

Abstract
All mammalian cells absolutely require polyamines (putrescine, spermidine, and spermine) for growth. Here we show that the overexpression of cDNA for S-adenosylmethionine decarboxylase (AdoMetDC), the main regulatory enzyme in the biosynthesis of higher polyamines, induces transformation of rodent fibroblasts when expressed in the sense or the antisense orientation. Both transformants were able to induce invasive tumors in nude mice. Neither transformation was associated with activation of the mitogen-activated protein kinases Erk1 and Erk2. Instead, the AdoMet DC sense, but not antisense, transformants displayed constitutive activation of the c-Jun NH2-terminal kinase (JNK) pathway. However, both transformations converged on persistent phosphorylation of endogenous c-Jun at Ser73. The phenotype of the AdoMetDC sense transformants was reversed by expression of dominant-negative mutants of SEK1 (MKK4), JNK1, and c-Jun (TAM-67), which were also found to impair cytokinesis. Similarly, TAM-67 reverted the morphology of the AdoMetDC-antisense expressors. This report is the first demonstration of a protein whose overexpression or block of synthesis can induce cell transformation. In addition, we show that the polyamine biosynthetic enzymes require c-Jun activation for eliciting their biological effects.
Keywords: cell transformation, c-Jun, JNK, S-adenosylmethionine decarboxylase, polyamines
Introduction
The polyamines putrescine, spermidine (spd), and spermine are essential for cell proliferation (Tabor and Tabor 1984; Pegg 1988; Heby and Persson 1990; Cohen 1998). In cells stimulated to grow and divide, the synthesis of polyamines is always rapidly induced. The precise physiological functions of polyamines have remained elusive, even though they have been shown to interact with and stabilize negatively charged macromolecules, such as nucleic acids and phospholipids, and to stimulate the synthesis of DNA, RNA, and proteins (Tabor and Tabor 1984; Pegg 1988; Heby and Persson 1990; Cohen 1998).
Polyamines also seem to be involved in the process of cell transformation. Ornithine decarboxylase (ODC), the first enzyme in polyamine biosynthesis, has thus far been the main focus of interest. ODC is known to become activated after treatment with chemical carcinogens and tumor promoters, as well as in cells transformed by various oncogenes, such as v-src, neu, myc, and ras (Pegg 1988; Auvinen et al. 1992). Furthermore, ODC is a direct transcriptional target of c-Myc (Bello-Fernandez et al. 1993) and has oncogenic potential when overexpressed (Auvinen et al. 1992, Auvinen et al. 1997; Clifford et al. 1995; O'Brien et al. 1997).
Here, we examined whether S-adenosylmethionine decarboxylase (AdoMetDC; EC 4.1.1.50), the other key regulatory enzyme in polyamine biosynthesis (Tabor and Tabor 1984; Pegg 1988; Heby and Persson 1990; Cohen 1998), might also be involved in cell transformation. AdoMetDC catalyzes the formation of decarboxylated S-adenosylmethionine, which serves as an aminopropyl donor in the biosynthesis of spd and spermine, which are vital for cell growth. Like ODC, AdoMetDC has a fast turnover rate, and is rapidly induced up to tenfold and higher in various normal and neoplastic growth processes (Cohen 1998). Furthermore, inhibition of AdoMetDC by various drugs has been found to have antiproliferative and antitumor activity (Pegg and McCann 1992; Regenass et al. 1994) and inhibits metastasis (Gutman et al. 1995). However, there has not been direct evidence for a specific role of AdoMetDC in transformation.
Cell transformation is thought to result from aberrant activation of signal transduction molecules that control cell proliferation (Weinberg 1996). Most studies have suggested that the Ras-Raf-mitogen-activated protein kinase (MAPK) pathway (Seger and Krebs 1995) plays a major role in transformation. Indeed, many oncogenes, including ras, src, raf, and mos, have been shown to exert their effect through the activation of MAPKs Erk1 and Erk2 (Howe et al. 1992; Samuels et al. 1993; Okazaki and Sagata 1995). However, recent studies have indicated that the activation of Erks is not invariable, and a parallel pathway, the c-Jun NH2-terminal kinase (JNK) pathway, may be activated instead (Raitano et al. 1995; Crespo et al. 1996; Clark et al. 1997; Rodrigues et al. 1997). The major target of JNKs is c-Jun, which becomes phosphorylated on Ser63 and Ser73 and thereby becomes activated (Su and Karin 1996; Ip and Davis 1998). Here, we report that overexpression of AdoMetDC causes full transformation of rodent fibroblasts, and it is capable of inducing highly invasive tumors in nude mice, and these transformed cells show constitutive activation of JNKs and phosphorylation of c-Jun on Ser73. Expression of dominant-negative mutants of the upstream kinases of c-Jun (DN SEK1 and DN JNK1) and the transcriptionally inactive mutant of c-Jun, TAM67, reversed the transformation and impaired cytokinesis, yielding polykaryotic cells. Paradoxically, tumorigenic transformation was also seen in cells expressing the AdoMetDC cDNA in the antisense orientation. This transformation, again, appeared to involve the phosphorylation of c-Jun, but did not involve JNK activation.
Materials and Methods
AdoMetDC Vector Constructions and Transfections
A 1.65-kb insert of human AdoMetDC cDNA was isolated from pSAMh1 (Pajunen et al. 1988) with PvuII/BamHI digestion, resulting in a shorter 5′-untranslated region than the full-length cDNA. The fragment was blunt-ended, ligated to SalI linkers, and inserted in both orientations into the SalI site of the pLTRpoly vector (American Type Culture Collection 77109). The sense and antisense constructs and the empty vector (control) were transfected together with a neo-selection marker (pSV2neo) into cells using LipofectAMINE (GIBCO BRL). The transfected cells were selected for resistance to G418 (GIBCO BRL) (400 μg/ml) for two weeks. The transfections were repeated four times. To avoid problems with clonal variation, the data are from a total pool of the transfectants.
Cell Culture
The NIH3T3 cells (American Type Culture Collection), their stable derivatives expressing neo and the empty pLTRpoly vector (4N), and AdoMetDC in sense (Amdc-s) or antisense (Amdc-as) orientations were cultured in DME supplemented with antibiotics and 5% newborn calf serum (GIBCO BRL) or FCS (Bioclear). Spermidine at a low concentration (1 μM), was added to parallel Amdc-as cell cultures (Amdc-as + spd) to prevent the counter selection of cells expressing the AdoMetDC-as mRNA at levels that would block the synthesis of vital higher polyamines. The corresponding Rat-1 cell transfectants were grown similarly.
Northern Blotting
Polyadenylated mRNA was isolated by oligo(dT) cellulose chromatography from 5–10 × 107 cells. RNA samples (8 μg) were size-fractionated in agarose gels, transferred to Hybond-N nylon filters (Amersham Pharmacia Biotech), and hybridized with specific probes, as described previously (Paasinen-Sohns and Hölttä 1997). [32P]dCTP-labeled probes for AdoMetDC (PvuII/BamHI fragment of pSAMh1) and ODC (HindIII fragment of pODC16) were generated using a multiprime DNA-labeling kit (Amersham Pharmacia Biotech).
Assay of AdoMetDC and ODC Activities
The activities of AdoMetDC and ODC were assayed by measuring the production of 14CO2 from S-adenosyl-l-(carboxyl-14C)methionine or l-(1-14CO2)ornithine (Cohen 1998), respectively.
Analysis of Polyamines
Polyamine concentrations in the cells were measured with a Hewlett-Packard HP 1090 liquid chromatograph with fluorescence detection (Hyvönen et al. 1992).
Staining of Actin Filaments
The cells grown on glass coverslips were fixed with 3.5% paraformaldehyde, permeabilized with 0.2% NP-40, and stained with rhodamine-conjugated phallacidin.
Soft Agar Growth
5 × 104 cells, in growth media, were mixed with agar, resulting in a 0.35% agar-containing mixture, and laid over a 0.7% bottom agar layer. The cells were grown for four weeks, and growth media was added on to the top agar twice a week.
Tumorigenicity Assay
Nude mice (nu/nu-BALB/cABom, female) were obtained from Bomholtgaard Breeding and Research Center, Ltd., and the tumorigenicity assays were performed at Orion-Farmos Experimental Cancer Research Center. The animals were maintained in a barrier unit, and all materials that came into direct contact with the mice were autoclaved. The control (4N), Amdc-s, Amdc-as, and Amdc-as+spd cells were injected subcutaneously into both flanks of the mice (107 cells/injection site, five mice/cell line). The occurrence of tumors was followed regularly for 62 d.
Cell Lysates, Nuclear Fractions, and Western Blots
Cell lysates and nuclear fractions were prepared essentially as described previously (Paasinen-Sohns and Hölttä 1997). In brief, to isolate detergent soluble proteins, the cells were lysed in 1% Triton X-100 containing buffer, and the nuclei were removed by centrifugation. For nuclear fractions, the cells were suspended in hypotonic lysis buffer containing 0.5% NP-40, the nuclei were collected by centrifugation at maximal speed in an Eppendorf microcentrifuge for 20 s, washed in lysis buffer, resuspended, and sonicated. After determining protein concentrations, the samples were suspended in Laemmli's sample buffer, resolved by SDS-PAGE, and immunoblotted, as detailed before (Paasinen-Sohns and Hölttä 1997). Erks were detected by mAbs to Erk1/Erk2 (Zymed Laboratories). Phosphorylated c-Jun, dominant-negative mutants of JNK1 and SEK1, and truncated c-Jun (TAM67) proteins were immunoblotted with pAbs to phospho-c-Jun(Ser73) (New England Biolabs, Inc.), JNK1(FL) (Santa Cruz Biotechnology, Inc.), SEK1/MKK4 (Sigma-Aldrich), and c-Jun/AP-1 (Ab-1) (Calbiochem-Novabiochem), respectively.
Assay of MAPK Activity
1 mg of soluble proteins was immunoprecipitated with α-rat MAPK R2 pAb (Upstate Biotechnology). The immunocomplex activity was measured in a 25-μl volume of kinase buffer (Paasinen-Sohns and Hölttä 1997) containing 50 μM ATP, 5 μCi [γ-32P]ATP (3,000 Ci/mmol) (NEN Life Science Products), and 25 μg MAPK substrate peptide (APRTPGGRR) (Upstate Biotechnology) at 30°C for 15 min (Paasinen-Sohns and Hölttä 1997).
Assay of JNK Activity
Immunocomplex assay of JNK activity was performed using the α-JNK1 (C-17; Santa Cruz Biotechnology, Inc.) antibody and c-Jun(1-169)–GST fusion protein (Upstate Biotechnology) as the substrate (Paasinen-Sohns and Hölttä 1997). For the solid-phase JNK kinase assay (Hibi et al. 1993), cells were lysed in buffer containing 20 mM Hepes, pH 7.7, 0.3 M NaCl, 2.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100, 0.5 mM DTT, 20 mM β-glycerophosphate, 0.1 mM Na3VO4, 2 μg/ml aprotinin/leupeptin, and 100 μM AEBSF, and the extracts were diluted to 75 mM NaCl. 1 mg of protein extracts was mixed with 2.5 μg of agarose-conjugated c-Jun(1-169)–GST fusion protein (Upstate Biotechnology) to affinity purify the JNKs. For the kinase reactions, the beads were suspended in 25 μl of the kinase assay buffer supplemented with 50 μM ATP and 5 μCi [γ-32P]ATP (3,000 Ci/mmol) (NEN Life Science Products). After a 20-min incubation at 30°C, the phosphorylated proteins were analyzed by 12.5% SDS-PAGE and autoradiography.
Dominant-Negative SEK/JNK/c-Jun Mutant Transfections
Normal 4N- and AdoMetDC-transformed Amdc-s cells were transfected with 3 μg of empty pcDNA3-vector, DN SEK1 (SEK1[AL]) (Yan et al. 1994), or DN JNK1 (FLAG-JNK1[APF]) plasmids (Dérijard et al. 1994), together with a pBabe Puro selection marker (Morgenstern and Land 1990) using LipofectAMINE PLUS (GIBCO BRL). Similarly, pCMV-vector or c-Jun-mutant pCMV-TAM67 (Brown et al. 1993), which was deleted at the NH2-terminal, together with a selection marker pZeoSV2/lacZ (Invitrogen), were transfected. 3 μg/ml puromycin (Sigma-Aldrich) or 250 μg/ml of Zeocin (Invitrogen) were added to the corresponding cultures 2 d after transfection.
Results
Overexpression of AdoMetDC cDNA in Sense or Antisense Orientation Causes Cell Transformation
To study the consequences of overexpression of AdoMetDC on growth regulation, we cloned the human AdoMetDC cDNA (truncated at the 5′-untranslated region to remove sequences that inhibit translation) into a pLTRpoly expression vector, both in sense and antisense orientations. The constructs were transfected with a neo selection marker (pSV2neo) into mouse NIH3T3 cells and Rat-1 fibroblasts, and stable cell lines were generated. The control NIH3T3 cells transfected with pLTRpoly and pSV2neo vectors (4N) exhibited an epithelioid morphology, stringent density-dependent growth (Fig. 1 A, a), normal actin filaments (Fig. 1 A, d), and did not grow in soft agar (only 1.7% of the cells formed tiny colonies) (Fig. 1 A, g). Transfection of NIH3T3 cells with the AdoMetDC sense construct resulted in a complete morphological transformation. The transfectants displayed an elongated morphology, grew without contact inhibition in a criss-cross manner, and formed innumerable foci in tissue culture (Fig. 1 A, b). They also showed disintegrated actin filaments (Fig. 1 A, e) and acquired the ability to grow in soft agar (15.2% of the cells formed large foci) (Fig. 1 A, h). Surprisingly, the same was true for the AdoMetDC antisense construct–expressing cells when they were cultured in the presence of spd (1 μM) (Fig. 1 A, c). Exogenous spermidine had to be added to keep alive the high Amdc-as expressors, which are blocked in the synthesis of spermidine. However, the Amdc-as+spd cells displayed a less elongated morphology than the Amdc-s cells, but showed a similar degree of disintegration of the actin filaments (Fig. 1 A, f) and growth in soft agar (19.1%). The Amdc-as cells grown in the absence of spermidine (except what is provided by serum in the growth media) exhibited a much lower degree of morphological transformation with partial disintegration of the actin filaments. Similar inductions of transformation by the AdoMetDC sense and antisense constructs were also seen in Rat-1 cells. Fig. 1 B, a shows the morphology of normal Rat-1 control cells, and Fig. 1 B, b and c, show the transformed phenotype induced by the AdoMetDC sense and antisense constructs (with spermidine supplementation), respectively. Spermidine had no effect on the morphology of normal 4N cells or Amdc-s cells. Overexpression of AdoMetDC was not associated with increased proliferation or cell death (data not shown), thus AdoMetDC is specifically associated with morphological transformation. The data presented below are mostly from experiments with NIH3T3 cells, but also hold true for Rat-1 cells.
Figure 1.
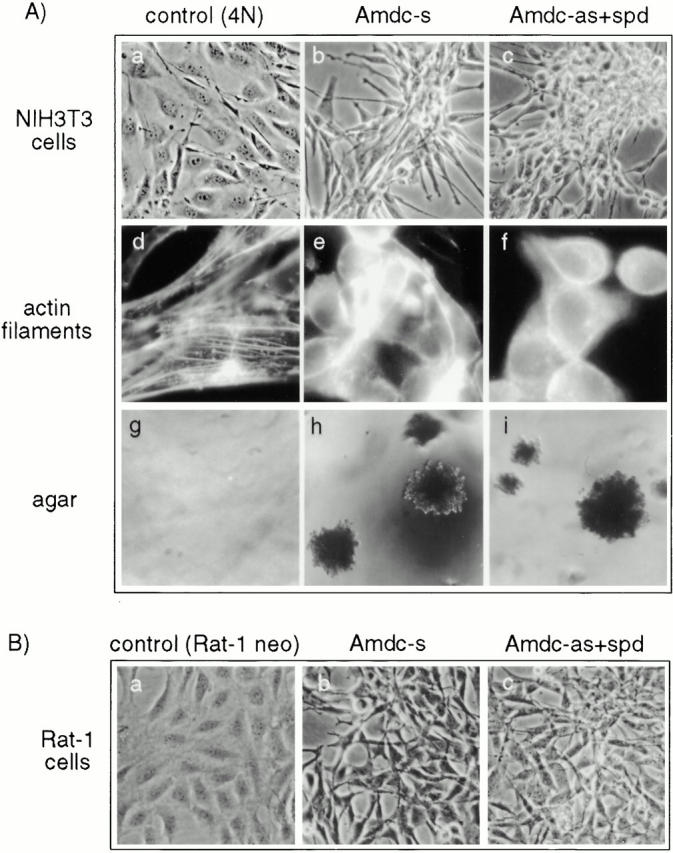
(A) Morphology, actin filaments, and soft agar growth of NIH3T3 cells overexpressing human AdoMetDC cDNA in sense (Amdc-s) and antisense (Amdc-as) orientations are shown. (a, d, and g) Parental NIH3T3 cells transfected with the neomycin resistance gene and the empty pLTRpoly vector (control, 4N), (b, e, and h) Amdc-s cells and (c, f, and i) Amdc-as cells grown with 1 μM spermidine (Amdc-as+spd) in tissue cultures (a–c), stained for actin filaments (d–f), and grown in soft agar (g–i). (B) Morphology of Rat-1 cell transfectants. (a–c) Rat-1 control cells and transfectants expressing AdoMetDC sense and antisense constructs. Rat-1 Amdc-as cells were grown in the presence of spermidine, as described above.
Human AdoMetDC Expression and Polyamine Levels in NIH3T3 Transfectants
Northern blot analyses showed that the normal 4N cells expressed low levels of two endogenous mouse Ado MetDC mRNAs (Pajunen et al. 1988). The 3.1-kb mRNA was expressed at a higher level than the 2.1-kb species, which was visible only upon longer exposure (Fig. 2 A). The Amdc-s cells carrying the human AdoMetDC sense construct expressed high levels of two AdoMetDC mRNAs of 1.65 kb and 2.65 kb, which are the sizes predicted for the use of polyadenylation signals in AdoMetDC cDNA (Pajunen et al. 1988) and in the pLTRpoly vector, respectively. The Amdc-as cells carrying the antisense construct also expressed the 2.65-kb mRNA of expected size, but the most prominent band was of a slightly smaller size (Fig. 2 A). This smaller mRNA likely arises as a result of splicing out 0.3-kb vector sequences, as there is a consensus splice donor site at the end of the AdoMetDC antisense cDNA and a splice acceptor site in the vector. The Amdc-as+spd cells had higher levels of AdoMetDC antisense mRNAs, as expected (see above). No appreciable changes in the expression level of the AdoMetDC mRNAs were detected in normal or Amdc-s cells cultured with spermidine.
Figure 2.
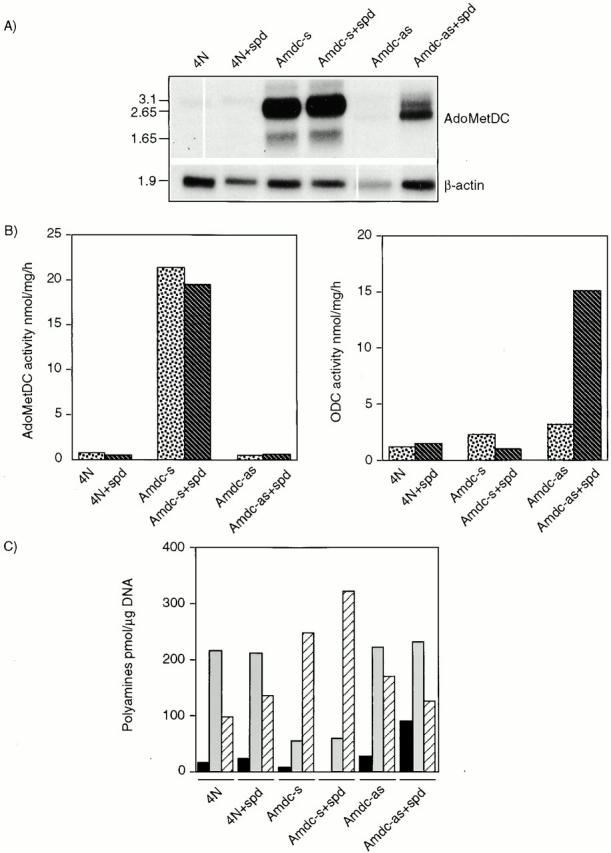
Expression of human AdoMetDC cDNA, the activities of AdoMetDC and ODC, and polyamine contents in NIH3T3 transfectants are shown. (A) The Northern blot shows the expression levels of AdoMetDC mRNA in transfected cells. The integrity and loading of RNA was controlled by hybridizing the blot with human β-actin c-DNA. Note, the consistent lower β-actin signal in the Amdc-as cells may also reflect their retarded growth rate. The size of the mRNAs are indicated in kb on the left. (B) AdoMetDC (left) and ODC (right) activities were measured as described in Materials and Methods. Dotted bars, cells grown without spermidine addition; striped bars, cells grown with spermidine. (C) The amount of polyamines is expressed as pmol/μg DNA. Acetylated polyamines were not detected. Black bars, putrescine; gray bars, spermidine; striped bars, spermine. The enzymatic activities and the polyamine contents were determined from parallel dishes after 2 d of culture. The results are representative of four independent experiments.
Amdc-s cells had a 30–40-fold higher constitutive AdoMetDC activity (ranging from a 15–60-fold increase in four independent transfection experiments), compared with the parental cell line (Fig. 2 B, left). In these experiments, the fold of increase in the AdoMetDC activity seemed to correlate with the degree of morphological transformation. Growing the cells with 1 μM spermidine had no marked effect on the AdoMetDC activity. The activity of ODC was not altered in the Amdc-s cells cultured with or without spermidine. Thus, there is no feed-back repression of polyamine biosynthetic enzymes (Tabor and Tabor 1984; Pegg 1988; Heby and Persson 1990; Cohen 1998) at this low concentration of spermidine. The Amdc-as cells had a slightly lower activity of AdoMetDC than normal cells, but showed an increase in ODC activity, especially in cultures supplemented with spermidine (∼12-fold) (Fig. 2 B, right). This increase in ODC activity, which appeared to be mainly posttranscriptional (data not shown), may be a compensatory mechanism. As AdoMetDC is the rate-limiting enzyme for the vital higher polyamines, the Amdc-as cells apparently try to keep alive by increasing ODC, and thus putrescine (a polyamine precursor), production (Fig. 2 C). No appreciable changes were observed in spermidine or spermine synthase activities in any of these transfectants (data not shown).
The amounts of spermidine and spermine were roughly the same in the 4N control and Amdc-as cells cultured for two or three days (Fig. 2 C). The Amdc-as+spd cells had increased putrescine levels due to ODC induction (see above), a block of the synthesis of spermidine from putrescine, or retroconversion of spermidine to putrescine. Other polyamines were not greatly affected. Generally, the polyamine pattern in Amdc-as+spd cells was similar to that of cells transformed by the overexpression of ODC (Auvinen et al. 1997). However, the increase in putrescine in the Amdc-as+spd cells was severalfold lower than that in the ODC-transformed cells, which, because of uninhibited AdoMetDC, also converted the excessive putrescine to spermidine. In the Amdc-s cells, the polyamine composition was very different. The level of spermidine was only one fourth, and the level of spermine was about twice as high, compared with normal or Amdc-as cells. The content of putrescine in the Amdc-s cells was very low or not detectable (Fig. 2 C).
Inoculation of AdoMetDC-overproducing Cells into Nude Mice Induces Rapidly Growing, Highly Invasive Tumors
To study the potential tumorigenicity, AdoMetDC-overexpressing cells were injected into athymic nude mice in both flanks. It should be noted that all extracellular fluids contain polyamines in low (micromolar) concentrations (Scalabrino and Ferioli 1982; Cohen 1998). All inoculations (10 out of 10 in each case) with the three transformed cell lines gave rise to tumors. Interestingly, the Amdc-s and Amdc-as+spd cells, particularly the former, induced aggressive growing, invasive tumors that were able to penetrate rapidly through the muscle and fat tissues into the peritoneal cavity. The mice exposed to the Amdc-s or Amdc-as+spd cells died or had to be killed 10–15 d after the inoculation. The Amdc-as cells, cultured in the absence of exogenous spermidine, also induced tumor formation, but only at the sites of inoculation and over a longer period of time (∼4 wk). The mice that received normal 4N cells, were followed to 62 d, and during this extended follow up, they developed, on occasion, small, noninvasive tumors (in 3 out of 10 inoculations).
Erk1 and Erk2 Are Not Constitutively Activated in AdoMetDC-transformed Cells
Numerous reports indicate that activation of the Erk1/Erk2 MAPK cascade is necessary for cell growth. Since MAPK is activated in cells transformed by many oncogenes (Howe et al. 1992; Samuels et al. 1993; Okazaki and Sagata 1995), we tested whether MAPK might also be constitutively activated and responsible for the transformed phenotype in Amdc-s and Amdc-as+spd transformants. To distinguish between the inactive and active states of Erk1 and Erk2 proteins, we starved the normal cells (4N and Rat-1) with 0.5% FBS for 20 h, and then stimulated the cells with PDGF-BB (30 ng/ml) for 15 min at 37°C. Exposure to PDGF-BB resulted in distinct shifts of Erk1 and Erk2 migration, as expected (Fig. 3 A). No shifts of Erks, indicative of constitutive activation, were detected in the transformants that were grown normally in the presence of serum (Fig. 3 A). Additionally, MAPK activities were measured by in vitro immunocomplex kinase assays, which used a synthetic peptide of myelic basic protein as a substrate. The normal 4N cells responded to PDGF-BB with a threefold activation, and Rat-1 cells exhibited a 15-fold increase in MAPK activity (Fig. 3 B). Again, neither the NIH3T3 nor Rat-1 transformants displayed a constitutive increase in MAPK activity (Fig. 3 B).
Figure 3.
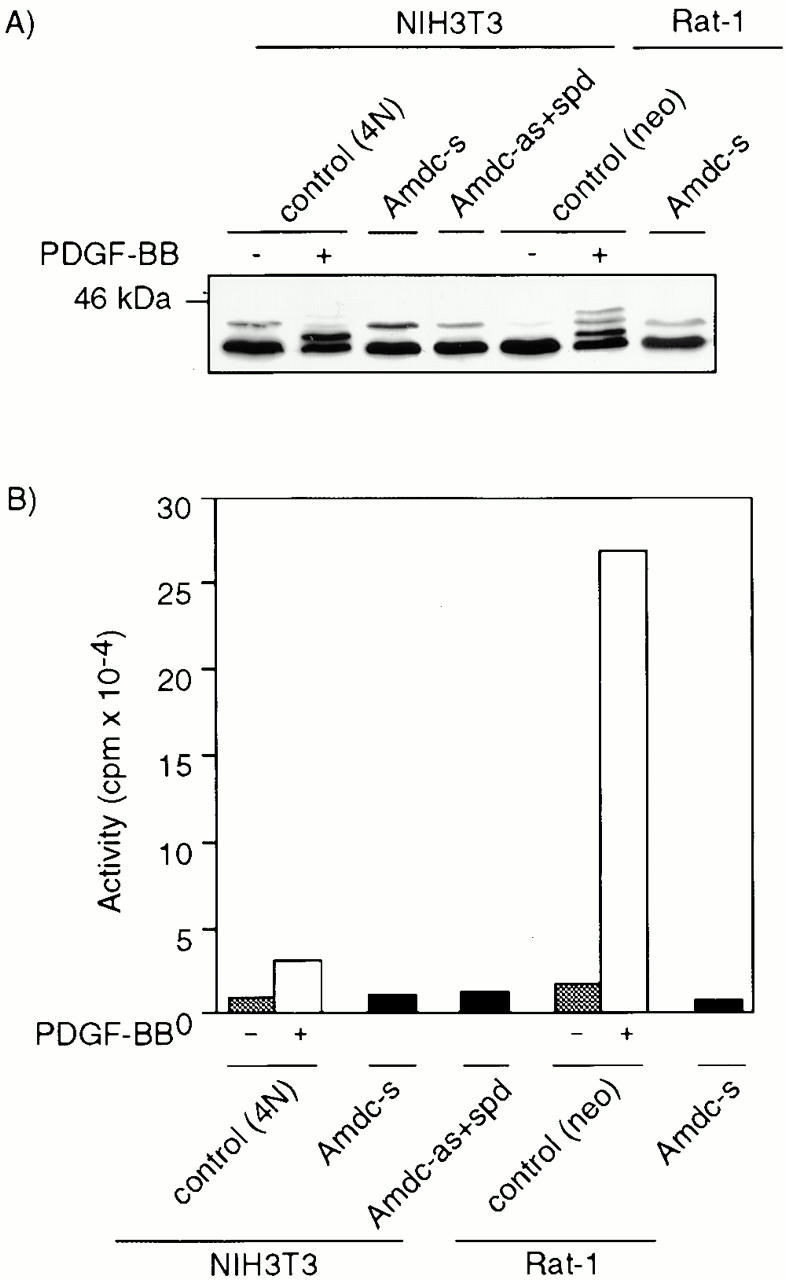
Erk1 and Erk2 are not constitutively activated in AdoMetDC transformants. (A) Western blot analysis of the phosphorylation status of Erks. The electrophoretic mobilities of Erk1 and Erk2 in Amdc-s or Amdc-as+spd cells did not display upshifts of protein bands (in 12.5% SDS-PAGE) that were seen in control cells (4N and Rat-1) stimulated with PDGF-BB. (B) In vitro immunocomplex kinase assays showed a clear stimulation of MAPK activity in PDGF-BB–stimulated control cells, but no constitutive upregulation was detected in AdoMetDC transformants. Gray bars, cells starved for 24 h in 0.5% serum; white bars, starved cells stimulated with PDGF-BB for 15 min at 37°C; black bars, cells grown normally in 5% serum.
Endogenous JNK Is Activated in the AdoMetDC Sense, but Not Antisense, Transformants
As AdoMetDC transformation appeared not to be linked to Erk1/Erk2 activation, we analyzed the activity of the JNKs. These kinases were originally described as stress-induced kinases, but have been recently connected to oncogenesis, as well (Raitano et al. 1995; Rodrigues et al. 1997). Interestingly, the JNK kinase activity was 5–9-fold higher in NIH3T3 Amdc-s cells than in control cells, with the Amdc-as+spd cells displaying no significant increase, as analyzed by the solid-phase assay (Fig. 4 A) or the immunocomplex kinase assay (data not shown). Also the transient transfection of AdoMetDC cDNA into NIH3T3 cells resulted in increased JNK activity (data not shown), indicating that the activation of JNK in Amdc-s cells is not just secondary to transformation. p38 MAPK was not activated in these cells, as confirmed by phosphospecific antibody blots (data not shown).
Figure 4.

Endogenous JNK is constitutively activated in AdoMetDC-overexpressing NIH3T3 and Rat-1 cells, resulting in increased phosphorylation of c-Jun at Ser73. (A) The JNKs were purified by virtue of their binding to agarose-conjugated GST-c-Jun. The autoradiograms show the phosphorylation status of GST-c-Jun used as the substrate in the solid-phase kinase assays. (B) Western blots from the nuclear extracts show a strong phosphorylation of c-Jun at Ser73 in AdoMetDC transformants. The bottom rows show the total amount of c-Jun in nuclear extracts. The levels of JunD and ATF-2 remained constant and were used as loading controls (data not shown).
AdoMetDC-transformed Cells Display a Constitutive Increase in Phosphorylation of c-Jun at Ser73
Transcriptional activity of c-Jun is regulated by phosphorylation of its NH2-terminal transactivation domain at Ser73 (and Ser63 as a minor site) (Binétruy et al. 1991; Smeal et al. 1991, Smeal et al. 1992). Both the Amdc-s and Amdc-as+spd cells, grown normally in the presence of serum, displayed a distinctive, constitutive phosphorylation of c-Jun on Ser73, whereas the normal control cells did not show any significant amount of phosphorylated c-Jun, as blotted with an antibody specific for c-Jun carrying phosphoserine at position 73 (Fig. 4 B). The cellular level of c-Jun was elevated to some extent in the AdoMetDC-transformed cells, compared with normal cells (Fig. 4 B). This could be explained by an increased expression of c-Jun through autoactivation (Su and Karin 1996) or by phosphorylation of c-Jun at Ser63 and Ser73, which stabilizes the protein (Musti et al. 1997).
The Dominant-Negative Mutants of the JNK Pathway Reverse the Transformed Morphology and Interfere with Cytokinesis of the AdoMetDC Sense Expressors
Previous studies with Ras-transformed cells have indicated that c-Jun becomes transcriptionally activated by the phosphorylation of Ser63 and Ser73 (Smeal et al. 1991), and the transactivation domain of c-Jun is required for transformation by ras in NIH3T3 cells (Westwick et al. 1994) and for cotransformation of rat embryo fibroblasts (Alani et al. 1991). The cascade leading to these phosphorylations includes consecutive activation of the kinases SEK1/MKK4 or MKK7 (immediate upstream activators of JNKs) and the JNKs 1 and 2 (Su and Karin 1996; Ip and Davis 1998). We tested the effects of dominant-negative mutants of SEK1, JNK1, and TAM-67, lacking the transactivation domain of c-Jun, on AdoMetDC-transformed cells. The plasmids containing the mutants or the control vectors were transfected together with a selection marker (zeo or puro) into the 4N cells and Amdc-s cells (Fig. 5 A, a). Transfections of the control vectors did not elicit any morphological changes in either cell type. In contrast, expression of dominant-negative mutants of SEK1 and JNK1 in Amdc-s cells led to a reversion towards the normal, epithelioid phenotype (Fig. 5 A, b and d) and produced multinucleated cells in subsequent cell divisions (Fig. 5 A, c and e). Expression of TAM67 resulted in the most efficient reversion of the transformed phenotype of Amdc-s cells to normal (Fig. 5 A, f) and formation of flat multinucleated cells, as well (Fig. 5 A, g). The reversal was not due to TAM67 interfering nonspecifically with the expression of AdoMetDC, as verified by the continued elevation in AdoMetDC activity. Also the phenotype of Amdc-as+spd cells was reverted by TAM67 expression (Fig. 5 B, a and b), but DN SEK1 and DN JNK1 mutants did not appear to be effective (data not shown). The mutants had no visible effect on the morphology of normal cells. The reversal of transformation by TAM67 expression was also seen in soft agar growth of the transfected cell lines (Fig. 5 C). In both Amdc-s (Fig. 5 C, a) and Amdc-as+spd cells (Fig. 5 C, c), TAM67 expression markedly reduced the size and amount of the colonies (Fig. 5 C, b and d). To confirm the expression of the mutants, cell lysates were analyzed for mutant proteins by Western blotting (Fig. 6A and Fig. B). It is also notable that the expression of DN JNK1 and DN SEK1 mutants in Amdc-s cells markedly inhibited c-Jun phosphorylation (Fig. 6 C).
Figure 5.
The transformed morphology of AdoMetDC-overexpressing cells is reversed by the expression of dominant-negative mutants of SEK1, JNK1, and c-Jun (TAM-67). (A) The morphology of (a) Amdc-s cells was changed to a more flattened and normal-looking phenotype by expressing (b) DN SEK1, (d) DN JNK1, and (f) TAM67 and resulted in the formation of multinucleated, growth-arrested cell populations, indicative of cytokinetic failure, in all three transfections (c, e, and g). The cells were transfected with mutant plasmids and selection markers, grown in the presence of zeosin or puromycin for 10 d (b, d, and f) or 17–21 d (c, e, and g), and photomicrographed. (B) The transformed morphology of Amdc-as+spd cells is reversed by c-Jun mutant TAM67. The cells were transfected as described above. (C) The ability of the Amdc-s and Amdc-as+spd cells to grow in soft agar is inhibited by TAM67 expression. In both cell lines the inhibition of soft agar growth by TAM67 was >90%, and the colonies formed were tiny in size.
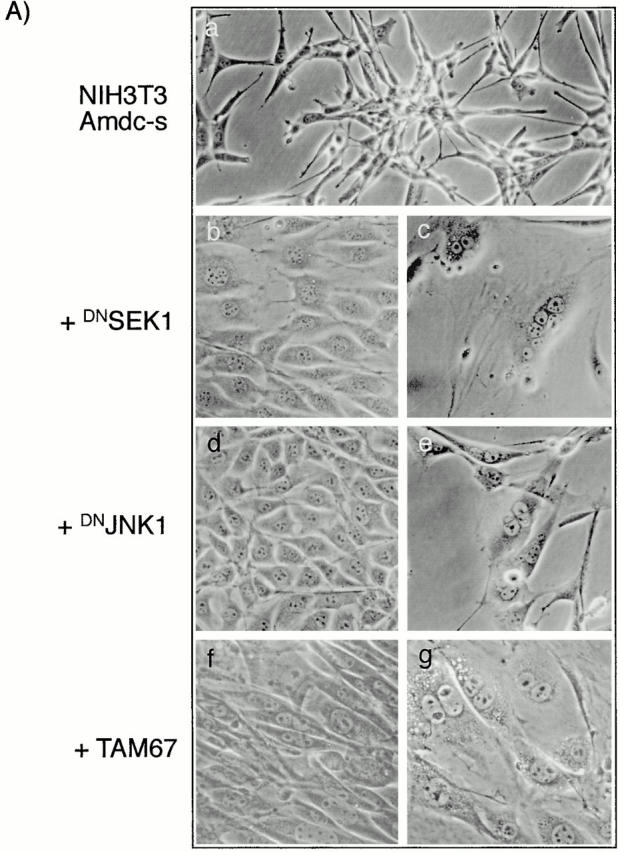
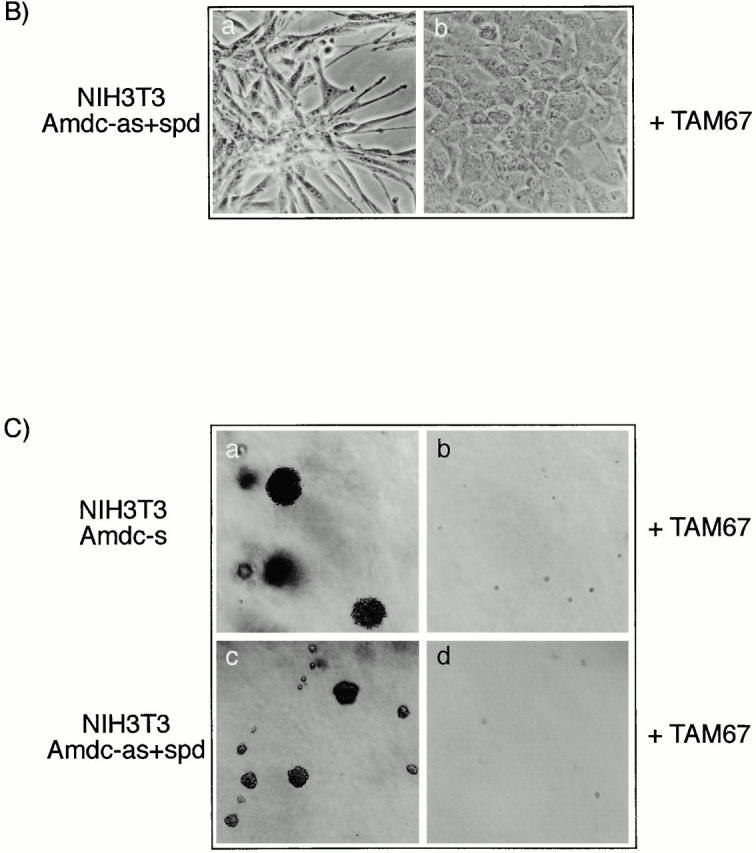
Figure 6.
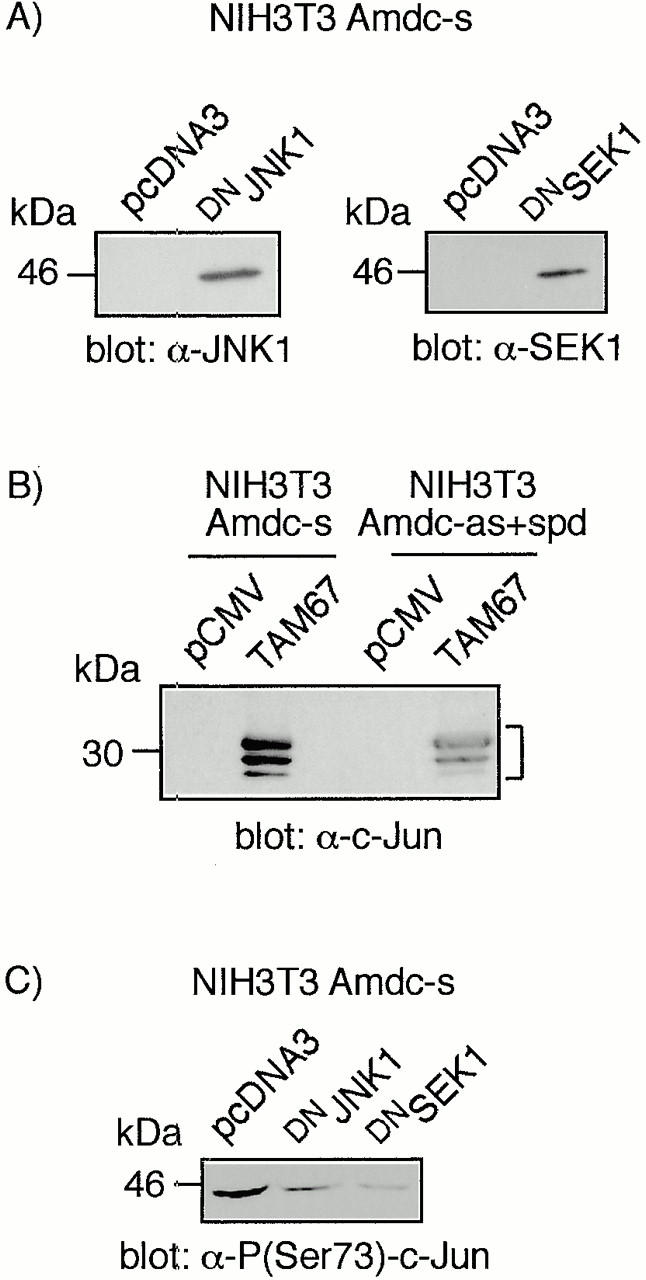
The expression of DN JNK1, DN SEK1, and c-Jun (TAM67) mutant proteins and inhibition of c-Jun phosphorylation by DN JNK1 and DN SEK1. (A and B) Western blot analyses of the cell lysates for the expression of the mutant proteins. Note that there are multiple c-Jun TAM67 bands (marked with a bracket), as reported earlier (Brown et al. 1994). (C) Western blot analysis showing the inhibition of c-Jun phosphorylation in Amdc-s cells transfected with DN JNK1 and DN SEK1.
Discussion
Here, we found that overexpression of human AdoMetDC induces transformation in NIH3T3 and Rat-1 cells, and it is tumorigenic in nude mice. Interestingly, AdoMetDC appears to be a more potent inducer of transformation than ODC (Auvinen et al. 1992) (this study and data not shown). Considering this, it is important to keep in mind that many human cancers show elevated AdoMetDC activity, though to a lesser level than ODC (Scalabrino and Ferioli 1982; Meyeskens and Gerner 1999). However, it is notable that activation of AdoMetDC in the cancer cells in vivo may be a much more common event than is currently thought by the in vitro enzyme assays performed in the presence of exogenous putrescine (the activator of mammalian AdoMetDC). Indeed, besides through enhanced expression, the AdoMetDC activity may be increased due to an elevation of endogenous putrescine, as a result of the activation of ODC (and other means). Therefore, we should perhaps reassess the earlier reported increases and possible role of AdoMetDC in cancer development.
Previous studies have suggested that an increase in putrescine is specifically connected to cell proliferation and malignant transformation (Clifford et al. 1995; Auvinen et al. 1997; O'Brien et al. 1997). This idea is in line with our results for the Amdc-as+spd transformants, which show an enhanced ODC activity and elevated putrescine content, that is, a similar polyamine pattern than the ODC-transformed cells (Auvinen et al. 1997). However, our results on AdoMetDC-induced transformation question the unconditional importance of putrescine in dysregulated growth, as the Amdc-s cells had very low levels of putrescine and the polyamine balance was strongly toward the end product, spermine. Considering the opposite patterns of total polyamines in the cells transformed by either AdoMetDC (this study) or ODC (Auvinen et al. 1997), it seems that an imbalanced ratio of polyamines, rather than a single polyamine change, may lead to cell transformation.
The tumorigenicity assays revealed that all three cell lines, Amdc-s, Amdc-as, and Amdc-as+spd with a transformed morphology in vitro, were also capable of producing tumors in nude mice. Intriguingly, the Amdc-s cells gave rise to highly invasive tumors. The tumor cells traversed rapidly from the site of inoculation into the peritoneal cavity. Thus, the AdoMetDC-overexpressing cells seem to possess a more invasive activity than the ODC-overexpressing cells that were found to invade only the neighboring striated muscle and fat tissues (Auvinen et al. 1997). The mechanisms behind the remarkable tumorigenicity and invasiveness of the AdoMetDC-overexpressing cells are under investigation. As single genes are not known to induce oncogenesis in primary embryonic cells or in vivo, it will be interesting to search for the possible oncogenes cooperating with AdoMetDC.
Intracellular signaling is mediated by networks of interacting proteins that govern various cellular processes. One of the best characterized signal transduction pathways is the Ras-Raf-MAPK route, which appears to be important in the regulation of cell growth, differentiation, and protection against apoptosis (Seger and Krebs 1995). Many oncogenes are known to encode proteins along this pathway or to require elevated Erk1/Erk2 activity for their transforming activity (Su and Karin 1996; Ip and Davis 1998), suggesting an important role for these MAPKs in transformation. However, we did not detect any constitutive activation of Erk1 and Erk2 in the AdoMetDC-overexpressing cells, indicating that they cannot be responsible for the maintenance of AdoMetDC-induced transformation. This is consistent with the idea that activation of Erks is not essential for the transformed phenotype or proliferation of all cell types, as recently suggested for the c-Ha-ras-, v-src-, and ODC-transformed cells (Paasinen-Sohns and Hölttä 1997), as well as other transformations (Samuels et al. 1993; Khosravi-Far et al. 1995; Raitano et al. 1995; Alessandrini et al. 1996; Attar et al. 1996; Greulich et al. 1996; Stofega et al. 1997).
Amdc-s cells showed a constitutive activation of JNKs, which belong to the MAPK superfamily (Su and Karin 1996; Ip and Davis 1998). The JNKs were first identified as protein kinases activated by various stress treatments, but a low level of JNK activation has been observed also after growth factor stimulation (Su and Karin 1996; Ip and Davis 1998). On the other hand, activation of JNK and concurrent inhibition of the Erk pathway have recently been associated with apoptosis (Xia et al. 1995), the induction of which is suggested to be determined by the duration of JNK activity (Chen et al. 1996). However, this is not the case with the AdoMetDC-transformed NIH3T3 and Rat-1 cells, which do not show any apoptotic features (data not shown) and are actively proliferating. Likewise, activation of JNK has been found to be associated with transformation by the ras, met, v-crk, and bcr-abl oncogenes (Raitano et al. 1995; Clark et al. 1997; Rodrigues et al. 1997; Tanaka et al. 1997). Constitutive activation of JNK in Amdc-s cells was accompanied by a persistent phosphorylation of the transactivation domain of c-Jun on Ser73. These results, together with the observed morphological reversion by the DN SEK1 and DN JNK1 mutants, which inhibit JNK activation and c-Jun phosphorylation, support an important role for the JNK-c-Jun pathway in transformation of these cells. In addition, the accumulation of multinucleated cells suggests that activation of c-Jun may also be of importance in the regulation of cytokinesis. Interestingly, the Amdc-as+spd cells also displayed a moderate increase in c-Jun phosphorylation, despite the lack of JNK activation.
Phosphorylation of c-Jun on Ser63 and Ser73 has been found to stimulate the transcriptional activity of c-Jun, without affecting its DNA-binding activity (Smeal et al. 1991, Smeal et al. 1992). Ser73 is the major site of phosphorylation and crucial for the transcriptional activity of c-Jun (Su and Karin 1996). Our finding of the constitutively elevated phosphorylation of c-Jun on its NH2-terminal Ser73 in vivo in the Amdc-s and Amdc-as+spd cells raises the possibility that transactivation by c-Jun, or some other function mediated by the transactivation domain, is important for the transforming ability of AdoMetDC. The transactivation domain of c-Jun was found to be essential for the transformed phenotype, as transfection of the dominant-negative Jun mutant TAM-67 reversed the morphology of Amdc-s and Amdc-as+spd cells to normal. The present data are the first to indicate that the polyamine biosynthetic enzymes, and thereby polyamines that are essential for growth, may require the c-Jun transcription factor for eliciting their cellular responses. It is now evident that the signal pathways initiated by many different oncogenes, such as ras, src, mos, fos, and myc (Rapp et al. 1994; Johnson et al. 1996), involve c-Jun activation, which may be mediated, at least in part, by its phosphorylation. As c-Jun is a component of the AP-1–transcription factor complex, which can also contain other Jun family members (JunB and JunD) and different Fos family members (c-Fos, Fra-1 and Fra-2), or form a complex with ATF-2 or Maf (Karin et al. 1997), it is possible that the complex formation or the possible phosphorylation of other proteins is of importance for transformation, as well. Besides, it has been demonstrated that c-Jun can recruit JNK to the complex to phosphorylate other substrates via heterodimerization (Kallunki et al. 1996). Furthermore, though the current opinion is that the Jun members are phosphorylated by JNKs and the Fos members by Erk1/Erk2 (Davis 1994; Su and Karin 1996), it is possible that there could be an unknown kinase capable of phosphorylating both family members. Indeed, we have detected an Erk- and JNK-independent phosphorylation of c-Jun in ras- (Paasinen-Sohns and Hölttä 1997) and Amdc-as- (this study) transformed cells. Correspondingly, an Erk- and JNK-independent, transformation-specific phosphorylation of FosB has been reported (Skinner et al. 1997).
Taken together, this study shows for the first time that expression of AdoMetDC, either in sense or antisense orientation, can induce tumorigenic transformation. To our knowledge, this is the first example of a protein whose overexpression or the block of synthesis can lead to transformation. These data suggest that the levels of AdoMetDC must be carefully regulated in order not to predispose the cells to transformation. An interesting finding is the observed high invasive capacity of the AdoMetDC-transformed cells, which may open a new avenue in the studies of cancer cell spreading. In fact, inhibition of AdoMetDC has been reported to inhibit metastasis of transplanted melanoma cells (Gutman et al. 1995). The AdoMetDC sense, but not the antisense, expressors exhibited activation of JNK. However, both transformations converged on phosphorylation of c-Jun on its transactivation domain. Since the expression of the dominant-negative mutant of Jun, TAM-67, reverted both the AdoMetDC sense– and antisense–induced transformations, it is tempting to speculate that the phosphorylation of c-Jun, or specific AP-1 components, would be an important point of convergence in the transforming action of AdoMetDC and many other oncogenes. This idea is further supported by our recent preliminary studies of Amdc-s and Amdc-as+spd cells transfected with c-Jun mutants with Ser63 and Ser73 mutated to alanines (our unpublished data together with Dr. D. Bohmann). These phosphorylations could lead to novel activating interactions with the basal transcription machinery or with other proteins, or change the affinity of the transcription factors for their cognate DNA motifs. Interestingly, the AdoMetDC-transformed cells did not exhibit a significant increase in transcription from a consensus tetradecanoyl phorbol acetate–responsive element (TRE)–containing reporter construct (col-TREx5/TK-CAT) (Angel et al. 1987) (our unpublished data). Whether this indicates that the number of genes relevant for transformation is smaller than the number of genes normally regulated by AP-1 or the transformation-specific genes having a somewhat different AP-1 sites than col-TREx5 remains to be seen. Significantly, a recent report (Young et al. 1999) supports this idea that only a subset of AP-1–dependent genes, inhibited by TAM67 expression, are involved in tumor promotion.
Acknowledgments
We would like to thank Roger J. Davis and James R. Woodgett for the DN JNK1 and DN SEK1 plasmids, and Hilkka Toivonen for technical assistance.
This work was supported by the University of Helsinki, the Finnish Cancer Organizations, the Academy of Finland, the Maud Kuistila, and the Emil Aaltonen Foundations.
Footnotes
Abbreviations used in this paper: AdoMetDC, S-adenosylmethionine decarboxylase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; ODC, ornithine decarboxylase; spd, spermidine; TRE, TPA-responsive element.
References
- Alani R., Brown P., Binétruy B., Dosaka H., Rosenberg R.K., Angel P., Karin M., Birrer M.J. The transactivating domain of the c-Jun proto-oncoprotein is required for cotransformation of rat embryo cells. Mol. Cell. Biol. 1991;11:6286–6295. doi: 10.1128/mcb.11.12.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandrini A., Greulich H., Huang W., Erikson R.L. Mek1 phosphorylation site mutants activate Raf-1 in NIH3T3 cells. J. Biol. Chem. 1996;271:31612–31618. doi: 10.1074/jbc.271.49.31612. [DOI] [PubMed] [Google Scholar]
- Angel P., Imagawa M., Chiu R., Stein B., Imbra R.J., Rahmsdorf H.J., Jonat C., Herrlich P., Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Attar B.M., Atten M.J., Holian O. MAPK activity is down-regulated in human colon adenocarcinomacorrelation with PKC activity. Anticancer Res. 1996;16:395–400. [PubMed] [Google Scholar]
- Auvinen M., Paasinen A., Andersson L.C., Hölttä E. Ornithine decarboxylase activity is critical for cell transformation. Nature. 1992;360:355–358. doi: 10.1038/360355a0. [DOI] [PubMed] [Google Scholar]
- Auvinen M., Laine A., Paasinen-Sohns A., Kangas A., Kangas L., Saksela O., Andersson L.C., Hölttä E. Human ornithine decarboxylase-overproducing NIH3T3 cells induce rapidly growing, highly vascularized tumors in nude mice. Cancer Res. 1997;57:3016–3025. [PubMed] [Google Scholar]
- Bello-Fernandez C., Packham G., Cleveland J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA. 1993;90:7804–7808. doi: 10.1073/pnas.90.16.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binétruy B., Smeal T., Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991;351:122–127. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- Brown P.H., Alani R., Preis L.H., Szabo E., Birrer M.J. Suppression of oncogene-induced transformation by a deletion mutant of c-jun . Oncogene. 1993;8:877–886. [PubMed] [Google Scholar]
- Brown P.H., Chen T.K., Birrer M.J. Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene. 1994;9:791–799. [PubMed] [Google Scholar]
- Chen Y.-R., Wang X., Tempelton D., Davis R.J., Tan T.-H. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. J. Biol. Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- Clark G.J., Westwick J.K., Der C.J. p120 GAP modulates Ras activation of Jun kinases and transformation. J. Biol. Chem. 1997;272:1677–1681. doi: 10.1074/jbc.272.3.1677. [DOI] [PubMed] [Google Scholar]
- Clifford A., Morgan D., Yuspa S.H., Peralta Soler A., Gilmour S. Role of ornithine decarboxylase in epidermal tumorigenesis. Cancer Res. 1995;55:1680–1686. [PubMed] [Google Scholar]
- Cohen S.S. A guide to the polyamines 1998. Oxford University Press; New York: pp. 595 pp [Google Scholar]
- Crespo P., Bustelo X.R., Aaronson D.S., Coso O.A., Lopez-Parahona M., Barbacid M., Gutkind J.S. Rac-1 dependent stimulation of the JNK/SAPK signaling pathway by Vav. Oncogene. 1996;13:455–460. [PubMed] [Google Scholar]
- Davis R.J. MAPKsnew JNK expands the group. Trends Biochem. Sci. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Dérijard B., Hibi M., Wu I.-H., Barrett T., Su B., Deng T., Karin M., Davis R.J. JNK1a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Greulich H., Reichman C., Hanafusa H. Delay in serum stimulation of Erk activity by oncogenic transformation. Oncogene. 1996;12:1689–1695. [PubMed] [Google Scholar]
- Gutman M., Beltran P.J., Fan D., Delworth M.G., Singh R.K., Wilson M.R., Fidler I.J. Treatment of nude mice with 4-amidinoidan-1-one2′-amidinohydrazone, a new S-adenosylmethionine decarboxylase inhibitor, delays growth and inhibits metastasis of human melanoma cells. Melanoma Res. 1995;5:147–154. doi: 10.1097/00008390-199506000-00002. [DOI] [PubMed] [Google Scholar]
- Heby O., Persson L. Molecular genetics of polyamine synthesis in eukaryotic cells. Trends Biochem. Sci. 1990;15:153–158. doi: 10.1016/0968-0004(90)90216-x. [DOI] [PubMed] [Google Scholar]
- Hibi M., Lin A., Smeal T., Minden A., Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2147. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Howe L.R., Leevers S.J., Gómez N., Nakielny S., Cohen P., Marshall C.J. Activation of the MAP kinase pathway by the protein kinase raf. Cell. 1992;71:335–342. doi: 10.1016/0092-8674(92)90361-f. [DOI] [PubMed] [Google Scholar]
- Hyvönen T., Keinänen T.A., Khomutov A.R., Khomutov R.M., Eloranta T.O. Monitoring of the uptake and metabolism of aminooxy analogues of polyamines in cultured cells by high-performance liquid chromatography. J. Chromatography. 1992;574:17–21. doi: 10.1016/0378-4347(92)80093-6. [DOI] [PubMed] [Google Scholar]
- Ip Y.T., Davis R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Johnson R., Spiegelman B., Hanahan D., Wisdom R. Cellular transformation and malignancy induced by ras require c-jun. Mol. Cell. Biol. 1996;16:4504–4511. doi: 10.1128/mcb.16.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallunki T., Deng T., Hibi M., Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- Karin M., Liu Z.-G., Zandi E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R., Solski P.A., Clark G.J., Kinch M.S., Der C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyeskens F.L.J., Gerner E.W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 1999;5:945–951. [PubMed] [Google Scholar]
- Morgenstern J.P., Land H. Advanced mammalian gene transferhigh titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musti A.M., Treier M., Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- O'Brien T.G., Megosh L.C., Gilliard G., Peralta Soler A. Ornithine decarboxylase overexpression is a sufficient condition for tumor promotion in mouse skin. Cancer Res. 1997;57:2630–2637. [PubMed] [Google Scholar]
- Okazaki K., Sagata N. MAP kinase activation is essential for oncogenic transformation of NIH3T3 cells by Mos. Oncogene. 1995;10:1149–1157. [PubMed] [Google Scholar]
- Paasinen-Sohns A., Hölttä E. Cells transformed by ODC, c-Ha-ras and v-src exhibit MAP kinase/Erk-independent constitutive phosphorylation of Sos, Raf and c-Jun activation domain, and reduced PDGF receptor expression. Oncogene. 1997;15:1953–1966. doi: 10.1038/sj.onc.1201366. [DOI] [PubMed] [Google Scholar]
- Pajunen A., Crozat A., Jänne O.A., Ihalainen R., Laitinen P.H., Stanley B., Madhubala R., Pegg A.E. Structure and regulation of mammalian S-adenosylmethionine decarboxylase. J. Biol. Chem. 1988;263:17040–17049. [PubMed] [Google Scholar]
- Pegg A.E. Polyamine metabolism and its importance in neoplastic growth and as a target for chemotherapy. Cancer Res. 1988;48:759–774. [PubMed] [Google Scholar]
- Pegg A.E., McCann P.P. S-adenosylmethionine decarboxylase as an enzyme target for therapy. Pharmac. Ther. 1992;56:359–377. doi: 10.1016/0163-7258(92)90025-u. [DOI] [PubMed] [Google Scholar]
- Raitano A.B., Halpern J.R., Hambuch T.M., Sawyers C.L. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc. Natl. Acad. Sci. USA. 1995;92:11746–11750. doi: 10.1073/pnas.92.25.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp U.R., Troppmair J., Beck T., Birrer M.J. Transformation by Raf and other oncogenes renders cells differentially sensitive to growth inhibition by a dominant negative c-jun mutant. Oncogene. 1994;9:3493–3498. [PubMed] [Google Scholar]
- Regenass U., Mett H., Stanek J., Mueller M., Kramer D., Porter C.W. CGP 48664, a new S-adenosylmethionine decarboxylase inhibitor with broad spectrum antiproliferative and antitumor activity. Cancer Res. 1994;54:3210–3217. [PubMed] [Google Scholar]
- Rodrigues G.A., Park M., Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2634–2645. doi: 10.1093/emboj/16.10.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels M.L., Weber M.J., Bishop J.M., McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human Raf-1 protein kinase. Mol. Cell. Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalabrino G., Ferioli M.E. Polyamines in mammalian tumors. Part II. Adv. Cancer Res. 1982;36:1–102. doi: 10.1016/s0065-230x(08)60422-4. [DOI] [PubMed] [Google Scholar]
- Seger R., Krebs E.G. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Skinner M., Qu S., Moore C., Wisdom R. Transcriptional activation and transformation by FosB protein require phosphorylation of the carboxyl-terminal activation domain. Mol. Cell. Biol. 1997;17:2372–2380. doi: 10.1128/mcb.17.5.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal T., Binétruy B., Mercola D.A., Birrer M., Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Smeal T., Binétruy B., Mercola D., Grover-Bardwick A., Heidecker G., Rapp U.R., Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of Serines 63 and 73. Mol. Cell. Biol. 1992;12:3507–3513. doi: 10.1128/mcb.12.8.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stofega M.R., Yu C.-L., Wu J., Jove R. Activation of extracellular signal-regulated kinase (ERK) by mitogenic stimuli is repressed in v-Src-transformed cells. Cell Growth Differ. 1997;8:113–119. [PubMed] [Google Scholar]
- Su B., Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Tabor C.W., Tabor H. Polyamines. Annu. Rev. Biochem. 1984;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Ouchi T., Hanafusa H. Downstream of Crk adaptor signaling pathwayactivation of Jun kinase by v-Crk through the guanine nucleotide exchange protein C3G. Proc. Natl. Acad. Sci. USA. 1997;94:2356–2361. doi: 10.1073/pnas.94.6.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R.A. How cancer arises. Sci. Am. 1996;275:32–40. doi: 10.1038/scientificamerican0996-62. [DOI] [PubMed] [Google Scholar]
- Westwick J.K., Cox A.D., Der C.J., Cobb M.H., Hibi M., Karin M., Brenner D.A. Oncogenic Ras activates c-Jun via a separate pathway from the activation of extracellular signal-regulated kinases. Proc. Natl. Acad. Sci. USA. 1994;91:6030–6034. doi: 10.1073/pnas.91.13.6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z., Dickens M., Raingeaud J., Davis R.J., Greenberg M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yan M., Dai T., Deak J.C., Kyriakis J.M., Zon L.I., Woodgett J.R., Templeton D.J. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- Young M.R., Li J.-J., Rincón M., Flavell R.A., Sathyanarayana B.K., Hunziker R., Colburn N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. USA. 1999;96:9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]