

Abstract
While the mechanisms that regulate actin dynamics in cellular motility are intensively studied, relatively little is known about signaling events that transmit outside-in signals and direct assembly and regulation of actin polymerization complexes at the cell membrane. The kidney podocyte provides a unique model for investigating these mechanisms since deletion of Nephrin or Neph1, two interacting components of the specialized podocyte intercellular junction, results in abnormal podocyte morphogenesis and junction formation. We provide evidence that extends the existing model by which the Nephrin-Neph1 complex transduces phosphorylation-mediated signals that assemble an actin polymerization complex at the podocyte intercellular junction. Upon engagement, Neph1 is phosphorylated on specific tyrosine residues by Fyn, which results in the recruitment of Grb2, an event that is necessary for Neph1-induced actin polymerization at the plasma membrane. Importantly, Neph1 and Nephrin directly interact and, by juxtaposing Grb2 and Nck1/2 at the membrane following complex activation, cooperate to augment the efficiency of actin polymerization. These data provide evidence for a mechanism reminiscent of that employed by vaccinia virus and other pathogens, by which a signaling complex transduces an outside-in signal that results in actin filament polymerization at the plasma membrane.
Precisely regulated actin polymerization provides the motive force necessary for intercellular junction formation and contributes to defining the shape and polarity of the cell. Similarly, directed actin polymerization proximate to the leading edge of the plasma membrane drives cell motility and is required in the complicated dynamics of lamellipodia, filopodia, and other specialized membrane structures including invadopodia and podosomes (6). While substantial progress has been made in understanding the cellular and molecular mechanisms that determine actin dynamics in these model systems (32), less is known about membrane-based proximal signaling events that transmit outside-in signals and direct assembly and regulation of actin polymerization complexes at these sites.
Kidney glomerular visceral epithelial cells or podocytes are necessary for maintaining the glomerular filtration barrier (reviewed in reference 20). When mature, these cells have a unique octopus-like structure comprised of a central cell body that gives off branching primary, secondary, and tertiary processes. The tertiary processes or “foot processes” attach the podocyte to the glomerular capillary basement membrane, where they surround the glomerular capillary and where they interdigitate to form a specialized intercellular junction called the “slit diaphragm.” Here foot processes provide a necessary element of the permeability-selective glomerular filter, allowing passage of water, solutes, and other small macromolecules from the capillary lumen to the urinary space while restricting the flux of cells and larger macromolecules.
The podocyte provides a unique model for investigating the molecular mechanisms that integrate actin dynamics and intercellular junction formation. The morphology of mature podocyte foot processes is defined by its actin cytoskeleton. Mature podocytes develop from cuboidal podocyte precursors that undergo reorganization of a classical adherens junctional complex to form a specialized intercellular junctional complex (20). Junctional reorganization appears to occur concurrently with the emergence of processes from the basolateral aspect of the precursor cell presumably driven by a motive force provided by the assembly of the cytoskeleton of podocyte processes (15). In a reciprocal fashion, podocyte injury observed in most types of kidney glomerular disease causes a simplification in podocyte cytoskeletal and intercellular junctional architecture to a state reminiscent of the cuboidal podocyte precursor (1). While the signaling mechanisms that integrate podocyte structure and filter integrity are incompletely defined, recent studies have revealed the importance of the protein complex located at the slit diaphragm in regulating the actin cytoskeleton and in determining the relationship between podocyte structure and maintenance of glomerular permselectivity.
Nephrin and Neph1 form a protein complex targeted to the foot process intercellular junction that appears to function as a transmembrane receptor. Absence of either of these receptor elements in humans carrying an inherited mutation, or in experimental mutant mice, causes proteinuria and developmental failure of foot process formation (5, 9, 23). Nephrin and Neph1 are structurally similar type I transmembrane proteins of the immunoglobulin superfamily. They directly interact in the plane of the plasma membrane via their cytoplasmic domains (4, 12, 25). In addition, the Nephrin extracellular domain interacts with itself and with Neph1 via a trans-interaction across the mature podocyte intercellular junction; for this reason, it has been presumed that these proteins mediate cell-cell adhesive interactions at this site (4, 10). The hypothesis that the Nephrin-Neph1 complex participates in regulating foot process actin cytoskeletal dynamics has been suggested by the observation that this complex interacts at its cytoplasmic face with known actin-associated proteins including α-actinin-4, synaptopodin, CD2ap, ZO-1, CASK, IQGAP1, β-arrestin, and Nck (2, 17, 18, 26, 28, 33). This hypothesis is strengthened by the observations that deletion or mutation of α-actinin-4, synaptopodin, CD2ap, Nck1, and Nck2 is associated with developmental abnormalities of podocyte cytoskeletal architecture and/or junction formation (17, 21, 22, 38).
Upon engagement of Nephrin's extracellular domain, the Src family protein kinase Fyn rapidly catalyzes the phosphorylation of Nephrin on multiple tyrosine residues (27, 42). Among these residues are three (Y1191, Y1208, and Y1232) necessary for mediating the interaction between Nephrin and adaptor proteins including Nck (21, 41). Phosphorylation at these sites occurs transiently during foot process formation and following podocyte injury. Importantly, recruitment of Nck to Nephrin is necessary for the induction of Nephrin-mediated actin polymerization in a cell culture model. Moreover, podocyte-specific deletion of Nck1/2 in mice results in developmental failure of foot process formation. Given these observations, it has been suggested that Nephrin participates in regulating podocyte cytoskeletal dynamics (21). Because deletion of either Nephrin or Neph1 results in an indistinguishable phenotype of abnormal foot process development and junction formation, we examined Neph1 function and report here that—like Nephrin—Neph1 serves to transmit Fyn-dependent signals that participate in regulating actin cytoskeletal dynamics. Moreover, our results indicate that Nephrin and Neph1 form a functional complex that, by recruiting Nck and Grb2, cooperates to transduce outside-in signals that induce actin polymerization.
MATERIALS AND METHODS
Antibodies.
Purified rabbit polyclonal antibodies against Nephrin (15), Neph1 (4), and phospho-Nephrin (41) were previously described. Antibodies against Grb2 and Nck (BD Biosciences), CD16 (clone 3G8) antibody (Beckman Coulter), rhodamine-conjugated goat anti-mouse immunoglobulin G (IgG; Pierce), anti-glutathione S-transferase (anti-GST) antibody (Santa Cruz), Fyn (Upstate), phosphotyrosine (clone PT-66), and Flag antibody (Sigma-Aldrich) were obtained commercially. 50A9 antibody was a gift from K. Tryggvason (25).
Plasmids.
A plasmid encoding GST-Neph1CD fusion protein that includes the entire cytoplasmic domain of mouse Neph1 was prepared in pGEX4T-1 (Amersham Biosciences) using standard techniques (16). GST-Grb2 was a gift from B. Margolis, University of Michigan, and GST-Grb2-SH2 was a gift from A. Pandey, Johns Hopkins University. Recombinant GST fusion proteins were prepared and purified from bacterial lysates as described previously (29). Where indicated, tyrosine-phosphorylated GST-Neph1CD was expressed in and purified from TKB1 cells (Stratagene). Mammalian plasmids encoding mouse Nephrin (15), Fyn (43), FynKD (K295M) (14), and human Nephrin (gift from K. Tryggvason) (25) were described previously. Mammalian expression plasmids encoding a human Nephrin point mutant (Y1191/1208F/1232F) and Neph1 point mutations (Y604F, Y637F, Y638F, and Y637/638F) were prepared in pcDNA3.1 (Invitrogen) using standard methods. Restriction digestion and DNA sequencing were used to confirm all construct sequences. The CD16-hemagglutinin (HA) construct was a gift from B. Mayer (University of Connecticut) (34). Constructs encoding fusion protein consisting of CD16 extracellular domain, CD7 transmembrane domain, and Nephrin or Neph1 cytoplasmic domain and their mutants were generated. Grb2-GFP, actin-cyan fluorescent protein (CFP), and N-WASp-CFP were a gift from L. E. Samelson (NIH).
In vitro phosphate labeling.
GST-Neph1CD (2 μg) was incubated with 1 μg recombinant active Fyn (Upstate Cell Signaling) in kinase buffer containing 25 μM ATP and 5 μCi [γ-32P]ATP (3,000 Ci/mmol), incubated at 25°C for 15 min, and processed as previously described (42).
Immunoprecipitation and immunoblotting.
Neph1 was extracted from plasma membranes in RIPA buffer (phosphate-buffered saline [PBS] containing 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet P-40, 0.5% sodium deoxycholate, and 100 mM potassium iodide). Endogenous immunoprecipitations were performed by extracting tissue in RIPA buffer containing 0.1% bovine serum albumin.
Cell culture.
Transient transfections were carried out in COS7 or HEK293T cells cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (Invitrogen Corp.) and 200 U/ml penicillin and streptomycin (Roche Applied Science). Transfections were performed using Fugene-6 (Roche Applied Science) per the manufacturer's directions. Where indicated, cells were treated with 50 μM pervanadate prior to harvesting cells. The maintenance of NIH 3T3 cells, SYF cells, and HEK293 cells stably expressing human Nephrin was described previously (41). Transfection in NIH 3T3 cells and SYF cells was performed with Lipofectamine 2000 per the manufacturer's directions.
Phosphotyrosine mapping by peptide array.
Oligopeptides (11- to 18-mers) and peptides with tyrosine-to-phenylalanine mutations encompassing each tyrosine residue in the Neph1 cytoplasmic domain were synthesized (Sigma Genosys PEPScreen). Tyrosine residues in the oligopeptides were flanked by five to six amino acid residues (Table 1). Oligopeptides were dissolved per the manufacturer's recommendation. Solutions containing equimolar amounts of peptides were made in 50 mM Tris buffer (pH 8.0). This mixture was then blotted onto polyvinylidene difluoride (PVDF) membranes using a dot blot apparatus. Membranes were blocked with 5% milk solution in Tris-buffered saline with 0.1% Tween. Subsequently the membrane was incubated in kinase assay buffer containing active Fyn (Upstate Cell Signaling) and 32P-labeled ATP for 3 h at room temperature. The membrane was washed extensively and then analyzed by phosphorimager analysis (Storm 860; Molecular Dynamics).
TABLE 1.
Peptide sequences with mutation
| Tyrosine position | Peptide sequence |
|---|---|
| Y604 WTa | VMKAIYSSFKD |
| Y604F | VMKAIFSSFKD |
| Y628 WT | ETREEYEMKDP |
| Y628F | ETREEFEMKDP |
| Y637/638 WT | DPTNGYYNVRAH |
| Y637F | DPTNGFYNVRAH |
| Y638F | DPTNGYFNVRAH |
| Y637/638F | DPTNGFFNVRAH |
| Y654/657 WT | SRAVLYADYRAPGP |
| Y654F | SRAVLFADYRAPGP |
| Y657F | SRAVLYADFRAPGP |
| Y654/657F | SRAVLFADFRAPGP |
| Y679 WT | SHSSGYAQLNT |
| Y679F | SHSSGFAQLNT |
| Y685 WT | AQLNTYSRAPA |
| Y685F | AQLNTFSRAPA |
| Y693 WT | APASDYGTEP |
| Y693F | APASDFGTEPT |
| Y716/719 WT | TSQLSYENYEKFNS |
| Y716F | TSQLSFENYEKFNS |
| Y719F | TSQLSYENFEKFNS |
| Y716/719F | TSQLSFENFEKFNS |
| Y733Y736/Y740 WT | PGAAGYPTYRLGYPQAPP |
| Y733F | PGAAGFPTYRLGYPQAPP |
| Y736F | PGAAGYPTFRLGYPQAPP |
| Y740F | PGAAGYPTYRLGFPQAPP |
| Y733/736/740F | PGAAGFPTFRLGFPQAPP |
| Y753/756 WT | LERTPYEAYDPIGK |
| Y753F | LERTPFEAYDPIGK |
| Y756F | LERTPYEAFDPIGK |
| Y753/756F | LERTPFEAFDPIGK |
| Y762 WT | DPIGKYATATRF |
| Y762F | DPIGKFATATRF |
| Y770 WT | ATRFSYTSQHS |
| Y770F | ATRFSFTSQHS |
| Y777 WT | SQHSDYGQRFQ |
| Y777F | SQHSDFGQRFQ |
| Nephrin Y1208 WT | AWGPLYDEVQMG |
| Nephrin Y1208F | AWGPLFDEVQMG |
| Neph1 GST | GST-Neph1 CD |
WT, wild type.
PAN nephrosis.
Female Sprague-Dawley rats weighing 200 to 250 g were injected with puromycin aminonucleoside (PAN) (Sigma-Aldrich) (10 mg/100 g of body weight) or PBS (vehicle) intraperitoneally. The urine albumin/creatinine ratio was determined from urine obtained in metabolic cages by direct competitive enzyme-linked immunosorbent assay for urine albumin (Nephrat II; Exocell Inc.) and Jaffé reaction for urine creatinine (Creatinine Companion; Exocell Inc.). Rats were sacrificed, and glomeruli were isolated using graded sieving as described previously (26).
Pull-down.
In some instances, GST-Neph1CD recombinant protein was expressed and purified from TKB1 Escherichia coli (Stratagene) to induce tyrosine phosphorylation. Purified GST fusion proteins bound to glutathione agarose were incubated with isolated mouse glomerular lysate extracted with RIPA buffer. After washing with PBS containing 0.1% Tween 20, 1 mM sodium orthovanadate, and 1 mM sodium fluoride, protein complexes were eluted with reduced glutathione. Elute was resolved by SDS-polyacrylamide gel electrophoresis (PAGE) in replicate prior to immunoblotting with the indicated antibodies.
CD16/CD7/Neph1 chimera, Grb2 recruitment, and actin polymerization experiments.
NIH 3T3 cells were transfected with CD16/CD7 chimeric constructs bearing HA, NephrinCD, or Neph1CD at the C-terminal end. Thirty hours following transfection, Dulbecco modified Eagle medium was removed and replaced with fresh medium containing 1 μg/ml CD16 antibody (clone 3G8; Beckman Coulter). Cells were maintained on ice for 1 h for Grb2/Nck recruitment experiments or at 37°C for actin experiments. At this point, cells were washed twice with PBS, 1 μg/ml rhodamine-conjugated anti-mouse IgG (Pierce Biotechnology) was added to the medium, and incubation was continued at 37°C for 20 min for recruitment experiments and for 1 h for actin experiments. Cells were washed three times with PBS and fixed with cytoskeleton buffer. The composition of cytoskeleton buffer stock was 10 mM 2-CN-morpholinoethanesulfonic acid, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, and sucrose to a final concentration of 0.32 M. On the day of use, 20% paraformaldehyde was added to cytoskeleton buffer stock to achieve a final concentration of 4%. Coverslips were mounted on glass slides using ProLong Gold antifade reagent (Invitrogen Corp.). Samples were analyzed by fluorescence confocal microscopy with an Olympus FV-500 microscope using a 100× oil-immersion objective lens and Fluoview software (version TIEMPO 4.3; Olympus). Images were processed using Adobe Photoshop software. All images were acquired at 1,024- by 1,024-pixel resolution.
Fyn-null mice and wild-type littermates used were described previously (42). All animal experiments were approved by the University Committee on the Use and Care of Animals Institutional Review Board at the University of Michigan Medical School.
RESULTS
It was previously reported that Neph1 can be tyrosine phosphorylated (18, 37). For this reason, we sought to identify proteins that interact with Neph1 in a tyrosine-dependent fashion. GST-Neph1CD was prepared in TKB1 E. coli cells expressing a tyrosine kinase that promiscuously catalyzes tyrosine phosphorylation of Neph1. This recombinant tyrosine-phosphorylated GST-Neph1 cytoplasmic domain was employed to pull down associated proteins from isolated rat glomerular lysate. After resolution by SDS-PAGE, associated proteins were identified using a panel of antibodies specific for known proteins that contain SH2 domains. Nonphosphorylated GST-Neph1CD and phosphorylated GST served as controls. By this approach, Grb2 and Fyn were identified as proteins that might interact with Neph1 (Fig. 1A).
FIG. 1.

Neph1 interacts with Grb2 in a tyrosine phosphorylation-dependent manner. (A) Rat glomerular lysate was incubated with GST-Neph1 expressed in tyrosine kinase-expressing E. coli TKB1 or E. coli BL21. Following pull-down and resolution of GST-Neph1-associated proteins on SDS-PAGE, blots were immunoblotted with indicated antibodies. (B) Neph1 and Grb2 associate in vivo. Coimmunoprecipitation experiments performed on cells from rat glomerular lysate using antibodies against Neph1 and Grb2 show endogenous association between these two proteins. (C) Direct interaction between Neph1 and Grb2. Purified recombinant GST-Neph1 and GST-Grb2 were mixed, and immunoprecipitation and immunoblotting were performed with the indicated antibodies. Only phosphorylated Neph1 (lane 5) associated with Grb2. Input recombinant proteins were identified by immunoblotting. Tyrosine phosphorylation of GST-Neph1 prepared in TKB1 was confirmed (data not shown). (D) Grb2 interacts with tyrosine-phosphorylated Neph1 via its SH2 domain. Lysates from COS7 cells expressing Flag-tagged wild-type Neph1 treated where indicated with pervanadate (PV) were mixed with GST fusion proteins containing full-length Grb2 and a truncated Grb2 containing only its SH2 domain. The Grb2 SH2 domain was sufficient to pull down tyrosine-phosphorylated Neph1. The Coomassie blue-stained SDS-polyacrylamide gel shows the purified GST recombinant protein input. Input COS7 cell lysates employed are shown. (E) GST-Grb2 overlay. Arrayed Neph1 oligopeptides synthesized with tyrosine phosphorylation at Y604, Y716, Y719, Y637, and Y638 were incubated with GST-Grb2 and then probed with anti-Grb2 antibody. Grb2 associated with peptides containing tyrosine phosphorylation at the Y637YNV motif. Relative molecular masses are in kDa. WB, Western blot; IP, immunoprecipitation.
Neph1 interacts directly with Grb2.
The interaction of endogenous Grb2 and endogenous Neph1 was investigated by coimmunoprecipitation from rat-isolated glomerular lysate; under the conditions of this experiment, Grb2 readily coimmunoprecipitated with Neph1 and Neph1 coimmunoprecipitated with Grb2 (Fig. 1B). A direct interaction between Neph1 and Grb2 protein was confirmed by coimmunoprecipitation of purified recombinant proteins that were combined in vitro (Fig. 1C). Notably, Grb2 coimmunoprecipitated with GST-Neph1CD only when GST-Neph1CD was prepared in TKB1 cells and was therefore tyrosine phosphorylated. To extend this analysis, pull-down experiments were used to demonstrate that the Grb2-SH2 domain alone is sufficient to mediate Grb2-Neph1 interaction (Fig. 1D); this interaction required tyrosine phosphorylation of Neph1.
Songyang et al. initially identified and characterized the Grb2 SH2 domain consensus binding motif, pYXNX (39). Extending this work, van der Geer and Hunter showed that phosphorylation of either tyrosine residue of a Y239YNX motif in ShcA was sufficient for ShcA-Grb2 interaction and demonstrated that simultaneous phosphorylation of the two tyrosine residues augmented the interaction affinity of ShcA and Grb2. These investigators also noted that a YYNX motif is conserved in several proteins that interact with Grb2 (40). With this in mind and using the Scansite algorithm, we examined the Neph1 sequence and identified a Y637YNV motif located within the Neph1 cytoplasmic domain that we hypothesized might represent a Grb2 SH2 binding motif necessary and sufficient for mediating an interaction between Neph1 and Grb2 (http://scansite.mit.edu). To begin to examine this hypothesis, synthetic tyrosine-phosphorylated oligopeptides blotted onto a PVDF membrane were incubated with GST-Grb2 and interactions were detected using a Grb2 antibody (Fig. 1E). As predicted from sequence analysis, peptides phosphorylated at Y637 or Y638 bound Grb2 while Grb2 did not associate with control phospho-oligopeptides. These data suggested the hypothesis that following tyrosine phosphorylation of Neph1 on Y637 and Y638, Grb2 is recruited to Neph1 at Y637YNV via an interaction that involves its SH2 domain.
Fyn is necessary for Neph1-Grb2 interaction.
Because Fyn was identified in our screen for Neph1-interacting proteins (Fig. 1A), we tested the hypothesis that Fyn-catalyzed tyrosine phosphorylation of Neph1 is necessary to induce Neph1-Grb2 interaction. A Neph1-Fyn interaction was confirmed by coimmunoprecipitation from isolated rat glomerular extract of endogenous Fyn with Neph1 (Fig. 2A). To examine whether Fyn was capable of directly catalyzing the phosphorylation of Neph1, recombinant GST-Neph1 cytoplasmic domain (GST-Neph1-CD) was incubated with active recombinant Fyn in the presence of [γ-32P]ATP in kinase assay buffer for 30 min at 30°C (Fig. 2B). Under these conditions, recombinant Neph1 was phosphorylated in the presence of active Fyn, a process that was attenuated by an inhibitor of Src family kinase (SFK) catalytic activity (PP2, 10 μM). The necessity of Fyn for mediating Neph1-Grb2 recruitment was confirmed in mice depleted of Fyn by gene targeting (Fig. 2C). Here, Neph1 was tyrosine phosphorylated and was associated with Grb2 when Neph1 was immunoprecipitated from glomerular lysate isolated from wild-type mice. In contrast, Neph1 tyrosine phosphorylation was markedly attenuated and Neph1-Grb2 association was not detected when Neph1 was obtained from Fyn−/− glomerular lysate. Therefore, Fyn is necessary for Neph1-Grb2 interaction in the podocyte in vivo. That Neph1 tyrosine phosphorylation is attenuated but not absent when Neph1 is obtained from Fyn−/− mice suggests that Neph1 tyrosine phosphorylation might be catalyzed by additional protein tyrosine kinases (18, 37).
FIG. 2.
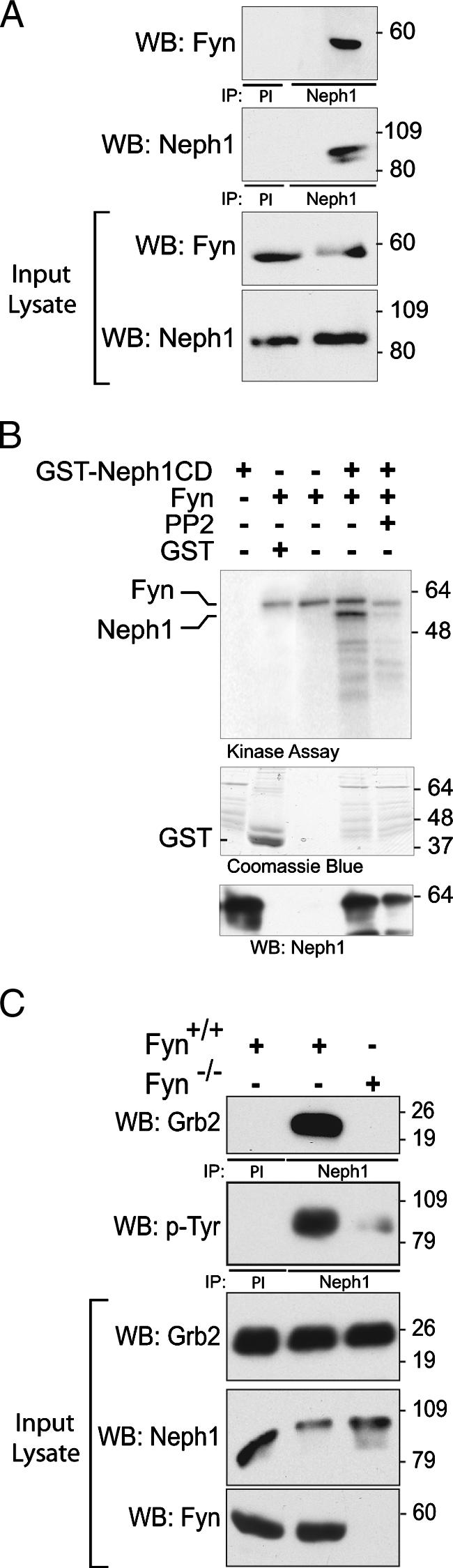
Fyn catalyzes Neph1 tyrosine phosphorylation and is necessary for Neph1-Grb2 interaction. (A) Coimmunoprecipitation experiments on cells from rat glomerular lysates showing interaction between endogenous Neph1 and Fyn (PI, preimmune serum). (B) In vitro kinase assay. GST-Neph1 was incubated with active recombinant His-tagged Fyn in the presence of [γ-32P]ATP for 20 min. Radiolabeled Fyn is observed as anticipated in lanes 2 to 4. Phosphorylation of both Neph1 and Fyn is attenuated in the presence of PP2, a Src family kinase inhibitor. A Coomassie blue-stained gel indicates expression of recombinant GST proteins. (C) Neph1-Grb2 interaction in Fyn-null mice. Glomerular lysates from kidneys of wild-type mice or mice with deletion of Fyn were immunoprecipitated with Neph1 antibody and blotted for Grb2. Grb2 coimmunoprecipitated with Neph1 in wild-type mice. Tyrosine phosphorylation of Neph1 was significantly attenuated in Fyn-null mice compared to control.
An unbiased peptide array screening method was used to identify tyrosine residues in Neph1 that are phosphorylated by Fyn to establish that Neph1 can be phosphorylated by Fyn on Y637 and Y638 (Fig. 3A). Synthetic 11- to 20-mer oligopeptides were synthesized that overlapped each of the 19 tyrosine residues present within the cytoplasmic domain of Neph1. As controls, corresponding oligopeptides were prepared with tyrosine-to-phenylalanine mutations. In some cases compound substitutions were required that allowed analysis of closely spaced tyrosine residues. Peptide arrays prepared on PVDF membranes were probed by incubation with activated recombinant Fyn. By this approach, four tyrosine residues were identified that became phosphorylated in the presence of Fyn (Y637, Y638, Y716, and Y719). Arrays not treated with Fyn or peptides containing Y-to-F mutations did not become phosphorylated. To confirm that Neph1 tyrosine residues identified by this screen are phosphorylated on Neph1 expressed in cells, tyrosine-to-phenylalanine mutations were introduced into full-length Flag-tagged Neph1 constructs and were expressed with Fyn in COS7 cells (Fig. 3B). Wild-type Neph1 or its tyrosine mutants were immunoprecipitated from these lysates and were immunoblotted with antiphosphotyrosine antibody. As anticipated from the initial screen, point substitutions at Y637, Y638, Y716, and Y719 attenuated Neph1 tyrosine phosphorylation relative to wild type. In a control experiment, substitution of F for Y604 (which was not predicted to be phosphorylated by Fyn in the initial screen) had no effect on Neph1 tyrosine phosphorylation. In summary, these data support the conclusion that Neph1 Y637, Y638, Y716, and Y719 can be tyrosine phosphorylated by Fyn. These data do not exclude the possibility that additional Neph1 residues are phosphorylated in vivo under various conditions. To integrate these in vitro observations with those above suggesting that phosphorylation at Y637 and Y638 is necessary for recruitment of Grb2, Neph1 or its mutants and Grb2 were expressed in COS7 cells and their interaction was assessed by coimmunoprecipitation (Fig. 3C). Here, Grb2 interacted with Neph1 only when catalytically active Fyn and not kinase-dead Fyn was simultaneously expressed. Moreover, Neph1 that had a substitution of phenylalanine either at Y637 or at Y638 coimmunoprecipitated Grb2 while Neph1 mutated at both residues failed to coimmunoprecipitate Grb2. Collectively, these data suggest that following tyrosine phosphorylation of Neph1 on Y637 and Y638 by Fyn, Grb2 is recruited to Neph1 via an interaction that involves its SH2 domain.
FIG. 3.

Mapping of Neph1 tyrosine residues phosphorylated by Fyn. (A) Peptide array screen. Oligopeptides (11- to 18-mers) were synthesized covering all 19 tyrosine residues in the Neph1 cytoplasmic domain and were arrayed. WT, wild type. Peptides with replacements of tyrosine by phenylalanine are indicated. Equal quantities of oligopeptides were blotted onto a PVDF membrane, incubated with or without active recombinant Fyn and [γ-32P]ATP, and evaluated by phosphorimager analysis. Four tyrosine residues, Y637, Y638, Y716, and Y719, were phosphorylated in the presence of active Fyn. (B) COS7 cells were transfected with plasmids encoding Fyn or kinase-dead Fyn (KD) and with wild-type Neph1 or its indicated tyrosine mutants. Neph1 was immunoprecipitated from cell lysates and immunoblotted for phosphotyrosine content (pY). The graph displays band intensity normalized to total immunoprecipitated Neph1; represented are means ± standard errors of the means. Results are representative of three independent experiments. (C) Both tyrosines Y637 and Y638 are essential for Neph1-Grb2 interaction. COS7 cells were transiently transfected with plasmids encoding full-length wild-type Neph1 (Neph1 WT) and its tyrosine mutants. Grb2-GFP, Fyn, and kinase-dead (Fyn KD) plasmids were cotransfected where indicated. Neph1-Grb2 association was abrogated when both Y637 and Y638 were replaced with phenylalanine simultaneously.
Grb2-Neph1 interaction is induced after podocyte injury.
The rat PAN model of podocyte injury (19, 35) was employed to examine the interaction of Neph1 and Grb2 in vivo. As described by other investigators (16, 30), we observed that proteinuria was first increased 3 days post-PAN injection (Fig. 4A). At the same time point, electron microscopy demonstrated foot process effacement in PAN-treated animals (Fig. 4B). The 72-h time point was chosen for investigation because we were interested in examining signaling events early in the evolution of PAN-induced podocyte injury. At this time point, Neph1 tyrosine phosphorylation in isolated glomeruli was increased compared to vehicle-treated control animals (Fig. 4C). Concomitantly, the association of Neph1 and Grb2 increased in injured podocytes relative to control. In a similar fashion, total Nephrin tyrosine phosphorylation and phosphorylation specifically on Nephrin Y1192/Y1208 increased and the association of Nephrin and Nck increased in PAN-treated glomeruli relative to control (Fig. 4D). These results are consistent with our previous observations of Nephrin Y1192/Y1208 phosphorylation in mice following protamine sulfate-induced foot process effacement (41). Taken together, these results support the conclusion that podocyte injury induces tyrosine phosphorylation of the Nephrin/Neph1 protein complex and subsequent recruitment of adaptor proteins that include Nck and Grb2.
FIG. 4.

Neph1 is tyrosine phosphorylated and recruits Grb2 following PAN-induced podocyte injury in vivo. (A) Time course of urine albumin/creatinine ratios of control or PAN-treated female rats (PAN-treated rats had increased proteinuria compared to controls; P < 0.05, day 3 and later). (B) Electron micrographs of kidneys from vehicle control and PAN-treated rats showing loss of normal foot process architecture at 72 h postinjection (CL, capillary lumen; GBM, glomerular basement membrane; P, podocyte). Bar, 1 μm. (C) Neph1 complexes immunoprecipitated from rat glomerular lysates treated with PAN or vehicle alone were evaluated by immunoblotting. (D) In similar experiments, Nephrin was immunoprecipitated from rat glomerular lysates prepared as indicated (PI, preimmune serum).
Clustered Neph1CD becomes tyrosine phosphorylated and recruits Grb2 to the plasma membrane.
We hypothesized that, like Nephrin, Neph1 might participate in signaling sufficient to induce actin polymerization. By employing a model system similar to that used previously by us and others to study Nephrin (20, 39), a series of fusion protein constructs were created in which the CD16 extracellular domain and the CD7 transmembrane domain were coupled to either the Neph1 cytoplasmic domain or the mutant Neph1 cytoplasmic domains; these fusion proteins were expressed in NIH 3T3 cells. After addition of mouse anti-CD16 antibody and a secondary anti-mouse IgG antibody to the culture medium of live cells, “clustering” of CD16/CD7 fusion proteins on the plasma membrane was readily visualized (Fig. 5A and additional control data not shown). This maneuver induced tyrosine phosphorylation on Neph1 within 5 min (Fig. 5B). To test the possibility that clustering of the Neph1 cytoplasmic domain resulted in the recruitment of Grb2, NIH 3T3 cells were cotransfected with plasmid encoding CD16/CD7/Neph1CD and Grb2-GFP and were examined by immunofluorescence confocal microscopy. In the presence of aggregating antibody, Grb2-GFP colocalized at the plasma membrane in clusters with CD16/CD7/Neph1CD (Fig. 5A). Consistent with results reported above, a similar pattern of Grb2-GFP clustering at the plasma membrane was not observed when Y-to-F point mutations were introduced at both Y637 and Y638 (Fig. 5A). Single substitutions either at Y637 or at Y638 did not attenuate Grb2 recruitment.
FIG. 5.

Grb2 is recruited to the CD16/CD7/Neph1CD cluster at the plasma membrane. (A) NIH 3T3 cells expressing indicated CD16/CD7 chimeric proteins (red) and Grb2-GFP (green) were treated with anti-CD16 antibody (primary) and rhodamine-labeled anti-IgG antibody (secondary) or secondary antibody only (top row, as control) and then fixed and examined by confocal microscopy. The farthest-right panels are images reconstructed in the yz plane. Colocalization of Grb2-GFP and CD16/CD7/Neph1CD appears yellow on merged images. Indicated CD16/CD7/Neph1CD chimeric proteins mutated at Y637 and/or Y638 were expressed with Grb2-GFP in NIH 3T3 cells. Complexes were clustered as described and evaluated by confocal microscopy. Data are representative of multiple experiments. Magnification, ×600. (B) NIH 3T3 cells expressing CD16/CD7/Neph1CD were treated with clustering antibodies (primary and secondary antibodies) for the time periods shown; following Neph1 immunoprecipitation, immune complexes were immunoblotted using antiphosphotyrosine antibody.
Fyn is necessary for Neph1-Grb2 interaction.
Mouse embryonic fibroblasts with deletions of Src, Yes, and Fyn (SYF cells) were employed to test whether Fyn is necessary for Neph1-Grb2 recruitment. In contrast to NIH 3T3 cells, clustered CD16/CD7/Neph1CD expressed in SYF cells failed to recruit Grb2-GFP (Fig. 6). However, Fyn expressed in SYF cells rescued Grb2 recruitment to clustered Neph1 chimeric protein, suggesting that SFKs are necessary and that Fyn is sufficient for Neph1-Grb2 recruitment.
FIG. 6.

Fyn is necessary for Neph1-Grb2 recruitment. SYF cells were transfected with CD16/CD7/Neph1CD and Grb2-GFP. Grb2 was recruited to Neph1 only in the presence of Fyn. The extreme right panels show the images reconstructed in the yz plane with recruitment at the apical surface of the cell.
Neph1-Grb2 interaction results in actin polymerization.
Previously published work suggested that Nephrin regulates actin polymerization by recruiting Nck following ligand engagement (21, 41). In an analogous fashion, it was hypothesized that Neph1 tyrosine phosphorylation and Grb2 recruitment are sufficient to induce localized actin polymerization. NIH 3T3 cells were transfected with plasmid encoding CD16/CD7/Neph1CD, Grb2-GFP, and actin-CFP (Fig. 7). Addition of aggregating antibodies to the culture medium of these cells induced actin tail formation at sites where CD16/CD7/Neph1CD was clustered at the plasma membrane. Actin tail formation was not seen when the clustering antibody was not used (not shown) or when Neph1CD was replaced with an HA epitope tag. When the Neph1 cytoplasmic tail of CD16/CD7/Neph1CD was mutated at both Y637 and Y638 with F, actin tail formation was not seen, indicating that Grb2 recruitment to Neph1 phosphorylated at these sites is necessary for actin polymerization.
FIG. 7.

Neph1 activation results in Grb2-dependent actin polymerization. CD16/CD7/Neph1CD or CD16/CD7/HA chimeric protein (red), Grb2-GFP, and actin-CFP were expressed in NIH 3T3 cells, clustered with anti-CD16 antibody (primary) and rhodamine-labeled anti-IgG antibody (secondary), and then fixed and examined by confocal microscopy. In the presence of the Y637/Y638 Neph1 mutant neither Grb2 recruitment nor actin polymerization was seen. The bottom panels show a schematic of actin pedestal and enlarged views of representative actin pedestals identified in cells coexpressing clustered CD16/CD7/Neph1CD, GFP-Grb2, and actin-CFP.
Nephrin and Neph1 form a complex that when engaged is tyrosine phosphorylated in the two proteins simultaneously and recruits both Nck and Grb2 to induce actin polymerization.
At the podocyte foot process intercellular junction, Nephrin and Neph1 directly interact via a cis-interaction in the plane of the plasma membrane (4, 12, 24). Based on our previous observations, these proteins are presumably activated simultaneously via trans-junctional interactions during junction formation (41, 42). As such, we hypothesized that upon binding their ligands, Nephrin and Neph1 simultaneously become tyrosine phosphorylated by Fyn and collaborate to regulate actin polymerization. Nephrin and Neph1 association was confirmed using the CD16 antibody-mediated clustering model. Here CD16/CD7/NephrinCD recruited and clustered with Neph1GFP (which lacked CD16) when clustering antibodies were present (Fig. 8A and B). Moreover, clustering of Nephrin with Neph1 in this system resulted in simultaneous tyrosine phosphorylation of Nephrin and Neph1 (Fig. 8B). These phosphorylation events were SFK dependent since pretreatment of cells with PP2 (10 μM) inhibited Nephrin and Neph1 tyrosine phosphorylation (Fig. 8B). Similar results were obtained in an alternate model system employed to exclude CD16 chimera-induced artifact. Full-length wild-type Nephrin and Neph1 were coexpressed in HEK293 cells. In this system, both Neph1 and Nephrin became tyrosine phosphorylated in an SFK-dependent fashion after addition to the medium of a monoclonal antibody (50A9) (25) directed against the extracellular domain of Nephrin (Fig. 8C).
FIG. 8.

Clustering of Nephrin cytoplasmic domain with Neph1 induces simultaneous tyrosine phosphorylation of both Nephrin and Neph1. (A) CD16/CD7/NephrinCD chimeric protein (red) and full-length wild-type Neph1-GFP were expressed in NIH 3T3 cells and were clustered with anti-CD16 antibody (primary) and rhodamine-labeled anti-IgG antibody (secondary) and then fixed and examined by confocal microscopy. Far right panels are images reconstructed in the yz plane. (B) CD16/CD7/NephrinCD chimeric protein and full-length Flag-tagged Neph1 were expressed in NIH 3T3 cells and clustered (right panel) or not clustered (left panel). Lysates were resolved with SDS-PAGE and probed with the indicated antibody. (C) HEK293 cells stably expressing human Nephrin were transfected with Flag-tagged Neph1 and Fyn. 50A9 antibody against human Nephrin ectodomain was used to induce Nephrin clustering. Lysates were immunoprecipitated with Neph1 antibody, resolved with SDS-PAGE, and probed with antiphosphotyrosine antibody.
Experiments were designed to test whether engagement of Nephrin or Neph1 when associated resulted in recruitment of Nck and Grb2. We examined cells expressing CD16/CD7/NephrinCD, wild-type Neph1, and Grb2-GFP or those expressing CD16/CD7/Neph1CD, wild-type Nephrin, and Nck-GFP. In these experiments, Nck was recruited to Nephrin when CD16/CD7/Neph1CD was clustered by addition to the medium of CD16 clustering antibodies. Reciprocally, Grb2 was recruited to Neph1 when CD16/CD7/NephrinCD was clustered. These interactions also resulted in actin tail formation at the assembled complexes (Fig. 9). In control experiments, clustered CD16/CD7/NephrinCD expressed alone did not recruit GFP-Grb2; reciprocally, clustered CD16/CD7/Neph1CD expressed alone did not recruit GFP-Nck (data not shown). These experiments suggest that, independently of the manner in which the Nephrin-Neph1 complex is clustered, the activated Nephrin-Neph1 complex recruits Grb2 and Nck and induces actin polymerization.
FIG. 9.

Engagement of either Nephrin or Neph1 when in association results in recruitment of Nck and Grb2. Indicated expression plasmids were combined, expressed in NIH 3T3 cells, clustered with anti-CD16 antibody (primary) and rhodamine-labeled anti-IgG antibody (secondary), and then fixed and examined by confocal microscopy. The farthest-right panels are images reconstructed in the yz plane. Insets in the bottom two panels represent fivefold enlargements of a portion of the micrograph demonstrating a key feature.
It has been proposed that pathogen-host interactions provide models useful for understanding the regulation of actin dynamics at the plasma membrane of eukaryotic cells. For example, the vaccinia virus protein A36R is inserted into the host plasma membrane where it is phosphorylated by host Fyn on two tyrosine residues. This phosphorylation event results in recruitment of host Nck and Grb2, which cooperate to induce localized actin filament polymerization (36). Importantly, while Nck recruitment in this system is capable of inducing actin polymerization independently of Grb2, Grb2 appears to augment Nck-dependent actin polymerization. In a fashion similar to that of vaccinia virus, we hypothesized that by forming a two-part receptor complex that juxtaposes docking motifs for Nck and Grb2, Nephrin and Neph1 cooperate to regulate localized actin polymerization. To examine this hypothesis, we used the chimeric CD16 model system to test the relative efficiency of actin tail formation induced by aggregated NephrinCD alone or aggregated Neph1CD alone in the presence of overexpressed Nck and Grb2. We also examined the relative efficiency of actin tail formation induced by expressing both NephrinCD and full-length Neph1 together in the presence of Nck and Grb2 (Fig. 10). Aggregation of chimeric NephrinCD recruited Nck but not Grb2, and aggregation of chimeric Neph1CD recruited Grb2 but not Nck, while aggregation of combined CD16/CD7/NephrinCD and Neph1 recruited both Nck and Grb2 to the plasma membrane (data not shown). Aggregation of chimeric NephrinCD resulted in slightly more efficient actin tail formation than Neph1CD aggregation. Remarkably, combining NephrinCD and Neph1 in this system resulted in substantially more efficient actin tail formation than that by either aggregated NephrinCD or Neph1CD expressed alone (Fig. 10). Importantly, substitution in this system of chimeric NephrinCD in which the Nck binding motifs were mutated [NephrinCD(TM)] or Neph1 in which the Grb2 binding motif was mutated [Neph1(DM)] attenuated actin tail formation efficiency to a level similar to that observed by aggregation of wild-type NephrinCD or wild-type Neph1CD alone. Taken together, these results are consistent with the conclusion that Nephrin and Neph1 form a receptor complex that when activated by Fyn recruits Nck and Grb2, which cooperate to regulate actin polymerization at the podocyte intercellular junction.
FIG. 10.

Nephrin and Neph1 cooperate to induce actin polymerization in a Nck- and Grb2-dependent fashion. Quantification of actin tail-forming efficiency. As described in the text, combinations of CD16/CD7/NephrinCD, full-length wild-type Neph1, CD16/CD7/NephrinCD(TM), or full-length wild-type Neph1(DM) were expressed with Nck and/or Grb2 in NIH 3T3 cells as indicated. The number of actin tails per individual cell was counted in 30 cells for each condition. Each experiment was repeated four times; the graph represents the mean number of tails per cell ± standard deviation. The table provides results of the single-tailed paired t test used to compare groups. Neph1(DM), Y-to-F mutation at Y637 and Y638; Nephrin(TM), Y-to-F mutation at Y1191, Y1208, and Y1232. NA, not applicable.
DISCUSSION
Nephrin and Neph1 physically interact in the plane of the membrane at the podocyte intercellular junction and also form counterligands that interact in a trans fashion across the intercellular junction at the same location. Although the molecular mechanisms by which Neph1 functions at the podocyte intercellular junction have not been previously described, the finding that deletion of Neph1 in mice results in a phenotype indistinguishable from that produced by deletion of Nephrin suggests that these transmembrane proteins are interdependent. The results reported here demonstrate that in a manner similar to that of Nephrin, Neph1 can transduce outside-in signals that result in Fyn-dependent tyrosine phosphorylation of Neph1, recruitment of Grb2, and subsequent induction of actin polymerization. Our results also predict that Nephrin and Neph1 form a functional receptor complex that is well placed to coordinate junctional and cytoskeletal dynamics during podocyte development and following podocyte injury.
The molecular and cellular mechanisms by which the structurally unique podocyte foot process and its specialized intercellular junction are assembled remain poorly understood. During podocyte differentiation, Nephrin is targeted in a polarized fashion to the nascent intercellular junction that forms on the tips of budding cellular processes that emerge from the lateral aspect of podocyte precursors (15). Given that the Nephrin-Neph1 complex assembles a protein complex capable of inducing polymerization and elongation of actin filaments, it is plausible that junction formation-induced activation of this complex at this site directs an actin filament elongation-derived motive force that is necessary for maturation of podocyte processes and their intercellular junctions.
The discovery that the activated Nephrin-Neph1 complex recruits Nck and Grb2 to initiate actin polymerization suggests a mechanism that provides for the integration of podocyte intercellular junction structure with cytoskeletal dynamics during podocyte process formation. In a similar fashion, recruitment of Nck and/or Grb2 to plasma membranes has been demonstrated in several systems in which actin polymerization is thought to drive cellular movement. In mammalian systems, for example, actin polymerization determines lamellipodial or filopodial dynamics at the cellular leading edge or during junction formation, drives invadopodium formation observed in transformed cells in culture, induces podosome formation in osteoclasts, or propels intracellular vesicles identified in a model of actin assembly induced by increased phosphatidylinositol 4,5-biphosphate levels in cell culture. In each of these systems, Nck serves as an adaptor protein that recruits to a membrane locus N-WASp, components of the Arp2/3 complex, and other components of the actin polymerization machinery sufficient to induce and regulate actin polymerization (3, 8, 11, 13, 34, 44). While Grb2 has been observed juxtaposed to the membrane in the phosphatidylinositol 4,5-biphosphate-induced model of actin comet-driven endocytic vesicles, its functional relevance there was not determined. Lacking in each of these models has been identification of a specific transmembrane protein that recruits Nck and/or Grb2 adaptor proteins and which subsequently assembles and regulates actin polymerization machinery at these sites.
Similar mechanisms employed by several bacterial and viral pathogens serve as useful models for examining the process of membrane-associated actin dynamics and are instructive when considering the function of the Nephrin-Neph1 complex (31). For example, vaccinia virus attaches to its host and induces host pedestal formation by inserting transmembrane protein A36R into the host cell membrane. In this system, A36R recruits Nck and Grb2 when tyrosine phosphorylated by host Fyn at two phosphotyrosine residues. Nck subsequently recruits WASP-interacting protein (WIP) and associated N-WASp. Importantly, Nck recruitment to A36R is necessary for generation of actin pedestals while Grb2 recruitment augments actin polymerization efficiency. Our results indicate that the Nephrin-Neph1 complex employs a similar mechanism to assemble and regulate an actin polymerization complex at the cell membrane, which may occur during foot process intercellular junction formation. By forming a protein complex that, when activated, recruits both Nck (and N-WASp) and Grb2, Nephrin and Neph1 might work synergistically to promote and/or regulate actin polymerization in the foot process.
The mechanism by which Grb2 determines actin polymerization efficiency in the Nephrin-Neph1 system requires additional investigation. In vitro experiments have suggested that Grb2 can interact with the proline-rich domain of N-WASp and augment N-WASp-associated Arp2/3 activity by overcoming N-WASp autoinhibition (7). It is reasonable to postulate that by juxtaposing the Nck-WIP-N-WASp complex and Grb2 in a manner similar to that proposed in the vaccinia virus A36R model (36), the Nephrin-Neph1 complex facilitates a Grb2-N-WASp interaction, which promotes actin polymerization. However, activated Neph1 can induce Grb2-dependent actin tail formation independently of Nephrin and Nck, albeit less efficiently than when in a complex with Nephrin. Unlike clustered Nephrin, which can recruit N-WASp, we have not found evidence that clustered Neph1 similarly recruits N-WASp (unpublished data). Therefore, Neph1 might employ an N-WASp-independent mechanism to induce actin polymerization. Whether the quality of the actin cytoskeleton created by Nephrin, or Neph1, or Nephrin-Neph1 in complex is distinct requires investigation.
Nephrin tyrosine phosphorylation on Nck-associating SH2-binding motifs occurs transiently during glomerular development, presumably during a period when podocyte foot process and junction formation takes place (41). The timing of this Nephrin phosphorylation event, and the additional observation that artificial ligation of Nephrin's extracellular domain also results in SFK activation and Nephrin tyrosine phosphorylation, is consistent with the hypothesis that Nephrin transmits an outside-in signal to induce actin polymerization at the forming junction. Nephrin phosphorylation on these tyrosine residues is not observed in the mature glomerulus (41). Since Neph1 forms a functional complex with Nephrin and like Nephrin transmits an outside-in signal, it is probable that Neph1 is also activated during junction formation. Of interest, tyrosine phosphorylation of Nephrin and Neph1 and recruitment of Nck and Grb2 are also induced following podocyte injury during a period when podocyte effacement is occurring. The observation that activation of the Nephrin-Neph1 complex occurs both during junction formation and again during effacement in response to injury might be considered paradoxical. However, alteration of podocyte cytoskeletal architecture and intercellular junction structure observed following injury likely requires increased actin filament turnover potentially regulated by tyrosine phosphorylation of the Nephrin-Neph1 complex. Whether Nephrin-Neph1 protein complex activation that results in alterations in Nephrin-Neph1-regulated actin dynamics following podocyte injury occurs as a result of inside-out signaling is an interesting possibility that requires further investigation.
Acknowledgments
This work was supported by an NIH-sponsored T32 research fellowship (P.G.), an NIH-sponsored K01 (D.N.), a Merit Review (L.B.H.) from the Department of Veterans Affairs, and an O'Brien Renal Center Grant from the NIH (L.B.H.).
Footnotes
Published ahead of print on 8 October 2007.
REFERENCES
- 1.Andrews, P. M. 1975. Scanning electron microscopy of the nephrotic kidney. Virchows Arch. B Cell Pathol. 17:195-211. [DOI] [PubMed] [Google Scholar]
- 2.Asanuma, K., K. Kim, J. Oh, L. Giardino, S. Chabanis, C. Faul, J. Reiser, and P. Mundel. 2005. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J. Clin. Investig. 115:1188-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barda-Saad, M., A. Braiman, R. Titerence, S. C. Bunnell, V. A. Barr, and L. E. Samelson. 2005. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat. Immunol. 6:80-89. [DOI] [PubMed] [Google Scholar]
- 4.Barletta, G. M., I. A. Kovari, R. K. Verma, D. Kerjaschki, and L. B. Holzman. 2003. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J. Biol. Chem. 278:19266-19271. [DOI] [PubMed] [Google Scholar]
- 5.Boute, N., O. Gribouval, S. Roselli, F. Benessy, H. Lee, A. Fuchshuber, K. Dahan, M. C. Gubler, P. Niaudet, and C. Antignac. 2000. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 24:349-354. [DOI] [PubMed] [Google Scholar]
- 6.Buccione, R., J. D. Orth, and M. A. McNiven. 2004. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 5:647-657. [DOI] [PubMed] [Google Scholar]
- 7.Carlier, M. F., P. Nioche, I. Broutin-L'Hermite, R. Boujemaa, C. Le Clainche, C. Egile, C. Garbay, A. Ducruix, P. Sansonetti, and D. Pantaloni. 2000. GRB2 links signaling to actin assembly by enhancing interaction of neural Wiskott-Aldrich syndrome protein (N-WASp) with actin-related protein (ARP2/3) complex. J. Biol. Chem. 275:21946-21952. [DOI] [PubMed] [Google Scholar]
- 8.Chen, M., H. She, A. Kim, D. T. Woodley, and W. Li. 2000. Nckβ adapter regulates actin polymerization in NIH 3T3 fibroblasts in response to platelet-derived growth factor bb. Mol. Cell. Biol. 20:7867-7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donoviel, D. B., D. D. Freed, H. Vogel, D. G. Potter, E. Hawkins, J. P. Barrish, B. N. Mathur, C. A. Turner, R. Geske, C. A. Montgomery, M. Starbuck, M. Brandt, A. Gupta, R. Ramirez-Solis, B. P. Zambrowicz, and D. R. Powell. 2001. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol. Cell. Biol. 21:4829-4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dworak, H. A., M. A. Charles, L. B. Pellerano, and H. Sink. 2001. Characterization of Drosophila hibris, a gene related to human nephrin. Development 128:4265-4276. [DOI] [PubMed] [Google Scholar]
- 11.Frischknecht, F., V. Moreau, S. Rottger, S. Gonfloni, I. Reckmann, G. Superti-Furga, and M. Way. 1999. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature 401:926-929. [DOI] [PubMed] [Google Scholar]
- 12.Gerke, P., T. B. Huber, L. Sellin, T. Benzing, and G. Walz. 2003. Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J. Am. Soc. Nephrol. 14:918-926. [DOI] [PubMed] [Google Scholar]
- 13.Gruenheid, S., R. DeVinney, F. Bladt, D. Goosney, S. Gelkop, G. D. Gish, T. Pawson, and B. B. Finlay. 2001. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 3:856-859. [DOI] [PubMed] [Google Scholar]
- 14.Haynes, M. P., L. Li, D. Sinha, K. S. Russell, K. Hisamoto, R. Baron, M. Collinge, W. C. Sessa, and J. R. Bender. 2003. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J. Biol. Chem. 278:2118-2123. [DOI] [PubMed] [Google Scholar]
- 15.Holzman, L. B., P. L. St. John, I. A. Kovari, R. Verma, H. Holthofer, and D. R. Abrahamson. 1999. Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int. 56:1481-1491. [DOI] [PubMed] [Google Scholar]
- 16.Hosoyamada, M., K. Yan, Y. Nishibori, Y. Takiue, A. Kudo, H. Kawakami, T. Shibasaki, and H. Endou. 2005. Nephrin and podocin expression around the onset of puromycin aminonucleoside nephrosis. J. Pharmacol. Sci. 97:234-241. [DOI] [PubMed] [Google Scholar]
- 17.Huber, T. B., C. Kwoh, H. Wu, K. Asanuma, M. Godel, B. Hartleben, K. J. Blumer, J. H. Miner, P. Mundel, and A. S. Shaw. 2006. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J. Clin. Investig. 116:1337-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huber, T. B., M. Schmidts, P. Gerke, B. Schermer, A. Zahn, B. Hartleben, L. Sellin, G. Walz, and T. Benzing. 2003. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J. Biol. Chem. 278:13417-13421. [DOI] [PubMed] [Google Scholar]
- 19.Inokuchi, S., I. Shirato, N. Kobayashi, H. Koide, Y. Tomino, and T. Sakai. 1996. Re-evaluation of foot process effacement in acute puromycin aminonucleoside nephrosis. Kidney Int. 50:1278-1287. [DOI] [PubMed] [Google Scholar]
- 20.Johnstone, D. B., and L. B. Holzman. 2006. Clinical impact of research on the podocyte slit diaphragm. Nat. Clin. Pract. Nephrol. 2:271-282. [DOI] [PubMed] [Google Scholar]
- 21.Jones, N., I. M. Blasutig, V. Eremina, J. M. Ruston, F. Bladt, H. Li, H. Huang, L. Larose, S. S. Li, T. Takano, S. E. Quaggin, and T. Pawson. 2006. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440:818-823. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan, J. M., S. H. Kim, K. N. North, H. Rennke, L. A. Correia, H. Q. Tong, B. J. Mathis, J. C. Rodriguez-Perez, P. G. Allen, A. H. Beggs, and M. R. Pollak. 2000. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24:251-256. [DOI] [PubMed] [Google Scholar]
- 23.Kestila, M., U. Lenkkeri, M. Mannikko, J. Lamerdin, P. McCready, H. Putaala, V. Ruotsalainen, T. Morita, M. Nissinen, R. Herva, C. E. Kashtan, L. Peltonen, C. Holmberg, A. Olsen, and K. Tryggvason. 1998. Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome. Mol. Cell 1:575-582. [DOI] [PubMed] [Google Scholar]
- 24.Khoshnoodi, J., K. Sigmundsson, L. G. Ofverstedt, U. Skoglund, B. Obrink, J. Wartiovaara, and K. Tryggvason. 2003. Nephrin promotes cell-cell adhesion through homophilic interactions. Am. J. Pathol. 163:2337-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lahdenpera, J., P. Kilpelainen, X. L. Liu, T. Pikkarainen, P. Reponen, V. Ruotsalainen, and K. Tryggvason. 2003. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. 64:404-413. [DOI] [PubMed] [Google Scholar]
- 26.Lehtonen, S., J. J. Ryan, K. Kudlicka, N. Iino, H. Zhou, and M. G. Farquhar. 2005. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc. Natl. Acad. Sci. USA 102:9814-9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, H., S. Lemay, L. Aoudjit, H. Kawachi, and T. Takano. 2004. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J. Am. Soc. Nephrol. 15:3006-3015. [DOI] [PubMed] [Google Scholar]
- 28.Liu, G., B. Kaw, J. Kurfis, S. Rahmanuddin, Y. S. Kanwar, and S. S. Chugh. 2003. Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J. Clin. Investig. 112:209-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mata, M., S. E. Merritt, G. Fan, G. G. Yu, and L. B. Holzman. 1996. Characterization of dual leucine zipper-bearing kinase, a mixed lineage kinase present in synaptic terminals whose phosphorylation state is regulated by membrane depolarization via calcineurin. J. Biol. Chem. 271:16888-16896. [DOI] [PubMed] [Google Scholar]
- 30.Matsui, I., T. Ito, H. Kurihara, E. Imai, T. Ogihara, and M. Hori. 2007. Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab. Investig. 87:273-283. [DOI] [PubMed] [Google Scholar]
- 31.Munter, S., M. Way, and F. Frischknecht. 2006. Signaling during pathogen infection. Sci. STKE 2006:re5. [DOI] [PubMed] [Google Scholar]
- 32.Pollard, T. D., and G. G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112:453-465. [DOI] [PubMed] [Google Scholar]
- 33.Quack, I., L. C. Rump, P. Gerke, I. Walther, T. Vinke, O. Vonend, T. Grunwald, and L. Sellin. 2006. Beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc. Natl. Acad. Sci. USA 103:14110-14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rivera, G. M., C. A. Briceno, F. Takeshima, S. B. Snapper, and B. J. Mayer. 2004. Inducible clustering of membrane-targeted SH3 domains of the adaptor protein Nck triggers localized actin polymerization. Curr. Biol. 14:11-22. [DOI] [PubMed] [Google Scholar]
- 35.Ryan, G. B., and M. J. Karnovsky. 1975. An ultrastructural study of the mechanisms of proteinuria in aminonucleoside nephrosis. Kidney Int. 8:219-232. [DOI] [PubMed] [Google Scholar]
- 36.Scaplehorn, N., A. Holmstrom, V. Moreau, F. Frischknecht, I. Reckmann, and M. Way. 2002. Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr. Biol. 12:740-745. [DOI] [PubMed] [Google Scholar]
- 37.Sellin, L., T. B. Huber, P. Gerke, I. Quack, H. Pavenstadt, and G. Walz. 2003. NEPH1 defines a novel family of podocin interacting proteins. FASEB J. 17:115-117. [DOI] [PubMed] [Google Scholar]
- 38.Shih, N. Y., J. Li, V. Karpitskii, A. Nguyen, M. L. Dustin, O. Kanagawa, J. H. Miner, and A. S. Shaw. 1999. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286:312-315. [DOI] [PubMed] [Google Scholar]
- 39.Songyang, Z., S. E. Shoelson, M. Chaudhuri, G. Gish, T. Pawson, W. G. Haser, F. King, T. Roberts, S. Ratnofsky, R. J. Lechleider, et al. 1993. SH2 domains recognize specific phosphopeptide sequences. Cell 72:767-778. [DOI] [PubMed] [Google Scholar]
- 40.van der Geer, P., and T. Hunter. 1993. Mutation of Tyr697, a GRB2-binding site, and Tyr721, a PI 3-kinase binding site, abrogates signal transduction by the murine CSF-1 receptor expressed in Rat-2 fibroblasts. EMBO J. 12:5161-5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma, R., I. Kovari, A. Soofi, D. Nihalani, K. Patrie, and L. B. Holzman. 2006. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Investig. 116:1346-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verma, R., B. Wharram, I. Kovari, R. Kunkel, D. Nihalani, K. K. Wary, R. C. Wiggins, P. Killen, and L. B. Holzman. 2003. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J. Biol. Chem. 278:20716-20723. [DOI] [PubMed] [Google Scholar]
- 43.Wary, K. K., A. Mariotti, C. Zurzolo, and F. G. Giancotti. 1998. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 94:625-634. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi, H., M. Lorenz, S. Kempiak, C. Sarmiento, S. Coniglio, M. Symons, J. Segall, R. Eddy, H. Miki, T. Takenawa, and J. Condeelis. 2005. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]