

Abstract
Members of the protein kinase C (PKC) family of signal transduction molecules have been widely implicated in regulation of cell growth and differentiation, although the underlying molecular mechanisms involved remain poorly defined. Using combined in vitro and in vivo intestinal epithelial model systems, we demonstrate that PKC signaling can trigger a coordinated program of molecular events leading to cell cycle withdrawal into G0. PKC activation in the IEC-18 intestinal crypt cell line resulted in rapid downregulation of D-type cyclins and differential induction of p21waf1/cip1 and p27kip1, thus targeting all of the major G1/S cyclin-dependent kinase complexes. These events were associated with coordinated alterations in expression and phosphorylation of the pocket proteins p107, pRb, and p130 that drive cells to exit the cell cycle into G0 as indicated by concomitant downregulation of the DNA licensing factor cdc6. Manipulation of PKC isozyme levels in IEC-18 cells demonstrated that PKCα alone can trigger hallmark events of cell cycle withdrawal in intestinal epithelial cells. Notably, analysis of the developmental control of cell cycle regulatory molecules along the crypt–villus axis revealed that PKCα activation is appropriately positioned within intestinal crypts to trigger this program of cell cycle exit–specific events in situ. Together, these data point to PKCα as a key regulator of cell cycle withdrawal in the intestinal epithelium.
Keywords: protein kinase C, cell cycle, intestinal mucosa, pocket proteins, cyclin-dependent kinase regulation
Introduction
Signaling pathways mediated by the protein kinase C (PKC) family of serine-threonine kinases are involved in the regulation of a wide variety of fundamental cellular processes, including cell growth and cell cycle progression, differentiation, and apoptosis (Nishizuka 1992; Dekker and Parker 1994; Fishman et al. 1998; Black 2000). The PKC family consists of at least 10 distinct isozymes (α, βΙ, βΙΙ, γ, δ, ε, η, θ, ζ, and ι) which share the same basic structure but differ with respect to activator and cofactor requirements, substrate specificity, tissue expression, and subcellular distribution (Dekker and Parker 1994). Phylogenetic conservation of individual PKCs and their widespread expression in different cell types underscore the central importance of members of this family in regulation of normal cellular functions. Furthermore, evidence of alterations in the expression/activity of PKC isozymes in neoplastic tissues (e.g., Guillem et al. 1987; Wali et al. 1991; Kahl-Rainer et al. 1996; Verstovsek et al. 1998), together with the identification of PKC as the major cellular receptor for tumor-promoting phorbol esters (e.g., PMA) (Castagna et al. 1982), suggests that disruption of the signaling pathways mediated by these molecules may contribute to uncontrolled cell growth and transformation.
Increasing evidence points to a role for PKC signaling in regulation of progression through the cell cycle, although the underlying molecular mechanisms remain unclear (Fishman et al. 1998; Black 2000). Control of G0/G1→S phase transit is accomplished primarily through regulation of the phosphorylation state of members of the pocket protein family of growth suppressor proteins, p107, pRb, and p130. In the hypophosphorylated (active) state, these molecules repress the activity of E2F transcription factors, which is required for expression of genes essential for DNA synthesis. In preparation for S phase entry, cyclin–cyclin-dependent kinase (cdk) complexes (cyclin D–cdk4/6 and cyclin E–cdk2) phosphorylate pRb, p107, and p130, inactivating their growth-suppressive function (Graña and Reddy 1995). An additional complex, cyclin A–cdk2, is critical for S phase entry/DNA synthesis, and is thought to maintain pocket protein phosphorylation during S phase (Li et al. 1993; Ludlow et al. 1993; Mayol et al. 1995). The activity of cyclin–cdk complexes is in turn regulated by cdk phosphorylation, expression and binding of cyclins, and association of cdk inhibitory proteins (CKIs) such as p21waf1/cip1 and p27kip1 (Cip/Kip family members) and p15 and p16 (Ink4 family members) (Pines 1995; Chellappan et al. 1998; Sherr and Roberts 1999). Pocket protein function has also been linked to regulation of cell cycle exit and onset of differentiation; cell cycle withdrawal into G0 involves coordinated regulation of pocket protein phosphorylation and expression, including hypophosphorylation of p107 and pRb, rapid downregulation of p107, and accumulation of the G0-specific phosphoforms 1 and 2 of p130 (Garriga et al. 1998; Graña et al. 1998). Emerging data from several studies implicate PKC isozyme(s) in either positive or negative regulation of G1→S progression, via alterations in the expression of cyclins and/or cdk inhibitors and modulation of the activity of specific cyclin–cdk complexes (Fishman et al. 1998; Black 2000).
In an effort to further understand the interplay between PKC signaling and regulation of the cell cycle machinery, our laboratory has used the intestinal epithelium as a model system. The unique architecture of this self-renewing tissue, with its well-defined regions of cell proliferation, differentiation, mature function, and senescence, has enabled correlation of changes in the expression and activation of PKC isozymes with specific stages of development. Using a combined morphological and biochemical approach, we have determined that several PKC isozymes (α, βΙΙ, δ, and ζ) are activated precisely at the point within intestinal crypts at which cells cease dividing (Saxon et al. 1994), suggesting that one or more of these molecules are involved in negative regulation of cell growth in this system. Consistent with these findings, direct activation of PKC (α, δ, and ε) in the nontransformed IEC-18 immature intestinal crypt cell line resulted in cell cycle arrest in G0/G1 phase (Frey et al. 1997). Differential downmodulation of individual PKC isozymes indicated that PKCα, in particular, is sufficient to inhibit cell cycle progression in this system. PKC-mediated cell cycle arrest in G0/G1 was shown to involve induction of Cip/Kip family cdk inhibitors and hypophosphorylation/activation of pRb, thus linking PKCα to control of cdk activity and the growth-suppressive function of pRb in the intestinal epithelium.
This study uses in vitro and in vivo model systems to investigate further the cell cycle–specific effects of PKC signaling in intestinal epithelial cells. The data demonstrate for the first time that PKC signaling can initiate a coordinated program of cell cycle withdrawal into G0 and that PKCα is sufficient to initiate this program of cell cycle exit–specific effects. Similar changes in the regulation of cell cycle regulatory molecules were observed coincident with PKC activation in the midcrypt region in situ, underscoring the physiological relevance of these findings. Together, the data implicate members of the PKC family as key regulators of cell cycle withdrawal in the midcrypt region of the intestinal epithelium.
Materials and Methods
Antibodies
Monoclonal anti–cyclin D1, polyclonal rabbit anti-p15, -p16, -cdk4, –cyclin A, –cyclin D (pan), –cyclin E, -pRb, -p107, -p130, -PKCδ, and -PKCε, and polyclonal goat anti-p15 and -cdk2 antibodies were obtained from Santa Cruz Biotechnology, Inc. Monoclonal against 5′-bromo-2′-deoxyuridine (BrdU) was purchased from Dako. mAbs recognizing p21waf1/cip1 and pRb were purchased from BD PharMingen, mABs anti-p27kip1 was obtained from BD Transduction Labs, and monoclonal anti–cyclin D1 was purchased from Sigma-Aldrich. Polyclonal and monoclonal PKCα-specific antibodies were purchased from GIBCO BRL and Upstate Biotechnology, respectively. HRP-conjugated goat anti–rabbit IgG and donkey anti–goat IgG antibodies, and unconjugated anti–green fluorescent protein (GFP) mAb were obtained from Boehringer. HRP-conjugated rat anti–mouse IgG, TRITC-conjugated donkey anti–rabbit IgG, and TRITC-conjugated goat anti–mouse IgG antibodies were purchased from Jackson ImmunoResearch Laboratories. TRITC-conjugated donkey anti–goat IgG and unconjugated goat anti–mouse IgG were obtained from Accurate Chemical & Scientific Corp.
Identification of Proliferating Crypt Cells In Situ
To label proliferating intestinal epithelial cells in S phase, an aqueous solution of 120 mg/kg of the thymidine analogue BrdU (Sigma-Aldrich) was injected intraperitoneally into Sprague-Dawley (CD) rats (Charles River Laboratories) 2 h before killing. Paraffin sections of formalin-fixed duodenum were heated (95°C) in 10 mM citrate buffer, pH 6.0, for 20 min for antigen retrieval, blocked in PBS containing 0.03% casein and 0.05% Tween 20, and immunostained with anti-BrdU antibody (1:10 dilution), followed by unconjugated goat anti–mouse secondary antibody (1:100 dilution) and HRP-conjugated donkey anti–goat tertiary antibody (1:100 dilution). Bound peroxidase was detected by incubation in 0.1% diaminobenzidine and 0.01% H2O2, and sections were counterstained with Harris hematoxylin.
Immunofluorescence Staining
Duodenal tissue from 200-g male Sprague-Dawley (CD) rats (Charles River Laboratories) was removed and flushed with ice-cold PBS. Tissue used for p15, p16, cyclin D1, and cdk2 immunostaining was immediately frozen in liquid nitrogen–cooled 2-methylbutane. Sections (4–6 μm) were cut on a cryostat microtome (Reichert-Jung), thaw-mounted onto gelatin-coated coverslips, and fixed in 2% formaldehyde/PBS for 15 min. Tissue used for p21waf1/cip1, p27kip1, cdk4, cyclin A, cyclin E, p107, pRb, p130, and PKCα immunostaining was fixed immediately after removal in 2% freshly depolymerized paraformaldehyde/PBS for 2 h at 4°C, washed three times (15 min) in 50 mM NH4Cl/PBS, cryoprotected in 30% sucrose/PBS for at least 5 h, and frozen as described above.
Nonspecific binding of antibodies to tissue sections was blocked by preincubation in blocking buffer (PBS containing 0.03% casein and 0.05% Tween 20) for 30 min at room temperature. Sections were then immunostained essentially as described previously (Saxon et al. 1994). In brief, sections were incubated with primary antibody in PBS containing 0.2% Triton X-100 (Sigma-Aldrich) for 60 min, followed by a 15-min PBS/Triton wash, 40 min in secondary antibody in PBS/Triton, and a 15-min PBS wash. Primary antibody dilutions were 1:10 for anti–cyclin D1, 1:25 for anti-cdk2 and -p21waf1/cip1, 1:50 for anti-p27kip1, 1:100 for anti-PKCα, -p16, –cyclin A, –cyclin E, and -p130, 1:200 for anti-cdk4, -p107, and -pRb, and 1:400 for anti-p15. TRITC-conjugated secondary antibodies were used at 1:100 dilution. Fluorescence was viewed with a ZEISS epifluorescence microscope equipped with the appropriate optics and filter modules.
Cell Culture, PKC Activation Protocols, and Subcellular Fractionation
The IEC-18 cell line (American Type Culture Collection), a nontransformed intestinal crypt cell line derived from rat ileal epithelium (Quaroni and May 1980), was maintained in DME (GIBCO BRL) supplemented with 10 μg/ml insulin, 4 mM glutamine, and 5% FCS (Intergen). PKC isozymes were activated in these cells by treatment with a panel of PKC activators including 100 nM PMA (Sigma-Aldrich), 100 nM 12-deoxyphorbol 13-phenylacetate (dPP; Alexis Corp.), 58 μM 1,2-dioctanoyl-sn-glycerol (DiC8; Sigma-Aldrich), 100 nM phorbol 12,13-dibutyrate (PDBu; Alexis Corp.), 200 nM 12-deoxyphorbol 13-phenylacetate 20-acetate (dPPA; Alexis Corp.), 20 nM thymeleatoxin (Thy; Alexis Corp.), or 10 μM resiniferatoxin (Res; Alexis Corp.).
Construction of a Retroviral Vector for Expression of PKCα-GFP Fusion Protein in IEC-18 Cells
To generate a retroviral vector for expression of PKCα fused at its COOH terminus to the NH2-terminal end of GFP, PKCα cDNA (American Type Culture Collection) was excised from pBluescript with EcoR1 and ligated into the EcoR1 sites of EGFP-N1 (CLONTECH Laboratories, Inc.). After correct orientation of the cDNA was confirmed, the stop codon of PKCα was mutated to an alanine codon by PCR-based mutagenesis (Cormack 1997), using the primers 5′-ATCCTTGTCCAAGGAGGCTGT-3′ and 3′-TGGATCCACTGCACTCTGTAAGAT-5′ to introduce the mutation. Presence of the required alteration and absence of other PCR-induced mutations was confirmed by sequencing. The PKCα-GFP fusion cDNA was excised from this construct by consecutive digestion with Not1 and EcoR1. Between these digestions, the Not1 cut was blunt-ended using the Klenow fragment of DNA polymerase. The cDNA was ligated into the EcoR1 and blunt-ended BamH1 sites of pLXSN (CLONTECH Laboratories, Inc.). This process generated the pLXSN-PKCα-GFP plasmid in which expression of PKCα-GFP fusion protein is under control of the Moloney murine leukemia virus long terminal repeat.
Transduction of IEC-18 Cells with PKCα-GFP
For packaging of retroviral particles, the Phoenix amphotropic cell line (American Type Culture Collection) was transfected with pLXSN or pLXSN-PKCα-GFP using the calcium phosphate precipitation method (Chen and Okayama 1987). Cells were exposed to precipitate for 12–18 h in the presence of 25 μM chloroquine before fresh medium was applied and cells were transferred to a 32°C incubator. After 24 h, medium containing retroviral particles was collected, cellular debris was removed by 0.45 μM filtration, and retroviral supernatants were stored at −70°C until use.
IEC-18 cells were plated at 5–6 × 105/100-mm dish. After 24 h, medium was removed and 3 ml of viral supernatants were applied in the presence of 7 ml complete medium and 4 μg/ml polybrene. Cells were exposed to retrovirus for 24 h at 37°C, 5% CO2, and then subcultured in complete medium at a 1:4 ratio. Geneticin (G418; GIBCO BRL) was added at 2 mg/ml for 4 d to select transduced cells. Cells expressing high levels of PKCα-GFP were collected by fluorescence-activated cell sorting on the basis of GFP fluorescence. The brightest 3% of cells were collected and expanded for use in experiments.
Flow Cytometric Analysis
Propidium iodide staining of cellular DNA was performed and quantified as described previously (Frey et al. 1997). In brief, cells were fixed in 70% ethanol and treated with 0.04 mg/ml RNase A (Sigma-Aldrich) in 20 mM Tris, pH 7.5, 250 mM sucrose, 5 mM MgCl2, and 0.37% NP-40 (Sigma-Aldrich). Cellular DNA was stained with 25 μg/ml propidium iodide (Sigma-Aldrich) in 0.05% sodium citrate and quantified by flow cytometry. Cell cycle analysis was performed using the Winlist and Modfit programs (Verity Software House). Doublets were gated out based on propidium iodide fluorescence pulse-width, but all other events (including any in the sub-2N region) were retained for analysis.
Rat Intestinal Epithelial Cell Isolation
Epithelial cells at different developmental stages were obtained by sequential release from rat intestine by timed incubations with EDTA-containing buffer as described previously (Weiser 1973; Saxon et al. 1994). Upper villus (fraction 1), low-to-mid villus (fraction 5), and crypt cells (fraction 9) were collected and washed twice with ice-cold PBS before further processing. Crypt cells were obtained as pouches of short columnar epithelium, consisting mainly (at least 60–70%) of proliferating (lower crypt) cells (Bach et al. 2000), but including some differentiating (upper crypt) cells. Isolated low-to-mid villus epithelium contained differentiating as well as some functionally mature cells, and upper villus fractions contained only functionally mature cells. Each epithelial fraction was characterized as described previously (Weiser 1973; Burgess, 1989).
Preparation of Whole Cell Lysates and Immunoprecipitates
For preparation of whole cell lysates, cell populations were solubilized in boiling SDS lysis buffer (10 mM Tris, pH 7.4, 1% SDS). Extracts were cleared by centrifugation (10 min, 10,000 g) and boiled in Laemmli sample buffer (Laemmli 1970) before being subjected to SDS-PAGE and Western blot analysis. For immunoprecipitation experiments, subconfluent IEC-18 cells were lysed in modified RIPA buffer (50 mM Tris, pH 7.5, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.5 mM Na3VO4, 50 mM NaF, 5 mM NaPPi, 10 mM β-glycerophosphate, 0.1 mM PMSF, 1 μg/ml leupeptin, and 1 μg/ml aprotinin). Lysates were cleared by centrifugation and diluted 1:1 with PBS to reduce detergent concentration. Immunoprecipitations (500 μg cellular protein per reaction) were performed overnight at 4°C with 2 μg anti–cyclin A, D, or E antibody followed by 60 min with an excess of protein A/G agarose beads (Santa Cruz Biotechnology, Inc.). Immune complexes were collected by centrifugation, washed five times in modified RIPA buffer, and either boiled in Laemmli sample buffer for Western blot analysis or used in kinase assays.
Western Blot Analysis
SDS-PAGE and Western blot analysis were performed as described previously (Saxon et al. 1994; Frey et al. 1997), using either 10% (PKC isozymes, pRb, p107, and p130) or 20% (cdks, cyclins, and CKIs) SDS-polyacrylamide minigels. 30 μg of protein, assayed in quadruplicate by the bicinchoninic acid (BCA) method (Pierce Chemical Co.), was loaded per lane, and blots were routinely stained with either 0.1% Fast green (Sigma-Aldrich) or 1% Amido black (Sigma-Aldrich) immediately after transfer to ensure equal loading and even transfer. Primary antibody dilutions were as follows: 1:500 for anti-Rb, –cyclin D1, and -p27kip1; 1:1,000 for anti-p15, -p16, -p21waf1/cip1, -p107, -p130, -cdk2, -cdk4, -PKCδ, -GFP, and –cyclin A; and 1:2,000 for anti–cyclin E, –cyclin D (pan), -PKCα, and -PKCε. Secondary antibodies were used at 1:2,000.
Immune Complex Kinase Assays
Immunoprecipitates were washed in RIPA buffer, resuspended in 40 μl kinase assay buffer (25 mM Hepes, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 5 mM MnCl2, 10% glycerol, 0.5 mM Na3VO4, 50 mM NaF, 5 mM NaPPi, 10 mM β-glycerophosphate), combined with substrate (2 μg GST-pRb; Santa Cruz Biotechnology, Inc.) or 10 μg purified histone H1 (Upstate Biotechnology) and 10 μCi [γ-32P]ATP, and incubated for 20 min at 30°C. Reaction products were separated by SDS-PAGE and dried gels were exposed to x-ray film. Autoradiograms were analyzed using a densitometer and ImageQuant Software (Molecular Dynamics).
Selective Downregulation of PKCδ and -ε from IEC-18 cells
Subconfluent IEC-18 cells were treated with 100 nM PMA for 15 min, rinsed twice in DME, and returned to complete medium for 24 h. This procedure has previously been demonstrated to selectively downregulate PKCδ and -ε, producing a population of cells expressing PKCα as the major agonist-responsive PKC isozyme (Frey et al. 1997).
Results
Previous results from this laboratory have established a strong link between PKC isozyme signaling and control of cell growth/cell cycle progression in intestinal epithelial cells. Several members of the PKC family (α, βΙΙ, δ, and ζ) undergo marked changes in expression and membrane association, parameters indicative of PKC activation, precisely as cells cease division in the crypts of the small intestine and colon in situ (Fig. 1, a–c) (Saxon et al. 1994; Verstovsek et al. 1998). In addition, direct activation of PKC (α, δ, and ε) in the IEC-18 intestinal crypt cell line with a panel of PKC agonists, including phorbol esters (PMA, dPP, PDBu, dPPA, Thy, and Res) and the diacylglycerol analogue DiC8, results in cell cycle arrest in G0/G1 (Table and Fig. 1 d) (Frey et al. 1997). Depletion of phorbol ester–responsive PKC isozymes (α, δ, and ε) by long-term treatment with PDBu abrogates the growth-inhibitory effects of these agents, pointing to PKC as a major mediator of this response (Frey et al. 1997). Furthermore, the duration of the effect is directly related to the presence of membrane-associated/active PKC in the cells. Treatment of asynchronously growing IEC-18 cells with PMA, which initially activates but subsequently downregulates PKCα, -δ, and -ε in this system, results in a transient cell cycle blockade (Fig. 1d and Fig. e) (Frey et al. 1997). Reversal of growth arrest at 12–14 h after addition of PMA correlates with depletion of PMA-responsive PKC isozymes, which occurs between 6 and 12 h of treatment (Fig. 1 e) (Frey et al. 1997). In contrast, treatment with agents such as DiC8 (Frey et al. 1997) and Res (Frey, M.R., J.A. Clark, and J.D. Black, unpublished data), which activate PKC isozymes in a sustained manner, results in sustained cell cycle arrest in these cells.
Figure 1.
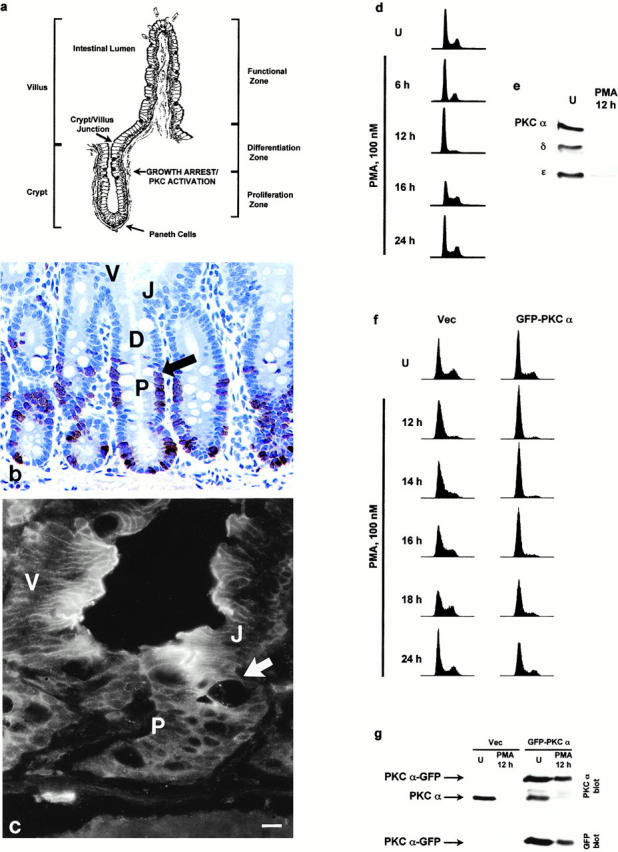
Relationship between PKC signaling and growth arrest in intestinal epithelial cells. (a) Diagram of intestinal epithelial crypt–villus architecture, indicating the point of growth arrest/PKC activation in the midcrypt region and the crypt–villus junction. (b) Immunodetection of BrdU incorporation to identify proliferating cells in small intestinal crypts. Arrow indicates point of growth arrest. P, proliferation zone; D, differentiation zone; V, villus/functional zone; J, crypt–villus junction. (c) Immunofluorescence localization of PKCα in rat duodenum. PKCα is diffusely distributed throughout the cytosol of proliferating lower crypt cells (P). Coincident with growth arrest in the midcrypt region (cell position 14–18 from the crypt base; arrow), levels of PKCα markedly increase and the protein becomes clearly detectable at the cell periphery, in the classical indication of PKC activation. (d) IEC-18 cells were treated with 100 nM PMA for the indicated times (U, control), and DNA content/cell cycle distribution was determined by flow cytometric analysis. PMA-induced cell cycle arrest is held for ∼12 h of treatment. (e) IEC-18 cells were treated with 100 nM PMA for 12 h and analyzed for expression of PKC isozymes by Western blot analysis. U, untreated. (f) Vector control (Vec) and PKCα-GFP–expressing IEC-18 cells were treated with 100 nM PMA for the indicated times, and cell cycle distribution was determined by flow cytometric analysis. Although vector control cells reenter S phase by 12–14 h, PKCα-GFP–expressing cells remain arrested until between 18 and 24 h. (g) Vector control and PKCα-GFP cells were treated with 100 nM PMA for 12 h and subjected to Western blot analysis using antibodies against PKCα and GFP. Note that, although endogenous PKCα is undetectable in vector-transduced cells at 12 h, PKCα-GFP–transduced cells still contain readily detectable, albeit diminished, levels of the fusion protein at this time point. Data are representative of at least four independent experiments (results of PKCα-GFP overexpression are representative of four separate transductions). Bar, 10 μm.
Table 1.
PKC Agonists Produce Cell Cycle Arrest in IEC-18 Cells
| Condition | G0/G1 | S | G2/M |
|---|---|---|---|
| Control | 58 | 30 | 12 |
| PMA 100 nM | 69 | 8 | 23 |
| dPP 100 nM | 69 | 9 | 22 |
| dPPA 200 nM | 67 | 15 | 18 |
| Thy 20 nM | 68 | 10 | 22 |
| Res 10 μM | 67 | 10 | 23 |
| DiC8 58 μM | 65 | 15 | 20 |
Asynchronously growing IEC-18 cells were treated with PKC agonists for 6 h and subjected to flow cytometric analysis to determine cell cycle distribution. PKC activation resulted in a marked decrease in cells transiting through S phase.
Previous studies using differential downmodulation of individual PKC isozymes have pointed to PKCα as being sufficient to mediate PMA-induced transient cell cycle blockade (Frey et al. 1997). To confirm the involvement of this isozyme in phorbol ester–induced cell cycle arrest, IEC-18 cells were retrovirally transduced to express a PKCα-GFP fusion protein, and effects of PMA on cell cycle progression and PKC expression were examined. Expression of PKCα-GFP significantly extended (∼6 h longer) phorbol ester–induced cell cycle arrest in this system (Fig. 1 f), and this effect correlated with increased (two- to threefold) and prolonged expression of PKCα proteins in PKCα-GFP–transduced cells (Fig. 1 g). Expression of other PKC isozymes (δ, ε, ζ, and ι) was unaltered in PKCα-GFP–expressing cells, and phorbol ester–induced downregulation of PKCδ/ε followed the same kinetics as seen in vector control cells (data not shown). Taken together with our previous findings, these data directly support the involvement of member(s) of the PKC family, and of PKCα in particular, in induction and maintenance of cell growth/cell cycle arrest in intestinal epithelial cells.
To investigate further the mechanisms involved in PKC-mediated inhibition of cell cycle progression in intestinal epithelial cells, PKC-induced alterations in the expression and activity of critical cell cycle regulatory molecules were determined in IEC-18 cells. The physiological relevance of these changes was then evaluated by comparing the developmental regulation of PKC isozymes and cell cycle control molecules along the crypt–villus axis in the unperturbed intestinal epithelium in situ.
PKC Signaling Regulates Pocket Protein Expression and Phosphorylation State in Intestinal Epithelial Cells
To investigate the role of pocket proteins in PKC-mediated cell cycle arrest in IEC-18 cells, the effects of PKC activation on the expression and phosphorylation state of these molecules were determined using Western blot analysis. This analysis was based on well-documented evidence that alterations in phosphorylation state of pocket proteins are reflected in characteristic changes in their migration patterns on SDS-PAGE gels (Ludlow et al. 1990; DeCaprio et al. 1992; Whyte and Eisenman 1992; Garriga et al. 1998; Smith et al. 1998; Thomas et al. 1998). As shown in Fig. 2, proliferating (untreated) IEC-18 cells express p107, pRb, and small amounts of p130; p107 and pRb are detected primarily in their hyperphosphorylated (i.e., slower-migrating), growth-permissive forms. Treatment with 100 nM PMA resulted in decreased expression and hypophosphorylation (i.e., appearance of characteristic faster-migrating forms) of p107 and pRb, and a marked accumulation of the faster-migrating forms 1/2 of p130 (which is characteristic of cell cycle exit into G0; see Mayol et al. 1995). These PMA-induced alterations were transient, reversing by 12–16 h after addition of agonist, coincident with release from cell cycle arrest as a consequence of PKC downregulation (see Fig. 1d and Fig. e).
Figure 2.

Altered expression and phosphorylation of the pocket proteins p107, pRb, and p130 after PKC activation in IEC-18 cells. IEC-18 cells were treated with 100 nM PMA for the indicated times (U, untreated), and expression and phosphorylation state of pocket proteins were determined by Western blot analysis. Altered phosphorylation of these molecules is reflected in characteristic changes in their migration patterns on SDS gels. p130 is detected as forms 1, 2, and 3; the accumulation of forms 1/2 after PMA treatment is a hallmark of cell cycle withdrawal into G0. Data are representative of at least three independent experiments.
To examine the physiological relevance of these data, a combined biochemical and morphological approach (described previously in Saxon et al. 1994) was used to compare the developmental regulation of pocket protein expression, subcellular distribution, and phosphorylation state along the crypt–villus axis in situ with changes in PKC activation/expression. Immunofluorescence analysis revealed that p107, pRb, and low levels of p130 are expressed in proliferating crypt cells (Fig. 3, a–c): p107 staining was predominantly nuclear, whereas pRb staining was detected both in the cytoplasm and the nucleus. Coincident with cell growth arrest and PKC activation in the midcrypt region (cell position 14–18 from the crypt base; Fig. 1b and Fig. c), all three pocket proteins underwent changes in expression and/or subcellular compartmentalization. p107 became barely detectable in postmitotic cells, whereas staining for pRb became restricted to the nucleus. Levels of p130, on the other hand, increased markedly with growth arrest and remained elevated on the villus, decreasing somewhat towards the villus tip. Consistent with these immunofluorescence data, Western blot analysis of pocket protein expression in isolated crypt, lower villus, and upper villus cells demonstrated marked decreases in levels of p107 and pRb in villus cells relative to crypt cells (Fig. 3 d). Similar levels of p130 were detected in the crypt and lower villus fractions, likely reflecting the presence of postmitotic upper crypt cells in the crypt sample and of some midvillus cells in the lower villus fraction (see Materials and Methods). Western blot analysis also demonstrated that pRb and p130 are primarily expressed in their underphosphorylated forms in postmitotic cells of the villus. Comparison of the data obtained from in vitro and in situ studies (see Fig. 2 above) demonstrates that direct activation of PKC isozymes in IEC-18 cells leads to alterations in pocket protein expression and phosphorylation state that closely parallel those seen coincident with PKC activation within intestinal crypts in situ. Notably, the data demonstrate that PKC activation in this system can initiate a coordinated program of pocket protein regulation that has been associated with cell cycle exit in several systems (Garriga et al. 1998).
Figure 3.
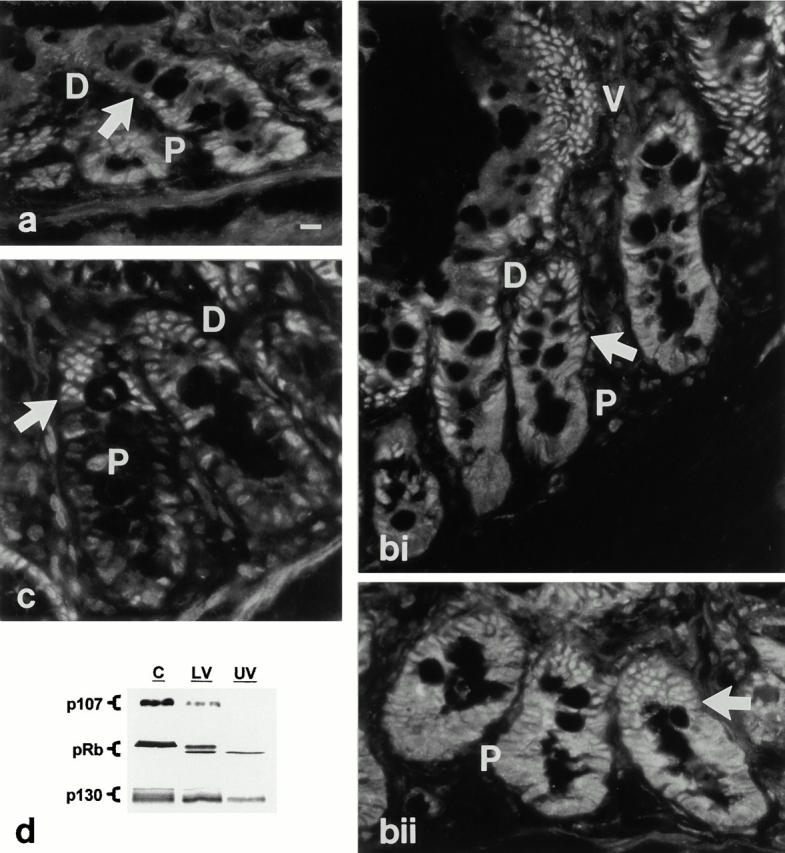
Developmental regulation of pocket protein expression and phosphorylation state in the intestinal epithelium in situ. (a–c) Immunofluorescence localization of pocket proteins. P, proliferation zone; D, differentiation zone; V, villus/functional zone. (a) p107 is readily detected in the nuclei of proliferating lower crypt cells (P). Coincident with growth arrest (arrow), p107 expression decreases to barely detectable levels. (bi and bii) pRb staining is evident in both nuclear and cytosolic compartments of proliferating crypt cells (P), and becomes predominantly nuclear in postmitotic cells of the upper crypt and villus (V). The arrow indicates the point of growth arrest. (c) p130 staining is low in proliferating crypt cells (P), but increases markedly coincident with growth arrest (arrow). (d) Whole cell lysates (30 μg protein) of isolated crypt (C), lower villus (LV), and upper villus (UV) cells were subjected to Western blot analysis for expression and migration/phosphorylation state of pocket proteins. Note that the crypt fraction includes some postmitotic cells of the upper crypt region. p130 form 3, which is only found in cycling cells, is only detected in the crypt fraction; the presence of forms 1 and 2 in this fraction reflects the postmitotic cells in this sample. Data are representative of at least three independent experiments. Bar, 10 μm.
PKC Activation in IEC-18 Cells Results in Decreased Cdk Activity
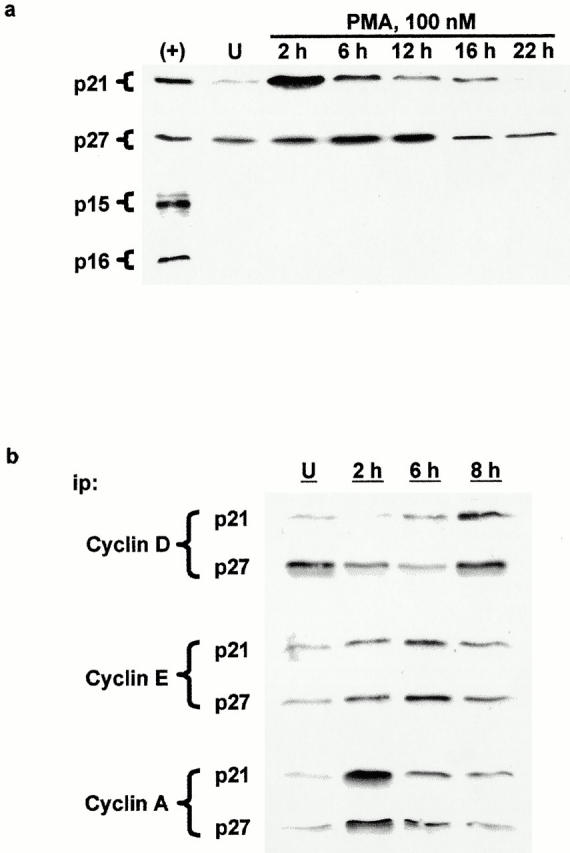
To examine the mechanisms underlying PKC-induced alterations in pocket protein phosphorylation, cyclin D–, E–, and A–cdk complexes were immunoprecipitated from control and PMA-treated cells, and their activity was determined in immune complex kinase assays (Fig. 4). Consistent with findings in other intestinal epithelial model systems (Tian and Quaroni 1999), cyclin D–associated kinase activity could not be detected in IEC-18 cells (data not shown), possibly as a result of the low levels of cyclin D expressed in these cells. On the other hand, kinase activity was readily detectable in cyclin E and cyclin A immunoprecipitates from untreated cells (Fig. 4), and the activity in both complexes was markedly inhibited after PMA treatment. Cyclin E–associated activity was decreased by 46% at 2 h and by 73% at 6 h. By 8 h, although still inhibited by 48%, cyclin E–cdk2 activity was beginning to rebound. Cyclin A–associated activity was decreased by 55% within 2 h of treatment, and was below the accurate detection limit of the assay at 6 and 8 h (see Fig. 5).
Figure 4.

PKC activation inhibits cyclin E– and cyclin A–associated kinase activity in IEC-18 cells. (a) Histone H1 kinase activity in cyclin E and cyclin A immunoprecipitates was determined by in vitro kinase assays and SDS-PAGE autoradiography as described in Materials and Methods. Results shown are representative of three independent assays. n/a, not applicable. (b) Bar graphs depict average activity in three replicate samples as determined by densitometric analysis of autoradiograms. U, untreated.
Figure 5.

Effect of PMA treatment on the expression of cyclins and cdks in IEC-18 cells. Cells were exposed to 100 nM PMA for the indicated times (U, untreated) and subjected to Western blot analysis using antibodies specific for cdk4, cdk2, and cyclins D (pan), D1, E, and A. Data are representative of three independent experiments.
Effects of PKC Activation on Cyclin–Cdk Expression Levels in Intestinal Epithelial Cells
Cdk activity can be affected by three major factors: expression of cdks and cyclins, positive and negative phosphorylation events, and association with CKIs (Chellappan et al. 1998; Sherr and Roberts 1999). To determine whether changes in expression or phosphorylation of cdks or cyclins could account for PKC-mediated inhibition of cdk activity in IEC-18 cells, Western blot analysis of whole cell lysates from control and PMA-treated IEC-18 cells was performed. As shown in Fig. 5, no detectable changes were observed in the expression or electrophoretic mobility (phosphorylation state) of cdks 4 and 2 in IEC-18 cells over the course of PMA treatment. In contrast, alterations in cyclin expression were observed after PKC activation in IEC-18 cells. The most rapid changes were seen with the D-type cyclins. As shown in Fig. 5, exposure to 100 nM PMA consistently resulted in depletion of cyclin D/D1 by 2 h of treatment. Expression of these molecules began to rebound by 6 h and, interestingly, reached markedly higher levels than those seen in untreated cells. Unlike the D-type cyclins, cyclins E and A were both readily detectable in proliferating IEC-18 cells. Although cyclin E expression changed little with PKC activation, cyclin A levels decreased markedly between 2 and 6 h of PMA treatment, returning to control levels by 16 h (Fig. 5). Notably, the changes in cyclin A expression occurred well after the observed decreases in associated cdk activity (by 2 h; see Fig. 4). Thus, although the depletion of D cyclins would be expected to result in loss of cdk4/6 activity, PKC-induced inhibition of cyclin E– and A–associated kinase activity (at early times) cannot be explained by decreased expression of either cdk or cyclin subunits.
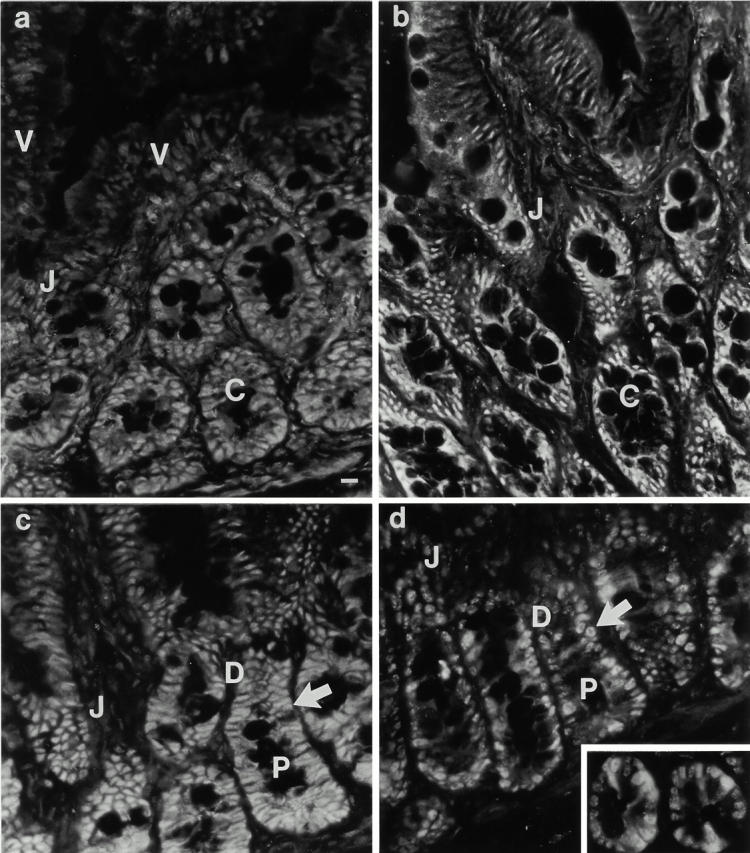
Immunofluorescence analysis of cdk and cyclin expression in the intestinal epithelium in situ revealed additional elements of the relationship between PKC signaling and cell cycle regulation, and framed the data obtained in IEC-18 cells in a physiological context. Although cyclin D/D1 was detected in proliferating crypt cells (data not shown), the developmental control of D-type cyclins along the crypt–villus axis could not be precisely discerned due to the weak signal produced by pan-D and anti–cyclin D1 antibodies in this tissue. Cdks 4 and 2 and cyclins E and A were all detected in the nuclei of proliferating intestinal crypt cells (Fig. 6, a–d). Levels of cyclin A expression varied from cell to cell, presumably reflecting different stages of the cell cycle (Fig. 6 d). Notably, major changes in the expression of these cyclins and cdks were observed only at the crypt–villus junction, well beyond the point of growth arrest and PKC activation in the midcrypt region. As cells exited the crypts, expression of cdks 4 and 2 and cyclins E and A decreased markedly, although low levels of these molecules remained detectable along the length of the villus. The regulation of cyclin and cdk expression in proliferating, differentiating, and functional intestinal epithelial cells was confirmed by Western blot analysis of isolated cell populations (Fig. 6 e). Together, these data suggest that decreased expression of cdks 4 and 2 and cyclins E and A in intestinal tissue is a downstream consequence rather than a cause of the accumulation of cells in G0/G1, and that the loss of cyclin E– and A–associated cdk activity observed shortly after PMA treatment of IEC-18 cells is not due to limited cyclin or cdk availability.
Figure 6.
Analysis of the developmental regulation of cdk and cyclin expression in the small intestinal epithelium. Immunofluorescence analysis of (a) cdk4, (b) cdk2, (c) cyclin E, and (d) cyclin A expression was performed on rat duodenal tissue. All four molecules are readily detectable in crypt cells (C), and undergo a marked decrease in expression at the crypt–villus junction (J), well beyond the point of growth arrest (arrow). Note the variable expression of cyclin A in proliferating cells (d, inset). P, proliferation zone; D, differentiation zone; V, villus. (e) Western blot analysis of cdk and cyclin expression in isolated crypt (C), lower villus (LV), and upper villus (UV) cell populations. Data are representative of at least three independent experiments. Bar, 10 μm.

PKC Signaling Regulates the Expression of Cip/Kip but Not Ink4 Family CKIs in Intestinal Epithelial Cells
Previous studies in this laboratory have demonstrated that PKC agonist treatment of IEC-18 cells results in induction of the Cip/Kip CKIs p21waf1/cip1 and p27kip1. This report extends these findings by (a) examining the extent and time course of p21waf1/cip1 and p27kip1 induction, (b) determining the association of p21waf1/cip1 and p27kip1 with cyclin–cdk complexes, and (c) expanding the analysis to include the Ink4 CKIs p15 and p16. As shown in Fig. 7 a, a substantial accumulation of p21waf1/cip1 was observed in IEC-18 cells by 2 h of PMA treatment, with levels remaining elevated for 6 h and returning to baseline by 12 h. The increased expression of p27kip1 exhibited delayed kinetics relative to p21waf1/cip1 induction, with peak levels sustained between 6 and 12 h of PMA treatment. (It should be noted that the level of induction of p27kip1 exhibited some variability at early time points, but was consistently high at later times.) In contrast to the Cip/Kip CKIs, the Ink4 CKIs p15 and p16 were not detected in either proliferating or PMA-arrested IEC-18 cells. Thus, PKC-mediated intestinal epithelial cell cycle arrest appears to involve specific induction of Cip/Kip CKIs, and does not require participation of Ink4 family members (Fig. 7 a).
Figure 7.
PKC activation in IEC-18 cells results in accumulation of Cip/Kip, but not Ink4, CKIs in cyclin–cdk complexes. (a) IEC-18 cells were exposed to 100 nM PMA for the indicated times (U, untreated) and examined for CKI expression by Western blot analysis (+, intestinal lower villus cell lysate as positive control). (b) Cyclin–cdk complexes were immunoprecipitated (ip) from control and PMA-treated IEC-18 cells using anti–cyclin D, E, and A antibodies and analyzed for p21waf1/cip1 and p27kip1 expression by Western blotting. Data are representative of at least three independent experiments.
To determine if PKC activation in IEC-18 cells resulted in increased association of Cip/Kip CKIs with cyclin–cdk complexes, immunoprecipitates were prepared from control and PMA-treated cells using antibodies directed against cyclin D, cyclin E, or cyclin A, and immunocomplexes were analyzed for the presence of p21waf1/cip1 and p27kip1 by Western blotting. Markedly increased levels of CKIs were detected in cyclin E and cyclin A immunocomplexes by 2 h (Fig. 7 b). CKI levels in cyclin E complexes peaked at 6 h and began to decline by 8 h, whereas those in cyclin A complexes decreased sharply by 6 h, paralleling the loss of cyclin A expression at this time point (see Fig. 5). Increased association of p21waf1/cip1 and p27kip1 with cyclin D was not observed at early times after addition of PMA, presumably reflecting the initial downregulation of D-type cyclins resulting from this treatment (see Fig. 5). However, significantly elevated levels of CKIs were seen in cyclin D complexes at later times, paralleling the resurgence of cyclin D seen with PKC downregulation. Interestingly, high levels of CKI recruitment to cyclin D immunocomplexes at 8 h were coincident with decreased association of p21waf1/cip1 and p27kip1 with cyclins E and A and restoration of cyclin E–cdk2 activity. The sequestration of these CKIs by cyclin D–cdk4/6 complexes has been suggested by some investigators to be a mechanism for relief of cdk2 inhibition, contributing to orderly progression through late G1 phase (Geng et al. 1999; Roberts 1999).
Immunofluorescence analysis revealed that PKC-mediated control of CKI expression in IEC-18 cells parallels CKI regulation along the crypt–villus axis in situ. Relatively low levels of p21waf1/cip1 were observed in the nuclei of proliferating lower crypt cells, whereas p27kip1 was undetectable in these cells (Fig. 8, a and b). Coincident with growth arrest, upregulation/activation of PKC, and alterations in pocket protein expression/phosphorylation, levels of both Cip/Kip CKIs increased markedly. Upregulation of p21waf1/cip1 was transient; expression remained high in the actively differentiating cells of the upper crypt and lower villus and decreased to barely detectable levels in functional cells of the upper villus. p27kip1 expression, on the other hand, remained relatively high in the entire postmitotic compartment. In contrast, changes in p15 and p16 expression did not correlate with growth arrest and PKC activation in the midcrypt region (Fig. 8c and Fig. d). Moderate levels of p15 and low amounts of p16 were detected in cells throughout the entire crypt. Increased p15 expression became apparent at the crypt–villus junction, which is well into the postmitotic compartment (Fig. 8 c), and levels of this molecule remained high along the length of the villus. Levels of p16, on the other hand, increased gradually with cell migration up the villus (Fig. 8 d). These patterns of Cip/Kip and Ink4 family CKI regulation along the crypt–villus unit were consistent with those observed by Western blot analysis of isolated crypt, lower villus, and upper villus cells (Fig. 8 e). Thus, upregulation of p21waf1/cip1 and p27kip1 expression in the midcrypt region coincides precisely with changes in PKC expression/activation, alterations in pocket protein expression/phosphorylation, and growth arrest. Changes in p15 and p16 do not correlate with PKC activation in this system.
Figure 8.
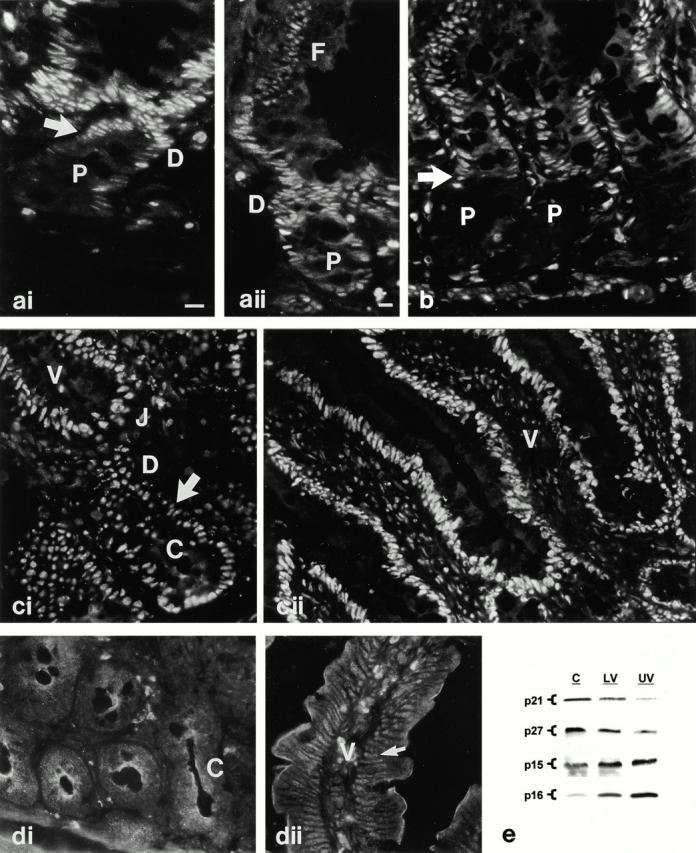
Developmental regulation of CKI expression in the small intestinal epithelium. (a–d) Immunofluorescence localization of CKIs in rat duodenum. (ai and aii) Expression of p21waf1/cip1 is weak in the nuclei of proliferating crypt cells (P), increases markedly coincident with growth arrest (arrow in ai), and declines in functional cells (F in aii) of the villus. D, differentiation zone. (b) p27kip1 staining is undetectable in cells of the lower crypt (P) and increases markedly with growth arrest, remaining high along the length of the villus. (ci and cii) p15 staining is readily detectable in nuclei of cells throughout the crypt and increases at the crypt–villus junction (J). V, villus; arrow, point of growth arrest. (di, dii) p16 staining is low in crypt cells (C) and increases gradually as cells migrate towards the villus tip. In dii, arrow points to a villus cell nucleus expressing p16. (e) Whole cell lysates (30 μg protein) of crypt (C), lower villus (LV), and upper villus (UV) populations were examined by Western blot analysis for expression of p21waf1/cip1, p27kip1, p15, and p16. Note that the crypt fraction includes some postmitotic cells of the upper crypt region which account for the p21waf1/cip1 and p27kip1 detected in this sample. Data are representative of at least three independent experiments. Bars, 10 μm.
PKC Activation in IEC-18 Cells Results in Downregulation of the DNA Licensing Factor Cdc6
The depletion of D-type cyclins and coordinated alterations in pocket protein expression/phosphorylation observed after PMA treatment of IEC-18 cells suggested that PKC activation in this system signals cell cycle withdrawal into G0. To further investigate this possibility, control and PMA-treated IEC-18 cells were analyzed for expression of the cdc6 DNA licensing factor, a molecule that is strictly associated with actively cycling populations and whose downregulation is indicative of cell cycle exit (Stillman 1996; Fujita 1999). PKC activation in IEC-18 cells resulted in marked downregulation of cdc6 by 2 h of treatment (Fig. 9), confirming cell cycle withdrawal into G0 or a G0-like state. Levels of this molecule recovered by 10–16 h, reflecting downregulation of PKC isozymes and reentry of IEC-18 cells into the cell cycle.
Figure 9.

PKC activation in IEC-18 cells leads to loss of cdc6 expression. Cells were treated with 100 nM PMA for the indicated times and subjected to Western blot analysis for cdc6 (U, untreated). Data are representative of three independent experiments.
PKCα Is Sufficient to Initiate a Program of Cell Cycle Withdrawal in IEC-18 Cells
Work from this and other laboratories has linked PKCα, in particular, to negative control of intestinal epithelial cell growth (Frey et al. 1997; Abraham et al. 1998; Scaglione-Sewell et al. 1998; also see Fig. 1). To investigate the ability of approximately physiological levels of this isozyme to signal cell cycle exit in intestinal epithelial cells, a population of IEC-18 cells expressing PKCα but not other phorbol ester–responsive isozymes was prepared by brief exposure to PMA as described in Materials and Methods (Fig. 10 a) (Frey et al. 1997). We have previously shown that activation of PKCα in this population by a second application of PMA is sufficient to induce cell cycle arrest (Frey et al. 1997). As shown in Fig. 10 b, retreatment of these PKCα-expressing cells (P) with 100 nM PMA for 2 h (R) resulted in hypophosphorylation and downregulation of p107 and pRb, accumulation of forms 1/2 of p130, loss of cyclin D, induction of p21waf1/cip1, and decreased expression of cdc6. Thus, PKCα activation alone is sufficient to recapitulate the conserved program of changes in CKIs, cyclins, and pocket proteins indicative of cell cycle withdrawal into G0 or a G0-like state. The observed responses were generally of slightly lesser magnitude than in control cells expressing the full profile of PMA-responsive isozymes, likely reflecting the slightly decreased amounts of PKCα remaining after PMA pulse treatment (Fig. 10 a). Although the participation of PKCδ and -ε in mediating negative cell cycle regulation in these cells cannot be formally excluded, their presence does not appear to be required to trigger a program of cell cycle exit in this system.
Figure 10.
PKCα activation is sufficient to initiate cell cycle withdrawal in IEC-18 cells. (a) IEC-18 cells were pulsed with PMA for 15 min to selectively downregulate PKCδ and -ε, and PKC isozyme expression was determined by Western blot analysis. (b) PKCδ- and -ε–depleted cells were retreated with PMA for 2 h and analyzed by Western blotting for expression and migration/phosphorylation state of cell cycle regulatory molecules. U, untreated; P, phorbol ester–pulsed cells expressing PKCα but not -δ or -ε; R, PKCα-expressing cells retreated with PMA. Data are representative of three independent experiments. (c) Model of PKCα-mediated intestinal epithelial cell cycle withdrawal (see text for details). (d) Time line of events involved in PKC-mediated cell cycle exit in IEC-18 cells.


Discussion
Through coordinate analysis of in vitro and in situ model systems, this study investigates the mechanisms by which PKC regulates cell cycle progression in intestinal epithelial cells. The data demonstrate for the first time that PKC activation can initiate a specific program of molecular events associated with cell cycle withdrawal into G0 or a G0-like state. Treatment of IEC-18 intestinal crypt cells with PKC agonists resulted in rapid downregulation of D-type cyclins, rapid and transient induction of p21waf1/cip1, delayed but sustained accumulation of p27kip1, inhibition of cyclin E– and cyclin A–associated kinase activity, coordinated alterations in pocket protein expression and phosphorylation, and downregulation of the DNA licensing factor cdc6. Manipulation of PKC isozyme levels in IEC-18 cells demonstrated that PKCα, in particular, is sufficient to produce this program of negative growth regulatory events. Furthermore, comparison of the developmental regulation of PKC isozymes and cell cycle control molecules in intestinal epithelial tissue revealed that (a) the program of events triggered by PKC activation in IEC-18 cells is representative of the changes seen in cell cycle regulation coincident with growth arrest in situ, and (b) PKC activation is appropriately positioned within intestinal crypts to initiate this program of cell cycle exit–associated events in situ.
PKC Activation in Intestinal Epithelial Cells Results in Coordinated Changes in Pocket Protein Expression and Phosphorylation
Data obtained from studies in multiple systems suggest a model for control of cell cycle exit which involves both alterations in pocket protein phosphorylation state and an orchestrated shift in relative levels of these molecules (Moberg et al. 1996; Smith et al. 1996; Mayol and Graña 1998). In this model, proliferating cells primarily express p107 and pRb as regulators of E2F function and thus G1/S progression; p130 is present at low levels in cycling cells. Cell cycle exit and differentiation are associated with hypophosphorylation of p107 and pRb, disappearance of p107 (and, in some systems, decreased expression of pRb), and marked accumulation of phosphoforms 1 and 2 of p130. p130 forms 1/2 are maintained in G0 cells as long-term binding partners and repressors of E2F-4. This program of pocket protein regulation has been shown to occur during in vitro skeletal muscle myogenesis (Garriga et al. 1998), granulocytic differentiation of mouse myeloid precursor cells (Garriga et al. 1998), and IFN-α–induced cell cycle exit of Daudi B cells (Thomas et al. 1998). Furthermore, similar patterns of pocket protein expression have been observed during epidermal differentiation in situ (Paramio et al. 1998).
In striking parallel to these findings, the data presented in the current study show that PKC-induced inhibition of cell cycle progression in IEC-18 cells is associated with hypophosphorylation and downregulation of p107 and pRb and with coordinated upregulation of p130 forms 1/2 (see Fig. 2). Although previous work in other systems has linked PKC to regulation of the function of pRb (for reviews see Fishman et al. 1998; Black 2000), the data presented here are the first to implicate PKC signaling in coordinated regulation of the expression and phosphorylation state of all three members of the pocket protein family. The ability of PKC signaling to induce this program of pocket protein control in IEC-18 cells points to a role for member(s) of this family in signaling cell cycle withdrawal in intestinal epithelial cells. This notion is strengthened by the finding that PKC activation within intestinal crypts in situ coincides precisely with similar changes in pocket protein regulation, i.e., decreased pocket protein phosphorylation, downregulation of p107, decreased expression and restriction of pRb to the nucleus, and increased expression of p130 (see Fig. 1 and Fig. 3). The disappearance of pRb from the cytoplasm of postmitotic cells (Fig. 3 b) (Chandrasekaran et al. 1996) suggests an additional mechanism of pocket protein regulation; however, the role of pocket protein subcellular distribution in regulating E2F function remains to be determined.
PKC Signaling Inhibits Cyclin–Cdk Activity in Intestinal Epithelial Cells
Inhibition of cyclin–cdk complex activity is critical for activation of pocket protein growth–suppressive function and cell cycle exit (Graña and Reddy 1995). PKC activation in IEC-18 cells appears to negatively regulate the function of all major cyclin–cdk complexes involved in G0/G1→S progression, i.e., cyclin D–cdk4/6, cyclin E–cdk2, and cyclin A–cdk2. Regulation of cyclin–cdk complexes by PKC signaling appears to involve two distinct mechanisms: changes in cyclin expression (modulating cyclin D complexes) and accumulation of CKIs (targeting cyclin E– and cyclin A–associated activity).
PKC activation in IEC-18 cells resulted in rapid disappearance of D-type cyclins, presumably leading to loss of any cdk4/6 activity present in these cells. Although delayed expression of cyclin D has been observed after growth factor stimulation of quiescent PKC-overexpressing cells in other systems (Fukumoto et al. 1997; Ashton et al. 1999), to our knowledge this study is the first to demonstrate the ability of PKC signaling to mediate the downregulation of this molecule in actively cycling cells. Several lines of evidence exclude a role for CKI accumulation in PKC-mediated modulation of cyclin D–associated activity. First, PKC has not been linked to induction of the cyclin D–cdk4/6-specific CKIs p15 or p16 in any system (Black 2000), including IEC-18 cells (see Fig. 7 a), and PKC activation in intestinal crypts in situ does not coincide with increased expression of either of these CKIs (see Fig. 8). Furthermore, recent evidence suggests that the Cip/Kip CKIs p21waf1/cip1 and p27kip1 are inefficient inhibitors of cyclin D–associated cdk activity (LaBaer et al. 1997), a notion supported by a study of Caco-2 colon adenocarcinoma cell differentiation in which association with p21waf1/cip1 was shown to be a critical component of cdk2 but not cdk4 inhibition (Ding et al. 1998). Together these data support a role for downregulation of D-type cyclins, rather than CKI association, in regulation of cdk4/6 activity by PKC-mediated signaling. Since levels of cyclin D remain relatively constant throughout the cell cycle in cycling cells and destruction of this molecule has been shown to be an early event in cell cycle withdrawal into G0 (Zwijsen et al. 1996), these data provide additional evidence for the ability of PKC signaling to induce a G0-like state in this system. The resurgence of cyclin D in IEC-18 cells after downregulation of PKC isozymes likely reflects reversal of cell cycle withdrawal and reentry of these cells from G0 into G1; similar hyperinduction of this molecule has been reported during serum-stimulated cell cycle entry from quiescence in several systems (e.g., Coppock et al. 1995; Fukumoto et al. 1997).
Inhibition of cdk2 complexes by PKC activation in IEC-18 cells appears to be accomplished through CKI binding rather than by destruction of cyclins. In this regard, cyclin E and cyclin A levels remained unchanged at times when their associated cdk activity was significantly inhibited (see Fig. 4 and Fig. 5). Downregulation of cyclin A was only observed at later time points (6 and 12 h), likely as a consequence of PKC-induced accumulation of cells in G0/G1 (cell cycle stages at which cyclin A is not normally expressed; Graña and Reddy 1995). These findings are consistent with data from other systems demonstrating PKC-induced inhibition of cdk2 activity under conditions in which levels of cyclins E or A were not limiting (Coppock et al. 1995; Livneh et al. 1996; Ashton et al. 1999). The notion that intestinal epithelial cell cycle withdrawal does not require the destruction of cyclins E and/or A was confirmed by the observation that, in intestinal tissue, downregulation of these molecules occurs at the crypt–villus junction, well after growth arrest in the midcrypt region (see Fig. 6). Although PKC activation in IEC-18 cells did not result in early alterations in cyclin E or A expression, increased levels of Cip/Kip CKIs were detected in both cyclin E and A complexes soon after PMA treatment, and could thus account for the observed inhibition of their associated kinase activity. The finding that p21waf1/cip1 and p27kip1 induction in the midcrypt region in situ coincides precisely with PKC activation (see Fig. 8 and Fig. 1 a) is consistent with this idea. A link between PKC signaling and increased Cip/Kip CKI expression has also been observed in a variety of other systems, including various epithelial cell types, leukemic cells, and cell lines overexpressing specific PKC isozymes (for review see Black 2000). Together with the absence of evidence for direct effects of PKC activation on cyclin or cdk expression or phosphorylation, these data suggest that increased association with Cip/Kip CKIs is the major mechanism for inhibition of cyclin E– and cyclin A–associated cdk activity by PKC signaling in intestinal epithelial cells.
PKC Signaling Differentially Regulates Cip/Kip CKI Expression in Intestinal Epithelial Cells
Although PKC signaling resulted in increased expression of both p21waf1/cip1 and p27kip1 in IEC-18 cells, marked differences were observed in the kinetics of induction of these molecules. Levels of p21waf1/cip1 peaked early after PMA treatment and rapidly returned to those seen in control cells. In contrast, induction of p27kip1 peaked later and increased levels were sustained as long as the cells remained in G0. Consistent with these results, PKC-mediated transient induction of p21waf1/cip1 has been noted in venous endothelial cells (Zezula et al. 1997), leukemic cell lines (Asiedu et al. 1995), and keratinocytes (Todd and Reynolds 1998), and sustained induction of p27kip1 has been observed during PMA-induced hematopoietic differentiation (Asiedu et al. 1997). Notably, these findings parallel the developmental regulation of Cip/Kip CKIs along the crypt–villus axis in situ. Although p21waf1/cip1 and p27kip1 are both upregulated coincident with growth arrest within intestinal crypts, elevated levels of p21waf1/cip1 are present only during early stages of differentiation (Fig. 8) (Gartel et al. 1996), whereas p27kip1 expression is sustained along the length of the villus. Similarly, increased p21waf1/cip1 expression was detected only during early stages of maturation of conditionally immortalized human fetal intestinal cells in vitro, whereas p27kip1 induction was delayed and sustained, coinciding with the appearance of morphological and functional markers of differentiation (Tian and Quaroni 1999). Recent findings suggest that the differential kinetics of induction of Cip/Kip CKIs reflect distinct roles for these molecules in the differentiation process, with p21waf1/cip1 playing a part in initiating irreversible growth arrest and p27kip1 functioning later to induce or maintain tissue-specific gene expression (Tian and Quaroni 1999; Yamamoto et al. 1999). Evidence that p21waf1/cip1 expression may in fact play an inhibitory role during late stages of differentiation (Di Cunto et al. 1998; Yamamoto et al. 1999) suggests that its downregulation is required for completion of the maturation process. Taken together, these findings demonstrate that PKC signaling can regulate Cip/Kip family CKI expression in a manner consistent with that seen in association with cell cycle exit and differentiation in a variety of biological systems.
PKCα Signals Cell Cycle Exit in Intestinal Epithelial Cells
To determine the specific PKC isozyme(s) involved in signaling cell cycle exit in intestinal epithelial cells, we took advantage of our previous finding that brief exposure to PMA, followed by extended incubation in the absence of drug, generates a population of IEC-18 cells expressing PKCα but not other phorbol ester–responsive isozymes (Frey et al. 1997). Activation of PKCα in these cells produced the same program of cell cycle exit–associated events observed after PKC agonist treatment of IEC-18 cells expressing the full panel of PKC isozymes. These data are consistent with the ability of PKCα to mediate cell cycle arrest and differentiation in a variety of in vitro systems (Gruber et al. 1992; Mischak et al. 1993; Murray et al. 1993; Abraham et al. 1998; Scaglione-Sewell et al. 1998; Desai et al. 1999; Slosberg et al. 1999), with the demonstration that PKCα overexpression can potentiate phorbol ester–mediated growth arrest in mammary (Slosberg et al. 1999) and intestinal epithelial cells (see Fig. 1), and with the observation that activation of this isozyme is maintained in all postmitotic cells of the intestinal epithelium in situ (Saxon et al. 1994) (see Fig. 1 a). These findings strongly support the involvement of PKCα, in particular, in mediating a program of cell cycle withdrawal, although it remains to be determined whether other members of the PKC family play a role in this process. For example, it is possible that downregulation of PKCε, which has been implicated in positive regulation of cell growth (e.g., Cacace et al. 1993), is also required for execution of this program. The continued proliferation of cells depleted of PKCα, -δ, and -ε, however, indicates that PKCε downregulation in the absence of active PKCα is not sufficient to produce growth arrest of these cells.
In summary, the data presented herein indicate that PKC signaling plays an important role in initiating a coordinated program of cell cycle exit in intestinal epithelial cells and suggest a model of PKC-mediated cell cycle exit (summarized in Fig. 10c and Fig. d). In this model, PKCα activation in IEC-18 cells leads to inhibition of G1/S cyclin–cdk activity via two distinct mechanisms: downregulation of D-type cyclins, which inhibits cdk4/6 activity, and accumulation of p21waf1/cip1 and p27kip1, which leads to inhibition of cyclin E– and cyclin A–associated cdk2. These effects result in hypophosphorylation/downregulation of p107 and pRb and accumulation of p130 forms 1/2, events which drive cells to exit the cell cycle into G0, as indicated by concomitant downregulation of cdc6. Sustained expression of p27kip1 and upregulation of p130 suggest that PKC-mediated cell cycle withdrawal is linked to the onset of a differentiation program. Analysis of the developmental regulation of pocket proteins, cyclins, cdks, and CKIs in intestinal epithelial tissue demonstrates that the program of cell cycle–specific events resulting from PKC activation in cultured cells closely parallels the changes seen coincident with PKC activation/growth arrest in the midcrypt region in situ. We also propose that sustained activation of PKC is required for maintenance of cell cycle exit in this system. As PKCα is downregulated from phorbol ester–treated IEC-18 cells, cyclin D is reexpressed and accumulates to high levels (see Fig. 5), sequestering Cip/Kip CKIs (see Fig. 7) to promote assembly of cyclin D–cdk4/6 complexes (Roberts 1999). Sequestration of p21waf1/cip1 and p27kip1 in cyclin D complexes, combined with decreased expression of p21waf1/cip1 (see Fig. 7), relieves inhibition of cdk2 (Roberts 1999) (see Fig. 4), allowing inactivation of pocket proteins and progression through the cell cycle.
The physiological importance of PKC-mediated negative control of cell cycle progression is underscored by evidence of decreased PKC expression or activity in colonic tumors (Guillem et al. 1987; Wali et al. 1991; Kahl-Rainer et al. 1996; Verstovsek et al. 1998), by the ability of phorbol ester treatment to inhibit growth in normal colonic tissue from biopsies (Assert et al. 1999), and by evidence that certain PKC isozymes can act as tumor suppressors in intestinal epithelial cells (Choi et al. 1990; Abraham et al. 1998; Scaglione-Sewell et al. 1998). Since PKC activation and/or overexpression have been shown to inhibit cell growth/cell cycle progression in a variety of systems, the results presented in this study may represent a conserved growth-inhibitory function for one or more members of the PKC family.
Acknowledgments
We wish to thank Deborah Ogden, Sulochana Dave, and Mary Vaughan for expert assistance with the immunofluorescence and immunohistochemistry studies.
This work was supported by a grant from the Crohn's and Colitis Foundation of America, by National Institutes of Health grants DK54909 and CA16056, by a grant from the Roswell Park Alliance Foundation, and by Developmental Funds from National Institutes of Health grant CA16056.
Footnotes
Abbreviations used in this paper: BrdU, 5′-bromo-2′-deoxyuridine; cdk, cyclin-dependent kinase; CKI, cdk inhibitor; DiC8, 1,2-dioctanoyl-sn-glycerol; dPP, 12-deoxyphorbol 13-phenylacetate; dPPA, 12-deoxyphorbol 13-phenylacetate 20-acetate; GFP, green fluorescent protein; PDBu, phorbol 12,13-dibutyrate; PKC, protein kinase C; Res, resiniferatoxin; Thy, thymeleatoxin.
References
- Abraham C., Scaglione-Sewell B., Skarosi S.F., Qin W., Bissonnette M., Brasitus T.A. Protein kinase C α modulates growth and differentiation in Caco-2 cells. Gastroenterology. 1998;114:503–509. doi: 10.1016/s0016-5085(98)70533-5. [DOI] [PubMed] [Google Scholar]
- Ashton A.W., Watanabe G., Albanese C., Harrington E.O., Ware J.A., Pestell R.G. Protein kinase C δ inhibition of S-phase transition in capillary endothelial cells involves the cyclin-dependent kinase inhibitor p27kip1 . J. Biol. Chem. 1999;274:20805–20811. doi: 10.1074/jbc.274.30.20805. [DOI] [PubMed] [Google Scholar]
- Asiedu C., Biggs J., Lilly M., Kraft A.S. Inhibition of leukemic cell growth by the protein kinase C activator bryostatin 1 correlates with the dephosphorylation of cyclin-dependent kinase 2. Cancer Re. 1995;s. 55:3716–3720. [PubMed] [Google Scholar]
- Asiedu C., Biggs J., Kraft A.S. Complex regulation of CDK2 during phorbol ester-induced hematopoietic differentiation. Blood. 1997;90:3430–3437. [PubMed] [Google Scholar]
- Assert R., Kotter R., Bisping G., Scheppach W., Stahlnecker E., Muller K.M., Dusel G., Schatz H., Pfeiffer A. Anti-proliferative activity of protein kinase C in apical compartments of human colonic cryptsevidence for a less activated protein kinase C in small adenomas. Int. J. Cancer. 1999;80:47–53. doi: 10.1002/(sici)1097-0215(19990105)80:1<47::aid-ijc10>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Bach S.P., Renehan A.G., Potten C.S. Stem cellsthe intestinal stem cell as a paradigm. Carcinogenesis. 2000;21:469–476. doi: 10.1093/carcin/21.3.469. [DOI] [PubMed] [Google Scholar]
- Black J.D. Protein kinase C-mediated regulation of the cell cycle. Front. Biosci. 2000;5:D406–D423. doi: 10.2741/black. [DOI] [PubMed] [Google Scholar]
- Burgess D.R., Jiang W., Mamajiwalla S., Kinsey W. Intestinal crypt stem cells possess high levels of cytoskeletal-associated phosphotyrosine-containing proteins and tyrosine kinase activity relative to differentiated enterocytes. J. Cell Biol. 1989;109:2139–2144. doi: 10.1083/jcb.109.5.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace A.M., Guadagno S.N., Krauss R.S., Fabbro D., Weinstein I.B. The epsilon isoform of protein kinase C is an oncogene when overexpressed in rat fibroblasts. Oncogene. 1993;8:2095–2104. [PubMed] [Google Scholar]
- Castagna M., Takai Y., Kaibuchi K., Sano K., Kikkawa U., Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- Chandrasekaran C., Coopersmith C.M., Gordon J.I. Use of normal and transgenic mice to examine the relationship between terminal differentiation of intestinal epithelial cells and accumulation of their cell cycle regulators. J. Biol. Chem. 1996;271:28414–28421. doi: 10.1074/jbc.271.45.28414. [DOI] [PubMed] [Google Scholar]
- Chellappan S.P., Giordano A., Fisher P.B. Role of cyclin-dependent kinases and their inhibitors in cellular differentiation and development. Curr. Top. Microbiol. Immunol. 1998;227:57–103. doi: 10.1007/978-3-642-71941-7_4. [DOI] [PubMed] [Google Scholar]
- Chen C., Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi P.M., Tchou-Wong K.M., Weinstein I.B. Overexpression of protein kinase C in HT29 colon cancer cells causes growth inhibition and tumor suppression. Mol. Cell. Biol. 1990;10:4650–4657. doi: 10.1128/mcb.10.9.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppock D., Buffolino P., Kopman C., Nathanson L. Inhibition of the melanoma cell cycle and regulation at the G1/S transition by 12-O-tetradecanoylphorbol-13-acetate (TPA) by modulation of cdk2 activity. Exp. Cell Res. 1995;221:92–102. doi: 10.1006/excr.1995.1356. [DOI] [PubMed] [Google Scholar]
- Cormack B. Introduction of point mutations by PCR Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Current Protocols in Molecular Biology. Vol. 1 1997. 8 John Wiley & Sons, Inc; New York: 5.5–8.5.10. [Google Scholar]
- DeCaprio J.A., Furukawa Y., Ajchenbaum F., Griffin J.D., Livingston D.M. The retinoblastoma-susceptibility gene product becomes phosphorylated in multiple stages during cell cycle entry and progression. Proc. Natl. Acad. Sci. 1992;USA. 89:1795–1798. doi: 10.1073/pnas.89.5.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker L.V., Parker P.J. PKC–a question of specificity. Trends Biochem. Sci. 1994;19:73–77. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Desai D.S., Hirai S., Karnes W.E., Jr., Niles R.M., Ohno S. Cloning and characterization of the murine PKCα promoteridentification of a retinoic acid response element. Biochem. Biophys. Res. Commun. 1999;263:28–34. doi: 10.1006/bbrc.1999.1307. [DOI] [PubMed] [Google Scholar]
- Di Cunto F., Topley G., Calautti E., Hsiao J., Ong L., Seth P.K., Dotto G.P. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science. 1998;280:1069–1072. doi: 10.1126/science.280.5366.1069. [DOI] [PubMed] [Google Scholar]
- Ding Q.M., Ko T.C., Evers B.M. Caco-2 intestinal cell differentiation is associated with G1 arrest and suppression of CDK2 and CDK4. Am. J. Physiol. 1998;275:C1193–C1200. doi: 10.1152/ajpcell.1998.275.5.C1193. [DOI] [PubMed] [Google Scholar]
- Fishman D.D., Segal S., Livneh E. The role of protein kinase C in G1 and G2/M phases of the cell cycle. Int. J. Oncol. 1998;12:181–186. doi: 10.3892/ijo.12.1.181. [DOI] [PubMed] [Google Scholar]
- Frey M.R., Saxon M.L., Zhao X., Rollins A., Evans S.S., Black J.D. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21waf1/cip1 and p27kip1 and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J. Biol. Chem. 1997;272:9424–9435. doi: 10.1074/jbc.272.14.9424. [DOI] [PubMed] [Google Scholar]
- Fujita M. Cell cycle regulation of DNA replication initiation proteins in mammalian cells. Front. Biosci. 1999;4:D816–D823. doi: 10.2741/fujita. [DOI] [PubMed] [Google Scholar]
- Fukumoto S., Nishizawa Y., Hosoi M., Koyama H., Yamakawa K., Ohno S., Morii H. Protein kinase C delta inhibits the proliferation of vascular smooth muscle cells by suppressing G1 cyclin expression. J. Biol. Chem. 1997;272:13816–13822. doi: 10.1074/jbc.272.21.13816. [DOI] [PubMed] [Google Scholar]
- Garriga J., Limon A., Mayol X., Rane S.G., Albrecht J.H., Reddy E.P., Andres V., Graña X. Differential regulation of the retinoblastoma family of proteins during cell proliferation and differentiation. Biochem. J. 1998;333:645–654. doi: 10.1042/bj3330645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel A.L., Serfas M.S., Gartel M., Goufman E., Wu G.S., el-Deiry W.S., Tyner A.L. p21 (WAF1/CIP1) expression is induced in newly nondividing cells in diverse epithelia and during differentiation of the Caco-2 intestinal cell line. Exp. Cell Res. 1996;227:171–181. doi: 10.1006/excr.1996.0264. [DOI] [PubMed] [Google Scholar]
- Geng Y., Whoriskey W., Park M.Y., Bronson R.T., Medema R.H., Li T., Weinberg R.A., Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Graña X., Reddy E.P. Cell cycle control in mammalian cellsrole of cyclins, cyclin-dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- Graña X., Garriga J., Mayol X. Role of the retinoblastoma protein family, pRb, p107, and p130, in the negative control of cell growth. Oncogene. 1998;17:3365–3383. doi: 10.1038/sj.onc.1202575. [DOI] [PubMed] [Google Scholar]
- Gruber J.R., Ohno S., Niles R.M. Increased expression of PKC alpha plays a key role in retinoic acid-induced melanoma differentiation. J. Biol. Chem. 1992;267:13356–13360. [PubMed] [Google Scholar]
- Guillem J.G., O'Brian C.A., Fitzer C.J., Forde K.A., LoGerfo P., Treat M., Weinstein I.B. Altered levels of protein kinase C and Ca2+-dependent protein kinases in human colon carcinomas. Cancer Res. 1987;47:2036–2039. [PubMed] [Google Scholar]
- Kahl-Rainer P., Sedivy R., Marian B. Protein kinase C tissue localization in human colonic tumors suggests a role for adenoma growth control. Gastroenterology. 1996;110:1753–1759. doi: 10.1053/gast.1996.v110.pm8964400. [DOI] [PubMed] [Google Scholar]
- LaBaer J., Garrett M.D., Stevenson L.F., Slingerland J.M., Sandhu C., Chou H.S., Fattaey A., Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Li Y., Graham C., Lacy S., Duncan A.M., Whyte P. The adenovirus E1A-associated 130-kD protein is encoded by a member of the retinoblastoma gene family and physically interacts with cyclins A and E. Genes Dev. 1993;7:2366–2377. doi: 10.1101/gad.7.12a.2366. [DOI] [PubMed] [Google Scholar]
- Livneh E., Shimon T., Bechor E., Doki Y., Schieren I., Weinstein I.B. Linking protein kinase C to the cell cycleectopic expression of PKC η in NIH 3T3 cells alters the expression of cyclins and Cdk inhibitors and induces adipogenesis. Oncogene. 1996;12:1545–1555. [PubMed] [Google Scholar]
- Ludlow J.W., Shon J., Pipas J.M., Livingston D.M., DeCaprio J.A. The retinoblastoma susceptibility gene product undergoes cell cycle-dependent dephosphorylation and binding to and release from SV40 large T. Cell. 1990;60:387–396. doi: 10.1016/0092-8674(90)90590-b. [DOI] [PubMed] [Google Scholar]
- Ludlow J.W., Glendening C.L., Livingston D.M., DeCaprio J.A. Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol. Cell. Biol. 1993;13:367–372. doi: 10.1128/mcb.13.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayol X., Graña X. The p130 pocket proteinkeeping order at cell cycle exit/re-entrance transitions. Front. Biosci. 1998;3:D11–D24. doi: 10.2741/a263. [DOI] [PubMed] [Google Scholar]
- Mayol X., Garriga J., Graña X. Cell cycle-dependent phosphorylation of the retinoblastoma-related protein p130. Oncogene. 1995;11:801–808. [PubMed] [Google Scholar]
- Mischak H., Goodnight J.A., Kolch W., Martiny-Baron G., Schaechtle C., Kazanietz M.G., Blumberg P.M., Pierce J.H., Mushinski J.F. Overexpression of protein kinase C-delta and -epsilon in NIH3T3 cells induces opposite effects on growth, morphology, anchorage dependance, and tumorigenicity. J. Biol. Chem. 1993;268:6090–6096. [PubMed] [Google Scholar]
- Moberg K., Starz M.A., Lees J.A. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell. Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray N.R., Baumgardner G.P., Burns D.J., Fields A.P. Protein kinase C isotypes in human erythroleukemia (K562) cell proliferation and differentiation. Evidence that beta II protein kinase C is required for proliferation. J. Biol. Chem. 1993;268:15847–15853. [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Paramio J.M., Lain S., Segrelles C., Lane E.B., Jorcano J.L. Differential expression and functionally co-operative roles for the retinoblastoma family of proteins in epidermal differentiation. Oncogene. 1998;17:949–957. doi: 10.1038/sj.onc.1202031. [DOI] [PubMed] [Google Scholar]
- Pines J. Cyclins, cdks, and cancer. Sem. Cancer Biol. 1995;6:63–72. doi: 10.1006/scbi.1995.0009. [DOI] [PubMed] [Google Scholar]
- Quaroni A., May R.J. Establishment and characterization of intestinal epithelial cell cultures. Methods Cell Biol. 1980;21B:403–427. [PubMed] [Google Scholar]
- Roberts J.M. Evolving ideas about cyclins. Cell. 1999;98:129–132. doi: 10.1016/s0092-8674(00)81007-7. [DOI] [PubMed] [Google Scholar]
- Saxon M.L., Zhao X., Black J.D. Activation of protein kinase C isozymes is associated with postmitotic events in intestinal epithelial cells in situ. J. Cell Biol. 1994;126:747–763. doi: 10.1083/jcb.126.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione-Sewell B., Abraham C., Bissonnette M., Skarosi S.F., Hart J., Davidson N.O., Wali R.K., Davis B.H., Sitrin M., Brasitus T.A. Decreased PKC-α expression increases cellular proliferation, decreases differentiation, and enhances the transformed phenotype of Caco-2 cells. Cancer Res. 1998;58:1074–1081. [PubMed] [Google Scholar]
- Sherr C.J., Roberts J.M. CDK inhibitorspositive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Slosberg E.D., Klein M.G., Yao Y., Han E.K., Schieren I., Weinstein I.B. The alpha isoform of protein kinase C mediates phorbol ester-induced growth inhibition and p21cip1 induction in HC11 mammary epithelial cells. Oncogene. 1999;18:6658–6666. doi: 10.1038/sj.onc.1203083. [DOI] [PubMed] [Google Scholar]
- Smith E.J., Leone G., DeGregori J., Jakoi L., Nevins J.R. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol. Cell. Biol. 1996;16:6965–6976. doi: 10.1128/mcb.16.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.J., Leone G., Nevins J.R. Distinct mechanisms control the accumulation of the Rb-related p107 and p130 proteins during cell growth. Cell Growth Differ. 1998;9:297–303. [PubMed] [Google Scholar]
- Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- Thomas N.S., Pizzey A.R., Tiwari S., Williams C.D., Yang J. p130, p107, and pRb are differentially regulated in proliferating cells and during cell cycle arrest by alpha-interferon. J. Biol. Chem. 1998;273:23659–23667. doi: 10.1074/jbc.273.37.23659. [DOI] [PubMed] [Google Scholar]
- Tian J.Q., Quaroni A. Involvement of p21(WAF1/Cip1) and p27(Kip1) in intestinal epithelial cell differentiation. Am. J. Physiol. 1999;276:C1245–C1258. doi: 10.1152/ajpcell.1999.276.6.C1245. [DOI] [PubMed] [Google Scholar]
- Todd C., Reynolds N.J. Up-regulation of p21waf1 by phorbol ester and calcium in human keratinocytes through a protein kinase C-dependent pathway. Am. J. Pathol. 1998;153:39–45. doi: 10.1016/S0002-9440(10)65543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek G., Byrd A., Frey M.R., Petrelli N.J., Black J.D. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology. 1998;115:75–85. doi: 10.1016/s0016-5085(98)70367-1. [DOI] [PubMed] [Google Scholar]
- Wali R.K., Baum C.L., Bolt M.J., Dudeja P.K., Sitrin M.D., Brasitus T.A. Down-regulation of protein kinase C activity in 1,2-dimethylhydrazine-induced rat colonic tumors. Biochim. Biophys. Acta. 1991;1092:119–123. doi: 10.1016/0167-4889(91)90185-z. [DOI] [PubMed] [Google Scholar]
- Weiser M.M. Intestinal epithelial cell surface membrane glycoprotein synthesis. I. An indicator of cellular differentiation. J. Biol. Chem. 1973;248:2536–2541. [PubMed] [Google Scholar]
- Whyte P., Eisenman R.N. Dephosphorylation of the retinoblastoma protein during differentiation of HL60 cells. Biochem. Cell Biol. 1992;70:1380–1384. doi: 10.1139/o92-186. [DOI] [PubMed] [Google Scholar]
- Yamamoto H., Soh J.W., Shirin H., Xing W.Q., Lim J.T., Yao Y., Slosberg E., Tomita N., Schieren I., Weinstein I.B. Comparative effects of overexpression of p27kip1 and p21Cip1/Waf1 on growth and differentiation in human colon carcinoma cells. Oncogene. 1999;18:103–115. doi: 10.1038/sj.onc.1202269. [DOI] [PubMed] [Google Scholar]
- Zezula J., Sexl V., Hutter C., Karel A., Schutz W., Freissmuth M. The cyclin-dependent kinase inhibitor p21cip1 mediates the growth inhibitory effect of phorbol esters in human venous endothelial cells. J. Biol. Chem. 1997;272:29967–29974. doi: 10.1074/jbc.272.47.29967. [DOI] [PubMed] [Google Scholar]
- Zwijsen R.M., Klompmaker R., Wientjens E.B., Kristel P.M., van der Burg B., Michalides R.J. Cyclin D1 triggers autonomous growth of breast cancer cells by governing cell cycle exit. Mol. Cell. Biol. 1996;16:2554–2560. doi: 10.1128/mcb.16.6.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]