasan et al., 2002; Rakoff-Nahoum et al., 2004). Treatment with the VMNA combination cured the mice of their colitis, and selective treatment with metronidazole alone cured the colitis as well, as demonstrated by colitis scores (Figure 6A). Conventional culturing of fecal pellets from VMNA antibiotic-treated and control mice, demonstrated relatively unchanged aerobic colony counts (1010.11 vs 1010.16), but anaerobic colony counts were dramatically decreased (109.74 vs < 104.01; log10CFU/gram dry weight of stool). Treatment for several months induced not only a 100,000-fold decrease in the culturable fecal anaerobes but also remission in all treated TRUC mice. These two observations drew attention to the potential pathogenicity of TRUC anaerobic commensals.
Figure 6. The TRUC niche generates a colitogenic microbial community which is transmissible to T-bet sufficient mice.
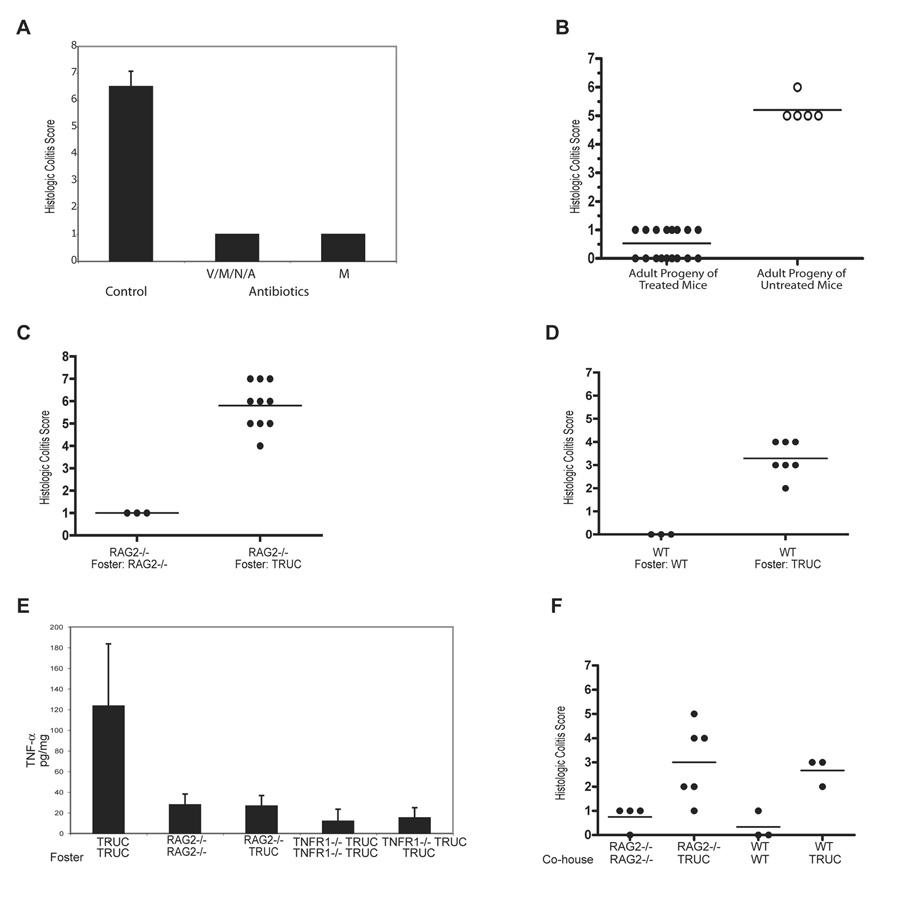
(A) Colitis scores for 6 wk TRUC treated with vancomycin (V), metronidazole (M), neomycin (N), and ampicillin (A) or metronidazole (M) alone for 6 wks. One representative exp of 3, n=4 per group. (B) Colitis scores for adult progeny of antibiotic-treated mice. Three separate litters are shown for antibiotic treated breeders and 1 litter is shown from untreated breeders, p<.0001.
Communicability of TRUC colitis. Colitis scores for cross-fostered RAG2-/- mice (C) and WT mice (D). Mice are from 3 exp (TRUC and RAG2-/-) and 2 exp (TRUC and WT). RAG2-/- reared by RAG2-/- female and WT reared by WT female shown for comparison. (E) TNF-a ELISA on colon explant cultures: RAG2-/- cross-fostered by TRUC, TRUC reared by TRUC, RAG2-/- reared by RAG2-/-, TRUC TNFR1-/- fostered by TRUC, and TRUC TNFR1-/- reared by TRUC TNFR1-/-. Samples were pooled, n=4–8 per group. (F) Colitis scores for RAG2-/- and WT co-housed with TRUC for 8 wks. RAG2-/- and WT co-housed with genotype-identical mice shown for comparison. Horizontal bars represent the mean.
We questioned the origin of the pathogenic TRUC microbial community. Did pathogenic anaerobic commensals expand anew at each generation in response to the developing TRUC colonic niche, or were colitogenic microbes propagated trans-generationally from founder TRUC mice? Genetic susceptibilities are heritable and microbial communities are also “heritable”, as they are passed from parent to offspring (Tannock, 2006). We asked whether the TRUC colitogenic microbial community was vertically transmissible. TRUC colitis is highly penetrant and develops at a young age. TRUC progeny had colitis, raising the issue whether the progeny of antibiotic-treated (Abx) TRUC mice would develop colitis. We bred Abx mice and examined their progeny, who had no exposure to antibiotics after weaning, after they matured to adulthood. The progeny had no evidence of colitis (Figure 6B). Thus, in TRUC mice, such treatment cures them of disease and prevents their progeny from developing colitis into adulthood. These experiments interested us in the communicability of TRUC colitis.
TRUC colitis is communicable to T-bet sufficient mice
We performed cross-fostering experiments to determine if TRUC intestines possessed a microbial community that was colitogenic in T-bet sufficient. We provided a mother different from the birth mother at the day of birth to RAG2-/- and WT mice and assessed the development of colitis in these cross-fostered (X-F) progeny. Adult RAG2-/- and WT mice X-F by TRUC mothers developed colitis that was histologically similar to TRUC (Figure 6C and D). Colitis scores from conventionally reared RAG2-/- and WT mice are shown for comparison. The colitis of X-F RAG2-/- was more severe than that of X-F WT. Hence, genetically “resistant” strains can vertically acquire colitis from association with the pathogenic microbiota of affected mice.
The vertical transmission of TRUC colitis raised the issue if TRUC colitis is horizontally transmissible. Perhaps, colitis transmission could only occur in the neonatal period at a time when the immune system is immature or that transmission required maternally transmitted factors such as breast milk in addition to commensal microbiota. To test these possibilities, adult RAG2-/- mice were co-housed at a 1:3 ratio with age and sex-matched adult TRUC mice for 8 wks. Remarkably, both RAG2-/- and WT co-housed mice had evidence of colitis with elevated colitis scores, demonstrating that TRUC colitis is horizontally transmissible. These results suggest that not only is TRUC colitis communicable but also that the T-bet deficient mucosal innate immune system creates a niche for a colitogenic microbial community. The question remained if the colitis contracted by T-bet sufficient mice was characterized by the high levels of TNF-α that are central to the pathophysiology of TRUC colitis and originate from T-bet deficiency in DCs. Perhaps, the transmitted colitogenic microbial community acted via a different mechanism to induce colitis in T-bet sufficient hosts. RAG2-/- and TRUC TNFR1-/- mice were fostered by TRUC mice and then explant cultures were performed on X-F and control mice. As expected, the RAG2-/- X-F mice displayed evidence of colitis. TNF-α levels were not elevated in TRUC X-F RAG2-/- or TRUC TNFR1-/- explant cultures (Figure 6F). TRUC mice harbor microbiota that drive a TNF-α mediated colitis in the absence of T-bet yet induce an etiologically distinct colitis in T-bet sufficient mice.
DISCUSSION
Our results reveal an unexpected role for the transcription factor T-bet in maintaining host-commensal relationships in the GI tract. T-bet fosters harmony between colonic eukaryotes and prokaryotes by establishing genetic programs in the immune system that affect the behavior of the colonic epithelium. T-bet functions in DCs to avoid pro-inflammatory responses to commensal microorganisms by regulating TNF-α production to ensure the integrity of the epithelial barrier. From the perspective of the microbial communities inhabiting the colon, the TRUC colonic environment facilitates the growth of a collection of colitogenic bacteria. Once generated, this community of bacteria drives intestinal inflammation even in immunosufficient mice that express T-bet. The pathophysiology of IBD has been attributed to mucosal immune system dysregulation or gut microbiota and/or abnormal epithelial barrier function to (Strober et al., 2007). Deletion of IKKβ or NEMO, solely in intestinal epithelium, for example, results in spontaneous colitis (Nenci et al., 2007; Zaph et al, 2007). The characteristics of TRUC colitis described here suggest a link between these two seemingly disparate mechanisms since T-bet expression impacts both the host’s mucosal immune system and barrier function as well as the commensal microbial community.
T-bet and the cytokine milieu
An altered mucosal cytokine milieu drives inflammation in both UC and CD (Bouma, 2003). We show that TNF-α is selectively elevated prior to and at the onset of pathologically diagnosable disease and treatment starting at 2 wks of age, prior to observable disease, and treatment at 8 wks of age, in the setting of fulminant disease, are both therapeutic (Supplemental data Panel B and C). Taken together, our studies have firmly established TNF-α as a key initiating cytokine in our model.
Neutralizing TNF-α Ab may act by binding TNF-α in the serum, sequestering it and preventing it from binding its receptor or by binding cell surface associated TNF-α to induce signaling events resulting in apoptosis of TNF-α-producing cells (Shen et al., 2006). Colonic DCs appear to produce the bulk of TNF-α in the TRUC colon but intestinal epithelial cells and colonic myofibroblasts also produce TNF-α (Lan et al., 2005; Rogler et al., 2001). Analysis of small intestinal biopsies from CD patients suggests that an imbalance in intestinal DC populations may initiate and perpetuate inflammation (Silva et al., 2004). How mucosal DCs decode environmental signals to mount appropriate immune responses is of great interest as its dysregulation is clearly a driving force in TRUC and in human IBD (Iwasaki, 2007).
Anti-TNF treated TRUC colons were indistinguishable from controls, with no evident DC accumulation consistent with the notion that anti-TNF-α therapy induces apoptosis of TNF-α producing cells in colitis. Our experiments also address a causality dilemma in the IBD field: that is, whether the epithelial barrier breach or the aberrant inflammation is the initial pathologic event. Elevated TNF-α in TRUC colonic DCs preceded remarkable epithelial discontinuities and antibiotic treatment reduced TNF-α concentrations in colon explant cultures. Thus, we favor the idea that bacterial-driven production of TNF-α from colonic DCs is an early, initiating event that triggers breakdown of the intestinal epithelium with consequent ongoing influx of pathogenic microbiota resulting in chronic inflammation. TNF-α blockade is an important therapeutic modality to treat IBD, but, as with many biologics, has limiting toxicities. Local augmentation of T-bet activity in intestinal mucosal DCs may be an alternative approach to regulating TNF-α production.
TRUC transmissibility
TRUC colitis is both vertically and horizontally transmissible to T-bet sufficient mice although the penetrance and severity of horizontally transmitted TRUC colitis was reduced relative to vertically transmitted colitis. Cellular, cytokine, or growth factor differences in TRUC milk may contribute to TRUC colitis but additional factors, aside from the absence of potentially inflammatory breast milk, may be involved. Bacterial colonization of mice and humans starts in the birth canal and continues after parturition. The adult mice used in the co-housing experiments have well-established microbial communities that may hinder the influx of colitogenic microbiota. Also, effective horizontal transmission depends on the coprophagic and grooming behaviors of individual mice and can vary greatly. The transmissibility experiments suggest that TRUC colitis is not only a microbial driven disease but also that colitogenic microbes have the ability to drive distinct colitides dependent upon different immunological mechanisms based on host genotype.
T-bet and the microbial flora
TRUC colitis is microbe-dependent and transmissible. Conventional culture data from antibiotic-treated vs untreated TRUC mice suggest that a set of anaerobic microbes may be pathogenic. We were unable to detect of Helicobacter hepaticus, bilis, or muridarum by PCR-based methods, and there were no marked differences in the culturable anaerobes, including Enterococcus and Prevotella species, or aerobes from fecal pellets from WT, T-bet-/-, RAG2-/-, and TRUC mice (data not shown); however, standard culturing techniques are capable of identifying only a small fraction of the known anaerobic microbiota. Genomic approaches using 16S rDNA enumerations are underway to identify this novel collection of colitogenic bacteria.
A genomic survey of mouse intestinal microbiota showed an anaerobic expansion of the microbiota in RAG2-/- mice (Suzuki et al., 2004). Deficiency of both T-bet-/- and RAG2 may result in microbial population shifts such that a host is exposed to an excessive dose of proinflammatory commensals and/or an insufficient dose of protective commensals. The cross-fostering experiments suggest that T-bet deficiency influences the nature of microbial communities resulting in a niche favorable for a colitogenic microbial population. These colitogenic bacteria can be transmitted regardless of the genotype of the recipient. Hence, T-bet sufficient pups fostered by a TRUC mother develop the disease. We envision the colitogenic process as instigated by T-bet deficiency but once established, perpetuated independently of T-bet. The molecular link between T-bet genetic programs enforced in the gut and the generation of colitogenic bacteria remains unclear. Intriguingly, vertically transmitted colitis is not characterized by high levels of TNF-α suggesting that other colitogenic mediators, perhaps IL-23 or IL-17, can be evoked by the transfer of colitogenic bacteria. Identification of the colitogenic bacteria present in these T-bet sufficient fostered pups should provide insight into microbial origins of mechanistically distinct colitides.
Control of T-bet initiated colitis by T regulatory cells
The difference in severity of TRUC X-F WT and RAG2-/- colitis mice coupled with the observation that immunosufficient T-bet-/- mice do not develop spontaneous colitis led to experiments demonstrating that T-regs prevented the development of colitis in T-bet-/- mice. The biology of T-regs and their contribution to promoting tolerance and preventing autoimmunity has burgeoned recently (Fontenot, 2005) with demonstrations that T-regs can down-regulate innate immunity-driven inflammation in both the H. hepaticus RAG2 infection model and the SCID T cell adoptive transfer model (Maloy et al., 2005; Uhlig et al., 2006). We found that infusion of T-regs was an effective treatment for TRUC colitis. Following T-reg transfer, we observed DCs in close contact with T-regs in the colon as described in NOD pancreatic LN(Tang et al., 2006). Clinical trials involving T-reg transfer for IBD patients are underway. Here we provide compelling evidence for an important role of T-regs in a novel model of innate-immune system-driven UC.
TRUC colitis mirrors much of the complexity of human UC. This colitis involves mucosal innate immune cell dysregulation, cytokine-driven inflammation, epithelial barrier breach, pathogenic commensal microbial communities, and invokes a role of T-regs in the maintenance of innate immune cell homeostasis in the enteric immune system. Our experiments suggest that there may be a window of vulnerability in early life that confers susceptibility to development of colitis. The TRUC model provides opportunities to study the pathogenesis of this inflammatory disease and more broadly to understand the biological programs that balance host-commensal relations at mucosal surfaces. T-bet is not only a key regulator of Type 1 immune responses but also a peacekeeper at the prokaryotic-eukaryotic interface.
Experimental Procedures
Animal Husbandry
Mice were housed in microisolator cages in a barrier facility and studied according to institutional and NIH guidelines. All mice used were specific pathogen free and negative for Helicobacter hepaticus, bilis, and muridarum. We thank Drs. K. Malloy (Oxford University) and T. Yatsunenko (Washington University) for carrying out the Hepaticus screening.
TNFR1p55-/- mice, a gift of Dr. R. Levy (University of Miami).
For cross-fostering, on the day of birth the mother was removed from the birthing cage and placed in a clean cage with the cross-foster litter. Pups were weaned between d21–28.
Co-housing of female, 8 wk RAG2-/- or WT with TRUC mice was at a ratio of 1:3 for 8 wks. By 12 wks of age, TRUC mice required euthanasia and were replaced with 8 wk TRUC for the duration of the co-housing.
For studies of the progeny of antibiotic treated mice, pups were weaned at d21 and examined 6 wks after weaning. There was no exposure to antibiotics after weaning.
Histology
Colons were cleaned with PBS or HBSS prior to fixation followed by routine paraffin embedding, sectioning, and staining with H&E.
Sections were examined and the degree of colitis scored by one of the authors (J.N.G.), who was blinded to the genotype and experimental protocols used. Each of 4 histologic parameters were scored as absent (0), mild (1), moderate (2), or severe (3): mononuclear cell infiltraton, polymorphonuclear cell infiltration, epithelial hyperplasia, and epithelial injury (Neurath et al., 2002). Colons from DSS treated mice were scored on a scale of 0–4 based on: % of colon involved by inflammation, % of crypt loss, presence of lymphoid follicles, edema, erosions, and density of inflammatory cells. The scores for the parameters were summed for a total severity score.
Barrier Function Assay
Mice were sedated with ketamine and xylazine. Fluorescein-dextran (MW 3000, Invitrogen, 0.6mg/gram mouse-weight) was delivered per rectum using a 3.5 Fr catheter. Serum fluorescence was measured, and pre-injection fluorescence was used as the blank. n=4–6 per group.
Preparation of Samples for EM
Samples were prepared for Epon embedding as per standard protocols. Blocks were sectioned and stained with toluidine blue for light microscopy. Thin sections were picked up on uncoated grids, stained with saturated aqueous uranyl acetate and acetone followed by lead citrate, then examined with a JEOL 1200 electron microscope.
Abs
Abs were obtained from BD Pharmingen unless otherwise noted: α-B220/CD45R (RA3-6B2), α-CD11c (HL3), α-CD11b (M1/70), α???MHC class II (AF6-120.1 and 39-10-8), α -CD45 (30-F11), α-Gr-1 (eBiosciences), α-F4/80 (BM8 eBiosciences), α-CD8 (53-6.7), α-CD49b (DX5), α-CD4 (RM4-5), α-CD3 (DAKO # AO452), α-S100 (Dako Inc.), α-CD62L (MEL-14), α-cytokeratin 5/8 (RCK102, Santa Cruz), α-TNFR1 (H-5, Santa Cruz) and α-CD25 (PC61).
Immunohistochemistry
Immunostaining for T-bet was performed on paraffin-embedded tissue sections as described (Dorfman et al., 2003). For double staining of human colonic biopsies with T-bet and S100 (Dako Inc). T-bet staining was as above with α-S100 using an ABC amplification kit (Vector Laboratories). For CD11c IHC, biotinylated α-CD11c and an amplification protocol were used.
For IF staining of colonic leukocytes, sections were stained with α-CD3 and biotin α-CD11c and amplification protocols were used. Sections were counterstained with DAPI (Sigma), viewed with an Olympus B40 microscope, and digitally photographed. Composites were assembled in Adobe Photoshop (Adobe Systems Inc.). TUNEL staining on the designated paraffin embedded tissues was performed using the fluorescein in situ cell death detection kit (Roche).
Anti-Cytokine Therapy
α-TNF-α (clone TN3-19.12), a hamster α-mouse TNF-α neutralizing IgG1, and control Ab, hamster α-GST IgG, were generous gifts of Dr. R. Schreiber. TN3-19.12 and control hamster IgG were purchased from Leinco Technologies, Inc. TN3-19.12 or control were given intra-peritoneally at a dose of 15 microgram/gm mouse weight every 7 days for 4 wks.
Colon Explant Culture
Explant cultures were carried out following a modification of described procedures (Rakoff-Nahoum et al., 2004). Media was collected after 4 hrs, centrifuged, and the supernatant was aliquoted and stored at −80°C.
Measurement of cytokines
Cytokines were measured in culture supernatants utilizing SearchLight high dynamic range (HDR) imaging and analysis. For TNF-α, the mouse BD OptEIA ELISA kit was used. Total supernatant protein concentration was measured and used to calculate the cytokine concentration in pg/mg.
Colonic epithelial cell culture
Epithelial cell culture and TUNEL staining was performed as described with the following modifications (Baumgart et al 1998). After 6 days, cells were harvested and stained with α–CD45, cytokeratin 5/8, and TUNEL reagent to label the apoptotic cells. After event acquisition using a LSRII, CD45- cytokeratin 5/8+ cells were gated and the % of TUNEL cells was quantitated using FACSDiva software.
Isolation of colonic DCs for Flow Cytometry and Intracytoplasmic Cytokine Staining
Mononuclear cells were isolated from the colon as described with modifications as noted (Camerini et al., 1993). Cell suspensions were layered onto Percoll gradients and the 30/70% interface, enriched for mononuclear cells, was harvested.
Methods for flow cytometry (FC) and intra-cytoplasmic cytokine staining of colonic leukocytes are as described with modifications noted (Rigby et al., 2005). Cells were stained with PE-conjugated Abs directed against TNF-α and the appropriate PE-conjugated isotype controls in parallel. For FC, samples were acquired using a Becton Dickinson LSRII and data were analyzed using FACS Diva software. For colonic DCs, acquisition gates were constructed such that an equal number of CD11c+ class II+ cells were collected from RAG2-/- and TRUC samples. Cell sorting was performed using a FACS Aria II at the BWH Center for Neurological Diseases Cell Sorting Core Facility.
Bone Marrow DC Culture
BMDCs were generated as described with the following modification: MACS bead depletion for class II, CD8, CD4, and B220 was employed and cells were cultured in the presence of GM-CSF (Inaba et al., 1992).
Human myeloid DCs
CD14+ cells were isolated from pooled human buffy coats and cultured in GM-CSF and IL-4 as has been previously described (Sallusto and Lanzavecchia, 1994).
ChIP and qPCR
20×106 DCs, harvested on day 6 and treated with LPS 100ng/ml for 24–30 hrs and IFNγ 20ng/ml, were used per IP. Cells were fixed for 30 min at RT for IP with the T-bet polyclonal Ab, 9856. ChIPs were performed as described (Ansel et al 2004). 1/20 vol of the ChIP sample was used per qPCR rxn. qPCR products were submitted to agarose gel electrophoresis using SyBR gold dye (Invitrogen) to verify amplification of products of the correct size. Primers sequences for qPCR are available upon request. qPCR reactions were carried out in an ABI Prism 7700 Sequencer Detector using SYBR green reagents.
Luciferase Assay
Constructs: 3kb mouse TNF-α promoter luciferase construct (generously provided by Dr. A. Goldfeld, Harvard University), a constitutively active Iκκβ mutant construct, a Renilla construct for normalization of transfection efficiency, and full length T-bet cDNA construct or control plasmid. Luciferase and Renilla activity were assayed using the Dual-Luciferase Reporter Assay System (Promega) and a Monolight 2010 luminometer.
Broad Spectrum antibiotic treatment of TRUC colitis
Mice were treated as described (Fagarasan et al., 2002; Rakoff-Nahoum et al., 2004). Alterations are as noted: ampicillin (1 g/L; Roche), vancomycin (1 gm/L; Henry Schein, Inc.(Hospira, Inc.)), neomycin sulfate (1 g/L; Sigma), and metronidazole (1 g/L; Sigma) were dissolved in autoclaved drinking water and fluid intake was monitored. Treatment was for 6 wks.
Bacterial Culture
Bacteria culture methods were performed as described (Rakoff-Nahoum et al., 2004). After 48 hrs for aerobes or 72 hrs for anaerobes at 37C, colonies were counted. Anaerobic cultures were grown in anaerobic chambers.
Adoptive Transfer Experiments
Peripheral LNs were harvested, cell suspension generated, and depleted of CD8+, CD11c+, B220+, Ter-119+, DX5+, classII+ populations using MACS beads depletion. The enriched cell population was stained with α-CD4(clone RM4-5), anti-CD62L(clone MEL-14), and anti-CD25(clone PC61) and subjected to FACS. CD4+, CD62L hi, and CD25+ and negative populations were collected, were resuspended in PBS, and 75,000 were injected per mouse. 1×106 CD4+CD62L+CD25− cells were injected per mouse as noted. For B cell adoptive transfer (AT), splenic B cells were purified using MACS positive selection and 1 ×106 CD19+ resuspended in PBS were injected per mouse. After 4 wks, mice were sacrificed except for mice receiving the na???ve T cells or B cells that were sacrificed after 2 wks due to morbidity. PBS was used for sham injection.
Statistical Analysis
All experiments were repeated 3 times unless otherwise noted. Statistical analysis was performed using the unpaired Student’s t-test. Error bars represent standard deviations.
Supplementary Material
(A) Anti-TNF-α treatment normalizes TRUC colon weights. Mice were treated with anti-TNF-α or isotype control from 4 wks of age through 8 wks of age on a weekly dosing schedule. Representative data from 1 of 3 independent experiments are shown. (B) There is increased apoptosis in the colonic epithelium of TRUC mice. TUNEL+ cells and the number of epithelial cells per crypt were counted across all 4 genotypes. 5 slides were generated from each genotype group (2–3 mice per genotype). Areas of the large bowel were randomly selected and epithelial cells, epithelial crypts, and TUNEL+ epithelial cells were counted. A minimum of 500 crypts per genotype were scored. (C) Anti-TNF-α treatment cures TRUC colitis when initiated prior to disease onset. Mice were treated with anti-TNF-α or isotype control from 2 wks of age through 6 wks of age, representative data from one of 2 independent experiments are shown, p =.002. (D) Anti-TNF-α treatment cures TRUC colitis when initiated during fulminate disease. Mice were treated with anti-TNF-α or isotype control from 8–12 wks of age, representative data from one of 2 independent experiments are shown, p = .012.
Acknowledgements
We thank D. Hu for expertise in histology and IHC; I. Jackson for assistance in phenotyping and FC; J. Ramirez for his care of our mice; Dr. J. Shim for advice and constructs for luciferase assays; Drs. S. Turley, T. Staton, M. Wein, A. Zullo, A. Kaser, and R. Blumberg for critical reading of the manuscript; Drs. W. Lencer, J. Gordon, D. Peterson, T. Yatsunenko, and members of the Glimcher and Grusby laboratories for helpful discussion. This work was supported by grants from the NIH (CA112663 and AI56296) and an Ellison Scholar Award to L.H.G. W.S.G. is a postdoctoral fellow of the Damon Runyon Cancer Research Foundation and received funding from the Irving Janock Fellowship. G.M.L. was supported by an MRC Clinician Scientist Grant (G108/380) and funding from the Department of Health via the NIHR comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London, UK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansel KM, Greenwald RJ, Agarwal S, Bassing CH, Monticelli S, Interlandi J, Djuretic IM, Lee DU, Sharpe AH, Alt FW, Rao A. Deletion of a conserved Il4 silencer impairs T helper type 1-mediated immunity. Nature Immunology. 2004;5:1251–1259. doi: 10.1038/ni1135. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Baumgart DC, Olivier WA, Reya T, Peritt D, Rombeau JL, Carding S. Mechanims of intestinal epithelial injury and colitis in IL-2 deficient mice. Cell Immunol. 1998;187:52–66. doi: 10.1006/cimm.1998.1307. [DOI] [PubMed] [Google Scholar]
- Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- Bruewer M, Samarin S, Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. 2006;1072:242–252. doi: 10.1196/annals.1326.017. [DOI] [PubMed] [Google Scholar]
- Camerini V, Panwala C, Kronenberg M. Regional specialization of the mucosal immune system. Intraepithelial lymphocytes of the large intestine have a different phenotype and function than those of the small intestine. J Immunol. 1993;151:1765–1776. [PubMed] [Google Scholar]
- Chieppa M, Rescigno M, Huang AY, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203:2841–2852. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman DM, van den Elzen P, Weng AP, Shahsafaei A, Glimcher LH. Differential expression of T-bet, a T-box transcription factor required for Th1 T-cell development, in peripheral T-cell lymphomas. Am J Clin Pathol. 2003;120:866–873. doi: 10.1309/MLUF-X0HR-5B96-GVAX. [DOI] [PubMed] [Google Scholar]
- Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- Glimcher L. Trawling for treasure: tales of T-bet. Nat Immunol. 2007;8:448–450. doi: 10.1038/ni0507-448. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A. Mucosal dendritic cells. Annu Rev Immunol. 2007;25:381–418. doi: 10.1146/annurev.immunol.25.022106.141634. [DOI] [PubMed] [Google Scholar]
- Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JG, Cruickshank SM, Singh JC, Farrar M, Lodge JP, Felsburg PJ, Carding SR. Different cytokine response of primary colonic epithelial cells to commensal bacteria. World J Gastroenterol. 2005;11:3375–3384. doi: 10.3748/wjg.v11.i22.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Geuking MB, McCoy KD. Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology. 2005;115:153–162. doi: 10.1111/j.1365-2567.2005.02159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes JG, Tattoli I, Girardin SE. The intestinal epithelial barrier: How to distinguish between the microbial flora and pathogens. Semin Immunol. 2007 doi: 10.1016/j.smim.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, Antonelli LR, Lefevre M, Powrie F. Cure of innate intestinal immune pathology by CD4+CD25+ regulatory T cells. Immunol Lett. 2005;97:189–192. doi: 10.1016/j.imlet.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niess JH, Reinecker HC. Dendritic cells: the commanders-in-chief of mucosal immune defenses. Curr Opin Gastroenterol. 2006;22:354–360. doi: 10.1097/01.mog.0000231807.03149.54. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Medzhitov R. Role of the innate immune system and host-commensal mutualism. Curr Top Microbiol Immunol. 2006;308:1–18. doi: 10.1007/3-540-30657-9_1. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rigby RJ, Knight SC, Kamm MA, Stagg AJ. Production of interleukin (IL)-10 and IL-12 by murine colonic dendritic cells in response to microbial stimuli. Clin Exp Immunol. 2005;139:245–256. doi: 10.1111/j.1365-2249.2004.02674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogler G, Gelbmann CM, Vogl D, Brunner M, Scholmerich J, Falk W, Andus T, Brand K. Differential activation of cytokine secretion in primary human colonic fibroblast/myofibroblast cultures. Scand J Gastroenterol. 2001;36:389–398. doi: 10.1080/003655201300051216. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, de Hertogh G, Bullens DM, Van Assche G, Geboes K, Rutgeerts P, Ceuppens JL. Remission-inducing effect of anti-TNF monoclonal Ab in TNBS colitis: Mechanisms beyond neutralization? Inflamm Bowel Dis. 2006 doi: 10.1002/ibd.20005. [DOI] [PubMed] [Google Scholar]
- Silva MA, Lopez CB, Riverin F, Oligny L, Menezes J, Seidman EG. Characterization and distribution of colonic dendritic cells in Crohn's disease. Inflamm Bowel Dis. 2004;10:504–512. doi: 10.1097/00054725-200409000-00003. [DOI] [PubMed] [Google Scholar]
- Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honja T, Fagarasan S. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci U S A. 2004;101:1981–1986. doi: 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock GW. What immunologists should know about bacterial communities of the human bowel. Semin Immunol. 2006 doi: 10.1016/j.smim.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Uhlig HH, Coombes J, Mottet C, Izcue A, Thompson C, Fanger A, Tannapfel A, Fontenot JD, Ramsdell F, Powrie F. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaph TA, Taylor BC, Berman-Booty LD, Guil KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, Eckman L, Kartin M, Artis D. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Anti-TNF-α treatment normalizes TRUC colon weights. Mice were treated with anti-TNF-α or isotype control from 4 wks of age through 8 wks of age on a weekly dosing schedule. Representative data from 1 of 3 independent experiments are shown. (B) There is increased apoptosis in the colonic epithelium of TRUC mice. TUNEL+ cells and the number of epithelial cells per crypt were counted across all 4 genotypes. 5 slides were generated from each genotype group (2–3 mice per genotype). Areas of the large bowel were randomly selected and epithelial cells, epithelial crypts, and TUNEL+ epithelial cells were counted. A minimum of 500 crypts per genotype were scored. (C) Anti-TNF-α treatment cures TRUC colitis when initiated prior to disease onset. Mice were treated with anti-TNF-α or isotype control from 2 wks of age through 6 wks of age, representative data from one of 2 independent experiments are shown, p =.002. (D) Anti-TNF-α treatment cures TRUC colitis when initiated during fulminate disease. Mice were treated with anti-TNF-α or isotype control from 8–12 wks of age, representative data from one of 2 independent experiments are shown, p = .012.