

Abstract
The proton translocation mechanism of the Escherichia coli cytochrome bo3 complex is intimately tied to the electron transfers within the enzyme. Herein we evaluate two models of proton translocation in this enzyme, a cytochrome c oxidase-type ion-pump and a Q-cycle mechanism, on the basis of the thermodynamics of electron transfer. We conclude that from a thermodynamic standpoint, a Q-cycle is the more favorable mechanism for proton translocation and is likely occurring in the enzyme.
In recent years, substantial progress has been made in our understanding of the action of the superfamily of enzymes known as the heme-copper oxidases (1). Composed of two families, the cytochrome c oxidases and the quinol oxidases, this superfamily of enzymes utilizes the redox energy from the transfer of electrons from cytochrome c or quinol to dioxygen to translocate protons across the cytoplasmic or mitochondrial membranes, ultimately generating the electrochemical gradient used in the production of ATP. Important structural breakthroughs have been made recently on the cytochrome c oxidases, with the crystal structures of the cytochromes aa3 from Paracoccus denitrificans (2) and beef heart (3, 4) determined to 2.8 Å, with putative proton channels delimited, and selected mutations shown to influence proton translocation (5). The quinol oxidases are not nearly as well characterized, however. While the sequencing of the cyo operon of Escherichia coli (coding for cytochrome bo3) has revealed strong sequence similarity of subunit I between the quinol oxidases and cytochrome c oxidases (6), it is not clear whether the similarity of structure necessarily requires an equivalent proton translocation mechanism or whether the chemical differences between the two substrates result in the formulation of two different mechanisms. These contrasting views of proton translocation in quinol oxidases have been discussed in prior literature (7–9). In this work we evaluate proposed mechanisms of proton translocation in quinol oxidases from a thermodynamic perspective, evaluating both the overall thermodynamics as well as the thermodynamics of possible individual electron transfers within the enzyme. The analysis will focus on the enzyme cytochrome bo3, as it is the best characterized of the quinol oxidases and it has been a focus of study in our laboratory for some time.
A Brief Overview of Cytochrome bo3
Cytochrome bo3 serves as the primary terminal oxidase in E. coli under conditions of high oxygen concentration. It is a membrane-bound, four-subunit enzyme coded by the cyoABCDE operon, with a molecular weight of 140,000 (6). As with the entire family of heme-copper oxidases, it contains a low-spin heme (heme b), a high-spin heme (heme o3), and a mononuclear copper center (CuB), of which the latter two constitute the dioxygen reduction site. It also contains a bound ubiquinone molecule, QB, in electronic contact with heme b. The substrate ubiquinol, QAH2, has been shown to bind at or near subunit II (10).
The overall reaction of this enzyme is shown in Eq. 1,
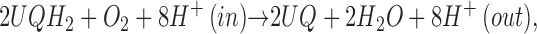 |
where UQ represents ubiquinone-8, and UQH2 is its reduced quinol form. The oxidation of ubiquinol and the concomitant reduction of dioxygen lead to the translocation of eight protons across the cytoplasmic membrane for every four electrons passed through the enzyme to dioxygen. The reduction of dioxygen occurs through the same series of intermediates as are observed in the aa3 oxidases (11–15). While the dioxygen chemistry is similar in the cytochrome c and quinol oxidases, the electron transfer chemistry is not rigorously identical, as we have shown the intermediacy of the QB semiquinone in the reaction of cytochrome bo3 with quinol (16). It is the electron transfer that we wish to explore from a thermodynamic basis.
The Overall Thermodynamics of Cytochrome bo3
The thermodynamics of the proton translocation are controlled by the reduction potentials of the quinone and dioxygen. The two-electron reduction potential of ubiquinone in a lipid bilayer has been reported as 60–70 mV at pH 7 (17, 18), while the four-electron reduction potential of dioxygen is 780 mV under a standard atmosphere of air at pH 7. Thus, the total energy available from the overall reaction catalyzed by the enzyme is around 2,860 meV under standard conditions. Bacteria under standard biochemical conditions maintain an electromotive force (Δψ − 2.3RTΔpH/F) of approximately 200 mV across the cytoplasmic membrane. As the quinones in the respiratory chain are membrane-bound, it can be assumed that proton translocation from the membrane to the extramembrane space goes against an electrochemical potential one-half of that for complete proton translocation across the membrane. One-half of the protons translocated by the quinol oxidases arise from quinol substrate that has been generated within the membrane by oxidoreductases operating earlier in the respiratory chain. These protons therefore travel against a 100-mV potential gradient, while the other half of the protons that are translocated by the quinol oxidases feel the full 200-mV gradient. So, for the complete reaction cycle of the quinol oxidases, an average energetic requirement of 150 meV per proton, rather than 200 meV, applies. Assuming a perfect coupling of redox energy to proton translocation, this then allows for a maximum proton translocation ratio of 19H+/4e− under standard conditions. To date, the highest stoichiometry observed for a quinol oxidase is 8H+/4e−. By way of comparison, cytochrome c oxidase uses cytochrome c, with a reduction potential of 250 mV, as its substrate, yielding around 2,120 meV of energy upon dioxygen reduction under standard conditions. This energy is used to translocate four protons across the mitochondrial membrane. Finally, in mitochondria, cytochrome bc1 is able to translocate protons with a stoichiometry of 8H+/4e− using the energy of the electron transfers from ubiquinol to cytochrome c. Under standard conditions, this free energy is 760 meV, and since 1,200 meV of work is performed, the cytochrome bc1 complex must operate under high electron pressure (high [ubiquinol]/[ubiquinone] ratio) and/or strongly oxidizing conditions (high [ferricytochrome c]/[ferrocytochrome c] ratio).
The Thermodynamics of Quinol Oxidation
Although ubiquinol is a two-electron donor, the quinol is oxidized in one-electron steps. Because of this, the thermodynamic values of interest are the one-electron reduction potentials, not the overall two-electron potential. As illustrated in Fig. 1, the stoichiometry of the reduction of quinone requires that the average of the Q/Q⋅ and Q⋅/QH2 potentials be equal to the two-electron Q/QH2 potential. The difference between the reduction potentials of the Q/Q⋅ and Q⋅/QH2 couples is a measure of the stability of the semiquinone. For a quinone in solution or in a lipid bilayer, the Q/Q⋅ potential is lower than the Q⋅/QH2 potential by a large amount, such that the semiquinone is highly unstable with respect to disproportionation. The absence of a semiquinone radical signal arising from the QA site suggests a similarly unstable semiquinone arising during oxidation of the substrate. This plus the fact that heme b, heme o3, and CuB are one-electron acceptors require that electron transfer from ubiquinol to cytochrome bo3 occurs via a branched electron transfer pathway. The first electron to leave the quinol is at a potential much higher (probably by 100 mV or more) than the 60- to 70-mV two-electron reduction potential of ubiquinol. Hence, the initial electron acceptor must have a potential that is at least as high. It should also be noted that any difference in the binding energies of ubiquinone vs. ubiquinol at the QA site will be reflected in a change in the two-electron reduction potential with respect to that of unbound ubiquinone. We expect the contribution to the overall reduction potential from this effect to be small.
Figure 1.

One- and two-electron reduction potentials of ubiquinone as a function of the stability of the ubisemiquinone. Higher values of Em are at the bottom of the plot.
The Ion-Pump and Q-Cycle Models
There are two published models of the reaction mechanism of cytochrome bo3 that take into account the involvement of all four redox centers during enzyme turnover (7, 19). We refer to them as the ion-pump and Q-cycle models. These models propose specific electron transfer pathways through the enzyme, account for the known substrate transformations, and predict the general mechanism of proton translocation, but they do not predict the intimate molecular details of proton translocation. The most distinguishing feature of the two models is the electron transfer. Reaction schemes for both the ion-pump and Q-cycle models are shown in Fig. 2. If the quinol oxidases act as ion-pumps analogous to the cytochrome c oxidases, one would predict a similar set of electron transfers within the enzyme. Specifically, one would expect the electrons from the substrate quinol (QAH2) to travel through the enzyme in the order QAH2 → heme b → (heme o3/CuB). Sato-Watanabe et al. (19) have proposed an electron transfer model in which QB serves merely as a means to couple the two-electron donor quinol with the one-electron acceptors heme b, heme o3, and CuB. In this model, QB and heme b serve as the initial electron acceptors from QAH2, after which the electrons are funneled, one at a time, to the heme-copper site for the reduction of dioxygen to water, with the associated pumping of two protons per electron occurring during the reduction of the peroxide intermediate to water. The release of four protons from the oxidation of quinol, along with the pumping of four protons, accounts for the correct proton/electron stoichiometry.
Figure 2.

Schematic view of the electron transfer pathways predicted by the ion-pump (Left) and Q-cycle (Right) models of proton translocation in cytochrome bo3. Dashed arrows denote electron transfers. The numbers in the ion-pump model denote the first and second electrons transferred during the oxidation of quinol. In the ion-pump model, QB is assumed to be bound at all times during the reaction cycle (figure adapted from ref. 16).
The Q-cycle model of proton translocation assumes a functional analogy between cytochrome bc1 and cytochrome bo3. In this model, electron transfer from QAH2 occurs via a branched electron transfer pathway, with one electron traveling to the high-potential heme-copper binuclear site, and the second electron traveling through heme b to QB, to form QB⋅. The second quinol molecule reacting at the QA site again donates an electron to the heme-copper site and passes the second to QB⋅ to form QBH2, which then dissociates, to be replaced by another molecule of quinone. The reaction of two more molecules of quinol at the QA site, with the formation of one more quinol molecule at the QB site and the concomitant reduction of peroxide to water at the heme-copper site, completes the reaction cycle. In this model, the release of eight protons during the oxidation of four quinol molecules accounts for the 8H+/4e− stoichiometry; no mechanical pumping of protons is necessary. The model does require that the quinone(ol) at the QB site be exchangeable. Studies of the inhibition of cytochrome bo3 by substrate, substrate analogs, and other inhibitors indicate that this is indeed the case (20, 21).
To date, sequence and structural information cannot give definitive information on the proton pathways in cytochrome bo3. Subunit I of cytochrome bo3 shares strong sequence similarity with the analogous subunit of the cytochrome c oxidases (6). Of several amino acid residues within this subunit that are highly conserved among the heme-copper oxidases, residues N124, D135, N142, and E286 of cytochrome bo3 have been implicated in the proton translocation process (22–24). Assuming structural similarity between cytochrome bo3 and the cytochrome c oxidases, residues N124, D135, and N142 are expected to lie on the inner side of the cytoplasmic membrane, while E286 should lie within the membrane. These residues are believed to be part of a proton channel within the enzyme that is not associated with the dioxygen reduction site. While this putative channel might span the entire cytochrome bo3 molecule and act as a proton-pumping conduit, in accord with an ion-pump model, a channel is also predicted in the Q-cycle model, since a source of protons is necessary for the reduction of quinone at the QB site of the enzyme. So, while mutagenesis studies undoubtedly will shed further light on the proton translocation process, current data cannot distinguish between an ion-pump or Q-cycle model.
In both the ion-pump and the Q-cycle mechanisms, the gross chemical features are identical. The overall stoichiometry and proton/electron ratios are the same, and the dioxygen chemistry is accommodated. Further, the energetics of proton translocation are identical, as in both cases four protons from membrane-bound quinol are transferred from the membrane to the extramembranous space, while there is a net transfer of four protons across the entire membrane. Both scenarios give rise to the 150 mV/proton ratio. The only difference between the two mechanisms is the manner in which the two-electron chemistry of the Q/QH2 couple is linked to the dioxygen chemistry to accomplish the vectorial proton translocation. Thus, one must analyze the individual electron transfers in the enzyme directly to differentiate the two proton translocation models.
Individual Electron Transfers Within the Enzyme
Understanding the thermodynamics of individual electron transfers in cytochrome bo3 requires a knowledge of the reduction potentials of the various redox centers in the enzyme. Several reports of the reduction potentials of the redox centers have been published, usually based on redox titrations, and often with conflicting results. For the semiquinone at pH 7.0, one-electron potentials of 22.5 mV and 115 mV have been measured for the QB/QB⋅ and QB⋅/QBH2 couples, respectively (25). The CuB center has a potential of at least 350 mV (26–28). Measurements of heme potentials have yielded a wide range of values (25–35). On the basis of the electron distribution in cytochrome bo3 upon the addition of substoichiometric amounts of substrate under rapid-mixing/freeze-quench conditions, we have calculated a reduction potential for heme b of approximately 50 mV (16). The mean value of the heme o3 potential, based on several redox titration studies, is around 200 mV. These values, along with approximate values for the one-electron potentials of QA (see below) and driving forces for selected electron transfers in cytochrome bo3, are listed in Table 1.
Table 1.
Reduction potentials of the redox centers in cytochrome bo3, approximate one-electron reduction potentials of ubiquinone at the quinol oxidation site (QA), and driving forces associated with possible electron transfers in cytochrome bo3
| Center/ redox couple | Em, mV, pH 7 | One-electron transfer | ΔE, mV |
|---|---|---|---|
| QA/QA⋅ | ≈−40 | QAH2 → QB | −138 |
| QA⋅/QAH2 | ≈160 | QAH2 → heme b | −110 |
| QB/QB⋅ | 22.5 | QAH2 → heme o3 | 40 |
| QB⋅/QBH2 | 115 | QAH2 → CuB | 190 |
| heme b | 50 | QA⋅ → QB | 62 |
| heme o3 | 200 | QA⋅ → heme b | 90 |
| CuB | 350 | QB⋅ → heme b | 28 |
| heme b → QB⋅ | 65 | ||
| heme b → heme o3 | 150 |
It should be noted that anticooperative redox behavior has been observed both in cytochrome c oxidase (36) and in cytochrome bo3 (26, 27). The magnitude of these interactions has been shown to be 60 mV or less. Further, heme b and QB, whose relative reduction potentials are especially important in the thermodynamic analysis, should be affected by the oxidation state of the heme o3/CuB binuclear center to the same extent. Thus, the effect of cooperativity should not affect the thermodynamic analysis in any significant fashion.
For the two models of proton translocation, there are various energetic requirements for the associated electron transfer pathways. Those for the ion-pump model are the following:
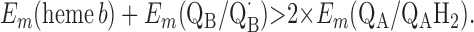 |
1 |
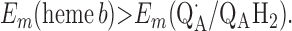 |
2 |
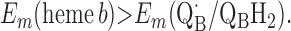 |
3 |
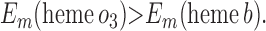 |
4 |
Requirement 1 states that the initial electron transfers from QAH2 to heme b and QB must be energetically favorable. Point 2 provides a more rigorous requirement. While the QA/QAH2 potential is ≈60 mV, the QA⋅/QAH2 potential will be significantly higher, although the absolute number is difficult to determine. The difference ΔEm = Em (Q⋅/QH2) − Em (Q/QH2) at physiological pH is 47 mV for QB (see above) and 60 mV for center N of cytochrome bc1 (18). These are highly stabilized semiquinones and provide a lower limit for ΔEm for QA. For this discussion, a ΔEm value of 100 mV will be assumed for QA. It is the QA⋅/QAH2 potential that determines whether the first single-electron transfer to heme b can occur. Point 3 is necessary to guarantee that upon electron transfer to cytochrome bo3, the reduced species formed are heme b2+ and QB⋅. If this condition is not met, the thermodynamics will favor the formation of QBH2, which is an unproductive reaction in this model. Point 4 again is necessary to guarantee the proper sequence of electron carriers.
The Q-cycle model predicts the following set of thermodynamic properties:
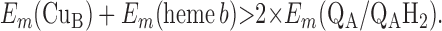 |
1' |
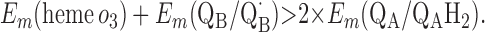 |
2' |
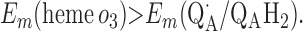 |
3' |
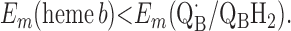 |
4' |
Points 1′ and 2′ represent the energetic requirements for the reactions of the enzyme with the first and second quinol molecules, respectively, during the reaction cycle. Point 3′ is the energetic requirement for the first electron transfer from the second substrate quinol molecule. The CuB potential is much higher than that of heme o3, so the electron transfer from QAH2 to CuB will be favorable if condition 3′ is met. Point 4′ is the requirement for the formation of QBH2 from QB⋅ and heme b2+.
The first postulate of the ion-pump model is that the electron transfer from the quinol substrate to the enzyme occurs by a branched pathway, with one electron going to heme b, and the other going to the QB site to form the QB semiquinone. Using the reported value of 60 mV for the two-electron reduction of membrane-bound quinones, this electron transfer will be unfavorable by at least 45 mV. Hence, requirement 1 is not met, and, by extension, point 2 is not met either. Assuming the formation of reduced heme b and QB⋅, the reduction potential of 82 mV for QB⋅ vs. ≈50 mV for heme b suggests that formation of QBH2 is the favored process. This observation is in conflict with point 3. Point 4 is satisfied, as the reduction potential of heme o3 is greater than that of heme b by at least 80 mV, even given the uncertainty in heme o potentials. Thus, in the absence of a kinetic barrier, electron transfer from heme b to heme o3 should be favored.
The Q-cycle model predicts a branched electron pathway, with electron transfer to the heme-copper binuclear site driving oxidation of QAH2. As the reduction potentials of CuB and heme o3 are much higher that those of QA, points 1′ and 2′ are satisfied easily. Point 3′ has not been tested rigorously, as the QA⋅/QAH2 and heme o3 potentials are not known to sufficient accuracy, and no experiments have been done to observe this electron transfer. However, it is known that the iron–sulfur protein of cytochrome bc1, with a reduction potential of 265 mV, is capable of promoting the quinol oxidation in the bc1 complex. This potential is somewhat higher than the mean value of the reported reduction potentials of heme o3 (≈200 mV). Heme o3 may have a sufficiently high potential to drive quinol oxidation on its own or may show an increase of potential upon interaction of the heme o3/CuB site with dioxygen. In cytochrome c oxidase, binding of dioxygen to the two-electron reduced heme-copper site raises the reduction potential of the site to more than 800 mV (37, 38); hence, oxidation of the third and fourth quinols by cytochrome bo3 is expected to be highly favored thermodynamically, according to this model. Point 4′, the reverse of Point 3, is satisfied on the basis of the low heme b potential.
The Effect of Membrane Potential and Electron Pressure
The presence of the membrane potential necessarily will retard electron transfer through cytochrome bo3 and all terminal oxidases. While it is difficult to predict the effect of the potential gradient on specific reduction potentials, both the Δψ and ΔpH terms should affect almost every electron transfer, as nearly every electron transfer step in either proposed proton translocation mechanism involves the uptake or release of protons. Oxidation of QAH2 results in the release of two protons, reduction of QB in the Q-cycle model requires proton uptake, and electron transfers to the binuclear heme-copper center have been shown to be coupled with proton uptake (39). Thus, any electron transfers that are nearly thermoneutral in the absence of a membrane potential will be disfavored in the presence of the potential. The effect of the electrochemical gradient also may be moderated somewhat by a buildup of reducing equivalents in the membrane. Regardless, the electron transfers that are least favored (the electron donation to heme b and QB in the ion-pump model) are likely to be the most hindered by the gradient.
Kinetic Limitations to Electron Transfer
Proton translocation by respiratory enzymes occurs through a careful kinetic control of electron transfers. As the thermodynamically most favored electron transfers are those that are not coupled to proton translocation, there must be small kinetic barriers that prevent electron transfer before the enzyme is in a state that is capable of translocating protons. However, in the opposite extreme, a large kinetic barrier to a coupled electron transfer allows the electron to travel through the enzyme by using an alternative pathway, bypassing the normal proton translocation apparatus. Thus, either extreme represents an electron leak. An optimal respiratory enzyme should have rapid electron transfers for those steps not directly involved in proton translocation, while those steps involved in proton translocation should be slowed only enough to provide efficient coupling of the proton and electron transfers. In the absence of precise measurements of distances between redox centers, the thermodynamics of the electron transfers are the best guide for estimating the rates of these transfers, because of the exponential dependence of rate on thermodynamic driving force predicted by Marcus theory. Proton-coupled electron transfers are expected to be slower than those predicted by Marcus theory, such that electron leaks will be of particular importance in situations in which an electron transfer coupled to proton translocation has a low driving force.
The ion-pump model of proton translocation by cytochrome bo3 assumes that the nature of the reducing substrate is not related to the proton translocation mechanism. Thus, the initial electron transfers from QAH2 into the enzyme should be rapid. Given an initial one-electron transfer from QAH2 to heme b, which could be unfavorable by as much as 150 meV, for a step that likely would have no bearing on the proton translocation apparatus, it appears that the inefficiency of this step would make the enzyme prone to electron leaks, or simply slow down enzyme turnover at the very least.
For the Q-cycle model, the low heme b potential makes it possible for such a mechanism to occur. Again, simply from the standpoint of relative energies of electron transfer, a favorable electron transfer from heme b to QB is necessary to promote the proton translocation associated with the cycling of electrons through the quinone pool, rather than allow the leak of electrons to heme o3. Because of the splitting of the QA/QA⋅ and QA⋅/QAH2 potentials, the electron donated to heme b arises from a very low potential center, allowing the low-potential heme to act as the electron acceptor in a kinetically facile manner.
To this point, the discussion has not dealt with the spatial orientation of the redox centers or the presence of kinetic barriers related to the protein structure. The influence of these can be determined only by experiment. Prior work from our laboratory has suggested that in the reaction of oxidized cytochrome bo3 with ubiquinol-2, CuB and heme b are the first two centers that are reduced and that QB is reduced to its semiquinone form more slowly than the former two centers (16). These kinetic observations favor a Q-cycle model. In addition, our work and that of others (40) have shown that, at least in certain states of the enzyme, electron transfer from heme b to heme o3 is hindered. As mentioned above, we consider this barrier to be crucial to the proton translocation in the enzyme, with rapid heme b→heme o3 electron transfer constituting an electron leak.
Allosteric Interactions in Electron Transfer
A key component of any Q-cycle mechanism is a branched electron transfer pathway in the oxidation of quinol. While it is possible to formulate a mechanism for this branched electron transfer solely on the basis of competition between the energetics of electron transfer and the distance dependence as described by semiclassical Marcus theory, issues of efficiency and irreversibility of the enzyme make allosteric interactions a necessary additional requirement. For an ion-pump, allosteric interactions are the entire basis for the proton translocation through the enzyme. Thus, the thermodynamic implications of this allostery have a direct bearing on proton translocation. In the Q-cycle, the one-electron oxidation of the quinol substrate to its semiquinone form must induce a conformational change in the enzyme that directs the second electron to a low-potential redox center. The practical effect of this structural change is to raise the Q⋅/QH2 potential relative to that which arises when electron transfer and proton transfer are decoupled. In cytochrome bo3, no direct measure of the stabilization of QA⋅ has been made to date, so there is no reliable estimate of the magnitude of the allosteric interaction occurring in quinol oxidation. Nevertheless, this additional energetic term provides further illustration of the need for a high-potential redox center to drive the oxidation of quinol.
As mentioned above, the view of cytochrome bo3 as an ion- pump usually assumes that neither QA nor QB is an integral part of the proton translocation apparatus. Under this assumption, allosteric interactions in this enzyme that are involved in proton pumping would involve only the other three redox centers in the enzyme. The analysis of the coupling of electron transfer and proton pumping thus would be identical to that for cytochrome c oxidases. As this analysis has been published previously for various models of proton translocation by the cytochrome c oxidases (41), no further mention will be made here. However, any adaptation of the ion-pump model to accommodate the possibility that the binding and reaction of quinol induce structural changes in the protein imposes similar limitations as described above for the Q-cycle, with the additional requirement that in an ion-pump, only the last two electron transfers through the enzyme are coupled to proton translocation.
A Hybrid Model for Proton Translocation
To date, detailed information on the electron input from ubiquinol to cytochrome bo3 has been limited to the first molecule of substrate that reacts with the enzyme in the reaction cycle. While the second molecule of quinol is expected to react in the same manner as the first, dioxygen binding to the enzyme after the reaction with the second quinol will dramatically change the thermodynamics of electron transfer. This change allows for a third possible mode of proton translocation by this complex, a hybrid model in which Q-cycle and ion-pump-type mechanisms occur during different stages of the enzyme-turnover cycle. In this model, the enzyme operates via a Q-cycle mechanism before the binding of dioxygen to the enzyme. After the heme-copper site is reduced by two electrons, dioxygen binds and the driving force for dioxygen reduction now controls electron transfer through the enzyme. At this point, an ion-pump proton translocation mechanism analogous to that of the cytochrome c oxidases is operative. For the first half of the reaction cycle, two molecules of ubiquinol are oxidized, releasing four protons to the outside of the cytoplasmic membrane, and one molecule of ubiquinone is reduced to ubiquinol. To accommodate the observed 8H+/4e− ratio, the second half of the reaction cycle must proceed through the oxidation of one molecule of ubiquinol, which leads to the release of two protons, and the translocation of two protons across the membrane via the ion-pump as the dioxygen is reduced from its peroxidic form to water. The net result is the oxidation of two molecules of ubiquinol, the reduction of dioxygen to water, the transfer of four protons from the membrane to the periplasm, and the translocation of four protons across the entire membrane, in accord with the overall stoichiometry.
The hybrid model has two drawbacks, however. First, the observed proton-pumping ratio requires that the enzyme pump only one proton across the membrane for each of the last two electrons passing through the enzyme during the reduction of dioxygen to water, rather than the 2H+/1e− ratio observed in the cytochrome c oxidases. A variant of the hybrid model, in which two protons are pumped for each of the last two electrons used to reduce dioxygen, would yield a stoichiometry of 10H+/4e−, higher than that shown experimentally. Second, the model introduces seemingly unnecessary complexity to the operation of the enzyme system. Having a single proton-translocation apparatus in an enzyme requires intricate machinery; the likelihood that an enzyme would evolve to perform two distinct types of proton translocation in an efficient manner with a given set of redox centers seems low. On the basis of these two factors, we feel that a hybrid model is unlikely for cytochrome bo3, even though there are currently no experimental data to rule it out.
Concluding Remarks
The above analysis provides a framework for understanding the coupling of electron transfers to proton translocation in E. coli cytochrome bo3 and, by extension, the entire family of quinol oxidases. Emphasis has been placed on the thermodynamic requirements of the proton translocation process, as these can be evaluated in the absence of detailed structural information concerning the enzyme. A quantitative analysis of other aspects of the proton translocation apparatus (e.g., allostery and the effects of membrane potential) must await further experimental data. In the analysis of the proton translocation in cytochrome bo3, it is important to take into account the chemical nature of the substrate ubiquinol, particularly the large split between the Q/Q⋅ and the Q⋅/QH2 potentials. Application of this and other observations to the models of proton translocation presented here suggests that the Q-cycle model is more favorable from a thermodynamic standpoint. The scope of this analysis is of necessity limited to proposed models of proton translocation that are in accord with the known characteristics of cytochrome bo3. So, for instance, this treatment cannot rigorously rule out an ion-pump mechanism in cytochrome bo3 with electron transfer pathways different from those in cytochrome c oxidase. However, the argument for an ion-pump mechanism in cytochrome bo3 rests on the assumption that the similarity of structure between cytochrome bo3 and cytochrome c oxidase implies similarity of function. Thus, a different electron transfer pathway calls into question this initial assumption. It should also be noted that no other ion-pump models for cytochrome bo3 have been presented.
The models presented in this work are based on mechanistic precedents from enzymes functionally similar to cytochrome bo3. While we doubt that a truly novel proton translocation mechanism exists for this enzyme, any variants of the models presented here, or any other proposed mechanisms of proton translocation, can be evaluated on the basis of the considerations discussed above. Respiratory quinol oxidases occur in a wide variety of organisms. It is to be expected that there will be variations of some sort between these organisms in the precise means by which the quinol oxidation is coupled to proton translocation. Ultimately, though, this coupling is constrained by the inherent chemistry of the enzyme substrates. Thus, the approach pursued in this work for evaluating proton translocation mechanisms is of general applicability to all of these enzymes.
Acknowledgments
This work was supported by Grant GM22432 from the National Institutes of Health.
ABBREVIATIONS
- Em
midpoint potential
- QA/QAH2
ubiquinone/ubiquinol interacting at the quinol oxidation site of cytochrome bo3
- QB/QBH2
ubiquinone/ubiquinol interacting at the high-affinity quinone-binding site of cytochrome bo3
- Q⋅
ubisemiquinone
References
- 1. García-Horsman J A, Barquera B, Rumbley J, Ma J, Gennis R B. J Bacteriol. 1994;176:5587–5600. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 3.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 4.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 5.Fetter J R, Qian J, Shapleigh J, Thomas J W, García-Horsman A, Schmidt E, Hosler J, Babcock G T, Gennis R B, Ferguson-Miller S. Proc Natl Acad Sci USA. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chepuri V, Lemieux L, Au D C-T, Gennis R B. J Biol Chem. 1990;265:11185–11192. [PubMed] [Google Scholar]
- 7.Musser S M, Stowell M H B, Chan S I. FEBS Lett. 1993;327:131–136. doi: 10.1016/0014-5793(93)80156-o. [DOI] [PubMed] [Google Scholar]
- 8.Haltia T. FEBS Lett. 1993;335:294–295. doi: 10.1016/0014-5793(93)80750-o. [DOI] [PubMed] [Google Scholar]
- 9.Musser S M, Stowell M H B, Chan S I. FEBS Lett. 1993;335:296–298. doi: 10.1016/0014-5793(93)80751-f. [DOI] [PubMed] [Google Scholar]
- 10.Welter R, Gu L-Q, Yu L, Yu C-A, Rumbley J, Gennis R B. J Biol Chem. 1994;269:28834–28838. [PubMed] [Google Scholar]
- 11.Babcock G T, Varotsis C. J Bioenerg Biomembr. 1993;25:71–80. doi: 10.1007/BF00762849. [DOI] [PubMed] [Google Scholar]
- 12.Svensson M, Nilsson T. Biochemistry. 1993;32:5442–5447. doi: 10.1021/bi00071a021. [DOI] [PubMed] [Google Scholar]
- 13.Hirota S, Mogi T, Ogura T, Hirano T, Anraku Y, Kitagawa T. FEBS Lett. 1994;352:67–70. doi: 10.1016/0014-5793(94)00919-8. [DOI] [PubMed] [Google Scholar]
- 14.Morgan J E, Verkhovsky M I, Puustinen A, Wikström M. Biochemistry. 1995;34:15633–15637. doi: 10.1021/bi00048a005. [DOI] [PubMed] [Google Scholar]
- 15.Verkhovsky M I, Morgan J E, Puustinen A, Wikström M. Biochemistry. 1996;35:16241–16246. doi: 10.1021/bi961433a. [DOI] [PubMed] [Google Scholar]
- 16.Schultz B E, Edmondson D E, Chan S I. Biochemistry. 1998;37:4160–4168. doi: 10.1021/bi971714y. [DOI] [PubMed] [Google Scholar]
- 17.Ohnishi T, Trumpower B L. J Biol Chem. 1980;255:3278–3284. [PubMed] [Google Scholar]
- 18.Rich P R. Biochim Biophys Acta. 1984;768:53–79. doi: 10.1016/0304-4173(84)90007-7. [DOI] [PubMed] [Google Scholar]
- 19.Sato-Watanabe M, Mogi T, Ogura T, Kitagawa T, Miyoshi H, Iwamura H, Anraku Y. J Biol Chem. 1994;269:28908–28912. [PubMed] [Google Scholar]
- 20.Sato-Watanabe M, Mogi T, Miyoshi H, Iwamura H, Matsushita K, Adachi O, Anraku Y. J Biol Chem. 1994;269:28899–28907. [PubMed] [Google Scholar]
- 21.Musser S M, Stowell M H B, Lee H K, Rumbley J N, Chan S I. Biochemistry. 1997;36:894–902. doi: 10.1021/bi961723r. [DOI] [PubMed] [Google Scholar]
- 22.Thomas J W, Puustinen A, Alben J O, Gennis R B, Wikström M. Biochemistry. 1993;32:10923–10928. doi: 10.1021/bi00091a048. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Horsman J A, Puustinen A, Gennis R B, Wikström M. Biochemistry. 1995;34:4428–4433. doi: 10.1021/bi00013a035. [DOI] [PubMed] [Google Scholar]
- 24.Verkhovskaya M L, Garcìa-Horsman A, Puustinen A, Rigaud J-L, Morgan J E, Verkhovsky M I, Wikström M. Proc Natl Acad Sci USA. 1997;94:10128–10131. doi: 10.1073/pnas.94.19.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ingledew W J, Ohnishi T, Salerno J C. Eur J Biochem. 1995;227:903–908. doi: 10.1111/j.1432-1033.1995.tb20217.x. [DOI] [PubMed] [Google Scholar]
- 26.Salerno J C, Bolgiano B, Ingledew W J. FEBS Lett. 1989;247:101–105. doi: 10.1016/0014-5793(89)81249-9. [DOI] [PubMed] [Google Scholar]
- 27.Salerno J C, Bolgiano B, Poole R K, Gennis R B, Ingledew W J. J Biol Chem. 1990;265:4364–4368. [PubMed] [Google Scholar]
- 28.Bolgiano B, Salmon I, Ingledew W J, Poole R K. Biochem J. 1991;274:723–730. doi: 10.1042/bj2740723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid G A, Ingledew W J. Biochem J. 1979;182:465–472. doi: 10.1042/bj1820465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hackett N R, Bragg P D. J Bacteriol. 1983;154:708–718. doi: 10.1128/jb.154.2.708-718.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kita K, Konishi K, Anraku Y. J Biol Chem. 1984;259:3368–3374. [PubMed] [Google Scholar]
- 32.Lorence R M, Green G N, Gennis R B. J Bacteriol. 1984;157:115–121. doi: 10.1128/jb.157.1.115-121.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Withers H K, Bragg P D. Biochem Cell Biol. 1990;68:83–90. doi: 10.1139/o90-010. [DOI] [PubMed] [Google Scholar]
- 34.Bolgiano B, Salmon I, Poole R K. Biochim Biophys Acta. 1993;1141:95–104. doi: 10.1016/0005-2728(93)90194-k. [DOI] [PubMed] [Google Scholar]
- 35.Sato-Watanabe M, Itoh S, Mogi T, Matsuura K, Miyoshi H, Anraku Y. FEBS Lett. 1995;374:265–269. doi: 10.1016/0014-5793(95)01125-x. [DOI] [PubMed] [Google Scholar]
- 36.Blair D F, Ellis W R, Jr, Wang H, Gray H B, Chan S I. J Biol Chem. 1986;261:11524–11537. [PubMed] [Google Scholar]
- 37.Wikström M. Chemica Scripta. 1988;28A:71–74. [Google Scholar]
- 38.Wikström M, Morgan J E. J Biol Chem. 1992;267:10266–10273. [PubMed] [Google Scholar]
- 39.Hallén S, Svennson M, Nilsson T. FEBS Lett. 1993;325:299–302. doi: 10.1016/0014-5793(93)81093-f. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Rumbley J, Ching Y-C, Takahashi S, Gennis R B, Rousseau D L. Biochemistry. 1995;34:15504–15511. doi: 10.1021/bi00047a016. [DOI] [PubMed] [Google Scholar]
- 41.Musser S, Stowell M H B, Chan S I. Adv Enzyme. 1995;71:79–208. doi: 10.1002/9780470123171.ch3. [DOI] [PubMed] [Google Scholar]