

Abstract
Homologs of the Saccharomyces cerevisiae Sir2 protein, sirtuins, promote longevity in many organisms. Studies of the sirtuin SIRT3 have so far been limited to cell culture systems. Here, we investigate the localization and function of SIRT3 in vivo. We show that endogenous mouse SIRT3 is a soluble mitochondrial protein. To address the function and relevance of SIRT3 in the regulation of energy metabolism, we generated and phenotypically characterized SIRT3 knockout mice. SIRT3-deficient animals exhibit striking mitochondrial protein hyperacetylation, suggesting that SIRT3 is a major mitochondrial deacetylase. In contrast, no mitochondrial hyperacetylation was detectable in mice lacking the two other mitochondrial sirtuins, SIRT4 and SIRT5. Surprisingly, despite this biochemical phenotype, SIRT3-deficient mice are metabolically unremarkable under basal conditions and show normal adaptive thermogenesis, a process previously suggested to involve SIRT3. Overall, our results extend the recent finding of lysine acetylation of mitochondrial proteins and demonstrate that SIRT3 has evolved to control reversible lysine acetylation in this organelle.
Conserved from bacteria to humans, the sirtuin family of NAD+-dependent protein deacetylase/mono-ADP-ribosyltransferase enzymes controls a variety of cellular processes such as aging, metabolism, and gene silencing (18, 24). It has been proposed that sirtuins mediate the longevity-promoting effects of calorie restriction (CR) in yeast, worms, flies, and mice (4, 17, 22, 24). Seven mammalian sirtuins (SIRT1 to -7) are known (11, 12, 18, 24). At least three sirtuins (SIRT3, SIRT4, and SIRT5) localize to mitochondria, suggesting the existence of sirtuin substrates in that organelle (19, 26, 28, 31-33). Several lines of evidence link SIRT3 to metabolism: SIRT3 is down-regulated in muscle from diabetic animals (37) and upregulated in white and brown adipose tissue in response to CR (33). Overexpression of SIRT3 in cells affects expression of genes involved in mitochondrial function (33). SIRT3 regulates the acetylation level and activity of acetyl-coenzyme A synthetase 2, a protein that may play a role in energy production in mammals under starvation conditions (20, 31). SIRT4 is an ADP-ribosyltransferase that has been implicated in regulating amino acid-stimulated insulin secretion in mice via modification of glutamate dehydrogenase (GDH) (19). No functions have been reported for SIRT5.
Although reversible lysine acetylation as a means of regulating protein function is well characterized for other cellular compartments, lysine acetylation of mitochondrial proteins has only recently been described (21, 31). The identities of the enzymes controlling mitochondrial protein acetylation are not known. Although a fraction of histone deacetylase 7 has been reported to localize to mitochondria, the implications of this finding are unclear (3). Among the mitochondrial sirtuins, only SIRT3 possesses robust NAD+-dependent deacetylase activity (27, 28, 31, 32).
To study the biological function of SIRT3 in vivo, we raised antibodies against mouse SIRT3 and generated SIRT3-deficient mice via homologous recombination. Here, we demonstrate that SIRT3-deficient mice show hyperacetylation of numerous mitochondrial proteins including the metabolic enzyme GDH. In contrast, SIRT4- and SIRT5-deficient mice do not exhibit globally increased mitochondrial protein acetylation levels. These results indicate that SIRT3 is a major mitochondrial protein deacetylase in mammals. However, the increase in mitochondrial lysine acetylation resulting from loss of SIRT3 did not translate into any metabolic abnormalities under standard laboratory housing conditions or in response to metabolic challenges such as fasting or cold exposure.
MATERIALS AND METHODS
Antibodies.
Antibodies used were anti-mitochondrial heat shock protein 70 (anti-mtHsp70; Affinity Bioreagents); anti-Hsp90α, anticalreticulin, and anti-manganese superoxide dismutase (anti-MnSOD; StressGen Biochemicals and Santa Cruz Biotechnology, Inc.); anti-RNA polymerase II and anti-histone H4 (Upstate); anti-Hsp60 (Santa Cruz Biotechnology, Inc.); anti-cytochrome c oxidase subunit IV (anti-COX-IV) and anti-F1F0-ATPase subunit a (Invitrogen Molecular Probes); anti-GDH (Biotrend Chemikalien GmbH); anti-uncoupling protein 1 (anti-UCP-1; Alpha Diagnostic Int. Inc.); anti-cytochrome c (clone 7H8.2C12; BD Pharmingen); and anti-acetylated-lysine polyclonal and monoclonal antibodies (Cell Signaling Technology). Antibodies against mitochondrial transcription factor A (TFAM) were a kind gift from N.-G. Larsson (Karolinska Institute, Stockholm, Sweden). Antibodies recognizing murine SIRT3 were raised against the C-terminal 15-amino-acid peptide (C)DLMQRERGKLDGQDR. The peptide was conjugated to carrier protein KLH via the added C residue and injected into rabbits at Covance Research Products, Inc. (Denver, PA). SIRT3 antisera were purified by immunoaffinity chromatography. Antibodies to murine SIRT5 were raised in rabbits using the C-terminal peptide GPCGKTLPEALAPHETERT (Covance Research Products, Inc.).
Subcellular fractionation, purification of mitochondria, and separation of mitochondrial protein complexes.
Murine liver mitochondria and nuclei were prepared and purified according to standard protocols (14-16). The postmitochondrial supernatant was centrifuged at 100,000 × g for 1 h at 4°C to obtain the light-membrane fraction (pellet) and cytosol (S-100; supernatant). Solubilization of purified mitochondria with n-dodecyl β-d-maltoside (Sigma) and separation of mitochondrial protein complexes by sucrose density gradient centrifugation were performed as previously described (34, 35).
Alkaline extraction of mitochondria.
Carbonate extraction of mitochondria was performed as described previously (13, 32).
SIRT3 gene targeting and SIRT3-deficient mice.
The murine SIRT3 gene was cloned from a 129Ola mouse genomic DNA library (kind gift from Raju Kucherlapati, Albert Einstein College of Medicine). Three overlapping λ phage clones were subcloned into pBluescript (Stratagene). A 5.8-kb genomic DNA fragment containing exon 1A, exon 1B, exon 2, and exon 3 was inserted flanking the pGK-Neo cassette of the pGEM7 vector. A 3-kb genomic DNA fragment containing exon 4 was inserted on the opposite side of the pGK-Neo cassette. A pGK-TK cassette was inserted adjacent to the 3-kb DNA fragment. LoxP sites were located flanking exons 2 and 4. The construct was electroporated into embryonic stem (ES) cells, and correctly targeted clones were isolated via positive and negative selection followed by Southern blotting. Chimeric mice were generated by injection of targeted ES clones into C57BL6/J blastocysts. Male chimeras were mated with 129Sv females to generate F1 heterozygous mice. Heterozygous animals were subsequently bred to the EIIA-Cre line to remove the Neor gene (23), and Neo-deleted heterozygotes were then interbred to generate homozygous knockout mice.
Immunoprecipitation.
Mitochondria were lysed in ice-cold LMIP buffer (1% n-dodecyl β-d-maltoside, 0.5 mM EDTA, 150 mM NaCl, 10 mM nicotinamide, 1 μM trichostatin A, 50 mM Tris-HCl, pH 7.4) containing the Complete EDTA-free protease inhibitor cocktail (Roche). Immunoprecipitations were performed according to standard procedures. Immune complexes were washed four times in NP1 buffer (1% NP-40, 300 mM NaCl, 0.5 mM EDTA, 50 mM Tris-HCl, pH 7.4).
In vitro deacetylation assays.
Equal amounts of purified recombinant SIRT3 and bovine GDH (Sigma) were incubated in SDAC deacetylation buffer (31) in the presence or absence of 1 mM NAD+ (Sigma) and 10 mM nicotinamide (Sigma) and in the presence of 500 nM trichostatin A (Wako Biochemicals) for 3 h at 32°C.
Dual-energy X-ray absorptiometry (DEXA).
To analyze body composition, mice were anesthetized with isoflurane and analyzed with a PixiMus2 scanner (GE Healthcare Lunar, Madison, WI).
Metabolic analysis.
Energy balance was determined in 4- to 6-month-old male mice fed a standard chow diet (5053 PicoLab diet; Purina, St. Louis, MO). Food intake, activity levels, oxygen consumption (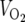 ), and respiratory exchange ratios (
), and respiratory exchange ratios (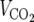 /
/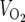 ) were measured using the Oxymax comprehensive laboratory animal monitoring system (Columbus Instruments, Columbus, OH).
) were measured using the Oxymax comprehensive laboratory animal monitoring system (Columbus Instruments, Columbus, OH). 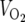 was normalized for lean body mass, as measured by DEXA scanning on the day before calorimetry studies. Briefly, mice were fasted for 4 h and anesthetized with isoflurane, and their body compositions were analyzed by DEXA. Data represent the means plus standard errors of the means (SEM) for six animals per genotype.
was normalized for lean body mass, as measured by DEXA scanning on the day before calorimetry studies. Briefly, mice were fasted for 4 h and anesthetized with isoflurane, and their body compositions were analyzed by DEXA. Data represent the means plus standard errors of the means (SEM) for six animals per genotype.
Adaptive thermogenesis.
Cold test experiments were performed as described previously (2). In brief, age-matched groups of male mice were housed individually in cages without food. Core body temperature was measured rectally with a digital thermometer (model 4600; Yellow Springs Instruments, Yellow Springs, OH). The results shown are representative of four separate assays.
RESULTS
SIRT3 is a soluble mitochondrial protein highly expressed in mitochondrion-rich tissues.
To determine the expression and localization of endogenous murine SIRT3, we generated and affinity purified antibodies against its C terminus. SIRT3 protein expression in a panel of murine tissues was analyzed (Fig. 1A). SIRT3 protein levels correlated with expression levels of the mitochondrial protein MnSOD and were particularly high in tissues rich in mitochondria: brain, heart, liver, and brown adipose tissue (BAT). Our results are in agreement with previous studies of SIRT3 mRNA expression (33).
FIG. 1.

SIRT3 is a soluble mitochondrial protein highly expressed in tissues rich in mitochondria. (A) Prominent SIRT3 protein expression in mitochondrion-rich tissues. Twenty micrograms of total protein extract from each tissue was analyzed by immunoblotting with antibodies to SIRT3 and the mitochondrial protein MnSOD. SKM, skeletal muscle (quadriceps); WAT, white adipose tissue. (B) Endogenous murine SIRT3 is a mitochondrial protein. Equal amounts of total liver homogenate (total), mitochondria (Mito), light membranes (LM), and cytosol (S-100) were separated by SDS-PAGE and analyzed by immunoblotting using antibodies against SIRT3 and marker proteins of known subcellular distribution. (C) Murine SIRT3 is a soluble mitochondrial protein. Purified mouse liver mitochondria were extracted with sodium carbonate (pH 11.5), and the distribution of SIRT3, the soluble mitochondrial protein GDH, and the integral membrane protein COX-IV was analyzed by immunoblotting. T, total; P, pellet (membranes); S, soluble. (D) SIRT3 is absent from the nucleus in liver. Equal amounts of nuclear and mitochondrial protein extracts were immunoblotted with antibodies against the indicated proteins. RNAP-II, RNA polymerase II; H4, histone H4.
Previously, ectopically expressed epitope-tagged murine SIRT3 was reported to localize to mitochondria in cultured cells (33). To determine the subcellular localization of endogenous murine SIRT3, subcellular fractions were prepared from the livers of wild-type mice and equal amounts of total homogenate, mitochondria, light membranes, and cytosol were probed with SIRT3-specific antibodies (Fig. 1B). SIRT3, like the mitochondrial marker protein MnSOD, was enriched in mitochondria in comparison to the total homogenate but was absent from other subcellular fractions. Immunoblotting of these fractions for the marker proteins calreticulin (endoplasmic reticulum) and Hsp90α (cytosol) confirmed the purity of the fractions. Thus, endogenous murine SIRT3 is a mitochondrial protein.
Next, we performed alkaline extraction experiments to address whether endogenous murine SIRT3 is soluble in mitochondria or bound to mitochondrial membranes. Previously, epitope-tagged murine SIRT3 overexpressed in cultured cells was reported to be an integral protein of the inner mitochondrial membrane (33). In contrast, human SIRT3 protein is a soluble mitochondrial matrix protein (31, 32). Sodium carbonate treatment (pH 11.5) of liver mitochondria released SIRT3 and the soluble matrix protein GDH, while the integral membrane protein COX-IV remained bound to the membrane (Fig. 1C). These results show that endogenous murine SIRT3, like its human homolog, is a soluble mitochondrial protein.
A fraction of full-length, unprocessed human SIRT3 has been localized to the nucleus (30). To test whether a fraction of endogenous murine SIRT3 might show nuclear localization, liver nuclei and mitochondria were purified according to standard procedures. Equal amounts of nuclear and mitochondrial protein extracts were analyzed by immunoblotting using antibodies against SIRT3 and organelle marker proteins. As expected, purified nuclei contained high levels of RNA polymerase II and histone H4 (Fig. 1D). Under basal or metabolic-stress conditions (overnight fast followed by cold exposure), SIRT3 was undetectable in nuclei while it was readily detectable in the mitochondria, along with the known mitochondrial proteins COX-IV and Hsp60 (Fig. 1D; see Fig. S1 in the supplemental material). We conclude that endogenous SIRT3 predominantly localizes to mitochondria in mouse liver under both basal and stress conditions.
Generation of SIRT3-deficient mice.
To explore the biological functions of SIRT3 in vivo, we inactivated the murine SIRT3 gene by homologous recombination in ES cells. The SIRT3 locus was mutated by deletion of exons 2 and 3, encoding the translational start site plus a portion of the catalytic domain (36) (Fig. 2A). Correctly targeted ES cell clones were used to obtain heterozygous mice, which were interbred to obtain homozygotes. Genotyping was performed by PCR (Fig. 2B) and by Southern blotting (not shown). Immunoblotting with SIRT3-specific antibodies confirmed the absence of SIRT3 protein in mitochondrial protein extracts from liver and brain of knockout mice (Fig. 2C). SIRT3-deficient mice were born at a Mendelian ratio (see Fig. S2A in the supplemental material), morphologically indistinguishable from the wild type (see Fig. S2B in the supplemental material), healthy until at least 1 year of age, and fertile as homozygotes.
FIG. 2.

Generation of SIRT3-deficient mice. (A) Gene targeting strategy for the SIRT3 locus. Arrows indicate loxP sites, and Neor indicates the neomycin resistance cassette used to select targeted ES cell clones. Restriction sites are as shown. WT, wild type. (B) SIRT3 PCR genotyping. KO, knockout; Het, heterozygous. (C) SIRT3 protein is absent from liver and brain mitochondria of knockout mice. Protein extracts were probed with antibodies to SIRT3 and F1F0-ATPase subunit α (loading control).
Hyperacetylation of mitochondrial proteins in SIRT3-deficient mice.
SIRT3 possesses potent NAD+-dependent deacetylase activity (27, 28, 31, 32). Recently, mitochondria have been shown to contain lysine-acetylated proteins (21, 31). Because SIRT3 is a mitochondrial protein deacetylase, we speculated that SIRT3 might affect the acetylation status of mitochondrial proteins. To address this hypothesis, global lysine acetylation levels in mitochondria from wild-type and SIRT3-deficient mice were assessed using commercially available pan-acetyl-lysine antibodies. Consistent with a role of SIRT3 as a mitochondrial deacetylase, mitochondria from SIRT3-deficient mice showed increased lysine acetylation of several distinct protein bands in liver and BAT (Fig. 3A and B), as well as in brain and heart (data not shown). Hyperacetylation in BAT was particularly striking (Fig. 3B). Different acetyl-lysine antibodies recognized distinct spectra of lysine-acetylated proteins (data not shown), but all antibodies tested showed higher levels of lysine acetylation in mitochondria from SIRT3-deficient mice. This phenotype was present irrespective of mouse gender.
FIG. 3.

SIRT3 is a major mitochondrial protein deacetylase in vivo. (A) Hyperacetylation of mitochondrial proteins in liver of SIRT3-deficient mice. SIRT3-deficient and littermate control mitochondrial extracts from two mice per genotype were fractionated by SDS-PAGE and immunoblotted with polyclonal antibodies to acetyl-lysine (Ac-K), SIRT3, or MnSOD as a loading control. (B) Hyperacetylation of mitochondrial proteins in BAT of SIRT3-deficient mice. Mitochondrial extracts from two SIRT3-deficient mice and two wild-type littermate controls were probed as in panel A except that COX-IV was used as a loading control. (C) Recombinant SIRT3 reverses mitochondrial hyperacetylation associated with SIRT3 deficiency. Mitochondrial extracts were treated with recombinant wild-type (WT) SIRT3 or a SIRT3 catalytic mutant (SIRT3-HY) as shown. NAD+, a cofactor required for sirtuin-mediated deacetylation, and nicotinamide (NAM), a sirtuin inhibitor, were added as indicated. Samples were separated by SDS-PAGE and probed with a monoclonal acetyl-lysine antibody. SIRT3 antibodies were used to demonstrate the presence of recombinant SIRT3; mtHsp70 served as a loading control. (D) SIRT3, but not SIRT4 or SIRT5, is responsible for global protein deacetylation in mitochondria. Liver mitochondrial extracts were generated from mice of the indicated genotypes and analyzed as in panel A.
To test whether the observed hyperacetylation of mitochondrial proteins is a direct consequence of SIRT3 deficiency, mitochondrial extracts from SIRT3-deficient mice were incubated with purified recombinant SIRT3 protein. In the presence of NAD+, recombinant SIRT3 reversed mitochondrial protein hyperacetylation (Fig. 3C, lane 2); this reaction was completely abolished in the presence of the sirtuin inhibitor nicotinamide (Fig. 3C, lane 3). Incubation of SIRT3-deficient mitochondrial extracts with catalytically inactive SIRT3 mutant protein (31, 32) did not reduce mitochondrial protein acetylation (Fig. 3C, lane 4), demonstrating that the exogenous recombinant SIRT3 but not some impurity present in the deacetylation reaction mixture mediated the deacetylation. These results indicate that mitochondrial protein hyperacetylation in SIRT3-deficient mice is a direct consequence of loss of the SIRT3 deacetylase.
In addition to SIRT3, SIRT4 and SIRT5 have been reported to localize to mitochondria (19, 26). To assess the role of these other proteins in mitochondrial protein deacetylation, we generated mice individually deficient in SIRT4 (19) or SIRT5 (see Fig. S3A in the supplemental material). SIRT5-deficient mice were born at a Mendelian ratio (see Fig. S3B in the supplemental material), fertile, and grossly healthy until at least 18 months of age. Immunoblot analysis of liver mitochondria confirmed the absence of SIRT5 protein in the knockout; incidentally, this represents the first demonstration of endogenous SIRT5 protein in the mitochondrion (Fig. 3D). No mitochondrial protein hyperacetylation was observed in liver extracts from mice lacking SIRT4 or SIRT5 (Fig. 3D). These results suggest that other mitochondrial sirtuins are not functionally redundant with SIRT3 with regard to regulation of global levels of lysine acetylation observed in mitochondria.
SIRT3 controls the acetylation status of GDH in vivo.
Liver mitochondria were used to identify hyperacetylated proteins present in SIRT3-deficient mice. Mitochondrial extracts prepared under conditions that preserve multiprotein complex integrity were fractionated by sucrose density gradient centrifugation followed by one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 4A) according to published methods (34, 35). Immunoblots of the fractions were probed with acetyl-lysine antibodies and were compared to gels stained with Coomassie blue prepared in parallel (Fig. 4A). Two acetyl-lysine antibody-reactive bands of approximately 50 kDa present in fractions 6 and 7 of the gradient (Fig. 4B) were analyzed by liquid chromatography-mass spectrometry. Peptides corresponding to GDH were found in both gel bands after in-gel digestion with trypsin, and three GDH peptides containing acetylated lysine residues were identified (see Fig. S4 and S5 and Table S1 in the supplemental material). One of these GDH acetylation sites (K527) has been reported previously (21).
FIG. 4.

SIRT3 deacetylates GDH in vivo. (A) Scheme for identifying putative SIRT3 substrates from mouse liver mitochondria. KO, knockout. (B) Representative immunoblot (IB) of protein fractions prepared as illustrated in panel A and probed with monoclonal acetyl-lysine antibody. *, acetyl-lysine antibody-reactive bands analyzed by mass spectrometry. (C and D) GDH is hyperacetylated in SIRT3-deficient mouse liver mitochondria. (C) Acetylated proteins from liver mitochondria of animals with the indicated genotypes were immunoprecipitated (IP) with monoclonal acetyl-lysine antibodies, fractionated by SDS-PAGE, and probed with GDH antibodies. Results shown are representative of five independent experiments. (D) GDH immune complexes from wild-type and SIRT3-deficient liver mitochondria were immunoblotted with monoclonal acetyl-lysine antibodies (Ac-GDH). Probing with GDH antibodies revealed total GDH levels. Results shown are representative of three independent experiments. (E) GDH is a bona fide SIRT3 substrate. Bovine GDH was incubated with recombinant SIRT3, NAD+, and nicotinamide (NAM) as indicated. Reactions were stopped by boiling in SDS sample buffer, and levels of total and acetylated GDH were analyzed by immunoblotting. The presence of recombinant SIRT3 was demonstrated by immunoblotting with antibodies to SIRT3.
To confirm that GDH is indeed a SIRT3 target, acetylated proteins were immunoprecipitated from wild-type and SIRT3-deficient mitochondrial extracts using acetyl-lysine antibodies and probed with an antibody against GDH (Fig. 4C). GDH was strongly enriched in the anti-acetyl-lysine immune complexes from SIRT3 knockout mitochondria, suggesting that GDH is indeed hyperacetylated in SIRT3-deficient mitochondria. To further confirm this observation, we immunoprecipitated GDH from mitochondrial extracts and analyzed the immune complexes with antibodies to acetyl-lysine (Fig. 4D). GDH from SIRT3 knockout mice exhibited higher levels of acetylation than GDH from wild-type mice, confirming GDH hyperacetylation in SIRT3 deficiency. To determine directly whether GDH can serve as a SIRT3 substrate, we incubated GDH derived from Bos taurus with recombinant SIRT3 (Fig. 4E). SIRT3 deacetylated GDH in the presence of NAD+ (Fig. 4E, lane 2); this reaction was completely blocked by nicotinamide (Fig. 4E, lane 3). GDHs from Bos taurus and Mus musculus show high homology (96% identity), and all but two lysine residues are conserved between both species (see Fig. S6 in the supplemental material). Our findings indicate that GDH is a target of SIRT3 in vivo.
SIRT3-deficient mice are metabolically unremarkable under both fed and fasted conditions.
In light of the mitochondrial protein hyperacetylation in SIRT3-deficient animals, these mice were examined for defects in metabolically relevant tissues. Histological examination of liver, BAT, heart, brain, kidney, and skeletal muscle from fed (not shown) and fasted animals did not reveal any morphological alterations in SIRT3-deficient mice (Fig. 5A and 6B; see Fig. S7 in the supplemental material). To assess overall mitochondrial number, levels of TFAM were assessed in total liver extracts from SIRT3-deficient and littermate control mice (Fig. 5B). TFAM levels were identical between genotypes, suggesting that mitochondrial numbers were not altered in the absence of SIRT3 and thereby arguing against the possibility that potential functional deficiencies of SIRT3-deficient mitochondria are compensated by an increase in total mitochondrial number.
FIG. 5.

SIRT3-deficient mice are viable and metabolically unremarkable under both fed and fasted conditions. (A) Normal liver histology in wild-type (WT) and SIRT3-deficient (KO) mice subject to an 18-hour fast. (B) SIRT3 deficiency does not affect mitochondrial content. Equal amounts of protein from total liver extracts generated from animals of the indicated genotypes representing two separate litters were probed with antibodies to TFAM to assess mitochondrial content and MnSOD, COX-IV, and cytochrome c as loading controls. (C) Body weight, adiposity, bone mineral density (BMD), and bone mineral content (BMC) are not affected by SIRT3 deficiency. (D) SIRT3-deficient and wild-type littermate control mice display similar measures of energy balance in both the fed and fasted states. Oxygen consumption (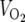 ), respiratory exchange ratios (
), respiratory exchange ratios (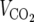 /
/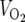 ), activity levels, and food intake were measured in metabolic cages. Data represent the means plus SEM for six animals per genotype.
), activity levels, and food intake were measured in metabolic cages. Data represent the means plus SEM for six animals per genotype.
FIG. 6.

SIRT3-deficient mice show normal adaptive thermogenesis. (A) SIRT3-deficient mice show wild-type levels of UCP-1. UCP-1 protein levels were assessed in BAT mitochondrial extracts from wild-type (WT) and SIRT3 knockout (KO) animals from three different litters. Levels of Hsp60 and MnSOD were measured as loading controls. (B) Normal BAT morphology in SIRT3 KO mice. Hematoxylin-eosin staining is shown. Original magnification, ×400. (C) Normal adaptive thermogenesis in SIRT3 KO mice. (Left) SIRT3 KO and WT cohorts are similar in both pre- and post-cold exposure body weights. (Middle) SIRT3 KO and WT mice lose equal percentages of body weight during 6 h of cold exposure (4°C). ns, not significant. (Right) Core body temperatures of 7- to 10-week-old male SIRT3 KO and WT mice during exposure to 4°C for 6 h. Data represent the means ± SEM for three animals per genotype.
Impaired mitochondrial β-oxidation or defects in the electron transport chain lead to hepatic steatosis in mice and humans (7), and altered mitochondrial function can translate into alterations in body composition (for example, see reference 6). To address this issue, SIRT3-deficient mice and controls were weighed and subjected to body composition analysis by DEXA analysis (Fig. 5C). On a chow diet, the body weight and body fat content of SIRT3-deficient mice were indistinguishable from those of wild-type mice. Furthermore, this analysis also demonstrated normal bone mineral density and overall bone mineral content in SIRT3-deficient animals (Fig. 5C).
Since perturbations in mitochondrial physiology can affect whole-body metabolism, we assessed energy balance in SIRT3-deficient mice during the last 6 h of a 24-hour fast and during the first 6 h of the transition to the fed state (Fig. 5D). SIRT3-deficient mice were indistinguishable from wild-type littermates with respect to oxygen consumption, respiratory exchange ratio, and activity and consumed similar amounts of food during the refeeding period (Fig. 5D). In summary, loss of SIRT3 did not affect overall metabolism under standard laboratory conditions.
SIRT3-deficient mice show normal adaptive thermogenesis.
The dramatic mitochondrial protein hyperacetylation in BAT of SIRT3-deficient animals prompted us to test the function of this tissue in these knockouts. In mice, one critical role of BAT is in adaptive thermogenesis (25). In this context, it has been proposed that SIRT3 affects adaptive thermogenesis via regulation of gene expression in BAT (33), in particular expression of UCP-1, a protein critical for cold tolerance (10). However, protein levels of UCP-1 in BAT mitochondria (Fig. 6A) and overall BAT morphology (Fig. 6B) were unaltered in SIRT3-deficient mice. In cold exposure studies, age-matched SIRT3-deficient and wild-type male littermates showed no difference in body weight at room temperature or after a 6-hour exposure to 4°C (Fig. 6C, left). Over the course of this assay, SIRT3-deficient animals were able to effectively mobilize body stores, as the percentages of body weight loss for wild-type and knockout mice, both in young mice (7 to 10 weeks old; Fig. 6C, middle) and in older mice (13 weeks old; see Fig. S8 in the supplemental material), were identical. Furthermore, both groups of mice showed effective maintenance of core body temperature for at least 6 hours under these conditions (Fig. 6C, right; see Fig. S8 in the supplemental material). Overall, our results suggest that SIRT3 is not essential for normal energy metabolism or short-term cold resistance in mice.
DISCUSSION
This study has established that murine SIRT3 is a soluble mitochondrial protein controlling global mitochondrial protein acetylation levels. SIRT3 represents the first factor known to affect protein acetylation in this compartment. In contrast, deletion of SIRT4 or SIRT5 is not associated with globally increased mitochondrial lysine acetylation, in agreement with previous reports showing that these factors possess little or no deacetylase activity (1, 19, 27, 31). We cannot exclude the possibility that SIRT4 and SIRT5 deacetylate specific mitochondrial factors not detected by our methodology. Surprisingly, despite accumulating hyperacetylated mitochondrial proteins, SIRT3-deficient mice are healthy under normal laboratory conditions and conditions of mild stress such as short-term food deprivation and show normal overall metabolism and cold resistance. We have focused on a metabolic characterization of SIRT3-deficient mice under basal conditions and conditions of mild nutritional stress, such as 24-h fasting; future studies are required to determine whether acute stressors such as oxidative insult, food deprivation, and conditions that require high levels of oxidative metabolism over a long period of time (e.g., high-fat diet) will uncover abnormalities in these mice. Along these lines, given that SIRT3 levels increase with CR in mice (33), SIRT3 may regulate mitochondrial protein function in response to CR. Of interest, it has been reported that polymorphisms in the human SIRT3 gene are linked to longevity (5, 29). In this context, formal life span analysis of SIRT3-deficient mice under various dietary regimens will be needed to establish a firm role for this protein in regulating life span overall and the CR response in particular. Alternatively, in light of the central role of mitochondria in many cellular processes, SIRT3 might have functions aside from metabolism. For example, given the importance of mitochondria in the regulation of cell survival, a role for SIRT3 in apoptosis is conceivable.
We have identified GDH as one target of SIRT3. The functional significance of GDH acetylation is unclear; chemical acetylation of GDH has been shown to reduce its enzymatic activity in vitro (8, 9). Future studies will be required to further address the role of reversible lysine acetylation in GDH regulation. Notably, GDH is ADP-ribosylated by SIRT4, leading to decreased GDH activity and decreased insulin secretion in response to amino acids (19). It will be of interest to determine whether SIRT3 and SIRT4 coregulate GDH and under which conditions this might occur. SIRT4 activity was proposed to decline with CR to permit higher levels of GDH activity (19). CR is associated with elevated SIRT3 expression (33) and potentially increased SIRT3 activity. Depending on the functional consequence of GDH acetylation, SIRT3 and SIRT4 may regulate GDH and potentially other proteins in a similar fashion during CR.
Given the pronounced hyperacetylation present in SIRT3-deficient mitochondria, the lack of an overt phenotype in these animals is puzzling. It is possible that, although mitochondrial protein hyperacetylation is dramatic in SIRT3 knockout mice at the level of immunoblot analysis, only a minor proportion of any particular factor is acetylated. Consequently, most proteins, e.g., metabolic enzymes such as GDH, would still be fully functional, explaining the absence of a strong metabolic phenotype in SIRT3-deficient mice. This possibility is supported by data showing that increased GDH acetylation in SIRT3-deficient mice does not result in grossly impaired GDH function, as indicated by normal tissue levels of the GDH substrate α-ketoglutarate (see Fig. S9 in the supplemental material). Presently, factors responsible for lysine acetylation of mitochondrial proteins are unknown. It might be necessary to overexpress the putative mitochondrial acetyltransferase(s) and/or delete putative redundant deacetylases in order to elicit a phenotype of SIRT3 deficiency. Indeed it is unclear whether lysine acetylation of mitochondrial proteins occurs in the mitochondrion itself or in the cytosol prior to mitochondrial import. Similarly, the biological function of mitochondrial protein acetylation is entirely unclear (21). This modification could a priori regulate enzymatic activity, intramitochondrial localization, protein-protein interactions, protein stability, or some combination of these. Overall, SIRT3 represents the first factor described to affect global mitochondrial lysine acetylation, and SIRT3-deficient mice should serve as a valuable tool to study reversible lysine acetylation biology in mitochondria in vivo.
Supplementary Material
Acknowledgments
We are grateful to C. B. Newgard, J. R. Bain, O. R. Ilkayeva, R. D. Stevens, B. R. Wenner, and L. C. Naliboff (Metabolomics Laboratory, Sarah W. Stedman Nutrition & Metabolism Center, Duke University Medical Center, Durham, NC) for metabolite analysis. We thank N.-G. Larsson (Karolinska Institute, Stockholm, Sweden) for his generous gift of the TFAM antibody and members of the Alt and Verdin laboratories, B. J. North (Harvard Medical School), and B. B. Lowell (BIDMC, Harvard Medical School) for helpful discussions.
This work was supported by Ellison Medical Foundation Senior Scholar Awards (to F.W.A. and E.V.), by funds from the Sandler Foundation Program in Basic Sciences (to E.V.), and an NIH National Center for Research Resources facilities grant (1C06RR18928) to the J. David Gladstone Institutes. F.W.A. is an Investigator of the Howard Hughes Medical Institute. D.B.L. is supported by a K08 award from NIA/NIH. B.S. is the recipient of a UCSF Sandler Postdoctoral Research Fellowship Award. J.B. is supported by a grant from the Carlsberg Foundation. CEBI is supported by a grant from the Danish National Research Foundation.
F.W.A. and E.V. are members of the scientific advisory board of Sirtris Pharmaceuticals. L.G. is a founder and board member of Elixir Pharmaceuticals. G.Y., D.V., and A.M. are employees and shareholders of Regeneron Pharmaceuticals.
All animal experiments carried out herein were performed under established protocols per institutional guidelines.
Footnotes
Published ahead of print on 8 October 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Ahuja, N., B. Schwer, S. Carobbio, D. Waltregny, B. J. North, V. Castronovo, P. Maechler, and E. Verdin. 22 August 2007, posting date. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. [Epub ahead of print.] doi: 10.1074/jbc.M705488200. [DOI] [PubMed]
- 2.Argmann, C. A., M. Champy, and J. Auwerx. 2006. Assessment of thermoregulation by the cold test, p. 29B1.6-29B1.17. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley & Sons, Inc., Hoboken, NJ.
- 3.Bakin, R. E., and M. O. Jung. 2004. Cytoplasmic sequestration of HDAC7 from mitochondrial and nuclear compartments upon initiation of apoptosis. J. Biol. Chem. 279:51218-51225. [DOI] [PubMed] [Google Scholar]
- 4.Baur, J. A., K. J. Pearson, N. L. Price, H. A. Jamieson, C. Lerin, A. Kalra, V. V. Prabhu, J. S. Allard, G. Lopez-Lluch, K. Lewis, P. J. Pistell, S. Poosala, K. G. Becker, O. Boss, D. Gwinn, M. Wang, S. Ramaswamy, K. W. Fishbein, R. G. Spencer, E. G. Lakatta, D. Le Couteur, R. J. Shaw, P. Navas, P. Puigserver, D. K. Ingram, R. de Cabo, and D. A. Sinclair. 2006. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellizzi, D., G. Rose, P. Cavalcante, G. Covello, S. Dato, F. De Rango, V. Greco, M. Maggiolini, E. Feraco, V. Mari, C. Franceschi, G. Passarino, and G. De Benedictis. 2005. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 85:258-263. [DOI] [PubMed] [Google Scholar]
- 6.Brown, L. J., R. A. Koza, C. Everett, M. L. Reitman, L. Marshall, L. A. Fahien, L. P. Kozak, and M. J. MacDonald. 2002. Normal thyroid thermogenesis but reduced viability and adiposity in mice lacking the mitochondrial glycerol phosphate dehydrogenase. J. Biol. Chem. 277:32892-32898. [DOI] [PubMed] [Google Scholar]
- 7.Caldwell, S. H., C. Y. Chang, R. K. Nakamoto, and L. Krugner-Higby. 2004. Mitochondria in nonalcoholic fatty liver disease. Clin. Liver Dis. 8:595-617. [DOI] [PubMed] [Google Scholar]
- 8.Colman, R. F., and C. Frieden. 1966. On the role of amino groups in the structure and function of glutamate dehydrogenase. I. Effect of acetylation on catalytic and regulatory properties. J. Biol. Chem. 241:3652-3660. [PubMed] [Google Scholar]
- 9.Colman, R. F., and C. Frieden. 1966. On the role of amino groups in the structure and function of glutamate dehydrogenase. II. Effect of acetylation on molecular properties. J. Biol. Chem. 241:3661-3670. [PubMed] [Google Scholar]
- 10.Enerback, S., A. Jacobsson, E. M. Simpson, C. Guerra, H. Yamashita, M. E. Harper, and L. P. Kozak. 1997. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387:90-94. [DOI] [PubMed] [Google Scholar]
- 11.Frye, R. A. 1999. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260:273-279. [DOI] [PubMed] [Google Scholar]
- 12.Frye, R. A. 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273:793-798. [DOI] [PubMed] [Google Scholar]
- 13.Fujiki, Y., A. L. Hubbard, S. Fowler, and P. B. Lazarow. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93:97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham, J. M. 2001. Isolation of mitochondria from tissues and cells by differential centrifugation, p. 3.3.1-3.3.15. In J. S. Bonifacino, M. Dasso, J. B. Harford, J. Lippincott-Schwartz, and K. M. Yamada (ed.), Current protocols in cell biology. Wiley Interscience, Hoboken, NJ. [DOI] [PubMed]
- 15.Graham, J. M. 2001. Isolation of nuclei and nuclear membranes from animal tissues, p. 3.10.1-3.10.19. In J. S. Bonifacino, M. Dasso, J. B. Harford, J. Lippincott-Schwartz, and K. M. Yamada (ed.), Current protocols in cell biology. Wiley Interscience, Hoboken, NJ. [DOI] [PubMed]
- 16.Graham, J. M. 2001. Purification of a crude mitochondrial fraction by density-gradient centrifugation, p. 3.4.1-3.4.22. In J. S. Bonifacino, M. Dasso, J. B. Harford, J. Lippincott-Schwartz, and K. M. Yamada (ed.), Current protocols in cell biology. Wiley Interscience, Hoboken, NJ. [DOI] [PubMed]
- 17.Guarente, L., and F. Picard. 2005. Calorie restriction—the SIR2 connection. Cell 120:473-482. [DOI] [PubMed] [Google Scholar]
- 18.Haigis, M. C., and L. P. Guarente. 2006. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20:2913-2921. [DOI] [PubMed] [Google Scholar]
- 19.Haigis, M. C., R. Mostoslavsky, K. M. Haigis, K. Fahie, D. C. Christodoulou, A. J. Murphy, D. M. Valenzuela, G. D. Yancopoulos, M. Karow, G. Blander, C. Wolberger, T. A. Prolla, R. Weindruch, F. W. Alt, and L. Guarente. 2006. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126:941-954. [DOI] [PubMed] [Google Scholar]
- 20.Hallows, W. C., S. Lee, and J. M. Denu. 2006. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 103:10230-10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim, S. C., R. Sprung, Y. Chen, Y. Xu, H. Ball, J. Pei, T. Cheng, Y. Kho, H. Xiao, L. Xiao, N. V. Grishin, M. White, X. J. Yang, and Y. Zhao. 2006. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23:607-618. [DOI] [PubMed] [Google Scholar]
- 22.Lagouge, M., C. Argmann, Z. Gerhart-Hines, H. Meziane, C. Lerin, F. Daussin, N. Messadeq, J. Milne, P. Lambert, P. Elliott, B. Geny, M. Laakso, P. Puigserver, and J. Auwerx. 2006. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 127:1109-1122. [DOI] [PubMed] [Google Scholar]
- 23.Lakso, M., J. G. Pichel, J. R. Gorman, B. Sauer, Y. Okamoto, E. Lee, F. W. Alt, and H. Westphal. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93:5860-5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Longo, V. D., and B. K. Kennedy. 2006. Sirtuins in aging and age-related disease. Cell 126:257-268. [DOI] [PubMed] [Google Scholar]
- 25.Lowell, B. B., and B. M. Spiegelman. 2000. Towards a molecular understanding of adaptive thermogenesis. Nature 404:652-660. [DOI] [PubMed] [Google Scholar]
- 26.Michishita, E., J. Y. Park, J. M. Burneskis, J. C. Barrett, and I. Horikawa. 2005. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16:4623-4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.North, B. J., B. L. Marshall, M. T. Borra, J. M. Denu, and E. Verdin. 2003. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11:437-444. [DOI] [PubMed] [Google Scholar]
- 28.Onyango, P., I. Celic, J. M. McCaffery, J. D. Boeke, and A. P. Feinberg. 2002. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 99:13653-13658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose, G., S. Dato, K. Altomare, D. Bellizzi, S. Garasto, V. Greco, G. Passarino, E. Feraco, V. Mari, C. Barbi, M. BonaFe, C. Franceschi, Q. Tan, S. Boiko, A. I. Yashin, and G. De Benedictis. 2003. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 38:1065-1070. [DOI] [PubMed] [Google Scholar]
- 30.Scher, M. B., A. Vaquero, and D. Reinberg. 2007. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 21:920-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwer, B., J. Bunkenborg, R. O. Verdin, J. S. Andersen, and E. Verdin. 2006. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 103:10224-10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwer, B., B. J. North, R. A. Frye, M. Ott, and E. Verdin. 2002. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 158:647-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi, T., F. Wang, E. Stieren, and Q. Tong. 2005. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 280:13560-13567. [DOI] [PubMed] [Google Scholar]
- 34.Taylor, S. W., E. Fahy, B. Zhang, G. M. Glenn, D. E. Warnock, S. Wiley, A. N. Murphy, S. P. Gaucher, R. A. Capaldi, B. W. Gibson, and S. S. Ghosh. 2003. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21:281-286. [DOI] [PubMed] [Google Scholar]
- 35.Taylor, S. W., D. E. Warnock, G. M. Glenn, B. Zhang, E. Fahy, S. P. Gaucher, R. A. Capaldi, B. W. Gibson, and S. S. Ghosh. 2002. An alternative strategy to determine the mitochondrial proteome using sucrose gradient fractionation and 1D PAGE on highly purified human heart mitochondria. J. Proteome Res. 1:451-458. [DOI] [PubMed] [Google Scholar]
- 36.Yang, Y. H., Y. H. Chen, C. Y. Zhang, M. A. Nimmakayalu, D. C. Ward, and S. Weissman. 2000. Cloning and characterization of two mouse genes with homology to the yeast Sir2 gene. Genomics 69:355-369. [DOI] [PubMed] [Google Scholar]
- 37.Yechoor, V. K., M. E. Patti, K. Ueki, P. G. Laustsen, R. Saccone, R. Rauniyar, and C. R. Kahn. 2004. Distinct pathways of insulin-regulated versus diabetes-regulated gene expression: an in vivo analysis in MIRKO mice. Proc. Natl. Acad. Sci. USA 101:16525-16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.