

Abstract
The ATM protein kinase is essential for cells to repair and survive genotoxic events. The activation of ATM's kinase activity involves acetylation of ATM by the Tip60 histone acetyltransferase. In this study, systematic mutagenesis of lysine residues was used to identify regulatory ATM acetylation sites. The results identify a single acetylation site at lysine 3016, which is located in the highly conserved C-terminal FATC domain adjacent to the kinase domain. Antibodies specific for acetyl-lysine 3016 demonstrate rapid (within 5 min) in vivo acetylation of ATM following exposure to bleomycin. Furthermore, lysine 3016 of ATM is a substrate in vitro for the Tip60 histone acetyltransferase. Mutation of lysine 3016 does not affect unstimulated ATM kinase activity but does abolish upregulation of ATM's kinase activity by DNA damage, inhibits the conversion of inactive ATM dimers to active ATM monomers, and prevents the ATM-dependent phosphorylation of the p53 and chk2 proteins. These results are consistent with a model in which acetylation of lysine 3016 in the FATC domain of ATM activates the kinase activity of ATM. The acetylation of ATM on lysine 3016 by Tip60 is therefore a key step linking the detection of DNA damage and the activation of ATM kinase activity.
Ataxia telangiectasia (A-T) is an inherited disease characterized by immune deficiencies, neurodegeneration, susceptibility to cancer, and sensitivity to ionizing radiation (28). The A-T gene product, the ATM protein kinase, is activated in response to DNA double-strand breaks (DSBs) (24, 35) and phosphorylates multiple DNA damage response proteins, including Nbs1, p53, chk2, and SMC1 (reviewed in reference 24). The phosphorylation of these proteins by ATM is essential for correct activation of cell cycle checkpoints and for the initiation of DNA repair. Consequently, cells lacking functional ATM protein exhibit defects in DNA repair and a loss of cell cycle checkpoints (24, 28, 35), resulting in increased sensitivity to ionizing radiation.
A central question in studying the ATM protein has been to determine the mechanism by which ATM's kinase activity is activated by DNA damage. Activation of ATM's kinase activity is associated with increased autophosphorylation of ATM at multiple sites (23), including serine 1981 (2). This autophosphorylation of ATM is proposed to initiate a dimer-monomer transition and the release of active ATM monomers (2), although the exact contribution of ATM autophosphorylation to ATM activation is still under debate (31). Several additional molecular events, including the Mre11-Rad50-Nbs1 (MRN) DNA binding complex (8, 11, 14, 41) and changes in chromatin structure (2, 4), also contribute to activation of ATM's kinase activity. Mutations in the individual protein components of the MRN complex can reduce or abolish the activation of ATM's kinase activity by DNA damage (8, 11, 14, 41). The recent characterization of a conserved C-terminal domain in Nbs1 which is essential for recruitment of ATM to the MRN complex and for the efficient activation of ATM's kinase activity (14, 42) further supports a key role for MRN in ATM activation. Finally, biochemical studies have shown that MRN can activate ATM's kinase activity in vitro (25, 26), suggesting that inactive ATM is recruited to DSBs by MRN prior to MRN-dependent activation of ATM's kinase activity. However, other studies indicate that disruption of chromatin structure in the absence of DNA damage can activate ATM's kinase activity (2) and that kinase-inactive ATM is not recruited to enzymatically induced DSBs (4), suggesting that ATM is activated in the nucleoplasm by DNA damage-induced changes in chromatin structure. In this case, the MRN complex may function as an adaptor complex which maintains ATM in an active form at the DSB (2, 4, 22). However, the mechanism by which interactions between ATM and MRN or structural changes in chromatin activate ATM's kinase activity is not known.
Previously, we demonstrated that the Tip60 histone acetyltransferase (HAT) is essential for the activation of ATM's kinase activity (19, 37). The HAT activity of Tip60 is increased by DNA damage, and the loss of Tip60 blocks the autophosphorylation and subsequent activation of ATM's kinase activity (37). However, although activation of Tip60's HAT activity is associated with an increase in the overall acetylation level of ATM, neither the acetylation site nor the functional significance of ATM acetylation is known. We now demonstrate that Tip60 directly acetylates ATM on lysine 3016 and that this acetylation is required for activation of ATM's kinase activity. The DNA damage-dependent acetylation of ATM on lysine 3016 by Tip60 is therefore the critical step which links the detection of DSBs to the activation of ATM kinase activity.
MATERIALS AND METHODS
Cells.
293T, HeLa (American Type Culture Collection, VA), and GM5849 A-T cells (Coriel Institute, NJ) were cultured according to the suppliers' recommendations. Transfection of cells (using Fugene-6 [Roche, IN]), establishment of stable cell lines by G418 selection, and clonogenic cell survival assays were done as previously described (15, 37). Small interfering RNAs (siRNAs) T3 (GGAAGCUGCUGAUCGAGUUUU) and T4 (GACGUAAGAACAAGAGUUAUU) and green fluorescent protein (GFP; Dharmacon, CO) were transfected into cells by using Lipofectamine 2000 (Invitrogen, CA) as previously described (37).
Mutagenesis.
Two overlapping fragments at the C terminus of ATM were subcloned from pcDNA3.1-ATM into pBluescript, creating pBS-SPBA (containing a 3,582-base-pair internal SpeI-BamHI fragment) and pBS-XBXH (containing an approximately 1,800-base-pair XbaI-XhoI fragment carrying the entire 3′ end of the cDNA, with the XhoI site located in pcDNA3.1). Mutagenesis was carried out using QuikChange II XL site-directed mutagenesis kits (Stratagene, CA) and the primers listed in Table S1 in the supplemental material. Following sequencing, mutations were released from pBS-SPBA and pBS-XBXH by digestion with SpeI-BlpI and BlpI-XhoI, respectively. The corresponding fragments were then excised from pcDNA3.1-ATM, and the mutated versions were ligated into the plasmid to create full-length ATM constructs.
Immunoprecipitation, Western blot analysis, HAT assay, and kinase assay.
General protocols for lysis buffers, immunoprecipitation, and Western blot analysis were done as described previously (19, 37). ATM immunoprecipitation was routinely performed using ATM antibody PC116 (EMD Biosciences, CA). Cells were lysed in ATM lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 0.2% Tween 20, 1.5 mM MgCl2, 1 mM EGTA, 2 mM dithiothreitol, 50 mM NaF, 500 μM NaVO4, 1 mM phenylmethylsulfonyl fluoride, 0.1 μg/ml aprotinin, 0.1 μg/ml leupeptin) and cleared by centrifugation. For kinase assays, ATM was immunoprecipitated and washed sequentially in ATM lysis buffer followed by ATM kinase buffer (10 mM HEPES, pH 7.4; 10 mM MgCl2; 50 mM NaCl; 10 mM MnCl2). Immunoprecipitates were incubated in kinase buffer (50 μl) supplemented with 50 μM ATP, p53 peptide (2 μg; EPPLSQEAFADLWKK), and 10 μCi [γ-32P]ATP for 15 min at 30°C (37). For HAT assays, ATM or Tip60 was immunoprecipitated and washed sequentially in ATM lysis buffer followed by HAT assay buffer (50 mM Tris, pH 8; 10% glycerol; 0.1 mM EDTA; 1 mM dithiothreitol). Immunoprecipitates were then incubated in HAT assay buffer (60 μl) supplemented with acetyl-coenzyme A (100 μM) and either biotinylated histone H4 peptide, biotinylated atmwt peptide (ERVLMRLQEKLKGVEEGT), or atmKR peptide (ERVLMRLQEALKGVEEGT) (0.5 μg) for 30 min at 30°C. An aliquot of the reaction mix was immobilized on a streptavidin plate, and acetylation was detected using a HAT enzyme-linked immunosorbent assay according to the manufacturer's instructions (Upstate Biotechnology, NY). For both kinase and HAT assays, each datum point represents the average for three separate immunoprecipitations, with each derived from approximately 3 × 107 cells and containing equivalent amounts of protein. Antibodies used were as follows: ATM antibodies PC116 (EMD Biosciences, CA), 5C2, 2C1 (both from Genetex, TX), and pS1981 ATM (Rockland Immunochemicals, PA) and antibodies to β-actin, chk2, hemagglutinin (HA), myc, phospho-chk2 (all from Cell Signaling, MA), Tip60, acetylated lysine (AcLys; Upstate Biotech, NY), p53, and pS15-p53 (EMD Biosciences, CA). A rabbit polyclonal antibody (AcK6) was raised against peptide CRVLMRLQE[KAc]LKG and affinity purified using immobilized acetylated peptide followed by absorption against immobilized nonacetylated peptide. The resulting antiserum was used at a 1:50 dilution for Western blot analysis.
Immunofluorescence.
Cells were seeded on LabTek II chamber slides (Nunc, NY). Cells were fixed as previously described (15) and stained with primary antibody ATM 5C2 (Genetex, TX) followed by immunoglobulin G (IgG)-fluorescein isothiocyanate (Santa Cruz, CA). Slides were mounted with Fluoromount-G (Southern Biotech, AL) and visualized with a Nikon Eclipse TE 2000 microscope.
RESULTS
Previously, we demonstrated that the Tip60 HAT plays a key role in the activation of ATM (19, 37) and that ATM is acetylated in cells exposed to agents which induce DNA DSBs. Here we report on studies designed to identify acetylation sites on the ATM protein. ATM acetylation can be monitored using a pan-specific acetyl-lysine antibody, previously characterized by us (37), which specifically detects the Tip60-dependent acetylation of ATM. The specificity of the pan-specific acetyl-lysine antibody is demonstrated in Fig. 1A and B. As shown in Fig. 1A, a previously validated siRNA was used to deplete cellular Tip60 protein levels (19, 37). As shown in Fig. 1B, cells exposed to the radiomimetic agent bleomycin displayed increased ATM acetylation as well as autophosphorylation of serine 1981, which can be used to monitor the intrinsic kinase activity of ATM (2). Depletion of Tip60 by siRNA reduced both the acetylation and autophosphorylation of ATM (Fig. 1B) and blocked the ability of ATM to phosphorylate downstream effector proteins (37). The pan-specific acetyl-lysine antibody can therefore detect Tip60-dependent acetylation of ATM. This antibody was then used in a focused, site-directed lysine mutagenesis study designed to identify ATM acetylation sites. Lysines were selected for mutagenesis based on three criteria. First, regulatory acetylations are predicted to occur in the conserved FAT/kinase domain/FATC region at the C terminus of ATM. Second, regulatory lysines should be conserved among ATMs from higher eukaryotes. Third, HATs frequently acetylate multiple lysines located within a few amino acids of each other. Based on these criteria, 17 regions within the C terminus (amino acids 1698 to 3056) containing closely spaced, conserved lysine residues were identified (see Table S1 in the supplemental material). Seventeen full-length ATM cDNA constructs, each containing two closely spaced lysine-to-alanine mutations, were then constructed and expressed in ATM-negative A-T fibroblasts. Each construct was then screened for DNA damage-induced acetylation by treating the cells with the radiomimetic agent bleomycin. Although mutation of several lysines in the FAT and kinase domains destabilized the ATM protein (see Table S2 in the supplemental material), only the ATM mutant ATMK17, containing the double mutation K3016/K3018A, lacked inducible acetylation (Fig. 1C). Lysines 3016 and 3018 were then mutated individually to arginine to determine which of these lysine residues was acetylated. Figure 1D demonstrates that ATMK3016R lacked both acetylation and autophosphorylation on serine 1981, whereas ATMK3018R was acetylated and autophosphorylated to the same extent as wild-type ATM. These results imply that lysine 3016 of ATM is acetylated when cells are exposed to DNA damage. Because mutations within the C termini of ATM and related proteins can destabilize ATM (see Table S2 in the supplemental material) (3, 29), lysine 3016 was mutated to alanine (to abolish the positive charge), to threonine or glutamic acid (to alter charge), or to the acetyl-lysine mimic glutamine. The mutants were expressed at similar levels to that of wild-type ATM protein (Fig. 1E). However, none of the mutants, including that containing the acetyl-lysine mimic glutamine, exhibited increased ATM acetylation or autophosphorylation in response to DNA damage. These results indicate that lysine 3016 is required for activation of ATM's kinase activity but does not influence the overall stability of the ATM protein. Furthermore, the acetyl-lysine mimic glutamine does not substitute for acetyl-lysine in ATM.
FIG. 1.

Lysine 3016 of ATM is acetylated after DNA damage. (A) 293T cells were transfected with siRNA targeting GFP or Tip60. Extracts were immunoprecipitated with either IgG (lane 1), Tip60 antibody (lanes 2 and 3, upper panel), or ATM antibody (lanes 2 and 3, lower panel). ATM and Tip60 were detected by Western blotting (WB). (B) 293T cells were transiently transfected with siRNA targeting either GFP (siGFP) or Tip60 (siTip60). Forty-eight hours later, cells were exposed to solvent (−) or bleomycin (5 μM for 30 min). Cell extracts were immunoprecipitated with IgG (lane 1) or anti-ATM antibody PC116 (lanes 2 to 5), and ATM protein levels (ATM), ATM acetylation (AcLys), and autophosphorylation (pS1981) of ATM were measured by Western blot analysis. (C) ATM-deficient GM5849 A-T cells transfected with vector, ATM, or ATM mutant K2 (K1772/1773A), K6 (K2204/2207A), or K17 (K3016/3018A) were exposed to bleomycin (+; 5 μM) for 30 min. ATM was immunoprecipitated with ATM antibody PC116, and ATM and ATM acetylation (AcLys) were measured by Western blotting. (D and E) GM5849 A-T cells transfected with vector, ATM, or ATMs with the indicated point mutations were exposed to bleomycin (+; 5 μM) for 30 min. ATM was immunoprecipitated, and Western blot analysis was used to monitor ATM levels, ATM acetylation (AcLys), and ATM autophosphorylation on serine 1981 (pS1981). (F) Location and sequence comparison of ATM acetylation sites.
Lysine 3016 is located within the conserved FATC domain at the C terminus of ATM and is conserved between ATMs from higher eukaryotes (Fig. 1F). The ATM acetylation site has sequence homology to previously identified Tip60 acetylation sites in the androgen receptor (ARKLK) (16) and the myc oncogene (SEKLA) (30). Overall, the data in Fig. 1 indicate that ATM is inducibly acetylated on lysine 3016 in response to DNA damage. However, we cannot exclude the possibility that there are additional acetylation sites within the ATM protein which are not detected by the pan-specific acetyl-lysine antibody used here. The ATMK3016A and ATMK3016R mutants were chosen for further analysis and yielded essentially identical results.
Previously, we demonstrated that the acetylation of ATM (detected using the pan-specific acetyl-lysine antibody used for Fig. 1) was abolished when Tip60 was inactivated using either siRNA (Fig. 1B) (37) or dominant-negative Tip60 proteins (37). Figure 1 demonstrates that this Tip60-dependent acetylation of ATM occurs on lysine 3016. To determine if Tip60 could directly acetylate lysine 3016 of ATM, a peptide corresponding to amino acids 3007 to 3024 of ATM (atmwt) and a control peptide in which lysine 3016 was replaced with arginine (atmKR) were synthesized. HA-Tip60 and HA-Tip60 with point mutations in the HAT domain (Tip60HD) were immunopurified from HeLa cells as described previously (37, 38). As shown in Fig. 2A, Tip60 acetylated both the histone H4 and atmwt peptides, and the ability of Tip60 to acetylate these peptides was increased following exposure to bleomycin (Fig. 2A). The higher levels of acetylation of the H4 peptide reflect the presence of three Tip60 acetylation sites in the histone H4 peptide (21), in contrast to one in the atmwt peptide. When lysine 3016 was replaced with arginine, the atmKR peptide was minimally acetylated by Tip60 and displayed no detectable increase in HAT activity in response to bleomycin. Furthermore, when a Tip60 protein with inactivating mutations in the HAT domain (Tip60HD) was used (18), minimal acetylation of either the H4, atmwt, or atmKR peptide was seen (Fig. 2A). These results demonstrate that Tip60 can directly acetylate a peptide containing lysine 3016 of ATM in vitro. In a further series of experiments, immunopurification of the ATM-Tip60 complex from cells also demonstrated that Tip60 acetylated lysine 3016 in vitro (see Fig. S1 in the supplemental material). Figure 2A therefore demonstrates that Tip60 can directly acetylate peptides containing lysine 3016 of ATM in vitro.
FIG. 2.
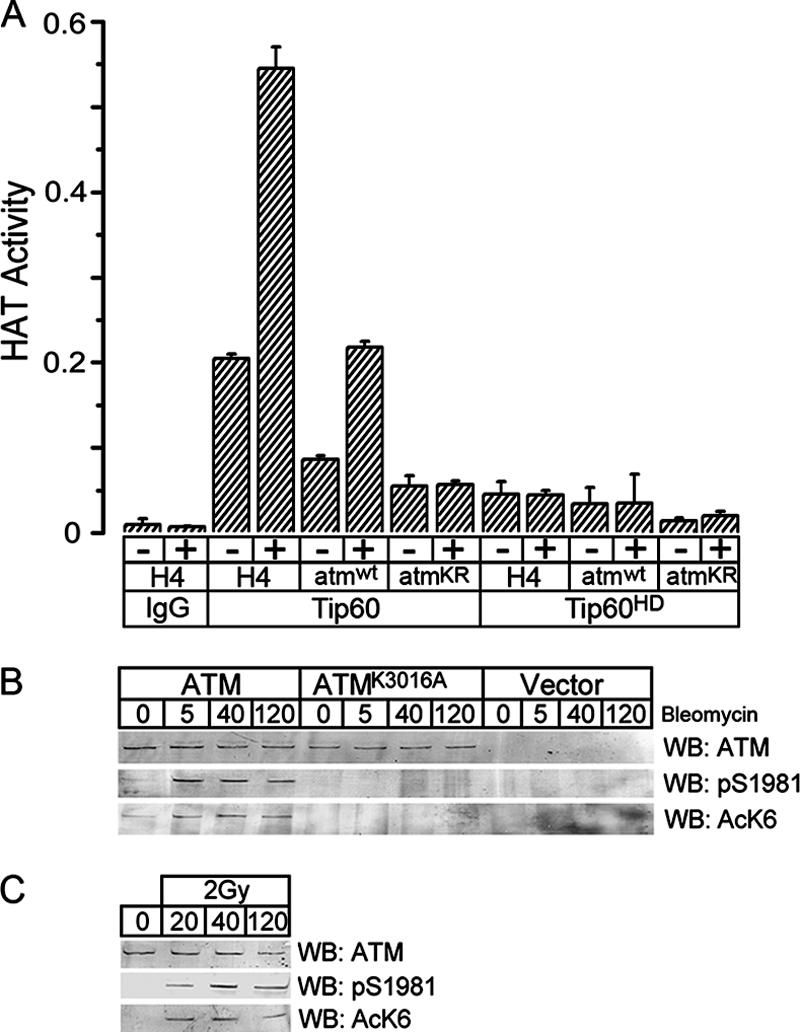
Lysine 3016 of ATM is acetylated in vivo. (A) Tip60 was immunopurified from HeLa cells expressing either HA-Tip60 (Tip60) or HAT-inactive HA-Tip60 (Tip60HD). Cell extracts were incubated with either HA antibody or IgG and washed as described in Materials and Methods to immunopurify Tip60. The ability of immunopurified Tip60 to acetylate peptides derived from either ATM or the N terminus of histone H4 was determined. Peptides used were as follows: amino acids 3007 to 3024 of ATM (atmwt; ERVLMRLQEKLKGVEEGT); amino acids 3007 to 3024 of ATM, with lysine 3016 replaced with arginine (atmKR; ERVLMRLQERLKGVEEGT); or amino acids 1 to 21 of histone H4 (SGRGKGGKGLGKGGAKRHRKV). All peptides contained a C-terminal biotin label. Cells were treated for 30 min with bleomycin (+; 5 μM), where indicated. Results are means ± standard deviations (SD) (n = 3). (B) GM5849 A-T cells transfected with vector, ATM, or ATMK3016A were exposed to bleomycin (+; 5 μM) for the indicated times (minutes). ATM was immunoprecipitated, and Western blot analysis (WB) was used to monitor ATM levels, ATM autophosphorylation on serine 1981 (pS1981), and ATM acetylation, using the K3016Ac-specific antiserum AcK6. (C) 293T cells were irradiated (2 Gy) and allowed to recover for the indicated times (minutes). ATM was immunoprecipitated, and Western blot analysis was used to monitor ATM levels, ATM-pS1981, and ATM acetylation, using the K3016Ac-specific antiserum AcK6.
Before examining the in vivo acetylation of ATM, immunofluorescence staining was used to confirm that ATMK3016R was located in the nucleus of the cell (see Fig. S2 in the supplemental material). To monitor the in vivo acetylation of lysine 3016 of ATM, a sequence-specific antibody was raised against acetylated lysine 3016. As shown in Fig. 2B, this antibody (AcK6) detected rapid (within 5 min) DNA damage-dependent acetylation of ATM. No significant acetylation or autophosphorylation of ATM was detected in either ATM-deficient A-T cells or A-T cells expressing ATMK3016A for up to 2 h following exposure to bleomycin, demonstrating that the AcK6 antibody specifically recognizes ATM acetylated on lysine 3016. To examine the acetylation of endogenous ATM, 293T cells were exposed to low doses of ionizing radiation. Figure 2C demonstrates the rapid acetylation of lysine 3016 of ATM (detected with the sequence-specific AcK6 antibody) and corresponding autophosphorylation of serine 1981 following exposure to ionizing radiation. Lysine 3016 of ATM is therefore rapidly acetylated in vivo in response to DNA damage, and this acetylation occurs with the same rapid kinetics as those for the previously described autophosphorylation of ATM on serine 1981 (2).
Previously, we demonstrated that ATM and Tip60 interact and that this interaction requires the FATC domain at the C terminus of ATM (19). Because lysine 3016 is located within the FATC domain of ATM (Fig. 1F), we determined if the mutation of lysine 3016 affected either ATM-Tip60 interactions or activation of Tip60's HAT activity by DNA damage. Immunoprecipitation studies demonstrate that mutation of lysine 3016 does not alter the interaction between ATM and Tip60 (see Fig. S3A in the supplemental material). As shown in Fig. 3A, ATM was immunoprecipitated from ATM-deficient A-T cells complemented with either vector, ATM, or ATMK3016A, and the associated Tip60-dependent HAT activity was measured. In the absence of ATM, minimal Tip60 HAT activity was measured, and this was not increased by bleomycin (Fig. 3A, vector). However, the HAT activities of Tip60 associated with both ATM and ATMK3016A were increased to similar levels in response to bleomycin treatment (Fig. 3A). Mutation of lysine 3016 therefore does not affect either the activation of Tip60's HAT activity or the interaction between ATM and Tip60.
FIG. 3.

Mutation of lysine 3016 of ATM blocks activation of ATM's kinase activity. (A) GM5849 A-T cells transfected with vector, ATM, or ATMK3016A were left untreated (−) or exposed to bleomycin (+; 5 μM) for 30 min, and ATM was immunoprecipitated. The HAT activity of the Tip60 associated with ATM was measured using histone H4 peptide as the substrate. Results are means ± SD (n = 3). (B) GM5849 A-T cells transfected with vector, ATM, ATMK3016R, or ATMK3018R were exposed to bleomycin (+; 5 μM) for 20 min. p53, phospho-p53 (pS15-p53), chk2, phospho-chk2 (pT68-chk2), and β-actin were detected by Western blotting (WB) of whole-cell extracts. (C) GM5849 A-T cells transfected with vector, ATM, ATMkd, or ATMK3016R were left untreated (−) or exposed to bleomycin (+; 5 μM) for 30 min, and ATM was immunoprecipitated. The intrinsic kinase activity of ATM was then measured using a p53 peptide (containing serine 15) as the substrate. Results are means ± SD (n = 3). (D) GM5849 A-T cells expressing ATM or ATMK3016R were preincubated with OA (0.5 μM) or dimethyl sulfoxide (−) for 20 min and then irradiated (IR; 2 Gy), and cell extracts were prepared 40 min later. ATM was immunoprecipitated, and the levels of ATM, acetylated ATM (AcLys), and ATM autophosphorylated on serine 1981 (pS1981) were determined. In addition, whole-cell extracts were probed by Western blot analysis to determine the levels of p53 and p53 phosphorylated on serine 15 (pS15-p53).
Next, we examined if lysine 3016 was required to activate ATM's kinase activity by monitoring the ability of ATMK3016R to phosphorylate p53 and chk2 in vivo. A potential problem in analyzing the ATMK3016R mutant is that although the introduced arginine maintains the overall charge, it may introduce structural changes which affect ATM function independently of the loss of the acetylation site. To address this point, the activity of ATMK3016R was compared with that of ATMK3018R, in which the adjacent lysine at amino acid 3018 was mutated to arginine (described in Fig. 1D). A-T cells expressing each construct were treated with bleomycin, and the ability of ATM, ATMK3016R, and ATMK3018R to phosphorylate p53 and chk2 was determined. Both wild-type ATM and ATMK3018R induced the ATM-dependent phosphorylation of p53 and chk2 in response to DNA damage (Fig. 3B), whereas ATMK3016R failed to elicit phosphorylation of either p53 or chk2, consistent with the failure of ATMK3016R to exhibit either autophosphorylation or acetylation (Fig. 1). Similar results were seen with ATMK3016A (see Fig. S3B in the supplemental material). Mutation of lysine 3016, but not of the adjacent residue lysine 3018, therefore blocks acetylation of ATM and the subsequent phosphorylation of p53 and chk2 by ATM. Furthermore, the ability of ATMK3018R to function like wild-type ATM indicates that this region of ATM can tolerate lysine-to-arginine substitutions without affecting function, suggesting that the loss of activity of ATMK3016R is due to a loss of acetylation rather than to structural effects.
The inability of ATMK3016R to phosphorylate p53 or chk2 suggests a defect in the kinase activity of ATMK3016R. The kinase activity of ATM was measured by immunoprecipitating ATM and then incubating it with a substrate peptide containing the phosphorylation site of the p53 protein. Immunoprecipitation with IgG or from cells expressing a kinase-inactive ATM (ATMkd) yielded minimal levels of ATM kinase activity (Fig. 3C). ATM had significant basal levels of ATM kinase activity, which were increased threefold following exposure to bleomycin. In contrast, whereas ATMK3016R had the same levels of basal kinase activity as ATM, the kinase activity of ATMK3016R was not increased by bleomycin exposure (Fig. 3C). This implies that the function of acetylation is to activate ATM's kinase activity. To further explore the functional properties of ATMK3016R, we took advantage of previous work demonstrating that ATM and protein phosphatase 2A exist as a complex in the cell (17). Inhibition of protein phosphatase 2A by okadaic acid (OA) leads to rapid accumulation of ATM autophosphorylated on serine 1981, a process requiring ATM's kinase activity. However, this form of ATM does not exhibit elevated kinase activity and is functionally inactive (17). Therefore, we examined if OA treatment of ATMK3016R could induce autophosphorylation of ATM on serine 1981. Figure 3D demonstrates that OA induced the autophosphorylation of both ATM and ATMK3016R on serine 1981. However, only wild-type ATM showed inducible acetylation and autophosphorylation in response to ionizing radiation or combined DNA damage and OA treatment. Mutation of lysine 3016 of ATM therefore does not affect the ability of ATM to undergo autophosphorylation on serine 1981, indicating that substitution of arginine for lysine at amino acid 3016 does not affect the intrinsic kinase activity of ATM. Autophosphorylation of ATM is postulated to be a key step in the production of active ATM (2, 23). This raises the possibility that sequential exposure of ATMK3016R-expressing cells to OA (to induce autophosphorylation of serine 1981) followed by ionizing radiation may bypass the need for acetylation, leading to the generation of an active ATM molecule able to phosphorylate key target proteins, including p53. A-T cells expressing wild-type ATM did not exhibit p53 phosphorylation following OA treatment but displayed robust p53 phosphorylation after either ionizing radiation or ionizing radiation plus OA (Fig. 3D), as previously shown (17). However, OA treatment of ATMK3016R cells, either alone or in combination with ionizing radiation, did not generate an active ATM molecule able to phosphorylate p53 in vivo (Fig. 3D). Figure 3D therefore clearly shows that ATMK3016R retains the ability to undergo autophosphorylation on serine 1981 in response to OA exposure but does not undergo autophosphorylation in response to DNA damage signals.
Next, we determined if the failure to activate the kinase activity of ATMK3016R was reflected in a loss of functional activity. First, the ability of ATMK3016R to complement the increased radiosensitivity of A-T cells was examined. As shown in Fig. 4A, the expression of ATM or ATMK3018R complemented the radiosensitivity of A-T cells, whereas expression of ATMK3016R had no effect, consistent with the lack of activation of the kinase activity of ATMK3016R and its inability to phosphorylate key target proteins. Autophosphorylation of ATM is proposed to initiate a dimer-monomer transition, releasing active ATM monomers (2, 23). To determine the contribution of acetylation to the dimer-monomer transition, the ability of ATM, ATMkd, and ATMK3016A to undergo dimer-monomer conversion was examined. A-T cells were stably transfected with equimolar ratios of HA-ATM and myc-ATM expression vectors, and the dimer-monomer transition was monitored by examining dissociation of the HA-ATM/myc-ATM dimer by coimmunoprecipitation techniques. Figure 4B demonstrates the coexpression of HA-ATM and myc-ATM in A-T cells. In some experiments, a nonspecific band of 340 kDa was detected for the ATM-negative A-T cells by the HA and myc antibodies (Fig. 4B and C); however, this band was not detected with ATM antibodies (Fig. 1 and 2). Following exposure to bleomycin, wild-type ATM, but not ATMkd or ATMK3016A, was autophosphorylated on serine 1981. Similarly, wild-type ATM, but not ATMK3016A, was acetylated. Interestingly, ATMkd, which does not possess kinase activity and does not autophosphorylate (40), was also acetylated. Immunoprecipitation with HA antibody brought down both HA-ATM and myc-ATM in all unstimulated cells (Fig. 4C). Following exposure to bleomycin, wild-type HA-ATM no longer interacted with myc-ATM, whereas both ATMkd and ATMK3016A remained in the dimeric form. Importantly, since ATMkd was still acetylated on lysine 3016, this indicates that acetylation on its own is not sufficient for initiating the dimer-monomer transition of the ATM protein, suggesting that additional events, such as autophosphorylation of ATM, may also be required (2, 23). The AcK6 anti-acetyl K3016 antiserum was not competent for immunofluorescence, precluding its use for monitoring recruitment of the acetylated ATM to DSBs.
FIG. 4.

Lysine 3016 of ATM is required for ATM to regulate radiosensitivity and dimer-monomer transition. (A) GM5849 A-T cells stably transfected with vector (•), ATM (○), ATMK3016R (▵), or ATMK3018R (▪) were irradiated, and cell survival was measured using a clonogenic cell survival assay. Results are means ± SD (n = 3). (B) A-T cells were stably cotransfected with HA- and myc-tagged ATM constructs, such that both HA-ATM and myc-ATM were expressed in the same cell. Cell lines coexpressing HA- and myc-tagged ATM, HA- and myc-tagged ATMkd, or HA- and myc-tagged ATMK3016A were exposed to bleomycin (5 μM for 30 min). Levels of HA-ATM, myc-ATM, pS1981, and acetylation of lysine K3016 (AcK6) were measured. (C) Cells were treated as described above and then immunoprecipitated with HA antibody. Interaction between HA-ATM and myc-ATM was then assessed by Western blot (WB) analysis to detect HA-ATM and coprecipitating myc-ATM.
DISCUSSION
Previously, we established that Tip60 was essential for activation of ATM's kinase activity (19, 37, 38) and that DNA damage increased the overall level of ATM acetylation in a Tip60-dependent manner. The results reported here have identified lysine 3016 of ATM as the amino acid acetylated by Tip60 (19, 37) and provide evidence that this acetylation of ATM is required for the activation of ATM's kinase activity. The acetylation of lysine 3016 is maximal within 5 min and is temporally indistinguishable from the time course of autophosphorylation of serine 1981 of ATM (Fig. 2) (2, 37). However, a kinase-inactive ATM protein was still acetylated on lysine 3016, demonstrating that ATM acetylation is independent of ATM's kinase activity. This implies that ATM acetylation precedes ATM autophosphorylation and defines acetylation of lysine 3016 of ATM as the earliest posttranslational modification of ATM identified to date.
The results indicate that mutation of lysine 3016 blocks the activation of ATM's kinase activity and subsequent autophosphorylation and dimer-monomer transition. Furthermore, ATMK3016R does not phosphorylate downstream target proteins or complement the increased radiosensitivity of A-T cells. These results are consistent with an essential role for acetylation in the activation of ATM kinase activity. A potentially confounding problem is that mutation of lysine 3016, within the FATC domain of ATM, may have a negative impact on the correct function of the kinase domain independent of the loss of acetylation. However, we carried out several controls which suggest that this is not the case, as follows: (i) ATMK3016R retains normal basal kinase activity and lacks only inducible activation; (ii) unlike many mutations in the kinase domain identified in A-T patients, substituting lysine 3016 for arginine does not destabilize ATM or alter its intrinsic kinase activity; (iii) introducing a lysine-to-arginine mutation at an adjacent lysine (amino acid 3018) does not affect ATM function, indicating that the region of ATM containing lysine 3016 can tolerate small changes in protein architecture; and (iv) ATMK3016R can still undergo autophosphorylation on serine 1981 following the addition of OA, indicating that mutation of lysine 3016 to arginine does not disrupt the critical intermolecular protein-protein interactions within the ATM dimer which are required for the autophosphorylation of ATM by the kinase domain (2). Taken together, we interpret these results to support a model in which the failure of ATMK3016R to increase its kinase activity in response to DNA damage is due to a loss of acetylation of lysine 3016 rather than to nonspecific effects related to the lysine-to-arginine substitution. Thus, the primary function of acetylation of lysine 3016 is to upregulate ATM's kinase activity.
Insights into how acetylation may regulate the intrinsic kinase activity of ATM are provided by recent structural studies on ATM and the structurally related DNA-PKcs protein. PIKK protein family members, including ATM and DNA-PKcs, contain a conserved FAT domain/kinase domain/FATC domain structure (6). Low-resolution structural modeling of the DNA-PKcs and ATM proteins indicates that the FAT and FATC domains protrude from either side of the kinase domain (27, 33, 36). The FAT domain is weakly conserved between family members, whereas the kinase and FATC domains are highly conserved (1). FATC domains are highly flexible α- helical structures (10, 33) and are required for the kinase activities of the mTor (39), DNA-PKcs (3, 32), and ATM (19) proteins. Studies indicate that the interaction between DNA-PK and DNA induces conformational changes in the FATC and FAT domains (33, 36), and these conformational changes are predicted to influence the catalytic activity of DNA-PK and ATM (33). Acetylation of lysine 3016 within the flexible FATC domain of ATM may therefore influence ATM kinase activity. This is further supported by reports that a tumor-derived mutation near the kinase domain of the ATM-like p110α phosphatidylinositol 3-kinase subunit results in constitutive kinase activity (20, 34). Sequence alignment of ATM and p110α indicates that one of these p110α mutations (at amino acid 1054) occurs in the same relative position as lysine 3016 of ATM. This implies that the C-terminal regions of p110α and ATM may be essential for regulating kinase activity. Based on the results presented here and the results of structural studies (10, 33, 36), we propose that acetylation of lysine 3016, which is located at the junction between the FATC and kinase domains, alters the conformation of the FATC domain. This altered conformation of the FATC domain could allow substrate proteins access to the kinase domain as well as positively regulating the intrinsic kinase activity of the kinase domain. Further structural studies are required to address this point.
A key question is to address the mechanistic links between ATM acetylation and autophosphorylation. The autophosphorylation of serine 1981 of ATM was originally proposed to be essential for ATM activation and to play a key role in initiating the dimer-monomer transition and the release of active ATM monomers (2). However, recent biochemical studies have shown that in vitro activation of ATM kinase activity can be achieved in the absence of significant serine 1981 autophosphorylation (12, 25). In addition, a mouse model expressing a mutation in the mouse equivalent of serine 1981 of ATM (serine 1987) displays normal ATM function. These observations indicate that autophosphorylation of serine 1981 is dispensable for ATM function under some conditions (31). Recently, several additional ATM autophosphorylation sites were identified (23). Mutation of these sites impaired the ability of ATM to phosphorylate downstream targets and to correct the radiosensitive phenotype of A-T cells (23). Therefore, although some studies demonstrate that autophosphorylation of serine 1981 is dispensable for ATM activity (12, 25, 31, 42), the identification of additional ATM autophosphorylation sites raises the possibility that, similar to the case for DNA-PKcs (5, 9), multiple ATM autophosphorylation sites contribute to ATM activation. The results presented here indicate that the main function of ATM acetylation is to activate ATM's kinase activity. In this model, acetylation of ATM precedes and is required for the activation of ATM's kinase activity. Once activated, ATM kinase activity induces autophosphorylation of ATM at multiple sites, including serine 1981 (2, 23), and subsequent conversion of ATM dimers to monomers. Thus, while ATM autophosphorylation and subsequent dimer-monomer transitions may be important for achieving full ATM activation, the results presented in this report indicate that acetylation of ATM, rather than autophosphorylation, is the key step in upregulating ATM's kinase activity.
The results reported here and in previous works from us (19, 37) and other groups (13) demonstrate that Tip60 plays a key role in the acetylation and activation of ATM and indicate a central role for Tip60 in the early events leading to activation of ATM's kinase activity. However, other significant contributors to ATM activation include both the MRN complex (8, 11, 22, 25, 41) and changes in chromatin structure (2). A wide range of experimental approaches have established that the MRN complex plays a key role in several aspects of ATM activation, including the recruitment of ATM to DSBs (14, 42) and modulation of the activation of ATM's kinase activity by DSBs (8, 22, 25, 26, 41). This raises the possibility that MRN may regulate ATM activation by influencing the HAT activity of Tip60, either through direct interactions between MRN and Tip60 or through positioning of the ATM-Tip60 complex at DSBs. Tip60 also contains a chromodomain, a protein domain with the potential to interact with methylated lysine residues on histones (7). Changes in chromatin structure following ionizing radiation (either localized to the DSB or generated over large chromatin domains) may expose methylated histones as potential binding sites for Tip60's chromodomain. Such interactions between Tip60 and altered chromatin structures adjacent to DSBs may be essential for upregulating Tip60's HAT activity and for the acetylation and activation of the ATM protein. Tip60 may therefore provide the key step which links previous observations on the relative contributions of the MRN complex, chromatin structural changes, and ATM acetylation with the activation of the ATM protein kinase by DNA damage.
Supplementary Material
Acknowledgments
This work was supported by grants to B.D.P. from the National Cancer Institute (CA93602 and CA64585). Y.S. and K.R. were supported by National Institutes of Health training grant T32 CA09078, and Y.X. was supported by NIAID center grant U19A1067751.
We thank Susan Lees-Miller and Sheng-Chung Lee for helpful discussions.
Footnotes
Published ahead of print on 8 October 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Abraham, R. T. 2004. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amsterdam) 3:883-887. [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. [DOI] [PubMed] [Google Scholar]
- 3.Beamish, H. J., R. Jessberger, E. Riballo, A. Priestley, T. Blunt, B. Kysela, and P. A. Jeggo. 2000. The C-terminal conserved domain of DNA-PKcs, missing in the SCID mouse, is required for kinase activity. Nucleic Acids Res. 28:1506-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkovich, E., R. J. Monnat, Jr., and M. B. Kastan. 2007. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 9:683-690. [DOI] [PubMed] [Google Scholar]
- 5.Block, W. D., Y. Yu, D. Merkle, J. L. Gifford, Q. Ding, K. Meek, and S. P. Lees-Miller. 2004. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Res. 32:4351-4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosotti, R., A. Isacchi, and E. L. Sonnhammer. 2000. FAT: a novel domain in PIK-related kinases. Trends Biochem. Sci. 25:225-227. [DOI] [PubMed] [Google Scholar]
- 7.Brehm, A., K. R. Tufteland, R. Aasland, and P. B. Becker. 2004. The many colours of chromodomains. Bioessays 26:133-140. [DOI] [PubMed] [Google Scholar]
- 8.Cerosaletti, K., J. Wright, and P. Concannon. 2006. Active role for nibrin in the kinetics of ATM activation. Mol. Cell. Biol. 26:1691-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, B. P., D. W. Chan, J. Kobayashi, S. Burma, A. Asaithamby, K. Morotomi-Yano, E. Botvinick, J. Qin, and D. J. Chen. 2005. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J. Biol. Chem. 280:14709-14715. [DOI] [PubMed] [Google Scholar]
- 10.Dames, S. A., J. M. Mulet, K. Rathgeb-Szabo, M. N. Hall, and S. Grzesiek. 2005. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J. Biol. Chem. 280:20558-20564. [DOI] [PubMed] [Google Scholar]
- 11.Difilippantonio, S., A. Celeste, O. Fernandez-Capetillo, H. T. Chen, B. Reina San Martin, F. Van Laethem, Y. P. Yang, G. V. Petukhova, M. Eckhaus, L. Feigenbaum, K. Manova, M. Kruhlak, R. D. Camerini-Otero, S. Sharan, M. Nussenzweig, and A. Nussenzweig. 2005. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 7:675-685. [DOI] [PubMed] [Google Scholar]
- 12.Dupre, A., L. Boyer-Chatenet, and J. Gautier. 2006. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 13:451-457. [DOI] [PubMed] [Google Scholar]
- 13.Eymin, B., P. Claverie, C. Salon, C. Leduc, E. Col, E. Brambilla, S. Khochbin, and S. Gazzeri. 2006. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol. Cell. Biol. 26:4339-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falck, J., J. Coates, and S. P. Jackson. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434:605-611. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes, N., Y. Sun, S. Chen, P. Paul, R. J. Shaw, L. C. Cantley, and B. D. Price. 2005. DNA damage-induced association of ATM with its target proteins requires a protein interaction domain in the N terminus of ATM. J. Biol. Chem. 280:15158-15164. [DOI] [PubMed] [Google Scholar]
- 16.Gaughan, L., I. R. Logan, S. Cook, D. E. Neal, and C. N. Robson. 2002. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 277:25904-25913. [DOI] [PubMed] [Google Scholar]
- 17.Goodarzi, A. A., J. C. Jonnalagadda, P. Douglas, D. Young, R. Ye, G. B. Moorhead, S. P. Lees-Miller, and K. K. Khanna. 2004. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 23:4451-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikura, T., V. V. Ogryzko, M. Grigoriev, R. Groisman, J. Wang, M. Horikoshi, R. Scully, J. Qin, and Y. Nakatani. 2000. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102:463-473. [DOI] [PubMed] [Google Scholar]
- 19.Jiang, X., Y. Sun, S. Chen, K. Roy, and B. D. Price. 2006. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 281:15741-15746. [DOI] [PubMed] [Google Scholar]
- 20.Kang, S., A. G. Bader, and P. K. Vogt. 2005. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 102:802-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura, A., and M. Horikoshi. 1998. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells 3:789-800. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa, R., C. J. Bakkenist, P. J. McKinnon, and M. B. Kastan. 2004. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 18:1423-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozlov, S. V., M. E. Graham, C. Peng, P. Chen, P. J. Robinson, and M. F. Lavin. 2006. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 25:3504-3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavin, M. F., G. Birrell, P. Chen, S. Kozlov, S. Scott, and N. Gueven. 2005. ATM signaling and genomic stability in response to DNA damage. Mutat. Res. 569:123-132. [DOI] [PubMed] [Google Scholar]
- 25.Lee, J. H., and T. T. Paull. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308:551-554. [DOI] [PubMed] [Google Scholar]
- 26.Lee, J. H., and T. T. Paull. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93-96. [DOI] [PubMed] [Google Scholar]
- 27.Llorca, O., A. Rivera-Calzada, J. Grantham, and K. R. Willison. 2003. Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene 22:3867-3874. [DOI] [PubMed] [Google Scholar]
- 28.Meyn, M. S. 1999. Ataxia-telangiectasia, cancer and the pathobiology of the ATM gene. Clin. Genet. 55:289-304. [DOI] [PubMed] [Google Scholar]
- 29.Nakada, D., Y. Hirano, Y. Tanaka, and K. Sugimoto. 2005. Role of the C terminus of mec1 checkpoint kinase in its localization to sites of DNA damage. Mol. Biol. Cell 16:5227-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel, J. H., Y. Du, P. G. Ard, C. Phillips, B. Carella, C. J. Chen, C. Rakowski, C. Chatterjee, P. M. Lieberman, W. S. Lane, G. A. Blobel, and S. B. McMahon. 2004. The c-Myc oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24:10826-10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellegrini, M., A. Celeste, S. Difilippantonio, R. Guo, W. Wang, L. Feigenbaum, and A. Nussenzweig. 2006. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 443:222-225. [DOI] [PubMed] [Google Scholar]
- 32.Priestley, A., H. J. Beamish, D. Gell, A. G. Amatucci, M. C. Muhlmann-Diaz, B. K. Singleton, G. C. Smith, T. Blunt, L. C. Schalkwyk, J. S. Bedford, S. P. Jackson, P. A. Jeggo, and G. E. Taccioli. 1998. Molecular and biochemical characterisation of DNA-dependent protein kinase-defective rodent mutant irs-20. Nucleic Acids Res. 26:1965-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rivera-Calzada, A., J. D. Maman, L. Spagnolo, L. H. Pearl, and O. Llorca. 2005. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure (Cambridge) 13:243-255. [DOI] [PubMed] [Google Scholar]
- 34.Samuels, Y., Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. Willson, S. Markowitz, K. W. Kinzler, B. Vogelstein, and V. E. Velculescu. 2004. High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554. [DOI] [PubMed] [Google Scholar]
- 35.Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3:155-168. [DOI] [PubMed] [Google Scholar]
- 36.Spagnolo, L., A. Rivera-Calzada, L. H. Pearl, and O. Llorca. 2006. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell 22:511-519. [DOI] [PubMed] [Google Scholar]
- 37.Sun, Y., X. Jiang, S. Chen, N. Fernandes, and B. D. Price. 2005. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 102:13182-13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun, Y., X. Jiang, S. Chen, and B. D. Price. 2006. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 580:4353-4356. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi, T., K. Hara, H. Inoue, Y. Kawa, C. Tokunaga, S. Hidayat, K. Yoshino, Y. Kuroda, and K. Yonezawa. 2000. Carboxyl-terminal region conserved among phosphoinositide-kinase-related kinases is indispensable for mTOR function in vivo and in vitro. Genes Cells 5:765-775. [DOI] [PubMed] [Google Scholar]
- 40.Turenne, G. A., P. Paul, L. Laflair, and B. D. Price. 2001. Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal serine residues of p53. Oncogene 20:5100-5110. [DOI] [PubMed] [Google Scholar]
- 41.Uziel, T., Y. Lerenthal, L. Moyal, Y. Andegeko, L. Mittelman, and Y. Shiloh. 2003. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22:5612-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You, Z., C. Chahwan, J. Bailis, T. Hunter, and P. Russell. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25:5363-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.